
Stem Cell Models and Gene Targeting for Human Motor Neuron Diseases


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Traditional Gene Targeting Methods
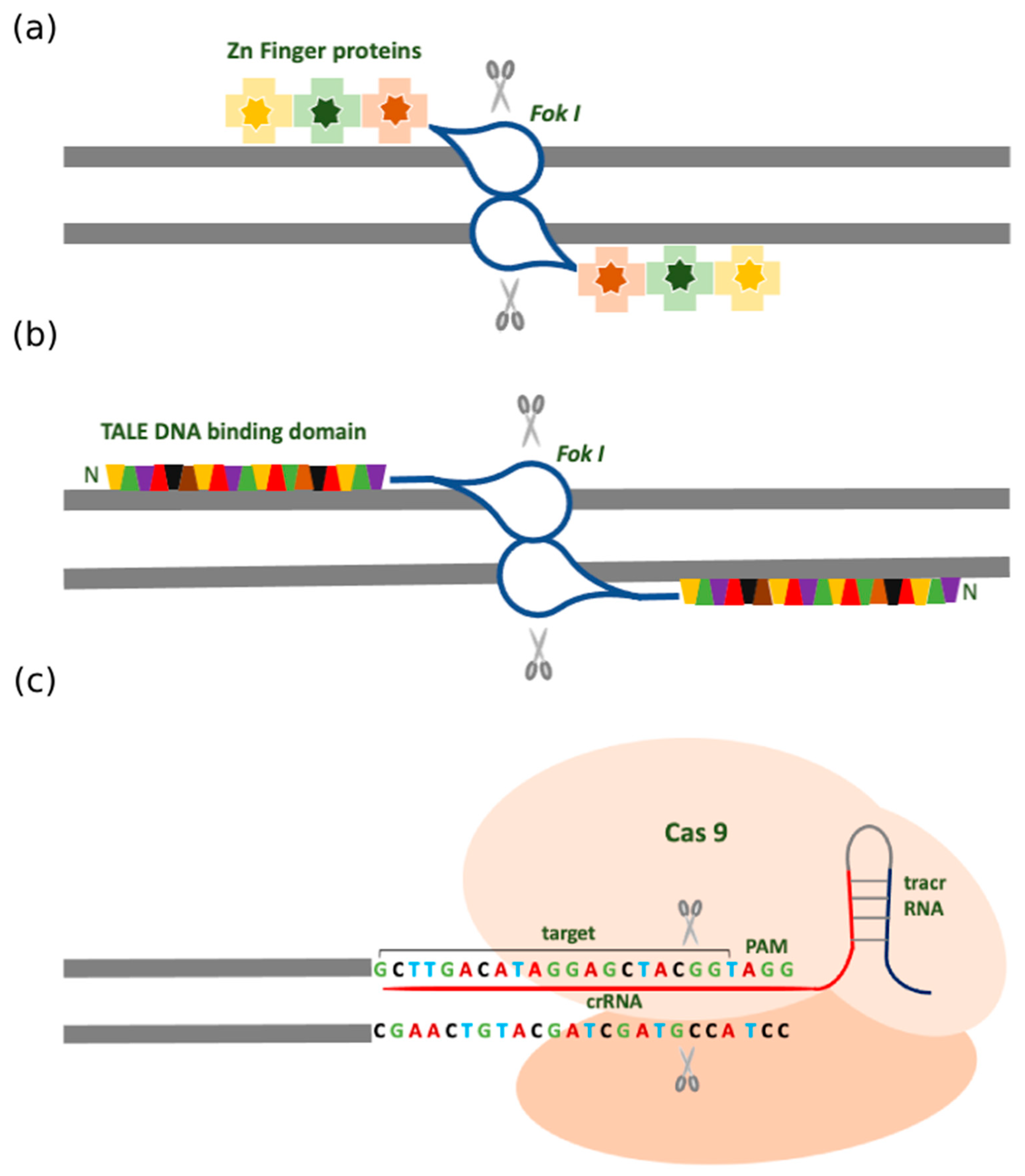
2.1. Zinc Finger Nuclease (ZFN)
2.2. Transcription Activator-Like Effector Nucleases (TALENs)
2.3. Clustered Regularly Interspersed Short Palindromic Repeats (CRISPR)
2.4. Different Forms of CRISPR-Cas Systems
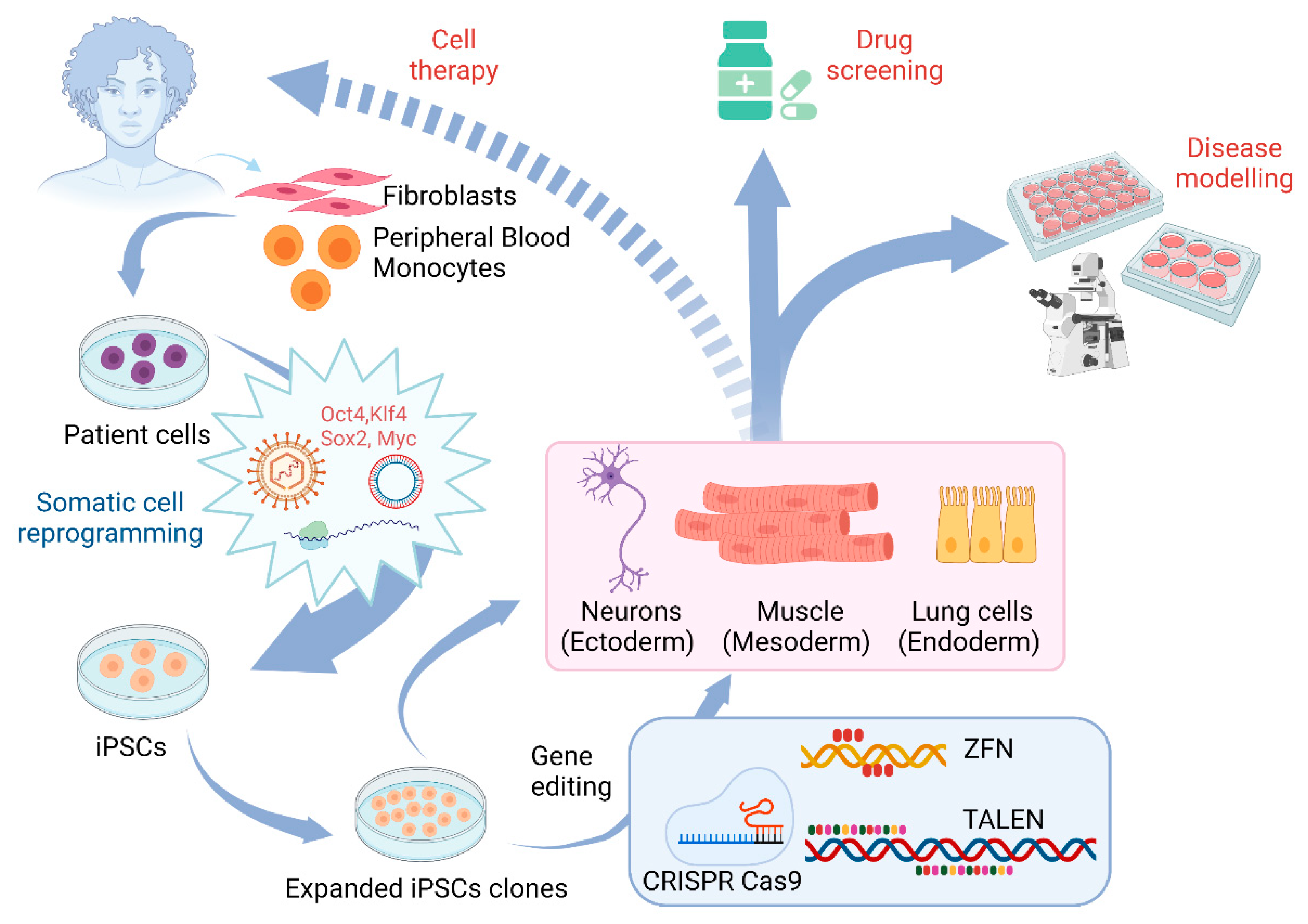
3. Application of Gene Editing in Stem Cell Models of Motor Neuron Diseases
3.1. The Use of Gene Editing for ALS
3.2. The Use of Gene Editing and Stem Cell Models for Studying SMA
3.3. Other Motor Neuron Diseases
4. Gene Therapy for Motor Neuron Diseases
5. Conclusions and Future Aspects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Amit, M.; Shariki, C.; Margulets, V.; Itskovitz-Eldor, J. Feeder Layer- and Serum-Free Culture of Human Embryonic Stem Cells1. Biol. Reprod. 2004, 70, 837–845. [Google Scholar] [CrossRef] [PubMed]
- Kolios, G.; Moodley, Y. Introduction to Stem Cells and Regenerative Medicine. Respiration 2013, 85, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Surani, M.A.; Durcova-Hills, G.; Hajkova, P.; Hayashi, K.; Tee, W.W. Germ Line, Stem Cells, and Epigenetic Reprogramming. Cold Spring Harb. Symp. Quant. Biol. 2008, 73, 9–15. [Google Scholar] [CrossRef][Green Version]
- Thomson, J.A. Embryonic Stem Cell Lines Derived from Human Blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced Pluripotent Stem Cell Lines Derived from Human Somatic Cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Hu, K.; Yu, J.; Suknuntha, K.; Tian, S.; Montgomery, K.; Choi, K.-D.; Stewart, R.; Thomson, J.A.; Slukvin, I.I. Efficient Generation of Transgene-Free Induced Pluripotent Stem Cells from Normal and Neoplastic Bone Marrow and Cord Blood Mononuclear Cells. Blood 2011, 117, e109–e119. [Google Scholar] [CrossRef]
- Schlaeger, T.M.; Daheron, L.; Brickler, T.R.; Entwisle, S.; Chan, K.; Cianci, A.; DeVine, A.; Ettenger, A.; Fitzgerald, K.; Godfrey, M.; et al. A Comparison of Non-Integrating Reprogramming Methods. Nat. Biotechnol. 2015, 33, 58–63. [Google Scholar] [CrossRef]
- Ban, H.; Nishishita, N.; Fusaki, N.; Tabata, T.; Saeki, K.; Shikamura, M.; Takada, N.; Inoue, M.; Hasegawa, M.; Kawamata, S.; et al. Efficient Generation of Transgene-Free Human Induced Pluripotent Stem Cells (IPSCs) by Temperature-Sensitive Sendai Virus Vectors. Proc. Natl. Acad. Sci. USA 2011, 108, 14234–14239. [Google Scholar] [CrossRef]
- Okita, K.; Matsumura, Y.; Sato, Y.; Okada, A.; Morizane, A.; Okamoto, S.; Hong, H.; Nakagawa, M.; Tanabe, K.; Tezuka, K.; et al. A More Efficient Method to Generate Integration-Free Human IPS Cells. Nat. Methods 2011, 8, 409–412. [Google Scholar] [CrossRef]
- Karagiannis, P.; Takahashi, K.; Saito, M.; Yoshida, Y.; Okita, K.; Watanabe, A.; Inoue, H.; Yamashita, J.K.; Todani, M.; Nakagawa, M.; et al. Induced Pluripotent Stem Cells and Their Use in Human Models of Disease and Development. Physiol. Rev. 2019, 99, 79–114. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.C.; Haggarty, S.J. Human Pluripotent Stem Cell–Derived Models and Drug Screening in CNS Precision Medicine. Ann. N. Y. Acad. Sci. 2020, 1471, 18–56. [Google Scholar] [CrossRef] [PubMed]
- Ming, G.-L.; Brüstle, O.; Muotri, A.; Studer, L.; Wernig, M.; Christian, K.M. Cellular Reprogramming: Recent Advances in Modeling Neurological Diseases. J. Neurosci. 2011, 31, 16070–16075. [Google Scholar] [CrossRef]
- Weick, J.P.; Meyer, J.S.; Ladewig, J.; Guo, W.; Liu, Y. Modeling CNS Development and Disease. Stem Cells Int. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Zhang, S.-C. Neural Subtype Specification From Human Pluripotent Stem Cells. Cell Stem Cell 2016, 19, 573–586. [Google Scholar] [CrossRef]
- Retinal Degenerative Diseases: Mechanisms and Experimental Therapy; Bowes Rickman, C., Grimm, C., Anderson, R.E., Ash, J.D., LaVail, M.M., Hollyfield, J.G., Eds.; Advances in Experimental Medicine and Biology; Springer International Publishing: Berlin/Heidelberg, Germany, 2019; Volume 1185, ISBN 978-3-030-27377-4. [Google Scholar]
- Li, X.-J.; Zhang, X.; Johnson, M.A.; Wang, Z.-B.; LaVaute, T.; Zhang, S.-C. Coordination of Sonic Hedgehog and Wnt Signaling Determines Ventral and Dorsal Telencephalic Neuron Types from Human Embryonic Stem Cells. Dev. Camb. Engl. 2009, 136, 4055–4063. [Google Scholar] [CrossRef]
- Li, X.-J.; Du, Z.-W.; Zarnowska, E.D.; Pankratz, M.; Hansen, L.O.; Pearce, R.A.; Zhang, S.-C. Specification of Motoneurons from Human Embryonic Stem Cells. Nat. Biotechnol. 2005, 23, 215–221. [Google Scholar] [CrossRef]
