
Kynurenines as a Novel Target for the Treatment of Malignancies



Abstract
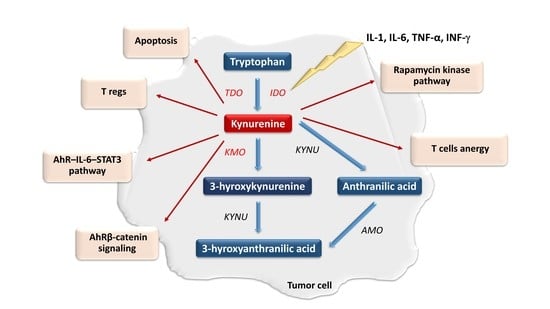
1. Introduction
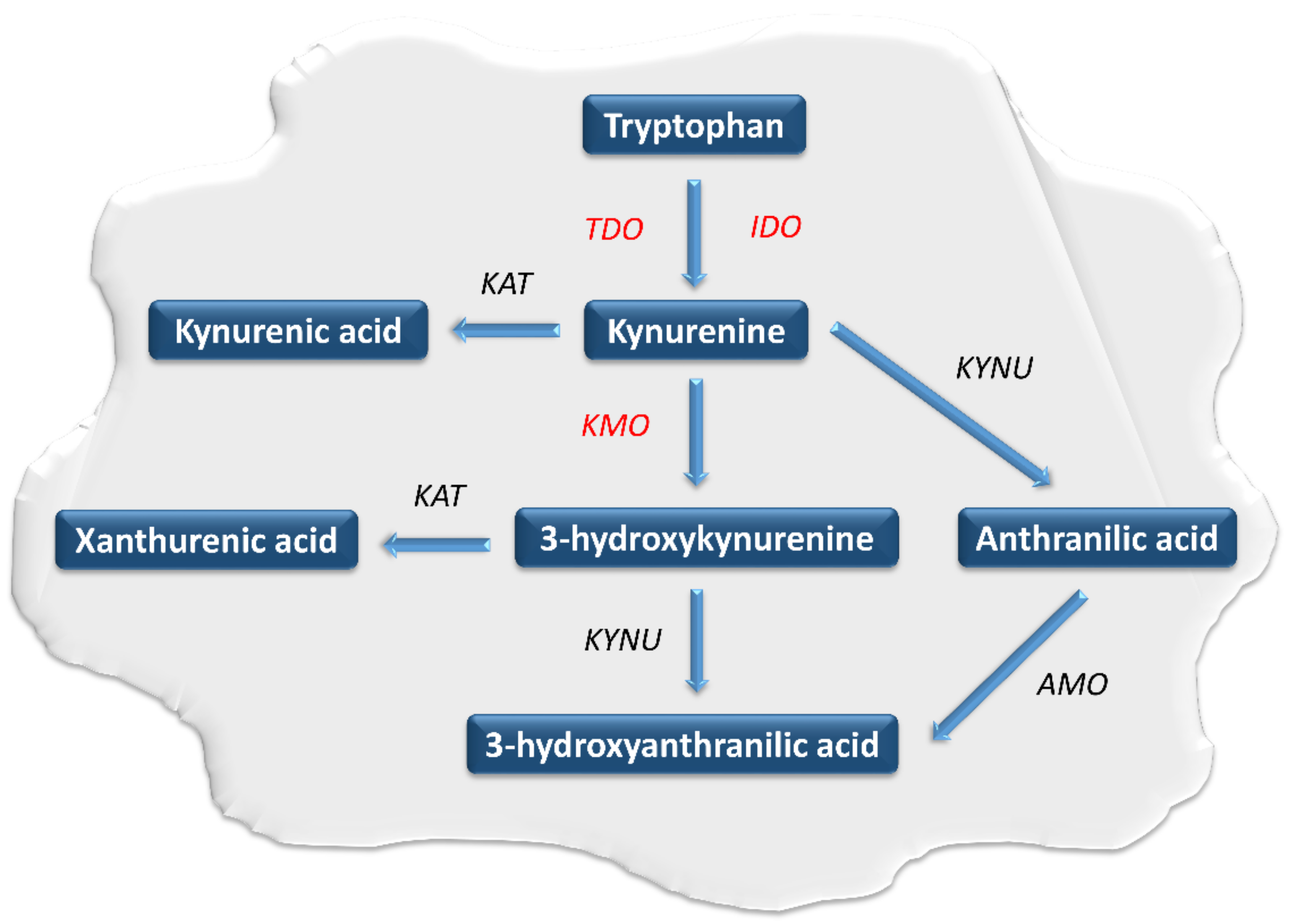
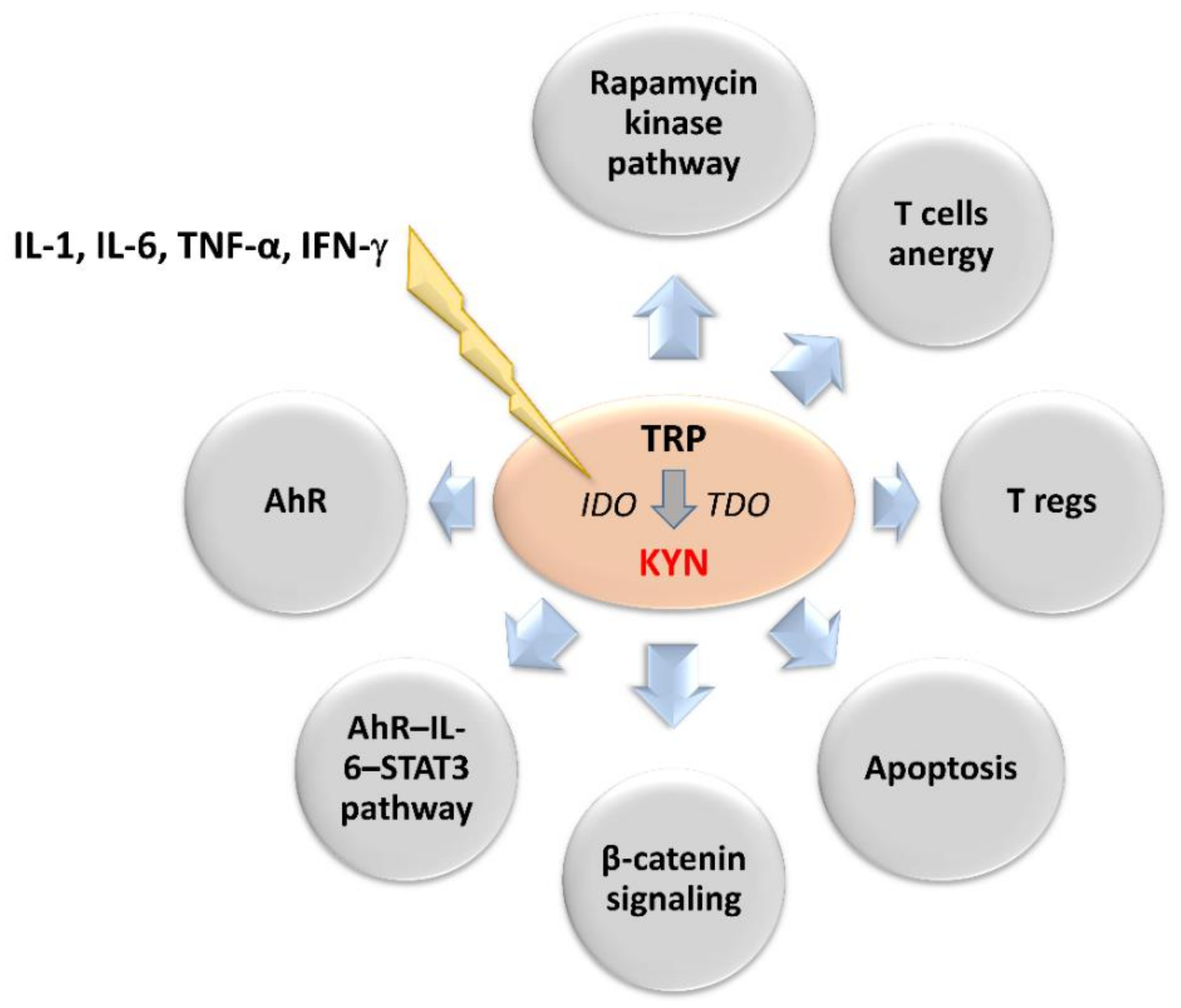
2. The Role of the Kynurenine Pathway in Neoplastic Processes
3. Modulation of Kynurenine Pathway Activity in the Management of Neoplastic Diseases
3.1. Pharmacological Modulation of IDO and TDO Activity
3.1.1. Lung Cancer
3.1.2. Breast Cancer
3.1.3. Leukemias and Lymphomas
3.1.4. Head and Neck Neoplasms
3.1.5. Melanoma
3.1.6. Ovarian and Cervical Cancer
3.1.7. Digestive System Cancers
3.1.8. Renal Cancer
3.1.9. Myelodysplastic Syndromes
3.1.10. Sarcomas
3.2. The Role of Intracellular Mechanisms of Kynurenine Pathway Modulation in Tumor Therapy
3.3. Angiogenesis Inhibition
3.4. Pharmacological Modulation Activity of KMO Activity
3.5. Gene Silencing
3.6. Blockade of the AhR
4. Summary and Future Directions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Perez-Gracia, E.; Blanco, R.; Carmona, M.; Carro, E.; Ferrer, I. Oxidative Stress Damage and Oxidative Stress Responses in the Choroid Plexus in Alzheimer’s Disease. Acta Neuropathol. 2009, 118, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Ocampo, J.R.; Huitrón, R.L.; González-Esquivel, D.; Ugalde-Muñiz, P.; Jiménez-Anguiano, A.; Pineda, B.; Pedraza-Chaverri, J.; Ríos, C.; de la Cruz, V.P. Kynurenines with Neuroactive and Redox Properties: Relevance to Aging and Brain Diseases. Oxid. Med. Cell. Longev. 2014, 2014, 646909. [Google Scholar] [CrossRef]
- Bender, D.A. Biochemistry of Tryptophan in Health and Disease. Mol. Asp. Med. 1983, 6, 101–197. [Google Scholar] [CrossRef]
- Badawy, A.A. Tryptophan Metabolism in Alcoholism. Adv. Exp. Med. Biol. 1999, 467, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Badawy, A.A.-B. Tryptophan Metabolism, Disposition and Utilization in Pregnancy. Biosci. Rep. 2015, 35, e00261. [Google Scholar] [CrossRef] [PubMed]
- Badawy, A.A.-B. Pellagra and Alcoholism: A Biochemical Perspective. Alcohol Alcohol. 2014, 49, 238–250. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, D.; Song, P.; Zou, M.-H. Deregulated Tryptophan-Kynurenine Pathway Is Linked to Inflammation, Oxidative Stress, and Immune Activation Pathway in Cardiovascular Diseases. Front. Biosci. Landmark Ed. 2015, 20, 1116–1143. [Google Scholar] [PubMed]
- Heyes, M.P.; Saito, K.; Crowley, J.S.; Davis, L.E.; Demitrack, M.A.; Der, M.; Dilling, L.A.; Elia, J.; Kruesi, M.J.; Lackner, A. Quinolinic Acid and Kynurenine Pathway Metabolism in Inflammatory and Non-Inflammatory Neurological Disease. Brain J. Neurol. 1992, 115 (Pt 5), 1249–1273. [Google Scholar] [CrossRef]
- Fallarino, F.; Grohmann, U.; Vacca, C.; Bianchi, R.; Orabona, C.; Spreca, A.; Fioretti, M.C.; Puccetti, P. T Cell Apoptosis by Tryptophan Catabolism. Cell Death Differ. 2002, 9, 1069–1077. [Google Scholar] [CrossRef]
- Topczewska-Bruns, J.; Pawlak, D.; Chabielska, E.; Tankiewicz, A.; Buczko, W. Increased Levels of 3-Hydroxykynurenine in Different Brain Regions of Rats with Chronic Renal Insufficiency. Brain Res. Bull. 2002, 58, 423–428. [Google Scholar] [CrossRef]
- Topczewska-Bruns, J.; Tankiewicz, A.; Pawlak, D.; Buczko, W. Behavioral Changes in the Course of Chronic Renal Insufficiency in Rats. Pol. J. Pharmacol. 2001, 53, 263–269. [Google Scholar]
- Forrest, C.M.; Mackay, G.M.; Oxford, L.; Stoy, N.; Stone, T.W.; Darlington, L.G. Kynurenine Pathway Metabolism in Patients with Osteoporosis after 2 Years of Drug Treatment. Clin. Exp. Pharmacol. Physiol. 2006, 33, 1078–1087. [Google Scholar] [CrossRef]
- Dayer, M.R.; Safari, I.; Dayer, M.S. New Evidence on Hypoglycemic Effect of Quinolinic Acid in Diabetic Rats. Pak. J. Biol. Sci. PJBS 2009, 12, 1025–1030. [Google Scholar] [CrossRef]
- Munipally, P.K.; Agraharm, S.G.; Valavala, V.K.; Gundae, S.; Turlapati, N.R. Evaluation of Indoleamine 2,3-Dioxygenase Expression and Kynurenine Pathway Metabolites Levels in Serum Samples of Diabetic Retinopathy Patients. Arch. Physiol. Biochem. 2011, 117, 254–258. [Google Scholar] [CrossRef]
- Prendergast, G.C. Cancer: Why Tumours Eat Tryptophan. Nature 2011, 478, 192–194. [Google Scholar] [CrossRef]
- Apalset, E.M.; Gjesdal, C.G.; Ueland, P.M.; Midttun, Ø.; Ulvik, A.; Eide, G.E.; Meyer, K.; Tell, G.S. Interferon (IFN)-γ-Mediated Inflammation and the Kynurenine Pathway in Relation to Bone Mineral Density: The Hordaland Health Study. Clin. Exp. Immunol. 2014, 176, 452–460. [Google Scholar] [CrossRef]
- Esquivel, D.G.; Ramírez-Ortega, D.; Pineda, B.; Castro, N.; Ríos, C.; de la Cruz, V.P. Kynurenine Pathway Metabolites and Enzymes Involved in Redox Reactions. Neuropharmacology 2017, 112, 331–345. [Google Scholar] [CrossRef]
- Kawajiri, K.; Fujii-Kuriyama, Y. The Aryl Hydrocarbon Receptor: A Multifunctional Chemical Sensor for Host Defense and Homeostatic Maintenance. Exp. Anim. 2017, 66, 75–89. [Google Scholar] [CrossRef]
- Gutiérrez-Vázquez, C.; Quintana, F.J. Regulation of the Immune Response by the Aryl Hydrocarbon Receptor. Immunity 2018, 48, 19–33. [Google Scholar] [CrossRef]
- Bessede, A.; Gargaro, M.; Pallotta, M.T.; Matino, D.; Servillo, G.; Brunacci, C.; Bicciato, S.; Mazza, E.M.C.; Macchiarulo, A.; Vacca, C.; et al. Aryl Hydrocarbon Receptor Control of a Disease Tolerance Defence Pathway. Nature 2014, 511, 184–190. [Google Scholar] [CrossRef]
- Kolachalama, V.B.; Shashar, M.; Alousi, F.; Shivanna, S.; Rijal, K.; Belghasem, M.E.; Walker, J.; Matsuura, S.; Chang, G.H.; Gibson, C.M.; et al. Uremic Solute-Aryl Hydrocarbon Receptor-Tissue Factor Axis Associates with Thrombosis after Vascular Injury in Humans. J. Am. Soc. Nephrol. JASN 2018, 29, 1063–1072. [Google Scholar] [CrossRef]
- Nguyen, N.T.; Nakahama, T.; Le, D.H.; Van Son, L.; Chu, H.H.; Kishimoto, T. Aryl Hydrocarbon Receptor and Kynurenine: Recent Advances in Autoimmune Disease Research. Front. Immunol. 2014, 5, 551. [Google Scholar] [CrossRef]
- King, N.J.C.; Thomas, S.R. Molecules in Focus: Indoleamine 2,3-Dioxygenase. Int. J. Biochem. Cell Biol. 2007, 39, 2167–2172. [Google Scholar] [CrossRef]
- Capece, L.; Lewis-Ballester, A.; Marti, M.A.; Estrin, D.A.; Yeh, S.-R. Molecular Basis for the Substrate Stereoselectivity in Tryptophan Dioxygenase. Biochemistry 2011, 50, 10910–10918. [Google Scholar] [CrossRef] [PubMed]
- Knox, W.E.; Greengard, O. The Regulation of Some Enzymes of Nitrogen Metabolism—an Introduction to Enzyme Physiology. Adv. Enzyme Regul. 1965, 3, 247–313. [Google Scholar] [CrossRef]
- Feigelson, M.; Feigelson, P. Metabolic Effects of Glucocorticoids as Related to Enzyme Induction. Adv. Enzyme Regul. 1965, 3, 11–27. [Google Scholar] [CrossRef]
- Schimke, R.T.; Sweeney, E.W.; Berlin, C.M. The Roles of Synthesis And Degradation in the Control of Rat Liver Tryptophan Pyrrolase. J. Biol. Chem. 1965, 240, 322–331. [Google Scholar] [CrossRef]
- Badawy, A.A.-B. Kynurenine Pathway of Tryptophan Metabolism: Regulatory and Functional Aspects. Int. J. Tryptophan Res. IJTR 2017, 10. [Google Scholar] [CrossRef]
- Badawy, A.A.-B.; Bano, S. Tryptophan Metabolism in Rat Liver After Administration of Tryptophan, Kynurenine Metabolites, and Kynureninase Inhibitors. Int. J. Tryptophan Res. IJTR 2016, 9, 51–65. [Google Scholar] [CrossRef]
- Brouns, R.; Verkerk, R.; Aerts, T.; De Surgeloose, D.; Wauters, A.; Scharpé, S.; De Deyn, P.P. The Role of Tryptophan Catabolism along the Kynurenine Pathway in Acute Ischemic Stroke. Neurochem. Res. 2010, 35, 1315–1322. [Google Scholar] [CrossRef]