- Li, X.-J.; Hu, B.-Y.; Jones, S.A.; Zhang, Y.-S.; Lavaute, T.; Du, Z.-W.; Zhang, S.-C. Directed Differentiation of Ventral Spinal Progenitors and Motor Neurons from Human Embryonic Stem Cells by Small Molecules. Stem Cells Dayt. Ohio 2008, 26, 886–893. [Google Scholar] [CrossRef]
- Langer, K.B.; Ohlemacher, S.K.; Phillips, M.J.; Fligor, C.M.; Jiang, P.; Gamm, D.M.; Meyer, J.S. Retinal Ganglion Cell Diversity and Subtype Specification from Human Pluripotent Stem Cells. Stem Cell Rep. 2018, 10, 1282–1293. [Google Scholar] [CrossRef]
- Barber, K.; Studer, L.; Fattahi, F. Derivation of enteric neuron lineages from human pluripotent stem cells. Nat. Protoc. 2019, 14, 1261–1279. [Google Scholar] [CrossRef]
- Zhao, X.; Bhattacharyya, A. Human Models Are Needed for Studying Human Neurodevelopmental Disorders. Am. J. Hum. Genet. 2018, 103, 829–857. [Google Scholar] [CrossRef]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-Homologous DNA End Joining and Alternative Pathways to Double-Strand Break Repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Wright, W.D.; Shah, S.S.; Heyer, W.-D. Homologous Recombination and the Repair of DNA Double-Strand Breaks. J. Biol. Chem. 2018, 293, 10524–10535. [Google Scholar] [CrossRef]
- Capecchi, M. Altering the Genome by Homologous Recombination. Science 1989, 244, 1288–1292. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.; McLachlan, A.D.; Klug, A. Repetitive Zinc-Binding Domains in the Protein Transcription Factor IIIA from Xenopus Oocytes. EMBO J. 1985, 4, 1609–1614. [Google Scholar] [CrossRef]
- Kim, Y.G.; Cha, J.; Chandrasegaran, S. Hybrid Restriction Enzymes: Zinc Finger Fusions to Fok I Cleavage Domain. Proc. Natl. Acad. Sci. USA 1996, 93, 1156–1160. [Google Scholar] [CrossRef] [PubMed]
- Bibikova, M.; Carroll, D.; Segal, D.J.; Trautman, J.K.; Smith, J.; Kim, Y.-G.; Chandrasegaran, S. Stimulation of Homologous Recombination through Targeted Cleavage by Chimeric Nucleases. Mol. Cell. Biol. 2001, 21, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Urnov, F.D.; Miller, J.C.; Lee, Y.-L.; Beausejour, C.M.; Rock, J.M.; Augustus, S.; Jamieson, A.C.; Porteus, M.H.; Gregory, P.D.; Holmes, M.C. Highly Efficient Endogenous Human Gene Correction Using Designed Zinc-Finger Nucleases. Nature 2005, 435, 646–651. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.C.; Holmes, M.C.; Wang, J.; Guschin, D.Y.; Lee, Y.-L.; Rupniewski, I.; Beausejour, C.M.; Waite, A.J.; Wang, N.S.; Kim, K.A.; et al. An Improved Zinc-Finger Nuclease Architecture for Highly Specific Genome Editing. Nat. Biotechnol. 2007, 25, 778–785. [Google Scholar] [CrossRef] [PubMed]
- Perez, E.E.; Wang, J.; Miller, J.C.; Jouvenot, Y.; Kim, K.A.; Liu, O.; Wang, N.; Lee, G.; Bartsevich, V.V.; Lee, Y.-L.; et al. Establishment of HIV-1 Resistance in CD4+ T Cells by Genome Editing Using Zinc-Finger Nucleases. Nat. Biotechnol. 2008, 26, 808–816. [Google Scholar] [CrossRef]
- Soldner, F.; Laganiere, J.; Cheng, A.W.; Hockemeyer, D.; Gao, Q.; Alagappan, R.; Khurana, V.; Golbe, L.I.; Myers, R.H.; Lindquist, S.; et al. Generation of Isogenic Pluripotent Stem Cells Differing Exclusively at Two Early Onset Parkinson Point Mutations. Cell 2011, 146, 318–331. [Google Scholar] [CrossRef]
- Cornu, T.I.; Cathomen, T. Quantification of Zinc Finger Nuclease-Associated Toxicity. In Engineered Zinc Finger Proteins; Mackay, J.P., Segal, D.J., Eds.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2010; Volume 649, pp. 237–245. ISBN 978-1-60761-752-5. [Google Scholar]
- Elliott, B.; Jasin, M. Human Genome and Diseases:¶Double-Strand Breaks and Translocations in Cancer. Cell. Mol. Life Sci. CMLS 2002, 59, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Richardson, C.; Jasin, M. Frequent Chromosomal Translocations Induced by DNA Double-Strand Breaks. Nature 2000, 405, 697–700. [Google Scholar] [CrossRef]
- DeKelver, R.C.; Choi, V.M.; Moehle, E.A.; Paschon, D.E.; Hockemeyer, D.; Meijsing, S.H.; Sancak, Y.; Cui, X.; Steine, E.J.; Miller, J.C.; et al. Functional Genomics, Proteomics, and Regulatory DNA Analysis in Isogenic Settings Using Zinc Finger Nuclease-Driven Transgenesis into a Safe Harbor Locus in the Human Genome. Genome Res. 2010, 20, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Hockemeyer, D.; Soldner, F.; Beard, C.; Gao, Q.; Mitalipova, M.; DeKelver, R.C.; Katibah, G.E.; Amora, R.; Boydston, E.A.; Zeitler, B.; et al. Efficient Targeting of Expressed and Silent Genes in Human ESCs and IPSCs Using Zinc-Finger Nucleases. Nat. Biotechnol. 2009, 27, 851–857. [Google Scholar] [CrossRef]
- Zou, J.; Maeder, M.L.; Mali, P.; Pruett-Miller, S.M.; Thibodeau-Beganny, S.; Chou, B.-K.; Chen, G.; Ye, Z.; Park, I.-H.; Daley, G.Q.; et al. Gene Targeting of a Disease-Related Gene in Human Induced Pluripotent Stem and Embryonic Stem Cells. Cell Stem Cell 2009, 5, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Didigu, C.A.; Wilen, C.B.; Wang, J.; Duong, J.; Secreto, A.J.; Danet-Desnoyers, G.A.; Riley, J.L.; Gregory, P.D.; June, C.H.; Holmes, M.C.; et al. Simultaneous Zinc-Finger Nuclease Editing of the HIV Coreceptors Ccr5 and Cxcr4 Protects CD4+ T Cells from HIV-1 Infection. Blood 2014, 123, 61–69. [Google Scholar] [CrossRef]
- Gupta, R.K.; Abdul-Jawad, S.; McCoy, L.E.; Mok, H.P.; Peppa, D.; Salgado, M.; Martinez-Picado, J.; Nijhuis, M.; Wensing, A.M.J.; Lee, H.; et al. HIV-1 Remission Following CCR5Δ32/Δ32 Haematopoietic Stem-Cell Transplantation. Nature 2019, 568, 244–248. [Google Scholar] [CrossRef]
- Schornack, S.; Meyer, A.; Römer, P.; Jordan, T.; Lahaye, T. Gene-for-Gene-Mediated Recognition of Nuclear-Targeted AvrBs3-like Bacterial Effector Proteins. J. Plant Physiol. 2006, 163, 256–272. [Google Scholar] [CrossRef]
- Boch, J.; Scholze, H.; Schornack, S.; Landgraf, A.; Hahn, S.; Kay, S.; Lahaye, T.; Nickstadt, A.; Bonas, U. Breaking the Code of DNA Binding Specificity of TAL-Type III Effectors. Science 2009, 326, 1509–1512. [Google Scholar] [CrossRef]
- Moscou, M.J.; Bogdanove, A.J. A Simple Cipher Governs DNA Recognition by TAL Effectors. Science 2009, 326, 1501. [Google Scholar] [CrossRef]
- Lamb, B.M.; Mercer, A.C.; Barbas, C.F. Directed Evolution of the TALE N-Terminal Domain for Recognition of All 5′ Bases. Nucleic Acids Res. 2013, 41, 9779–9785. [Google Scholar] [CrossRef] [PubMed]
- Christian, M.; Cermak, T.; Doyle, E.L.; Schmidt, C.; Zhang, F.; Hummel, A.; Bogdanove, A.J.; Voytas, D.F. Targeting DNA Double-Strand Breaks with TAL Effector Nucleases. Genetics 2010, 186, 757–761. [Google Scholar] [CrossRef]
- Miller, J.C.; Tan, S.; Qiao, G.; Barlow, K.A.; Wang, J.; Xia, D.F.; Meng, X.; Paschon, D.E.; Leung, E.; Hinkley, S.J.; et al. A TALE Nuclease Architecture for Efficient Genome Editing. Nat. Biotechnol. 2011, 29, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Ren, X.; Xu, X.; Zhang, X.; Hui, Y.; Liu, Z.; Shi, L.; Fang, Y.; Ma, L.; Liu, Y.; et al. Genetic Engineering of Human Embryonic Stem Cells for Precise Cell Fate Tracing during Human Lineage Development. Stem Cell Rep. 2018, 11, 1257–1271. [Google Scholar] [CrossRef] [PubMed]
- Shinkuma, S.; Guo, Z.; Christiano, A.M. Site-Specific Genome Editing for Correction of Induced Pluripotent Stem Cells Derived from Dominant Dystrophic Epidermolysis Bullosa. Proc. Natl. Acad. Sci. USA 2016, 113, 5676–5681. [Google Scholar] [CrossRef] [PubMed]
- Bultmann, S.; Morbitzer, R.; Schmidt, C.S.; Thanisch, K.; Spada, F.; Elsaesser, J.; Lahaye, T.; Leonhardt, H. Targeted Transcriptional Activation of Silent Oct4 Pluripotency Gene by Combining Designer TALEs and Inhibition of Epigenetic Modifiers. Nucleic Acids Res. 2012, 40, 5368–5377. [Google Scholar] [CrossRef] [PubMed]
- Qasim, W.; Zhan, H.; Samarasinghe, S.; Adams, S.; Amrolia, P.; Stafford, S.; Butler, K.; Rivat, C.; Wright, G.; Somana, K.; et al. Molecular Remission of Infant B-ALL after Infusion of Universal TALEN Gene-Edited CAR T Cells. Sci. Transl. Med. 2017, 9, eaaj2013. [Google Scholar] [CrossRef] [PubMed]
- Hockemeyer, D.; Wang, H.; Kiani, S.; Lai, C.S.; Gao, Q.; Cassady, J.P.; Cost, G.J.; Zhang, L.; Santiago, Y.; Miller, J.C.; et al. Genetic Engineering of Human ES and IPS Cells Using TALE Nucleases. Nat. Biotechnol. 2011, 29, 731–734. [Google Scholar] [CrossRef]
- Ishino, Y.; Shinagawa, H.; Makino, K.; Amemura, M.; Nakata, A. Nucleotide Sequence of the Iap Gene, Responsible for Alkaline Phosphatase Isozyme Conversion in Escherichia Coli, and Identification of the Gene Product. J. Bacteriol. 1987, 169, 5429–5433. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Makarova, K.S.; Wolf, Y.I.; Alkhnbashi, O.S.; Costa, F.; Shah, S.A.; Saunders, S.J.; Barrangou, R.; Brouns, S.J.J.; Charpentier, E.; Haft, D.H.; et al. An Updated Evolutionary Classification of CRISPR–Cas Systems. Nat. Rev. Microbiol. 2015, 13, 722–736. [Google Scholar] [CrossRef]
- Makarova, K.S.; Haft, D.H.; Barrangou, R.; Brouns, S.J.J.; Charpentier, E.; Horvath, P.; Moineau, S.; Mojica, F.J.M.; Wolf, Y.I.; Yakunin, A.F.; et al. Evolution and Classification of the CRISPR-Cas Systems. Nat. Rev. Microbiol. 2011, 9, 467–477. [Google Scholar] [CrossRef]
- Deng, D.; Yan, C.; Pan, X.; Mahfouz, M.; Wang, J.; Zhu, J.-K.; Shi, Y.; Yan, N. Structural Basis for Sequence-Specific Recognition of DNA by TAL Effectors. Science 2012, 335, 720–723. [Google Scholar] [CrossRef]