- Amori, L.; Guidetti, P.; Pellicciari, R.; Kajii, Y.; Schwarcz, R. On the Relationship between the Two Branches of the Kynurenine Pathway in the Rat Brain in Vivo. J. Neurochem. 2009, 109, 316–325. [Google Scholar] [CrossRef]
- Barone, P. The “Yin” and the “Yang” of the Kynurenine Pathway: Excitotoxicity and Neuroprotection Imbalance in Stress-Induced Disorders. Behav. Pharmacol. 2019, 30, 163–186. [Google Scholar] [CrossRef]
- Walsh, H.A.; Botting, N.P. Purification and Biochemical Characterization of Some of the Properties of Recombinant Human Kynureninase. Eur. J. Biochem. 2002, 269, 2069–2074. [Google Scholar] [CrossRef]
- Okuda, S.; Nishiyama, N.; Saito, H.; Katsuki, H. 3-Hydroxykynurenine, an Endogenous Oxidative Stress Generator, Causes Neuronal Cell Death with Apoptotic Features and Region Selectivity. J. Neurochem. 1998, 70, 299–307. [Google Scholar] [CrossRef]
- Stone, T.W. Neuropharmacology of Quinolinic and Kynurenic Acids. Pharmacol. Rev. 1993, 45, 309–379. [Google Scholar]
- Schwarcz, R.; Pellicciari, R. Manipulation of Brain Kynurenines: Glial Targets, Neuronal Effects, and Clinical Opportunities. J. Pharmacol. Exp. Ther. 2002, 303, 1–10. [Google Scholar] [CrossRef]
- Muller, A.J.; DuHadaway, J.B.; Donover, P.S.; Sutanto-Ward, E.; Prendergast, G.C. Inhibition of Indoleamine 2,3-Dioxygenase, an Immunoregulatory Target of the Cancer Suppression Gene Bin1, Potentiates Cancer Chemotherapy. Nat. Med. 2005, 11, 312–319. [Google Scholar] [CrossRef]
- Prendergast, G.C.; Malachowski, W.P.; DuHadaway, J.B.; Muller, A.J. Discovery of IDO1 Inhibitors: From Bench to Bedside. Cancer Res. 2017, 77, 6795–6811. [Google Scholar] [CrossRef]
- Schramme, F.; Crosignani, S.; Frederix, K.; Hoffmann, D.; Pilotte, L.; Stroobant, V.; Preillon, J.; Driessens, G.; Van den Eynde, B.J. Inhibition of Tryptophan-Dioxygenase Activity Increases the Antitumor Efficacy of Immune Checkpoint Inhibitors. Cancer Immunol. Res. 2020, 8, 32–45. [Google Scholar] [CrossRef]
- Platten, M.; von Knebel Doeberitz, N.; Oezen, I.; Wick, W.; Ochs, K. Cancer Immunotherapy by Targeting IDO1/TDO and Their Downstream Effectors. Front. Immunol. 2014, 5, 673. [Google Scholar] [CrossRef]
- Lee, G.K.; Park, H.J.; Macleod, M.; Chandler, P.; Munn, D.H.; Mellor, A.L. Tryptophan Deprivation Sensitizes Activated T Cells to Apoptosis Prior to Cell Division. Immunology 2002, 107, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Apetoh, L.; Quintana, F.J.; Pot, C.; Joller, N.; Xiao, S.; Kumar, D.; Burns, E.J.; Sherr, D.H.; Weiner, H.L.; Kuchroo, V.K. The Aryl Hydrocarbon Receptor Interacts with C-Maf to Promote the Differentiation of Type 1 Regulatory T Cells Induced by IL-27. Nat. Immunol. 2010, 11, 854–861. [Google Scholar] [CrossRef] [PubMed]
- Holmgaard, R.B.; Zamarin, D.; Munn, D.H.; Wolchok, J.D.; Allison, J.P. Indoleamine 2,3-Dioxygenase Is a Critical Resistance Mechanism in Antitumor T Cell Immunotherapy Targeting CTLA-4. J. Exp. Med. 2013, 210, 1389–1402. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Mellor, A.L. IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation, and Tolerance. Trends Immunol. 2016, 37, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Creelan, B.C.; Antonia, S.; Bepler, G.; Garrett, T.J.; Simon, G.R.; Soliman, H.H. Indoleamine 2,3-Dioxygenase Activity and Clinical Outcome Following Induction Chemotherapy and Concurrent Chemoradiation in Stage III Non-Small Cell Lung Cancer. Oncoimmunology 2013, 2, e23428. [Google Scholar] [CrossRef]
- Okamoto, A.; Nikaido, T.; Ochiai, K.; Takakura, S.; Saito, M.; Aoki, Y.; Ishii, N.; Yanaihara, N.; Yamada, K.; Takikawa, O.; et al. Indoleamine 2,3-Dioxygenase Serves as a Marker of Poor Prognosis in Gene Expression Profiles of Serous Ovarian Cancer Cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 11, 6030–6039. [Google Scholar] [CrossRef] [PubMed]
- Sim, S.H.; Ahn, Y.-O.; Yoon, J.; Kim, T.M.; Lee, S.-H.; Kim, D.-W.; Heo, D.S. Influence of Chemotherapy on Nitric Oxide Synthase, Indole-Amine-2,3-Dioxygenase and CD124 Expression in Granulocytes and Monocytes of Non-Small Cell Lung Cancer. Cancer Sci. 2012, 103, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, R.; Urade, Y.; Tokuda, M.; Hayaishi, O. Induction of Indoleamine 2,3-Dioxygenase in Mouse Lung during Virus Infection. Proc. Natl. Acad. Sci. USA 1979, 76, 4084–4086. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Li, L.; Klonowski, K.D.; Tompkins, S.M.; Tripp, R.A.; Mellor, A.L. Induction and Role of Indoleamine 2,3 Dioxygenase in Mouse Models of Influenza a Virus Infection. PLoS ONE 2013, 8, e66546. [Google Scholar] [CrossRef]
- Robinson, C.M.; Hale, P.T.; Carlin, J.M. The Role of IFN-Gamma and TNF-Alpha-Responsive Regulatory Elements in the Synergistic Induction of Indoleamine Dioxygenase. J. Interferon Cytokine Res. 2005, 25, 20–30. [Google Scholar] [CrossRef]
- Uyttenhove, C.; Pilotte, L.; Théate, I.; Stroobant, V.; Colau, D.; Parmentier, N.; Boon, T.; Van den Eynde, B.J. Evidence for a Tumoral Immune Resistance Mechanism Based on Tryptophan Degradation by Indoleamine 2,3-Dioxygenase. Nat. Med. 2003, 9, 1269–1274. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhang, R.; Li, S.; Liu, J. IDO1: An Important Immunotherapy Target in Cancer Treatment. Int. Immunopharmacol. 2017, 47, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Le Naour, S.; Knol, A.-C.; Pandolfino, M.-C.; Khammari, A.; Dréno, B. Effect of Indoleamine 2,3 Dioxygenase Inhibitor on the Cytotoxic Activity of Tumour-Infiltrating Lymphocytes. Acta Derm. Venereol. 2019, 99, 1186–1187. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Sharma, M.D.; Baban, B.; Harding, H.P.; Zhang, Y.; Ron, D.; Mellor, A.L. GCN2 Kinase in T Cells Mediates Proliferative Arrest and Anergy Induction in Response to Indoleamine 2,3-Dioxygenase. Immunity 2005, 22, 633–642. [Google Scholar] [CrossRef]
- McGaha, T.L. IDO-GCN2 and Autophagy in Inflammation. Oncotarget 2015, 6, 21771–21772. [Google Scholar] [CrossRef]
- Metz, R.; Rust, S.; Duhadaway, J.B.; Mautino, M.R.; Munn, D.H.; Vahanian, N.N.; Link, C.J.; Prendergast, G.C. IDO Inhibits a Tryptophan Sufficiency Signal That Stimulates MTOR: A Novel IDO Effector Pathway Targeted by D-1-Methyl-Tryptophan. Oncoimmunology 2012, 1, 1460–1468. [Google Scholar] [CrossRef]
- Zaher, S.S.; Germain, C.; Fu, H.; Larkin, D.F.P.; George, A.J.T. 3-Hydroxykynurenine Suppresses CD4+ T-Cell Proliferation, Induces T-Regulatory-Cell Development, and Prolongs Corneal Allograft Survival. Investig. Ophthalmol. Vis. Sci. 2011, 52, 2640–2648. [Google Scholar] [CrossRef]
- Turner, M.S.; Kane, L.P.; Morel, P.A. Dominant Role of Antigen Dose in CD4+Foxp3+ Regulatory T Cell Induction and Expansion. J. Immunol. 2009, 183, 4895–4903. [Google Scholar] [CrossRef]
- Kohlhaas, S.; Garden, O.A.; Scudamore, C.; Turner, M.; Okkenhaug, K.; Vigorito, E. Cutting Edge: The Foxp3 Target MiR-155 Contributes to the Development of Regulatory T Cells. J. Immunol. 2009, 182, 2578–2582. [Google Scholar] [CrossRef]
- Chen, W.; Liang, X.; Peterson, A.J.; Munn, D.H.; Blazar, B.R. The Indoleamine 2,3-Dioxygenase Pathway Is Essential for Human Plasmacytoid Dendritic Cell-Induced Adaptive T Regulatory Cell Generation. J. Immunol. 2008, 181, 5396–5404. [Google Scholar] [CrossRef]
- Pallotta, M.T.; Orabona, C.; Volpi, C.; Vacca, C.; Belladonna, M.L.; Bianchi, R.; Servillo, G.; Brunacci, C.; Calvitti, M.; Bicciato, S.; et al. Indoleamine 2,3-Dioxygenase Is a Signaling Protein in Long-Term Tolerance by Dendritic Cells. Nat. Immunol. 2011, 12, 870–878. [Google Scholar] [CrossRef]
- Cribbs, A.P.; Kennedy, A.; Penn, H.; Read, J.E.; Amjadi, P.; Green, P.; Syed, K.; Manka, S.W.; Brennan, F.M.; Gregory, B.; et al. Treg Cell Function in Rheumatoid Arthritis Is Compromised by Ctla-4 Promoter Methylation Resulting in a Failure to Activate the Indoleamine 2,3-Dioxygenase Pathway. Arthritis Rheumatol. 2014, 66, 2344–2354. [Google Scholar] [CrossRef]
- Bishnupuri, K.S.; Alvarado, D.M.; Khouri, A.N.; Shabsovich, M.; Chen, B.; Dieckgraefe, B.K.; Ciorba, M.A. IDO1 and Kynurenine Pathway Metabolites Activate PI3K-Akt Signaling in the Neoplastic Colon Epithelium to Promote Cancer Cell Proliferation and Inhibit Apoptosis. Cancer Res. 2019, 79, 1138–1150. [Google Scholar] [CrossRef]
- Thaker, A.I.; Rao, M.S.; Bishnupuri, K.S.; Kerr, T.A.; Foster, L.; Marinshaw, J.M.; Newberry, R.D.; Stenson, W.F.; Ciorba, M.A. IDO1 Metabolites Activate β-Catenin Signaling to Promote Cancer Cell Proliferation and Colon Tumorigenesis in Mice. Gastroenterology 2013, 145, 416–425.e4. [Google Scholar] [CrossRef]
- Wu, H.; Gong, J.; Liu, Y. Indoleamine 2, 3-Dioxygenase Regulation of Immune Response (Review). Mol. Med. Rep. 2018, 17, 4867–4873. [Google Scholar] [CrossRef]
- Brandacher, G.; Perathoner, A.; Ladurner, R.; Schneeberger, S.; Obrist, P.; Winkler, C.; Werner, E.R.; Werner-Felmayer, G.; Weiss, H.G.; Göbel, G.; et al. Prognostic Value of Indoleamine 2,3-Dioxygenase Expression in Colorectal Cancer: Effect on Tumor-Infiltrating T Cells. Clin. Cancer Res. 2006, 12, 1144–1151. [Google Scholar] [CrossRef]
- Ninomiya, S.; Hara, T.; Tsurumi, H.; Hoshi, M.; Kanemura, N.; Goto, N.; Kasahara, S.; Shimizu, M.; Ito, H.; Saito, K.; et al. Indoleamine 2,3-Dioxygenase in Tumor Tissue Indicates Prognosis in Patients with Diffuse Large B-Cell Lymphoma Treated with R-CHOP. Ann. Hematol. 2011, 90, 409–416. [Google Scholar] [CrossRef]
- Sansone, P.; Bromberg, J. Targeting the Interleukin-6/Jak/Stat Pathway in Human Malignancies. J. Clin. Oncol. 2012, 30, 1005–1014. [Google Scholar] [CrossRef]
- Yu, H.; Pardoll, D.; Jove, R. STATs in Cancer Inflammation and Immunity: A Leading Role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Litzenburger, U.M.; Opitz, C.A.; Sahm, F.; Rauschenbach, K.J.; Trump, S.; Winter, M.; Ott, M.; Ochs, K.; Lutz, C.; Liu, X.; et al. Constitutive IDO Expression in Human Cancer Is Sustained by an Autocrine Signaling Loop Involving IL-6, STAT3 and the AHR. Oncotarget 2014, 5, 1038–1051. [Google Scholar] [CrossRef]
- D’Amato, N.C.; Rogers, T.J.; Gordon, M.A.; Greene, L.I.; Cochrane, D.R.; Spoelstra, N.S.; Nemkov, T.G.; D’Alessandro, A.; Hansen, K.C.; Richer, J.K. A TDO2-AhR Signaling Axis Facilitates Anoikis Resistance and Metastasis in Triple-Negative Breast Cancer. Cancer Res. 2015, 75, 4651–4664. [Google Scholar] [CrossRef]
- Labadie, B.W.; Bao, R.; Luke, J.J. Reimagining IDO Pathway Inhibition in Cancer Immunotherapy via Downstream Focus on the Tryptophan-Kynurenine-Aryl Hydrocarbon Axis. Clin. Cancer Res. 2019, 25, 1462–1471. [Google Scholar] [CrossRef]
- Pantouris, G.; Mowat, C.G. Antitumour Agents as Inhibitors of Tryptophan 2,3-Dioxygenase. Biochem. Biophys. Res. Commun. 2014, 443, 28–31. [Google Scholar] [CrossRef]
- Santhanam, S.; Alvarado, D.M.; Ciorba, M.A. Therapeutic Targeting of Inflammation and Tryptophan Metabolism in Colon and Gastrointestinal Cancer. Transl. Res. 2016, 167, 67–79. [Google Scholar] [CrossRef]
- Pilotte, L.; Larrieu, P.; Stroobant, V.; Colau, D.; Dolusic, E.; Frédérick, R.; De Plaen, E.; Uyttenhove, C.; Wouters, J.; Masereel, B.; et al. Reversal of Tumoral Immune Resistance by Inhibition of Tryptophan 2,3-Dioxygenase. Proc. Natl. Acad. Sci. USA 2012, 109, 2497–2502. [Google Scholar] [CrossRef]
- Pei, Z.; Mendonca, R.; Gazzard, L.; Pastor, R.; Goon, L.; Gustafson, A.; VanderPorten, E.; Hatzivassiliou, G.; Dement, K.; Cass, R.; et al. Aminoisoxazoles as Potent Inhibitors of Tryptophan 2,3-Dioxygenase 2 (TDO2). ACS Med. Chem. Lett. 2018, 9, 417–421. [Google Scholar] [CrossRef]
- Prendergast, G.C.; Malachowski, W.J.; Mondal, A.; Scherle, P.; Muller, A.J. Indoleamine 2,3-Dioxygenase and Its Therapeutic Inhibition in Cancer. Int. Rev. Cell Mol. Biol. 2018, 336, 175–203. [Google Scholar] [CrossRef]
- Röhrig, U.F.; Majjigapu, S.R.; Caldelari, D.; Dilek, N.; Reichenbach, P.; Ascencao, K.; Irving, M.; Coukos, G.; Vogel, P.; Zoete, V.; et al. 1,2,3-Triazoles as Inhibitors of Indoleamine 2,3-Dioxygenase 2 (IDO2). Bioorg. Med. Chem. Lett. 2016, 26, 4330–4333. [Google Scholar] [CrossRef]
- Ball, H.J.; Yuasa, H.J.; Austin, C.J.D.; Weiser, S.; Hunt, N.H. Indoleamine 2,3-Dioxygenase-2; a New Enzyme in the Kynurenine Pathway. Int. J. Biochem. Cell Biol. 2009, 41, 467–471. [Google Scholar] [CrossRef]
- Pantouris, G.; Serys, M.; Yuasa, H.J.; Ball, H.J.; Mowat, C.G. Human Indoleamine 2,3-Dioxygenase-2 Has Substrate Specificity and Inhibition Characteristics Distinct from Those of Indoleamine 2,3-Dioxygenase-1. Amino Acids 2014, 46, 2155–2163. [Google Scholar] [CrossRef]
- Liu, M.; Wang, X.; Wang, L.; Ma, X.; Gong, Z.; Zhang, S.; Li, Y. Targeting the IDO1 Pathway in Cancer: From Bench to Bedside. J. Hematol. Oncol. 2018, 11, 100. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, T.; Moritake, K.; Yamada, K.; Hara, N.; Osago, H.; Shibata, T.; Akiyama, Y.; Tsuchiya, M. Indoleamine 2,3-Dioxygenase as a New Target for Malignant Glioma Therapy. J. Neurosurg. 2009, 111, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Chen, C.; Ju, R.; Wang, Q.; Li, J.; Guo, L.; Ye, C.; Zhang, D. Carboxyamidotriazole Combined with IDO1-Kyn-AhR Pathway Inhibitors Profoundly Enhances Cancer Immunotherapy. J. Immunother. Cancer 2019, 7, 246. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Shafizadeh, E.; Attwood, J.T.; Bondarev, I.; Pashine, A.; Mellor, A.L. Inhibition of T Cell Proliferation by Macrophage Tryptophan Catabolism. J. Exp. Med. 1999, 189, 1363–1372. [Google Scholar] [CrossRef] [PubMed]
- Hwu, P.; Du, M.X.; Lapointe, R.; Do, M.; Taylor, M.W.; Young, H.A. Indoleamine 2,3-Dioxygenase Production by Human Dendritic Cells Results in the Inhibition of T Cell Proliferation. J. Immunol. 2000, 164, 3596–3599. [Google Scholar] [CrossRef]
- Munn, D.H.; Sharma, M.D.; Lee, J.R.; Jhaver, K.G.; Johnson, T.S.; Keskin, D.B.; Marshall, B.; Chandler, P.; Antonia, S.J.; Burgess, R.; et al. Potential Regulatory Function of Human Dendritic Cells Expressing Indoleamine 2,3-Dioxygenase. Science 2002, 297, 1867–1870. [Google Scholar] [CrossRef]
- Mellor, A.L.; Baban, B.; Chandler, P.; Marshall, B.; Jhaver, K.; Hansen, A.; Koni, P.A.; Iwashima, M.; Munn, D.H. Cutting Edge: Induced Indoleamine 2,3 Dioxygenase Expression in Dendritic Cell Subsets Suppresses T Cell Clonal Expansion. J. Immunol. 2003, 171. [Google Scholar] [CrossRef]
- Lob, S.; Konigsrainer, A.; Schafer, R.; Rammensee, H.-G.; Opelz, G.; Terness, P. Levo- but Not Dextro-1-Methyl Tryptophan Abrogates the IDO Activity of Human Dendritic Cells. Blood 2008, 111, 2152–2154. [Google Scholar] [CrossRef]
- Li, J.; Li, Y.; Yang, D.; Hu, N.; Guo, Z.; Kuang, C.; Yang, Q. Establishment of a human indoleamine 2, 3-dioxygenase 2 (hIDO2) bioassay system and discovery of tryptanthrin derivatives as potent hIDO2 inhibitors. Eur. J. Med. Chem. 2016, 123, 171–179. [Google Scholar] [CrossRef]
- Zakharia, Y.; Johnson, T.S.; Colman, H.; Vahanian, N.N.; Link, C.J.; Kennedy, E.; Sadek, R.F.; Kong, F.M.; Vender, J.; Munn, D.; et al. A Phase I/II Study of the Combination of Indoximod and Temozolomide for Adult Patients with Temozolomide-Refractory Primary Malignant Brain Tumors. J. Clin. Oncol. 2014, 32, TPS2107. [Google Scholar] [CrossRef]