- Sternberg, S.H.; Redding, S.; Jinek, M.; Greene, E.C.; Doudna, J.A. DNA Interrogation by the CRISPR RNA-Guided Endonuclease Cas9. Nature 2014, 507, 62–67. [Google Scholar] [CrossRef]
- Cox, D.B.T.; Platt, R.J.; Zhang, F. Therapeutic Genome Editing: Prospects and Challenges. Nat. Med. 2015, 21, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Biagioni, A.; Laurenzana, A.; Margheri, F.; Chillà, A.; Fibbi, G.; so, M. Delivery Systems of CRISPR/Cas9-Based Cancer Gene Therapy. J. Biol. Eng. 2018, 12. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-Guided Human Genome Engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef]
- Doudna, J.A.; Charpentier, E. The New Frontier of Genome Engineering with CRISPR-Cas9. Science 2014, 346. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhang, L.; Liu, H.; Cheng, K. Delivery Strategies of the CRISPR-Cas9 Gene-Editing System for Therapeutic Applications. J. Control. Release Off. J. Control. Release Soc. 2017, 266, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Lin, C.-Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double Nicking by RNA-Guided CRISPR Cas9 for Enhanced Genome Editing Specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.Q.; Wyvekens, N.; Khayter, C.; Foden, J.A.; Thapar, V.; Reyon, D.; Goodwin, M.J.; Aryee, M.J.; Joung, J.K. Dimeric CRISPR RNA-Guided FokI Nucleases for Highly Specific Genome Editing. Nat. Biotechnol. 2014, 32, 569–576. [Google Scholar] [CrossRef]
- Safari, F.; Zare, K.; Negahdaripour, M.; Barekati-Mowahed, M.; Ghasemi, Y. CRISPR Cpf1 Proteins: Structure, Function and Implications for Genome Editing. Cell Biosci. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Chen, X.; Jin, Y.; Ge, W.; Wang, W.; Kong, L.; Ji, J.; Guo, X.; Huang, J.; Feng, X.-H.; et al. Small Molecules Promote CRISPR-Cpf1-Mediated Genome Editing in Human Pluripotent Stem Cells. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.; Joung, J.; van der Oost, J.; Regev, A.; et al. Cpf1 Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR-Cas System. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef]
- Renton, A.E.; Chiò, A.; Traynor, B.J. State of Play in Amyotrophic Lateral Sclerosis Genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar] [CrossRef]
- Taylor, J.P.; Brown, R.H.; Cleveland, D.W. Decoding ALS: From Genes to Mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef]
- Vucic, S.; Lin, C.S.-Y.; Cheah, B.C.; Murray, J.; Menon, P.; Krishnan, A.V.; Kiernan, M.C. Riluzole Exerts Central and Peripheral Modulating Effects in Amyotrophic Lateral Sclerosis. Brain 2013, 136, 1361–1370. [Google Scholar] [CrossRef]
- Nagai, M.; Re, D.B.; Nagata, T.; Chalazonitis, A.; Jessell, T.M.; Wichterle, H.; Przedborski, S. Astrocytes Expressing ALS-Linked Mutated SOD1 Release Factors Selectively Toxic to Motor Neurons. Nat. Neurosci. 2007, 10, 615–622. [Google Scholar] [CrossRef]
- Ng Kee Kwong, K.C.; Harbham, P.K.; Selvaraj, B.T.; Gregory, J.M.; Pal, S.; Hardingham, G.E.; Chandran, S.; Mehta, A.R. 40 Years of CSF Toxicity Studies in ALS: What Have We Learnt About ALS Pathophysiology? Front. Mol. Neurosci. 2021, 14. [Google Scholar] [CrossRef]
- Evans, C.S.; Holzbaur, E.L.F. Autophagy and Mitophagy in ALS. Neurobiol. Dis. 2019, 122, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Petrov, D.; Mansfield, C.; Moussy, A.; Hermine, O. ALS Clinical Trials Review: 20 Years of Failure. Are We Any Closer to Registering a New Treatment? Front. Aging Neurosci. 2017, 9. [Google Scholar] [CrossRef]
- Dimos, J.T.; Rodolfa, K.T.; Niakan, K.K.; Weisenthal, L.M.; Mitsumoto, H.; Chung, W.; Croft, G.F.; Saphier, G.; Leibel, R.; Goland, R.; et al. Induced Pluripotent Stem Cells Generated from Patients with ALS Can Be Differentiated into Motor Neurons. Science 2008, 321, 1218–1221. [Google Scholar] [CrossRef] [PubMed]
- Hawrot, J.; Imhof, S.; Wainger, B.J. Modeling Cell-Autonomous Motor Neuron Phenotypes in ALS Using IPSCs. Neurobiol. Dis. 2020, 134, 104680. [Google Scholar] [CrossRef] [PubMed]
- Fujimori, K.; Ishikawa, M.; Otomo, A.; Atsuta, N.; Nakamura, R.; Akiyama, T.; Hadano, S.; Aoki, M.; Saya, H.; Sobue, G.; et al. Modeling Sporadic ALS in IPSC-Derived Motor Neurons Identifies a Potential Therapeutic Agent. Nat. Med. 2018, 24, 1579–1589. [Google Scholar] [CrossRef]
- Chang, C.-Y.; Ting, H.-C.; Liu, C.-A.; Su, H.-L.; Chiou, T.-W.; Lin, S.-Z.; Harn, H.-J.; Ho, T.-J. Induced Pluripotent Stem Cell (IPSC)-Based Neurodegenerative Disease Models for Phenotype Recapitulation and Drug Screening. Molecules 2020, 25, 2000. [Google Scholar] [CrossRef]