- Colman, H.; Mott, F.; Spira, A.I.; Johnson, T.S.; Zakharia, Y.; Vahanian, N.N.; Link, C.J.; Kennedy, E.P.; Sadek, R.F.; Munn, D.; et al. A Phase 1b/2 Study of the Combination of the IDO Pathway Inhibitor Indoximod and Temozolomide for Adult Patients with Temozolomide-Refractory Primary Malignant Brain Tumors: Safety Analysis and Preliminary Efficacy of the Phase 1b Component. J. Clin. Oncol. 2015, 33, 2070. [Google Scholar] [CrossRef]
- Bakmiwewa, S.M.; Fatokun, A.A.; Tran, A.; Payne, R.J.; Hunt, N.H.; Ball, H.J. Identification of Selective Inhibitors of Indoleamine 2,3-Dioxygenase 2. Bioorg. Med. Chem. Lett. 2012, 22, 7641–7646. [Google Scholar] [CrossRef]
- Moreno, A.C.R.; Porchia, B.F.M.M.; Pagni, R.L.; Souza, P.d.C.; Pegoraro, R.; Rodrigues, K.B.; Barros, T.B.; Aps, L.R.d.M.M.; de Araújo, E.F.; Calich, V.L.G.; et al. The Combined Use of Melatonin and an Indoleamine 2,3-Dioxygenase-1 Inhibitor Enhances Vaccine-Induced Protective Cellular Immunity to HPV16-Associated Tumors. Front. Immunol. 2018, 9, 1914. [Google Scholar] [CrossRef]
- Fu, R.; Zhang, Y.-W.; Li, H.-M.; Lv, W.-C.; Zhao, L.; Guo, Q.-L.; Lu, T.; Weiss, S.J.; Li, Z.-Y.; Wu, Z.-Q. LW106, a Novel Indoleamine 2,3-Dioxygenase 1 Inhibitor, Suppresses Tumour Progression by Limiting Stroma-Immune Crosstalk and Cancer Stem Cell Enrichment in Tumour Micro-Environment. Br. J. Pharmacol. 2018, 175, 3034–3049. [Google Scholar] [CrossRef]
- Grobben, Y.; de Man, J.; van Doornmalen, A.M.; Muller, M.; Willemsen-Seegers, N.; Vu-Pham, D.; Mulder, W.R.; Prinsen, M.B.W.; de Wit, J.; Sterrenburg, J.G.; et al. Targeting Indoleamine 2,3-Dioxygenase in Cancer Models Using the Novel Small Molecule Inhibitor NTRC 3883-0. Front. Immunol. 2020, 11, 609490. [Google Scholar] [CrossRef]
- Yang, L.; Chen, Y.; He, J.; Njoya, E.M.; Chen, J.; Liu, S.; Xie, C.; Huang, W.; Wang, F.; Wang, Z.; et al. 4,6-Substituted-1H-Indazoles as Potent IDO1/TDO Dual Inhibitors. Bioorg. Med. Chem. 2019, 27, 1087–1098. [Google Scholar] [CrossRef]
- Bogdanova, L.A.; Morozkova, T.S.; Amitina, S.A.; Mazhukin, D.G.; Nikolin, V.P.; Popova, N.A.; Kaledin, V.I. Dual Effects of Indoleamine 2,3-Dioxygenase Inhibitors on the Therapeutic Effects of Cyclophosphamide and Cycloplatam on Ehrlich Ascites Tumor in Mice. Bull. Exp. Biol. Med. 2014, 157, 506–509. [Google Scholar] [CrossRef]
- Ogawa, K.; Hara, T.; Shimizu, M.; Nagano, J.; Ohno, T.; Hoshi, M.; Ito, H.; Tsurumi, H.; Saito, K.; Seishima, M.; et al. (-)-Epigallocatechin Gallate Inhibits the Expression of Indoleamine 2,3-Dioxygenase in Human Colorectal Cancer Cells. Oncol. Lett. 2012, 4, 546–550. [Google Scholar] [CrossRef]
- Ogawa, K.; Hara, T.; Shimizu, M.; Ninomiya, S.; Nagano, J.; Sakai, H.; Hoshi, M.; Ito, H.; Tsurumi, H.; Saito, K.; et al. Suppression of Azoxymethane-Induced Colonic Preneoplastic Lesions in Rats by 1-Methyltryptophan, an Inhibitor of Indoleamine 2,3-Dioxygenase. Cancer Sci. 2012, 103, 951–958. [Google Scholar] [CrossRef]
- Ye, Z.; Yue, L.; Shi, J.; Shao, M.; Wu, T. Role of IDO and TDO in Cancers and Related Diseases and the Therapeutic Implications. J. Cancer 2019, 10, 2771–2782. [Google Scholar] [CrossRef]
- Cheong, J.E.; Sun, L. Targeting the IDO1/TDO2-KYN-AhR Pathway for Cancer Immunotherapy—Challenges and Opportunities. Trends Pharmacol. Sci. 2018, 39, 307–325. [Google Scholar] [CrossRef]
- Poncelet, L.; Ait-Belkacem, R.; Marillier, R.; Gomes, B.; Stauber, J. Target Exposure and Pharmacodynamics Study of the Indoleamine 2,3-Dioxygenase-1 (IDO-1) Inhibitor Epacadostat in the CT26 Mouse Tumor Model. J. Pharm. Biomed. Anal. 2019, 170, 220–227. [Google Scholar] [CrossRef]
- Balachandran, V.P.; Cavnar, M.J.; Zeng, S.; Bamboat, Z.M.; Ocuin, L.M.; Obaid, H.; Sorenson, E.C.; Popow, R.; Ariyan, C.; Rossi, F.; et al. Imatinib Potentiates Antitumor T Cell Responses in Gastrointestinal Stromal Tumor through the Inhibition of Ido. Nat. Med. 2011, 17, 1094–1100. [Google Scholar] [CrossRef]
- Li, M.; Bolduc, A.R.; Hoda, M.N.; Gamble, D.N.; Dolisca, S.-B.; Bolduc, A.K.; Hoang, K.; Ashley, C.; McCall, D.; Rojiani, A.M.; et al. The Indoleamine 2,3-Dioxygenase Pathway Controls Complement-Dependent Enhancement of Chemo-Radiation Therapy against Murine Glioblastoma. J. Immunother. Cancer 2014, 2, 21. [Google Scholar] [CrossRef]
- Söderlund, J.; Erhardt, S.; Kast, R.E. Acyclovir Inhibition of IDO to Decrease Tregs as a Glioblastoma Treatment Adjunct. J. Neuroinflamm. 2010, 7, 44. [Google Scholar] [CrossRef]
- Riess, C.; Schneider, B.; Kehnscherper, H.; Gesche, J.; Irmscher, N.; Shokraie, F.; Classen, C.F.; Wirthgen, E.; Domanska, G.; Zimpfer, A.; et al. Activation of the Kynurenine Pathway in Human Malignancies Can Be Suppressed by the Cyclin-Dependent Kinase Inhibitor Dinaciclib. Front. Immunol. 2020, 11, 55. [Google Scholar] [CrossRef]
- Adams, S.; Braidy, N.; Bessede, A.; Bessesde, A.; Brew, B.J.; Grant, R.; Teo, C.; Guillemin, G.J. The Kynurenine Pathway in Brain Tumor Pathogenesis. Cancer Res. 2012, 72, 5649–5657. [Google Scholar] [CrossRef]
- Tourino, M.C.; de Oliveira, E.M.; Bellé, L.P.; Knebel, F.H.; Albuquerque, R.C.; Dörr, F.A.; Okada, S.S.; Migliorini, S.; Soares, I.S.; Campa, A. Tryptamine and Dimethyltryptamine Inhibit Indoleamine 2,3 Dioxygenase and Increase the Tumor-Reactive Effect of Peripheral Blood Mononuclear Cells. Cell Biochem. Funct. 2013, 31, 361–364. [Google Scholar] [CrossRef]
- Du, L.; Xing, Z.; Tao, B.; Li, T.; Yang, D.; Li, W.; Zheng, Y.; Kuang, C.; Yang, Q. Both IDO1 and TDO Contribute to the Malignancy of Gliomas via the Kyn-AhR-AQP4 Signaling Pathway. Signal Transduct. Target. Ther. 2020, 5, 10. [Google Scholar] [CrossRef]
- Takada, K.; Kohashi, K.; Shimokawa, M.; Haro, A.; Osoegawa, A.; Tagawa, T.; Seto, T.; Oda, Y.; Maehara, Y. Co-Expression of IDO1 and PD-L1 in Lung Squamous Cell Carcinoma: Potential Targets of Novel Combination Therapy. Lung Cancer 2019, 128, 26–32. [Google Scholar] [CrossRef]
- Volaric, A.; Gentzler, R.; Hall, R.; Mehaffey, J.H.; Stelow, E.B.; Bullock, T.N.; Martin, L.W.; Mills, A.M. Indoleamine-2,3-Dioxygenase in Non-Small Cell Lung Cancer: A Targetable Mechanism of Immune Resistance Frequently Coexpressed with PD-L1. Am. J. Surg. Pathol. 2018, 42, 1216–1223. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Barsoumian, H.B.; Schoenhals, J.E.; Cushman, T.R.; Caetano, M.S.; Wang, X.; Valdecanas, D.R.; Niknam, S.; Younes, A.I.; Li, G.; et al. Indoleamine 2,3-Dioxygenase 1 Inhibition Targets Anti-PD1-Resistant Lung Tumors by Blocking Myeloid-Derived Suppressor Cells. Cancer Lett. 2018, 431, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Stone, E.; Marshall, N.; Donkor, M.; Triplett, K.; Blazek, J.; Triplett, T.; Ehrlich, L.; Georgiou, G. Abstract LB-226: Depletion of Kynurenine Using an Engineered Therapeutic Enzyme Potently Inhibits Cancer Immune Checkpoints Both as a Monotherapy and in Combination with Anti-PD-1. Cancer Res. 2015, 75, 226. [Google Scholar] [CrossRef]
- Nguyen, D.J.M.; Theodoropoulos, G.; Li, Y.-Y.; Wu, C.; Sha, W.; Feun, L.G.; Lampidis, T.J.; Savaraj, N.; Wangpaichitr, M. Targeting the Kynurenine Pathway for the Treatment of Cisplatin-Resistant Lung Cancer. Mol. Cancer Res. 2020, 18, 105–117. [Google Scholar] [CrossRef]
- Luo, B.; Que, Z.-J.; Zhou, Z.-Y.; Wang, Q.; Dong, C.-S.; Jiang, Y.; Hu, B.; Shi, H.; Jin, Y.; Liu, J.-W.; et al. Feiji Recipe Inhibits the Growth of Lung Cancer by Modulating T-Cell Immunity through Indoleamine-2,3-Dioxygenase Pathway in an Orthotopic Implantation Model. J. Integr. Med. 2018, 16, 283–289. [Google Scholar] [CrossRef]
- Meng, Y.; Wang, W.; Chen, M.; Chen, K.; Xia, X.; Zhou, S.; Yang, H. GBP1 Facilitates Indoleamine 2,3-Dioxygenase Extracellular Secretion to Promote the Malignant Progression of Lung Cancer. Front. Immunol. 2020, 11, 622467. [Google Scholar] [CrossRef]