- Sances, S.; Bruijn, L.; Chandran, S.; Eggan, K.; Ho, R.; Klim, J.; Livesey, M.; Lowry, E.; Macklis, J.; Rushton, D.; et al. Modeling ALS Using Motor Neurons Derived from Human Induced Pluripotent Stem Cells. Nat. Neurosci. 2016, 19, 542–553. [Google Scholar] [CrossRef]
- Burkhardt, M.F.; Martinez, F.J.; Wright, S.; Ramos, C.; Volfson, D.; Mason, M.; Garnes, J.; Dang, V.; Lievers, J.; Shoukat-Mumtaz, U.; et al. A Cellular Model for Sporadic ALS Using Patient-Derived Induced Pluripotent Stem Cells. Mol. Cell. Neurosci. 2013, 56, 355–364. [Google Scholar] [CrossRef]
- Wang, L.; Yi, F.; Fu, L.; Yang, J.; Wang, S.; Wang, Z.; Suzuki, K.; Sun, L.; Xu, X.; Yu, Y.; et al. CRISPR/Cas9-Mediated Targeted Gene Correction in Amyotrophic Lateral Sclerosis Patient IPSCs. Protein Cell 2017, 8, 365–378. [Google Scholar] [CrossRef]
- Bhinge, A.; Namboori, S.C.; Zhang, X.; VanDongen, A.M.J.; Stanton, L.W. Genetic Correction of SOD1 Mutant IPSCs Reveals ERK and JNK Activated AP1 as a Driver of Neurodegeneration in Amyotrophic Lateral Sclerosis. Stem Cell Rep. 2017, 8, 856–869. [Google Scholar] [CrossRef]
- Chen, H.; Qian, K.; Du, Z.; Cao, J.; Petersen, A.; Liu, H.; Blackbourn, L.W.; Huang, C.-L.; Errigo, A.; Yin, Y.; et al. Modeling ALS with IPSCs Reveals That Mutant SOD1 Misregulates Neurofilament Balance in Motor Neurons. Cell Stem Cell 2014, 14, 796–809. [Google Scholar] [CrossRef] [PubMed]
- Yun, Y.; Hong, S.-A.; Kim, K.-K.; Baek, D.; Lee, D.; Londhe, A.M.; Lee, M.; Yu, J.; McEachin, Z.T.; Bassell, G.J.; et al. CRISPR-Mediated Gene Correction Links the ATP7A M1311V Mutations with Amyotrophic Lateral Sclerosis Pathogenesis in One Individual. Commun. Biol. 2020, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.W.; Ryu, J.; Jeong, Y.E.; Kim, J.; Martin, L.J. Human Motor Neurons With SOD1-G93A Mutation Generated From CRISPR/Cas9 Gene-Edited IPSCs Develop Pathological Features of Amyotrophic Lateral Sclerosis. Front. Cell. Neurosci. 2020, 14. [Google Scholar] [CrossRef] [PubMed]
- Imamura, K.; Izumi, Y.; Watanabe, A.; Tsukita, K.; Woltjen, K.; Yamamoto, T.; Hotta, A.; Kondo, T.; Kitaoka, S.; Ohta, A.; et al. The Src/c-Abl Pathway Is a Potential Therapeutic Target in Amyotrophic Lateral Sclerosis. Sci. Transl. Med. 2017, 9, eaaf3962. [Google Scholar] [CrossRef] [PubMed]
- Kramer, N.J.; Haney, M.S.; Morgens, D.W.; Jovičić, A.; Couthouis, J.; Li, A.; Ousey, J.; Ma, R.; Bieri, G.; Tsui, C.K.; et al. CRISPR–Cas9 Screens in Human Cells and Primary Neurons Identify Modifiers of C9ORF72 Dipeptide-Repeat-Protein Toxicity. Nat. Genet. 2018, 50, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Egawa, N.; Kitaoka, S.; Tsukita, K.; Naitoh, M.; Takahashi, K.; Yamamoto, T.; Adachi, F.; Kondo, T.; Okita, K.; Asaka, I.; et al. Drug Screening for ALS Using Patient-Specific Induced Pluripotent Stem Cells. Sci. Transl. Med. 2012, 4, 145ra104. [Google Scholar] [CrossRef]
- Gaj, T.; Ojala, D.S.; Ekman, F.K.; Byrne, L.C.; Limsirichai, P.; Schaffer, D.V. In Vivo Genome Editing Improves Motor Function and Extends Survival in a Mouse Model of ALS. Sci. Adv. 2017, 3. [Google Scholar] [CrossRef]
- Lim, C.K.W.; Gapinske, M.; Brooks, A.K.; Woods, W.S.; Powell, J.E.; Zeballos, C.M.A.; Winter, J.; Perez-Pinera, P.; Gaj, T. Treatment of a Mouse Model of ALS by In Vivo Base Editing. Mol. Ther. 2020, 28, 1177–1189. [Google Scholar] [CrossRef]
- Duan, W.; Guo, M.; Yi, L.; Liu, Y.; Li, Z.; Ma, Y.; Zhang, G.; Liu, Y.; Bu, H.; Song, X.; et al. The Deletion of Mutant SOD1 via CRISPR/Cas9/SgRNA Prolongs Survival in an Amyotrophic Lateral Sclerosis Mouse Model. Gene Ther. 2020, 27, 157–169. [Google Scholar] [CrossRef]
- Crawford, T.O.; Pardo, C.A. The Neurobiology of Childhood Spinal Muscular Atrophy. Neurobiol. Dis. 1996, 3, 97–110. [Google Scholar] [CrossRef]
- Bergin, A.; Kim, G.; Price, D.L.; Sisodia, S.S.; Lee, M.K.; Rabin, B.A. Identification and Characterization of a Mouse Homologue of the Spinal Muscular Atrophy-Determining Gene, Survival Motor Neuron. Gene 1997, 204, 47–53. [Google Scholar] [CrossRef]
- Lorson, C.L.; Hahnen, E.; Androphy, E.J.; Wirth, B. A Single Nucleotide in the SMN Gene Regulates Splicing and Is Responsible for Spinal Muscular Atrophy. Proc. Natl. Acad. Sci. USA 1999, 96, 6307–6311. [Google Scholar] [CrossRef] [PubMed]
- Monani, U.R. The Human Centromeric Survival Motor Neuron Gene (SMN2) Rescues Embryonic Lethality in Smn-/- Mice and Results in a Mouse with Spinal Muscular Atrophy. Hum. Mol. Genet. 2000, 9, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Ebert, A.D.; Yu, J.; Rose, F.F.; Mattis, V.B.; Lorson, C.L.; Thomson, J.A.; Svendsen, C.N. Induced Pluripotent Stem Cells from a Spinal Muscular Atrophy Patient. Nature 2009, 457, 277–280. [Google Scholar] [CrossRef]
- Hester, M.E.; Murtha, M.J.; Song, S.; Rao, M.; Miranda, C.J.; Meyer, K.; Tian, J.; Boulting, G.; Schaffer, D.V.; Zhu, M.X.; et al. Rapid and Efficient Generation of Functional Motor Neurons From Human Pluripotent Stem Cells Using Gene Delivered Transcription Factor Codes. Mol. Ther. 2011, 19, 1905–1912. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.; Zheng, W.; Tsark, W.; Bates, S.; Huang, H.; Lin, R.-J.; Yee, J.-K. Brief Report: Phenotypic Rescue of Induced Pluripotent Stem Cell-Derived Motoneurons of a Spinal Muscular Atrophy Patient. Stem Cells 2011, 29, 2090–2093. [Google Scholar] [CrossRef]
- Corti, S.; Nizzardo, M.; Simone, C.; Falcone, M.; Nardini, M.; Ronchi, D.; Donadoni, C.; Salani, S.; Riboldi, G.; Magri, F.; et al. Genetic Correction of Human Induced Pluripotent Stem Cells from Patients with Spinal Muscular Atrophy. Sci. Transl. Med. 2012, 4, 165ra162. [Google Scholar] [CrossRef]
- McGivern, J.V.; Patitucci, T.N.; Nord, J.A.; Barabas, M.-E.A.; Stucky, C.L.; Ebert, A.D. Spinal Muscular Atrophy Astrocytes Exhibit Abnormal Calcium Regulation and Reduced Growth Factor Production. Glia 2013, 61, 1418–1428. [Google Scholar] [CrossRef] [PubMed]
- Frattini, E.; Ruggieri, M.; Salani, S.; Faravelli, I.; Zanetta, C.; Nizzardo, M.; Simone, C.; Magri, F.; Corti, S. Pluripotent Stem Cell-Based Models of Spinal Muscular Atrophy. Mol. Cell. Neurosci. 2015, 64, 44–50. [Google Scholar] [CrossRef]
- Xu, C.-C.; Denton, K.R.; Wang, Z.-B.; Zhang, X.; Li, X.-J. Abnormal Mitochondrial Transport and Morphology as Early Pathological Changes in Human Models of Spinal Muscular Atrophy. Dis. Model. Mech. 2016, 9, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Li, J.-J.; Qian, W.-J.; Zhang, Q.-J.; Wang, Z.-F.; Lu, Y.-Q.; Dong, E.-L.; He, J.; Wang, N.; Ma, L.-X.; et al. Modeling the Differential Phenotypes of Spinal Muscular Atrophy with High-Yield Generation of Motor Neurons from Human Induced Pluripotent Stem Cells. Oncotarget 2017, 8, 42030–42042. [Google Scholar] [CrossRef]
- Yoshida, M.; Kitaoka, S.; Egawa, N.; Yamane, M.; Ikeda, R.; Tsukita, K.; Amano, N.; Watanabe, A.; Morimoto, M.; Takahashi, J.; et al. Modeling the Early Phenotype at the Neuromuscular Junction of Spinal Muscular Atrophy Using Patient-Derived IPSCs. Stem Cell Rep. 2015, 4, 561–568. [Google Scholar] [CrossRef]
- Lin, C.-Y.; Yoshida, M.; Li, L.-T.; Ikenaka, A.; Oshima, S.; Nakagawa, K.; Sakurai, H.; Matsui, E.; Nakahata, T.; Saito, M.K. IPSC-Derived Functional Human Neuromuscular Junctions Model the Pathophysiology of Neuromuscular Diseases. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-B.; Zhang, X.; Li, X.-J. Recapitulation of Spinal Motor Neuron-Specific Disease Phenotypes in a Human Cell Model of Spinal Muscular Atrophy. Cell Res. 2013, 23, 378–393. [Google Scholar] [CrossRef]
- Sison, S.L.; Patitucci, T.N.; Seminary, E.R.; Villalon, E.; Lorson, C.L.; Ebert, A.D. Astrocyte-Produced MiR-146a as a Mediator of Motor Neuron Loss in Spinal Muscular Atrophy. Hum. Mol. Genet. 2017, 26, 3409–3420. [Google Scholar] [CrossRef]
- Krencik, R.; Weick, J.P.; Liu, Y.; Zhang, Z.-J.; Zhang, S.-C. Specification of Transplantable Astroglial Subtypes from Human Pluripotent Stem Cells. Nat. Biotechnol. 2011, 29, 528–534. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Diaz, A.; Efe, G.; Kabra, K.; Patel, A.; Lowry, E.R.; Shneider, N.A.; Corneo, B.; Wichterle, H. Standardized Reporter Systems for Purification and Imaging of Human Pluripotent Stem Cell-Derived Motor Neurons and Other Cholinergic Cells. Neuroscience 2020, 450, 48–56. [Google Scholar] [CrossRef]
- Pagliarini, V.; Guerra, M.; Rosa, V.D.; Compagnucci, C.; Sette, C. Combined Treatment with the Histone Deacetylase Inhibitor LBH589 and a Splice-Switch Antisense Oligonucleotide Enhances SMN2 Splicing and SMN Expression in Spinal Muscular Atrophy Cells. J. Neurochem. 2020, 153, e14935. [Google Scholar] [CrossRef]
- Naryshkin, N.A.; Weetall, M.; Dakka, A.; Narasimhan, J.; Zhao, X.; Feng, Z.; Ling, K.K.Y.; Karp, G.M.; Qi, H.; Woll, M.G.; et al. SMN2 Splicing Modifiers Improve Motor Function and Longevity in Mice with Spinal Muscular Atrophy. Science 2014, 345, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Son, Y.S.; Choi, K.; Lee, H.; Kwon, O.; Jung, K.B.; Cho, S.; Baek, J.; Son, B.; Kang, S.-M.; Kang, M.; et al. A SMN2 Splicing Modifier Rescues the Disease Phenotypes in an In Vitro Human Spinal Muscular Atrophy Model. Stem Cells Dev. 2019, 28, 438–453. [Google Scholar] [CrossRef]