- Noh, K.T.; Chae, S.H.; Chun, S.H.; Jung, I.D.; Kang, H.K.; Park, Y.-M. Resveratrol Suppresses Tumor Progression via the Regulation of Indoleamine 2,3-Dioxygenase. Biochem. Biophys. Res. Commun. 2013, 431, 348–353. [Google Scholar] [CrossRef]
- Meng, X.; Du, G.; Ye, L.; Sun, S.; Liu, Q.; Wang, H.; Wang, W.; Wu, Z.; Tian, J. Combinatorial Antitumor Effects of Indoleamine 2,3-Dioxygenase Inhibitor NLG919 and Paclitaxel in a Murine B16-F10 Melanoma Model. Int. J. Immunopathol. Pharmacol. 2017, 30, 215–226. [Google Scholar] [CrossRef]
- Sandri, S.; Watanabe, L.R.M.; Oliveira, E.A.d.; Faião-Flores, F.; Migliorini, S.; Tiago, M.; Felipe-Silva, A.; Vazquez, V.d.L.; da Costa Souza, P.; Consolaro, M.E.L.; et al. Indoleamine 2,3-Dioxygenase in Melanoma Progression and BRAF Inhibitor Resistance. Pharmacol. Res. 2020, 159, 104998. [Google Scholar] [CrossRef]
- Pour, S.R.; Morikawa, H.; Kiani, N.A.; Gomez-Cabrero, D.; Hayes, A.; Zheng, X.; Pernemalm, M.; Lehtiö, J.; Mole, D.J.; Hansson, J.; et al. Immunometabolic Network Interactions of the Kynurenine Pathway in Cutaneous Malignant Melanoma. Front. Oncol. 2020, 10, 51. [Google Scholar] [CrossRef]
- Chevolet, I.; Schreuer, M.; Speeckaert, R.; Neyns, B.; Hoorens, I.; van Geel, N.; Krüse, V.; Hennart, B.; Allorge, D.; Van Gele, M.; et al. Systemic Immune Changes Associated with Adjuvant Interferon-A2b-Therapy in Stage III Melanoma Patients: Failure at the Effector Phase? Melanoma Res. 2015, 25, 357–361. [Google Scholar] [CrossRef]
- Jia, H.; Ren, W.; Feng, Y.; Wei, T.; Guo, M.; Guo, J.; Zhao, J.; Song, X.; Wang, M.; Zhao, T.; et al. The Enhanced Antitumour Response of Pimozide Combined with the IDO Inhibitor L-MT in Melanoma. Int. J. Oncol. 2018, 53, 949–960. [Google Scholar] [CrossRef]
- Hanafi, L.-A.; Gauchat, D.; Godin-Ethier, J.; Possamaï, D.; Duvignaud, J.-B.; Leclerc, D.; Grandvaux, N.; Lapointe, R. Fludarabine Downregulates Indoleamine 2,3-Dioxygenase in Tumors via a Proteasome-Mediated Degradation Mechanism. PLoS ONE 2014, 9, e99211. [Google Scholar] [CrossRef]
- Komrokji, R.S.; Wei, S.; Mailloux, A.W.; Zhang, L.; Padron, E.; Sallman, D.; Lancet, J.E.; Tinsley, S.; Nardelli, L.A.; Pinilla-Ibarz, J.; et al. A Phase II Study to Determine the Safety and Efficacy of the Oral Inhibitor of Indoleamine 2,3-Dioxygenase (IDO) Enzyme INCB024360 in Patients with Myelodysplastic Syndromes. Clin. Lymphoma Myeloma Leuk. 2019, 19, 157–161. [Google Scholar] [CrossRef]
- Liu, X.; Shin, N.; Koblish, H.K.; Yang, G.; Wang, Q.; Wang, K.; Leffet, L.; Hansbury, M.J.; Thomas, B.; Rupar, M.; et al. Selective Inhibition of IDO1 Effectively Regulates Mediators of Antitumor Immunity. Blood 2010, 115, 3520–3530. [Google Scholar] [CrossRef]
- Zhang, P.; Jiang, G.; Gao, J.; Li, L.; Du, J.; Jiao, X. SAHA Down-Regulates the Expression of Indoleamine 2,3-Dioxygenase via Inhibition of the JAK/STAT1 Signaling Pathway in Gallbladder Carcinoma Cells. Oncol. Rep. 2013, 29, 269–275. [Google Scholar] [CrossRef]
- Jiang, G.-M.; Wang, H.-S.; Du, J.; Ma, W.-F.; Wang, H.; Qiu, Y.; Zhang, Q.-G.; Xu, W.; Liu, H.-F.; Liang, J.-P. Bortezomib Relieves Immune Tolerance in Nasopharyngeal Carcinoma via STAT1 Suppression and Indoleamine 2,3-Dioxygenase Downregulation. Cancer Immunol. Res. 2017, 5, 42–51. [Google Scholar] [CrossRef]
- Hui, K.F.; Lam, B.H.; Ho, D.N.; Tsao, S.W.; Chiang, A.K. Bortezomib and SAHA synergistically induce ROS-driven caspase-dependent apoptosis of nasopharyngeal carcinoma and block replication of Epstein-Barr virus. Mol. Cancer Ther. 2013, 12, 747–758. [Google Scholar] [CrossRef]
- Ma, H.; Qin, Q.; Mi, J.; Feng, Q. 1-MT Inhibits the Invasion of CBP-Resistant Ovarian Cancer Cells via down-Regulating IDO Expression and Re-Activating Immune Cells Function. BMC Pharmacol. Toxicol. 2020, 21, 67. [Google Scholar] [CrossRef]
- Cherney, E.C.; Zhang, L.; Nara, S.; Zhu, X.; Gullo-Brown, J.; Maley, D.; Lin, T.-A.; Hunt, J.T.; Huang, C.; Yang, Z.; et al. Discovery and Preclinical Evaluation of BMS-986242, a Potent, Selective Inhibitor of Indoleamine-2,3-Dioxygenase 1. ACS Med. Chem. Lett. 2021, 12, 288–294. [Google Scholar] [CrossRef]
- Gomes, B.; Driessens, G.; Bartlett, D.; Cai, D.; Cauwenberghs, S.; Crosignani, S.; Dalvie, D.; Denies, S.; Dillon, C.P.; Fantin, V.R.; et al. Characterization of the Selective Indoleamine 2,3-Dioxygenase-1 (IDO1) Catalytic Inhibitor EOS200271/PF-06840003 Supports IDO1 as a Critical Resistance Mechanism to PD-(L)1 Blockade Therapy. Mol. Cancer Ther. 2018, 17, 2530–2542. [Google Scholar] [CrossRef]
- Blair, A.B.; Kleponis, J.; Thomas, D.L.; Muth, S.T.; Murphy, A.G.; Kim, V.; Zheng, L. IDO1 Inhibition Potentiates Vaccine-Induced Immunity against Pancreatic Adenocarcinoma. J. Clin. Investig. 2019, 129, 1742–1755. [Google Scholar] [CrossRef]
- Crosignani, S.; Bingham, P.; Bottemanne, P.; Cannelle, H.; Cauwenberghs, S.; Cordonnier, M.; Dalvie, D.; Deroose, F.; Feng, J.L.; Gomes, B.; et al. Discovery of a Novel and Selective Indoleamine 2,3-Dioxygenase (IDO-1) Inhibitor 3-(5-Fluoro-1H-Indol-3-Yl)Pyrrolidine-2,5-Dione (EOS200271/PF-06840003) and Its Characterization as a Potential Clinical Candidate. J. Med. Chem. 2017, 60, 9617–9629. [Google Scholar] [CrossRef]
- Yamahira, A.; Narita, M.; Iwabuchi, M.; Uchiyama, T.; Iwaya, S.; Ohiwa, R.; Nishizawa, Y.; Suzuki, T.; Yokoyama, Y.; Hashimoto, S.; et al. Activation of the Leukemia Plasmacytoid Dendritic Cell Line PMDC05 by Toho-1, a Novel IDO Inhibitor. Anticancer Res. 2014, 34, 4021–4028. [Google Scholar]
- Trott, J.F.; Kim, J.; Abu Aboud, O.; Wettersten, H.; Stewart, B.; Berryhill, G.; Uzal, F.; Hovey, R.C.; Chen, C.-H.; Anderson, K.; et al. Inhibiting Tryptophan Metabolism Enhances Interferon Therapy in Kidney Cancer. Oncotarget 2016, 7, 66540–66557. [Google Scholar] [CrossRef]
- Nafia, I.; Toulmonde, M.; Bortolotto, D.; Chaibi, A.; Bodet, D.; Rey, C.; Velasco, V.; Larmonier, C.B.; Cerf, L.; Adam, J.; et al. IDO Targeting in Sarcoma: Biological and Clinical Implications. Front. Immunol. 2020, 11, 274. [Google Scholar] [CrossRef]
- Nakamura, N.; Hara, T.; Shimizu, M.; Mabuchi, R.; Nagano, J.; Ohno, T.; Kochi, T.; Kubota, M.; Shirakami, Y.; Goto, N.; et al. Effects of Indoleamine 2,3-Dioxygenase Inhibitor in Non-Hodgkin Lymphoma Model Mice. Int. J. Hematol. 2015, 102, 327–334. [Google Scholar] [CrossRef]
- Chen, J.-Y.; Li, C.-F.; Kuo, C.-C.; Tsai, K.K.; Hou, M.-F.; Hung, W.-C. Cancer/Stroma Interplay via Cyclooxygenase-2 and Indoleamine 2,3-Dioxygenase Promotes Breast Cancer Progression. Breast Cancer Res. 2014, 16, 410. [Google Scholar] [CrossRef] [PubMed]
- Basu, G.D.; Tinder, T.L.; Bradley, J.M.; Tu, T.; Hattrup, C.L.; Pockaj, B.A.; Mukherjee, P. Cyclooxygenase-2 Inhibitor Enhances the Efficacy of a Breast Cancer Vaccine: Role of IDO. J. Immunol. 2006, 177, 2391–2402. [Google Scholar] [CrossRef] [PubMed]
- Flick, H.E.; Lalonde, J.M.; Malachowski, W.P.; Muller, A.J. The Tumor-Selective Cytotoxic Agent β-Lapachone Is a Potent Inhibitor of IDO1. Int. J. Tryptophan Res. 2013, 6, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Ren, J.; Ma, Y.; Liu, H.; Rong, Q.; Feng, Y.; Wang, Y.; Cheng, Y.; Ge, R.; Li, Z.; et al. Discovery of Cyanopyridine Scaffold as Novel Indoleamine-2,3-Dioxygenase 1 (IDO1) Inhibitors through Virtual Screening and Preliminary Hit Optimisation. J. Enzyme Inhib. Med. Chem. 2019, 34, 250–263. [Google Scholar] [CrossRef]
- Vasilyeva, E.D.; Kaledin, V.; Nikolin, V.P.; Popova, N.A.; Kirilyuk, I.A.; Grigor’ev, I.A. Accelerated Rejection of the Second Transplants of Immunogenic Tumor in Mice under Inhibition of Indoleamine 2,3-Dioxygenase Activity by Ethyl Pyruvate. Exp. Oncol. 2012, 34, 66–68. [Google Scholar]