- Li, J.-J.; Lin, X.; Tang, C.; Lu, Y.-Q.; Hu, X.; Zuo, E.; Li, H.; Ying, W.; Sun, Y.; Lai, L.-L.; et al. Disruption of Splicing-Regulatory Elements Using CRISPR/Cas9 to Rescue Spinal Muscular Atrophy in Human IPSCs and Mice. Natl. Sci. Rev. 2020, 7, 92–101. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, C.; Ma, L.; Mou, Y.; Zhang, B.; Zhou, S.; Tian, Y.; Trinh, J.; Zhang, X.; Li, X.-J. Drug Screening with Human SMN2 Reporter Identifies SMN Protein Stabilizers to Correct SMA Pathology. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef]
- Denton, K.R.; Lei, L.; Grenier, J.; Rodionov, V.; Blackstone, C.; Li, X.-J. Loss of Spastin Function Results in Disease-Specific Axonal Defects in Human Pluripotent Stem Cell-Based Models of Hereditary Spastic Paraplegia: Modeling Axonal Defects of HSP in Human Neurons. Stem Cells 2014, 32, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Havlicek, S.; Kohl, Z.; Mishra, H.K.; Prots, I.; Eberhardt, E.; Denguir, N.; Wend, H.; Plötz, S.; Boyer, L.; Marchetto, M.C.N.; et al. Gene Dosage-Dependent Rescue of HSP Neurite Defects in SPG4 Patients’ Neurons. Hum. Mol. Genet. 2014, 23, 2527–2541. [Google Scholar] [CrossRef]
- Zhu, P.-P.; Denton, K.R.; Pierson, T.M.; Li, X.-J.; Blackstone, C. Pharmacologic Rescue of Axon Growth Defects in a Human IPSC Model of Hereditary Spastic Paraplegia SPG3A. Hum. Mol. Genet. 2014, 23, 5638–5648. [Google Scholar] [CrossRef]
- Pérez-Brangulí, F.; Mishra, H.K.; Prots, I.; Havlicek, S.; Kohl, Z.; Saul, D.; Rummel, C.; Dorca-Arevalo, J.; Regensburger, M.; Graef, D.; et al. Dysfunction of Spatacsin Leads to Axonal Pathology in SPG11-Linked Hereditary Spastic Paraplegia. Hum. Mol. Genet. 2014, 23, 4859–4874. [Google Scholar] [CrossRef]
- Pozner, T.; Regensburger, M.; Engelhorn, T.; Winkler, J.; Winner, B. Janus-Faced Spatacsin (SPG11): Involvement in Neurodevelopment and Multisystem Neurodegeneration. Brain 2020, 143, 2369–2379. [Google Scholar] [CrossRef]
- Denton, K.; Mou, Y.; Xu, C.-C.; Shah, D.; Chang, J.; Blackstone, C.; Li, X.-J. Impaired Mitochondrial Dynamics Underlie Axonal Defects in Hereditary Spastic Paraplegias. Hum. Mol. Genet. 2018, 27, 2517–2530. [Google Scholar] [CrossRef] [PubMed]
- Mou, Y.; Dong, Y.; Chen, Z.; Denton, K.R.; Duff, M.O.; Blackstone, C.; Zhang, S.-C.; Li, X.-J. Impaired Lipid Metabolism in Astrocytes Underlies Degeneration of Cortical Projection Neurons in Hereditary Spastic Paraplegia. Acta Neuropathol. Commun. 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Mishra, H.K.; Prots, I.; Havlicek, S.; Kohl, Z.; Perez-Branguli, F.; Boerstler, T.; Anneser, L.; Minakaki, G.; Wend, H.; Hampl, M.; et al. GSK3ß-dependent Dysregulation of Neurodevelopment in SPG11-patient Induced Pluripotent Stem Cell Model. Ann. Neurol. 2016, 79, 826–840. [Google Scholar] [CrossRef]
- Mou, Y.; Li, X.-J. Rescue Axonal Defects by Targeting Mitochondrial Dynamics in Hereditary Spastic Paraplegias. Neural Regen. Res. 2019, 14, 574–577. [Google Scholar] [CrossRef] [PubMed]
- Durymanov, M.; Reineke, J. Non-Viral Delivery of Nucleic Acids: Insight Into Mechanisms of Overcoming Intracellular Barriers. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.F.; Swayze, E.E. RNA Targeting Therapeutics: Molecular Mechanisms of Antisense Oligonucleotides as a Therapeutic Platform. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 259–293. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A.; Miller, T.M.; Yamanaka, K.; Monia, B.P.; Condon, T.P.; Hung, G.; Lobsiger, C.S.; Ward, C.M.; McAlonis-Downes, M.; Wei, H.; et al. Antisense Oligonucleotide Therapy for Neurodegenerative Disease. J. Clin. Investig. 2006, 116, 2290–2296. [Google Scholar] [CrossRef]
- Jiang, J.; Zhu, Q.; Gendron, T.F.; Saberi, S.; McAlonis-Downes, M.; Seelman, A.; Stauffer, J.E.; Jafar-nejad, P.; Drenner, K.; Schulte, D.; et al. Gain of Toxicity from ALS/FTD-Linked Repeat Expansions in C9ORF72 Is Alleviated by Antisense Oligonucleotides Targeting GGGGCC-Containing RNAs. Neuron 2016, 90, 535–550. [Google Scholar] [CrossRef]
- Donnelly, C.J.; Zhang, P.-W.; Pham, J.T.; Heusler, A.R.; Mistry, N.A.; Vidensky, S.; Daley, E.L.; Poth, E.M.; Hoover, B.; Fines, D.M.; et al. RNA Toxicity from the ALS/FTD C9ORF72 Expansion Is Mitigated by Antisense Intervention. Neuron 2013, 80, 415–428. [Google Scholar] [CrossRef]
- Rüger, J.; Ioannou, S.; Castanotto, D.; Stein, C.A. Oligonucleotides to the (Gene) Rescue: FDA Approvals 2017–2019. Trends Pharmacol. Sci. 2020, 41, 27–41. [Google Scholar] [CrossRef]
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1723–1732. [Google Scholar] [CrossRef]
- Singh, N.N.; Howell, M.D.; Androphy, E.J.; Singh, R.N. How the Discovery of ISS-N1 Led to the First Medical Therapy for Spinal Muscular Atrophy. Gene Ther. 2017, 24, 520–526. [Google Scholar] [CrossRef]