- Liu, Q.; Hua, S.; Wang, X.; Chen, F.; Gou, S. The Introduction of Immunosuppressor (TDO Inhibitor) Significantly Improved the Efficacy of Irinotecan in Treating Hepatocellular Carcinoma. Cancer Immunol. Immunother. 2021, 70, 497–508. [Google Scholar] [CrossRef]
- Luo, Q.; Yan, L.; Xu, P.; Xiong, C.; Yang, Z.; Hu, P.; Hu, H.; Hong, R. Discovery of a Polysaccharide from the Fruiting Bodies of Lepista Sordida as Potent Inhibitors of Indoleamine 2, 3-Dioxygenase (IDO) in HepG2 Cells via Blocking of STAT1-Mediated JAK-PKC-δ Signaling Pathways. Carbohydr. Polym. 2018, 197, 540–547. [Google Scholar] [CrossRef]
- Ray, A.; Song, Y.; Du, T.; Tai, Y.-T.; Chauhan, D.; Anderson, K.C. Targeting Tryptophan Catabolic Kynurenine Pathway Enhances Antitumor Immunity and Cytotoxicity in Multiple Myeloma. Leukemia 2020, 34, 567–577. [Google Scholar] [CrossRef]
- Moyer, B.J.; Rojas, I.Y.; Murray, I.A.; Lee, S.; Hazlett, H.F.; Perdew, G.H.; Tomlinson, C.R. Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitors Activate the Aryl Hydrocarbon Receptor. Toxicol. Appl. Pharmacol. 2017, 323, 74–80. [Google Scholar] [CrossRef]
- Jung, K.H.; LoRusso, P.; Burris, H.; Gordon, M.; Bang, Y.-J.; Hellmann, M.D.; Cervantes, A.; de Olza, M.O.; Marabelle, A.; Hodi, F.S.; et al. Phase I Study of the Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitor Navoximod (GDC-0919) Administered with PD-L1 Inhibitor (Atezolizumab) in Advanced Solid Tumors. Clin. Cancer Res. 2019, 25, 3220–3228. [Google Scholar] [CrossRef]
- Ebata, T.; Shimizu, T.; Fujiwara, Y.; Tamura, K.; Kondo, S.; Iwasa, S.; Yonemori, K.; Shimomura, A.; Kitano, S.; Koyama, T.; et al. Phase I Study of the Indoleamine 2,3-Dioxygenase 1 Inhibitor Navoximod (GDC-0919) as Monotherapy and in Combination with the PD-L1 Inhibitor Atezolizumab in Japanese Patients with Advanced Solid Tumours. Investig. New Drugs 2020, 38, 468–477. [Google Scholar] [CrossRef]
- Iachininoto, M.G.; Nuzzolo, E.R.; Bonanno, G.; Mariotti, A.; Procoli, A.; Locatelli, F.; De Cristofaro, R.; Rutella, S. Cyclooxygenase-2 (COX-2) Inhibition Constrains Indoleamine 2,3-Dioxygenase 1 (IDO1) Activity in Acute Myeloid Leukaemia Cells. Molecules 2013, 18, 10132–10145. [Google Scholar] [CrossRef]
- Hanihara, M.; Kawataki, T.; Oh-Oka, K.; Mitsuka, K.; Nakao, A.; Kinouchi, H. Synergistic Antitumor Effect with Indoleamine 2,3-Dioxygenase Inhibition and Temozolomide in a Murine Glioma Model. J. Neurosurg. 2016, 124, 1594–1601. [Google Scholar] [CrossRef]
- Li, A.; Barsoumian, H.B.; Schoenhals, J.E.; Caetano, M.S.; Wang, X.; Menon, H.; Valdecanas, D.R.; Niknam, S.; Younes, A.I.; Cortez, M.A.; et al. IDO1 Inhibition Overcomes Radiation-Induced “Rebound Immune Suppression” by Reducing Numbers of IDO1-Expressing Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Int. J. Radiat. Oncol. Biol. Phys. 2019, 104, 903–912. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Desjardins, A.; Rixe, O.; Cloughesy, T.; Alekar, S.; Williams, J.H.; Li, R.; Taylor, C.T.; Lassman, A.B. A Phase 1 Study of PF-06840003, an Oral Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitor in Patients with Recurrent Malignant Glioma. Investig. New Drugs 2020, 38, 1784–1795. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Liu, Y.; Wang, S.; Wang, T.; Zhang, G.; Chen, X.; Li, Y.; Cui, H.; Lai, F.; Sheng, L. Design and Synthesis of Indoleamine 2,3-Dioxygenase 1 Inhibitors and Evaluation of Their Use as Anti-Tumor Agents. Molecules 2019, 24, 2124. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.-J.; Kim, T.M.; et al. Epacadostat (E) plus Pembrolizumab (P) versus Pembrolizumab Alone in Patients (Pts) with Unresectable or Metastatic Melanoma: Results of the Phase 3 ECHO-301/KEYNOTE-252 Study. J. Clin. Oncol. 2018, 36, 108. [Google Scholar] [CrossRef]
- Yentz, S.; Smith, D. Indoleamine 2,3-Dioxygenase (IDO) Inhibition as a Strategy to Augment Cancer Immunotherapy. BioDrugs 2018, 32, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-X.; Sun, S.-Y.; Dong, Q.-Q.; Wu, X.-X.; Tang, W.; Xing, Y.-Q. Recent Advances in the Discovery of Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitors. MedChemComm 2019, 10, 1740–1754. [Google Scholar] [CrossRef]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.-J.; Kim, T.M.; et al. Epacadostat plus Pembrolizumab versus Placebo plus Pembrolizumab in Patients with Unresectable or Metastatic Melanoma (ECHO-301/KEYNOTE-252): A Phase 3, Randomised, Double-Blind Study. Lancet Oncol. 2019, 20, 1083–1097. [Google Scholar] [CrossRef]
- Ferdinande, L.; Decaestecker, C.; Verset, L.; Mathieu, A.; Lopez, X.M.; Negulescu, A.-M.; Van Maerken, T.; Salmon, I.; Cuvelier, C.A.; Demetter, P. Clinicopathological Significance of Indoleamine 2,3-Dioxygenase 1 Expression in Colorectal Cancer. Br. J. Cancer 2012, 106, 141–147. [Google Scholar] [CrossRef]
- Park, J.-H.; Lee, J.-M.; Lee, E.-J.; Kim, D.-J.; Hwang, W.-B. Kynurenine Promotes the Goblet Cell Differentiation of HT-29 Colon Carcinoma Cells by Modulating Wnt, Notch and AhR Signals. Oncol. Rep. 2018, 39, 1930–1938. [Google Scholar] [CrossRef]
- Hacking, S.; Vitkovski, T.; Jain, S.; Jin, C.; Chavarria, H.; Wu, D.; Nasim, M. Clinical Significance of Program Death Ligand-1 and Indoleamine-2,3-Dioxygenase Expression in Colorectal Carcinoma. Appl. Immunohistochem. Mol. Morphol. AIMM 2021, 29, 201–208. [Google Scholar] [CrossRef]
- Gao, Y.-F.; Peng, R.-Q.; Li, J.; Ding, Y.; Zhang, X.; Wu, X.-J.; Pan, Z.-Z.; Wan, D.-S.; Zeng, Y.-X.; Zhang, X.-S. The Paradoxical Patterns of Expression of Indoleamine 2,3-Dioxygenase in Colon Cancer. J. Transl. Med. 2009, 7, 71. [Google Scholar] [CrossRef]
- Takamatsu, M.; Hirata, A.; Ohtaki, H.; Hoshi, M.; Ando, T.; Ito, H.; Hatano, Y.; Tomita, H.; Kuno, T.; Saito, K.; et al. Inhibition of Indoleamine 2,3-Dioxygenase 1 Expression Alters Immune Response in Colon Tumor Microenvironment in Mice. Cancer Sci. 2015, 106, 1008–1015. [Google Scholar] [CrossRef]
- Liu, X.; Zhou, W.; Zhang, X.; Ding, Y.; Du, Q.; Hu, R. 1-L-MT, an IDO Inhibitor, Prevented Colitis-Associated Cancer by Inducing CDC20 Inhibition-Mediated Mitotic Death of Colon Cancer Cells. Int. J. Cancer 2018, 143, 1516–1529. [Google Scholar] [CrossRef]
- Günther, J.; Däbritz, J.; Wirthgen, E. Limitations and Off-Target Effects of Tryptophan-Related IDO Inhibitors in Cancer Treatment. Front. Immunol. 2019, 10, 1801. [Google Scholar] [CrossRef]
- Zambetti, N.A.; Ping, Z.; Chen, S.; Kenswil, K.J.G.; Mylona, M.A.; Sanders, M.A.; Hoogenboezem, R.M.; Bindels, E.M.J.; Adisty, M.N.; Van Strien, P.M.H.; et al. Mesenchymal Inflammation Drives Genotoxic Stress in Hematopoietic Stem Cells and Predicts Disease Evolution in Human Pre-Leukemia. Cell Stem Cell 2016, 19, 613–627. [Google Scholar] [CrossRef]
- Mailloux, A.W.; Sugimori, C.; Komrokji, R.S.; Yang, L.; Maciejewski, J.P.; Sekeres, M.A.; Paquette, R.; Loughran, T.P.; List, A.F.; Epling-Burnette, P.K. Expansion of Effector Memory Regulatory T Cells Represents a Novel Prognostic Factor in Lower Risk Myelodysplastic Syndrome. J. Immunol. 2012, 189, 3198–3208. [Google Scholar] [CrossRef]
- Epling-Burnette, P.K.; List, A.F. Advancements in the Molecular Pathogenesis of Myelodysplastic Syndrome. Curr. Opin. Hematol. 2009, 16, 70–76. [Google Scholar] [CrossRef]
- Yang, L.; Qian, Y.; Eksioglu, E.; Epling-Burnette, P.K.; Wei, S. The Inflammatory Microenvironment in MDS. Cell. Mol. Life Sci. 2015, 72, 1959–1966. [Google Scholar] [CrossRef]
- Berthon, C.; Fontenay, M.; Corm, S.; Briche, I.; Allorge, D.; Hennart, B.; Lhermitte, M.; Quesnel, B. Metabolites of Tryptophan Catabolism Are Elevated in Sera of Patients with Myelodysplastic Syndromes and Inhibit Hematopoietic Progenitor Amplification. Leuk. Res. 2013, 37, 573–579. [Google Scholar] [CrossRef]
- Toulmonde, M.; Italiano, A. PD-1 Inhibition in Sarcoma Still Needs Investigation. Lancet Oncol. 2018, 19, e6. [Google Scholar] [CrossRef]
- Toulmonde, M.; Penel, N.; Adam, J.; Chevreau, C.; Blay, J.-Y.; Le Cesne, A.; Bompas, E.; Piperno-Neumann, S.; Cousin, S.; Grellety, T.; et al. Use of PD-1 Targeting, Macrophage Infiltration, and IDO Pathway Activation in Sarcomas: A Phase 2 Clinical Trial. JAMA Oncol. 2018, 4, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Duarte, D.; Amaro, F.; Silva, I.; Silva, D.; Fresco, P.; Oliveira, J.C.; Reguengo, H.; Gonçalves, J.; Vale, N. Carbidopa Alters Tryptophan Metabolism in Breast Cancer and Melanoma Cells Leading to the Formation of Indole-3-Acetonitrile, a Pro-Proliferative Metabolite. Biomolecules 2019, 9, 409. [Google Scholar] [CrossRef] [PubMed]
- Yuan, A.; Yang, P.C.; Yu, C.J.; Lee, Y.C.; Yao, Y.T.; Chen, C.L.; Lee, L.N.; Kuo, S.H.; Luh, K.T. Tumor Angiogenesis Correlates with Histologic Type and Metastasis in Non-Small-Cell Lung Cancer. Am. J. Respir. Crit. Care Med. 1995, 152, 2157–2162. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, H.; Saga, Y.; Fujiwara, H.; Akimoto, H.; Yamada, A.; Kagawa, S.; Takei, Y.; Machida, S.; Takikawa, O.; Suzuki, M. Indoleamine 2,3-Dioxygenase Promotes Peritoneal Dissemination of Ovarian Cancer through Inhibition of Natural Killercell Function and Angiogenesis Promotion. Int. J. Oncol. 2011, 38, 113–120. [Google Scholar]
- Su, C.; Zhang, P.; Liu, J.; Cao, Y. Erianin Inhibits Indoleamine 2, 3-Dioxygenase -Induced Tumor Angiogenesis. Biomed. Pharmacother. 2017, 88, 521–528. [Google Scholar] [CrossRef]
- Gong, Y.-Q.; Fan, Y.; Wu, D.-Z.; Yang, H.; Hu, Z.-B.; Wang, Z.-T. In Vivo and in Vitro Evaluation of Erianin, a Novel Anti-Angiogenic Agent. Eur. J. Cancer 2004, 40, 1554–1565. [Google Scholar] [CrossRef]
- Phacharapiyangkul, N.; Wu, L.-H.; Lee, W.-Y.; Kuo, Y.-H.; Wu, Y.-J.; Liou, H.-P.; Tsai, Y.-E.; Lee, C.-H. The Extracts of Astragalus Membranaceus Enhance Chemosensitivity and Reduce Tumor Indoleamine 2, 3-Dioxygenase Expression. Int. J. Med. Sci. 2019, 16, 1107–1115. [Google Scholar] [CrossRef]
- Chauhan, D.; Singh, A.V.; Brahmandam, M.; Carrasco, R.; Bandi, M.; Hideshima, T.; Bianchi, G.; Podar, K.; Tai, Y.-T.; Mitsiades, C.; et al. Functional Interaction of Plasmacytoid Dendritic Cells with Multiple Myeloma Cells: A Therapeutic Target. Cancer Cell 2009, 16, 309–323. [Google Scholar] [CrossRef]
- Zheng, X.; Koropatnick, J.; Li, M.; Zhang, X.; Ling, F.; Ren, X.; Hao, X.; Sun, H.; Vladau, C.; Franek, J.A.; et al. Reinstalling Antitumor Immunity by Inhibiting Tumor-Derived Immunosuppressive Molecule IDO through RNA Interference. J. Immunol. 2006, 177, 5639–5646. [Google Scholar] [CrossRef]
- Zheng, X.; Koropatnick, J.; Chen, D.; Velenosi, T.; Ling, H.; Zhang, X.; Jiang, N.; Navarro, B.; Ichim, T.E.; Urquhart, B.; et al. Silencing IDO in Dendritic Cells: A Novel Approach to Enhance Cancer Immunotherapy in a Murine Breast Cancer Model. Int. J. Cancer 2013, 132, 967–977. [Google Scholar] [CrossRef]
- Klar, R.; Michel, S.; Schell, M.; Hinterwimmer, L.; Zippelius, A.; Jaschinski, F. A Highly Efficient Modality to Block the Degradation of Tryptophan for Cancer Immunotherapy: Locked Nucleic Acid-Modified Antisense Oligonucleotides to Inhibit Human Indoleamine 2,3-Dioxygenase 1/Tryptophan 2,3-Dioxygenase Expression. Cancer Immunol. Immunother. 2020, 69, 57–67. [Google Scholar] [CrossRef]
- Campesato, L.F.; Budhu, S.; Tchaicha, J.; Weng, C.-H.; Gigoux, M.; Cohen, I.J.; Redmond, D.; Mangarin, L.; Pourpe, S.; Liu, C.; et al. Blockade of the AHR Restricts a Treg-Macrophage Suppressive Axis Induced by L-Kynurenine. Nat. Commun. 2020, 11, 4011. [Google Scholar] [CrossRef]
- Liu, Y.; Liang, X.; Yin, X.; Lv, J.; Tang, K.; Ma, J.; Ji, T.; Zhang, H.; Dong, W.; Jin, X.; et al. Blockade of IDO-Kynurenine-AhR Metabolic Circuitry Abrogates IFN-γ-Induced Immunologic Dormancy of Tumor-Repopulating Cells. Nat. Commun. 2017, 8, 15207. [Google Scholar] [CrossRef]
- Amobi, A.; Qian, F.; Lugade, A.A.; Odunsi, K. Tryptophan Catabolism and Cancer Immunotherapy Targeting IDO Mediated Immune Suppression. Adv. Exp. Med. Biol. 2017, 1036, 129–144. [Google Scholar] [CrossRef]
- Brochez, L.; Chevolet, I.; Kruse, V. The Rationale of Indoleamine 2,3-Dioxygenase Inhibition for Cancer Therapy. Eur. J. Cancer 2017, 76, 167–182. [Google Scholar] [CrossRef]
- Ricciuti, B.; Leonardi, G.C.; Puccetti, P.; Fallarino, F.; Bianconi, V.; Sahebkar, A.; Baglivo, S.; Chiari, R.; Pirro, M. Targeting Indoleamine-2,3-Dioxygenase in Cancer: Scientific Rationale and Clinical Evidence. Pharmacol. Ther. 2019, 196, 105–116. [Google Scholar] [CrossRef]
- Davar, D.; Bahary, N. Modulating Tumor Immunology by Inhibiting Indoleamine 2,3-Dioxygenase (IDO): Recent Developments and First Clinical Experiences. Target. Oncol. 2018, 13, 125–140. [Google Scholar] [CrossRef]
- Le Naour, J.; Galluzzi, L.; Zitvogel, L.; Kroemer, G.; Vacchelli, E. Trial Watch: IDO Inhibitors in Cancer Therapy. Oncoimmunology 2020, 9, 1777625. [Google Scholar] [CrossRef]
- Soliman, H.H.; Jackson, E.; Neuger, T.; Dees, E.C.; Harvey, R.D.; Han, H.; Ismail-Khan, R.; Minton, S.; Vahanian, N.N.; Link, C.; et al. A First in Man Phase I Trial of the Oral Immunomodulator, Indoximod, Combined with Docetaxel in Patients with Metastatic Solid Tumors. Oncotarget 2014, 5, 8136–8146. [Google Scholar] [CrossRef]
- Hou, D.-Y.; Muller, A.J.; Sharma, M.D.; DuHadaway, J.; Banerjee, T.; Johnson, M.; Mellor, A.L.; Prendergast, G.C.; Munn, D.H. Inhibition of Indoleamine 2,3-Dioxygenase in Dendritic Cells by Stereoisomers of 1-Methyl-Tryptophan Correlates with Antitumor Responses. Cancer Res. 2007, 67, 792–801. [Google Scholar] [CrossRef]
- Kudo, Y.; Boyd, C.A. Human Placental Indoleamine 2,3-Dioxygenase: Cellular Localization and Characterization of an Enzyme Preventing Fetal Rejection. Biochim. Biophys. Acta 2000, 1500, 119–124. [Google Scholar] [CrossRef]
- Lewis, H.C.; Chinnadurai, R.; Bosinger, S.E.; Galipeau, J. The IDO Inhibitor 1-Methyl Tryptophan Activates the Aryl Hydrocarbon Receptor Response in Mesenchymal Stromal Cells. Oncotarget 2017, 8, 91914–91927. [Google Scholar] [CrossRef] [PubMed]
- Siu, L.L.; Gelmon, K.; Chu, Q.; Pachynski, R.; Alese, O.; Basciano, P.; Walker, J.; Mitra, P.; Zhu, L.; Phillips, P.; et al. Abstract CT116: BMS-986205, an Optimized Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitor, Is Well Tolerated with Potent Pharmacodynamic (PD) Activity, Alone and in Combination with Nivolumab (Nivo) in Advanced Cancers in a Phase 1/2a Trial. Cancer Res. 2017, 77, CT116. [Google Scholar] [CrossRef]
- Mautino, M.R.; Jaipuri, F.A.; Waldo, J.; Kumar, S.; Adams, J.; Allen, C.V.; Marcinowicz-Flick, A.; Munn, D.; Vahanian, N.; Link, C.J. Abstract 491: NLG919, a Novel Indoleamine-2,3-Dioxygenase (IDO)-Pathway Inhibitor Drug Candidate for Cancer Therapy. Cancer Res. 2013, 73, 491. [Google Scholar] [CrossRef]
- Ma, S.; Suchomel, J.; Yanez, E.; Yost, E.; Liang, X.; Zhu, R.; Le, H.; Siebers, N.; Joas, L.; Morley, R.; et al. Investigation of the Absolute Bioavailability and Human Mass Balance of Navoximod, a Novel IDO1 Inhibitor. Br. J. Clin. Pharmacol. 2019, 85, 1751–1760. [Google Scholar] [CrossRef]
- Krähenbühl, L.; Goldinger, S.M.; Mangana, J.; Kerl, K.; Chevolet, I.; Brochez, L.; Horak, C.; Levesque, M.; Dummer, R.; Cheng, P.F. A Longitudinal Analysis of IDO and PDL1 Expression during Immune- or Targeted Therapy in Advanced Melanoma. Neoplasia 2018, 20, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Heng, B.; Bilgin, A.A.; Lovejoy, D.B.; Tan, V.X.; Milioli, H.H.; Gluch, L.; Bustamante, S.; Sabaretnam, T.; Moscato, P.; Lim, C.K.; et al. Differential Kynurenine Pathway Metabolism in Highly Metastatic Aggressive Breast Cancer Subtypes: Beyond IDO1-Induced Immunosuppression. Breast Cancer Res. 2020, 22, 113. [Google Scholar] [CrossRef] [PubMed]
- Ci, C.; Wu, C.; Lyu, D.; Chang, X.; He, C.; Liu, W.; Chen, L.; Ding, W. Downregulation of Kynureninase Restrains Cutaneous Squamous Cell Carcinoma Proliferation and Represses the PI3K/AKT Pathway. Clin. Exp. Dermatol. 2020, 45, 194–201. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumor | Target | Mechanism of Action and Agent | Suppression of Tumor Growth | Comments | References |
|---|---|---|---|---|---|
| Colon cancer | IDO1 | Direct enzyme inhibition | In-vitro SW837 cell line culture In-vivo Balb/c mice bearing CT26 tumor | [64,96,97,98,99,100,101,102] | |
| 1-MT | 7% | ||||
| 4,6-substituted-1H-indazole derivatives | 38% | ||||
| Amidoxime | N/A | ||||
| Epacadostat | 56% | ||||
| epigallocatechin gallate, Navoximod | N/A 75% (in co-treatment with oxaliplatin) | ||||
| 4-Bromophenylhydrazinyl benzenesulfonylphenylureas | 25% | ||||
| Ehrlich ascites carcinoma | IDO1 | Direct enzyme inhibition | In-vivo ICR mice bearing Erlich tumor | [97] | |
| Ethyl pyruvate | 78% (in co-treatment with cyclophosphamide); 79% (in co-treatment with cycloplatam) | ||||
| Amidoxime | 88% (in co-treatment with cyclophosphamid); 85% (in co-treatment with cycloplatam) | ||||
| Gastrointestinal stromal tumors | IDO1 | Downregulation of oncogenic KIT tyrosine-protein kinase signaling | In-vivo C57Bl/6J mice bearing GIST-T1 tumor | [103] | |
| Imatinib | 70% | ||||
| Glioblastoma | IDO1 | Direct enzyme inhibition | In-vivo C57BL/6 mice bearing GL261 glioblastoma | [83,104,105,106,107] | |
| 1-MT | 63% (in conjunction with two-fraction radiotherapy) | ||||
| Dinaciclib | N/A | ||||
| Acyclovir | N/A | ||||
| Glioma | IDO1 | Direct enzyme inhibition | In-vitro A172 human glioma cell line culture In-vivo C57BL/6 mice bearing LN229 glioma | [107,108,109] | |
| 1-MT | 87% (in co-treatment with temozonlomide) | ||||
| Tryptamine | N/A | ||||
| N-dimethyltryptamine | 64% (in combiantion with carboxyamidotriazole and cytotoxic T lymphocytes therapy) | ||||
| HPV-associated tumors | IDO1 | Direct enzyme inhibition | In-vivo 57BL/6 mice bearing TC-1 tumor | [93] | |
| 1-MT | 60% (in combiantion with gDE7-based immunotherapy) | ||||
| Lung cancer | IDO1 | Direct enzyme inhibition | In-vitro NCI-H460 and A549 cell line cultures In-vivo C57BL6/N mice bearing LLC, LL24 tumor lines, Balb/c nude mice bearing NCI-H1299 tumor | [94,95,110,111,112,113,114,115,116] | |
| 1-MT | 47% | ||||
| Navoximod | 65% | ||||
| EOS200271 | 45% | ||||
| LW106 | 68% | ||||
| Epacadostat | 51% | ||||
| INCB023843 | 22% | ||||
| Downregualtion of enzyme expression | |||||
| Feiji Recipe | 60% | ||||
| Inhibition of the extracellular IDO1 secretion | |||||
| Stragaloside IV | N/A | ||||
| Astragaloside IV | 72% (in combination with anti-PD1 antibody) | ||||
| Thymona | Downregualtion of enzyme expression | In-vivo C57BL/6 mice bearnng EG7 tumor line | [117] | ||
| Resveratrol | 51% | ||||
| Melanoma | IDO1 | Direct enzyme inhibition | In-vitro 624.38mel cell line culture In-vivo BALB/c mice bearing B16F10 melanoma | [94,95,113,118,119,120,121,122,123] | |
| 1-MT | 52% (in co-treatment with cyclophosphamide) 48% (in co-treatment with pimozide) 46% (in monotherapy), | ||||
| Navoximod | 95% (in co-treatment with pmel-1 T cells and gp100 peptide vaccination) | ||||
| Epacadostat | 50% | ||||
| DX-03-12 | 72.2% | ||||
| LW106 | 65% | ||||
| NTRC 3883-0 | 20% | ||||
| Increased proteasomal degradation | |||||
| Fludarabine | N/A | ||||
| Myelodysplastic syndrome | IDO1 | Direct enzyme inhibition | In-vivo C57BL/6N mice bearing MDS92 tumor | [124,125] | |
| Epacadostat | 50% | ||||
| Nasolaryngeal carcinoma | IDO1 | STAT1 acetylation | In-vitro CNE2 and CNE1 cell line cultures In-vivo BALB/C (NU/NU) mice bearing C666-1 tumor | [126,127,128] | |
| Bortezomib | 23% | ||||
| Suberoylanilide hydroxamic acid | 41% | ||||
| Combination of bortezomib with suberoylanilide hydroxamic acid | 76% | ||||
| Sodium butyrate | N/A | ||||
| Ovarian cancer | IDO1 | Direct enzyme inhibition | In-vitro SKOV3 cell line, patient-derived ovarian cancer cell culture In-vivo Balb/c NU/NU mice bearing SKOV3 tumor | [46,94,95,129,130] | |
| 1-MT | 10% | ||||
| NTRC 3883-0 | N/A | ||||
| LW106 | N/A | ||||
| BMS-986242 | N/A | ||||
| Pancreatic Ductal Adenocarcinoma | IDO1 | Direct enzyme inhibition | In-vitro Human PDAC specimens | [124,131,132,133] | |
| Epacadostat | 55% | ||||
| EOS200271 | 32% | ||||
| Plasmacytoid dendritic cell leukemia | IDO1 | Direct enzyme inhibition | In-vitro PMDC05 cell line culture | [134] | |
| Toho-1 | N/A | ||||
| Renal Cancer | IDO1 | Direct enzyme inhibition | In-vivo Balb/cJ mice bearing 786-O tumor | [135] | |
| Methyl-thiohydantoin-DL-TRP | 45% (in co-treatment with IFN-α) | ||||
| Sarcomas | IDO1 | Direct enzyme inhibition | In-vivo C57BL/6N mice bearing MCA205 tumor | [136] | |
| Navoximod | 0% (did not show antitumoral activity, but has a potential impact on the NK cell functions). | ||||
| Non-Hodgkin Lymphoma | IDO2 | Direct enzyme inhibition Indoximod | 77% (in co-treatment with cyclophosphamide) | In-vivo BalB/c NU/NU mice bearing A20 B-cell lymphoma | [137] |
| Breast cancer | IDO 1 | Direct enzyme inhibition | In-vitro MDA-231and BT549 cell lines In-vivo C57BL/6 mice bearing MDA-231 tumor | [71,123,131,138,139] | |
| EOS200271 | 21% | ||||
| Downregulation of enzyme expression | |||||
| Nimesulide | 54% | ||||
| Celecoxib | 52% | ||||
| Increased proteasomal degradation | |||||
| Fludarabine | N/A | ||||
| TDO2 | Direct enzyme inhibition | ||||
| 680C91 | 85% | ||||
| Cervical cancer | IDO1 | Direct enzyme inhibition | In-vitro HeLa cell line | [140,141] | |
| LBJ-10 | N/A | ||||
| IDO1 and TDO | Direct enzyme inhibition | ||||
| β-lapachone | N/A | ||||
| Liver cancer | IDO1 | Direct enzyme inhibition | In-vitro HepG2 cell line In-vivo BALB/c mice bearing H22 tumor C3HA mice bearing H-29 tumor | [142,143,144] | |
| Ethyl pyruvate | 39% | ||||
| JAK/STAT1 pathway inhibition | |||||
| Polysaccharide from Lepista sordid | N/A | ||||
| TDO | Direct enzyme inhibition | ||||
| 3-(2-(Pyridyl)ethenyl) indole derivatives | 65% (in combination with irinotecan) | ||||
| Multiple Myeloma | KMO | Direct enzyme inhibition | In-vitro Patient-derived multiple myeloma cell culture | [145] | |
| Ro-61–8048 | N/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mor, A.; Tankiewicz-Kwedlo, A.; Pawlak, D. Kynurenines as a Novel Target for the Treatment of Malignancies. Pharmaceuticals 2021, 14, 606. https://doi.org/10.3390/ph14070606
Mor A, Tankiewicz-Kwedlo A, Pawlak D. Kynurenines as a Novel Target for the Treatment of Malignancies. Pharmaceuticals. 2021; 14(7):606. https://doi.org/10.3390/ph14070606
Chicago/Turabian StyleMor, Adrian, Anna Tankiewicz-Kwedlo, and Dariusz Pawlak. 2021. "Kynurenines as a Novel Target for the Treatment of Malignancies" Pharmaceuticals 14, no. 7: 606. https://doi.org/10.3390/ph14070606
APA StyleMor, A., Tankiewicz-Kwedlo, A., & Pawlak, D. (2021). Kynurenines as a Novel Target for the Treatment of Malignancies. Pharmaceuticals, 14(7), 606. https://doi.org/10.3390/ph14070606