- Kantor, B.; Bailey, R.M.; Wimberly, K.; Kalburgi, S.N.; Gray, S.J. Methods for Gene Transfer to the Central Nervous System. Adv. Genet. 2014, 87, 125–197. [Google Scholar] [CrossRef] [PubMed]
- Tanguy, Y.; Biferi, M.G.; Besse, A.; Astord, S.; Cohen-Tannoudji, M.; Marais, T.; Barkats, M. Systemic AAVrh10 Provides Higher Transgene Expression than AAV9 in the Brain and the Spinal Cord of Neonatal Mice. Front. Mol. Neurosci. 2015, 8. [Google Scholar] [CrossRef] [PubMed]
- Foust, K.D.; Salazar, D.L.; Likhite, S.; Ferraiuolo, L.; Ditsworth, D.; Ilieva, H.; Meyer, K.; Schmelzer, L.; Braun, L.; Cleveland, D.W.; et al. Therapeutic AAV9-Mediated Suppression of Mutant SOD1 Slows Disease Progression and Extends Survival in Models of Inherited ALS. Mol. Ther. 2013, 21, 2148–2159. [Google Scholar] [CrossRef]
- Iannitti, T.; Scarrott, J.M.; Likhite, S.; Coldicott, I.R.P.; Lewis, K.E.; Heath, P.R.; Higginbottom, A.; Myszczynska, M.A.; Milo, M.; Hautbergue, G.M.; et al. Translating SOD1 Gene Silencing toward the Clinic: A Highly Efficacious, Off-Target-Free, and Biomarker-Supported Strategy for FALS. Mol. Ther. Nucleic Acids 2018, 12, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, B.; Qiu, L.; Yang, C.; Kramer, J.; Su, Q.; Guo, Y.; Brown, R.H.; Gao, G.; Xu, Z. Widespread Spinal Cord Transduction by Intrathecal Injection of RAAV Delivers Efficacious RNAi Therapy for Amyotrophic Lateral Sclerosis. Hum. Mol. Genet. 2014, 23, 668–681. [Google Scholar] [CrossRef]
- Borel, F.; Gernoux, G.; Cardozo, B.; Metterville, J.P.; Toro Cabreja, G.C.; Song, L.; Su, Q.; Gao, G.P.; Elmallah, M.K.; Brown, R.H.; et al. Therapeutic RAAVrh10 Mediated SOD1 Silencing in Adult SOD1G93A Mice and Nonhuman Primates. Hum. Gene Ther. 2016, 27, 19–31. [Google Scholar] [CrossRef]
- Borel, F.; Gernoux, G.; Sun, H.; Stock, R.; Blackwood, M.; Brown, R.H.; Mueller, C. Safe and Effective Superoxide Dismutase 1 Silencing Using Artificial MicroRNA in Macaques. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Biferi, M.G.; Cohen-Tannoudji, M.; Cappelletto, A.; Giroux, B.; Roda, M.; Astord, S.; Marais, T.; Bos, C.; Voit, T.; Ferry, A.; et al. A New AAV10-U7-Mediated Gene Therapy Prolongs Survival and Restores Function in an ALS Mouse Model. Mol. Ther. 2017, 25, 2038–2052. [Google Scholar] [CrossRef]
- Peters, O.M.; Cabrera, G.T.; Tran, H.; Gendron, T.F.; McKeon, J.E.; Metterville, J.; Weiss, A.; Wightman, N.; Salameh, J.; Kim, J.; et al. Expression of Human C9ORF72 Hexanucleotide Expansion Reproduces RNA Foci and Dipeptide Repeat Proteins but Not Neurodegeneration in BAC Transgenic Mice. Neuron 2015, 88, 902–909. [Google Scholar] [CrossRef]
- Foust, K.D.; Wang, X.; McGovern, V.L.; Braun, L.; Bevan, A.K.; Haidet, A.M.; Le, T.T.; Morales, P.R.; Rich, M.M.; Burghes, A.H.M.; et al. Rescue of the Spinal Muscular Atrophy Phenotype in a Mouse Model by Early Postnatal Delivery of SMN. Nat. Biotechnol. 2010, 28, 271–274. [Google Scholar] [CrossRef]
- Bevan, A.K.; Duque, S.; Foust, K.D.; Morales, P.R.; Braun, L.; Schmelzer, L.; Chan, C.M.; McCrate, M.; Chicoine, L.G.; Coley, B.D.; et al. Systemic Gene Delivery in Large Species for Targeting Spinal Cord, Brain, and Peripheral Tissues for Pediatric Disorders. Mol. Ther. 2011, 19, 1971–1980. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Al-Zaidy, S.; Shell, R.; Arnold, W.D.; Rodino-Klapac, L.R.; Prior, T.W.; Lowes, L.; Alfano, L.; Berry, K.; Church, K.; et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.-W.; Chen, H.; Liu, H.; Lu, J.; Qian, K.; Huang, C.T.-. Ling.; Zhong, X.; Fan, F.; Zhang, S.-C. Generation and Expansion of Highly-Pure Motor Neuron Progenitors from Human Pluripotent Stem Cells. Nat. Commun. 2015, 6, 6626. [Google Scholar] [CrossRef] [PubMed]
- Chu, V.T.; Weber, T.; Wefers, B.; Wurst, W.; Sander, S.; Rajewsky, K.; Kühn, R. Increasing the Efficiency of Homology-Directed Repair for CRISPR-Cas9-Induced Precise Gene Editing in Mammalian Cells. Nat. Biotechnol. 2015, 33, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Staahl, B.T.; Alla, R.K.; Doudna, J.A. Enhanced Homology-Directed Human Genome Engineering by Controlled Timing of CRISPR/Cas9 Delivery. eLife 2014, 3. [Google Scholar] [CrossRef]
- Dasgupta, I.; Flotte, T.R.; Keeler, A.M. CRISPR/Cas-Dependent and Nuclease-Free In Vivo Therapeutic Gene Editing. Hum. Gene Ther. 2021, 32, 275–293. [Google Scholar] [CrossRef]
- Chuang, Y.-F.; Phipps, A.J.; Lin, F.-L.; Hecht, V.; Hewitt, A.W.; Wang, P.-Y.; Liu, G.-S. Approach for in Vivo Delivery of CRISPR/Cas System: A Recent Update and Future Prospect. Cell. Mol. Life Sci. 2021, 78, 2683–2708. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karpe, Y.; Chen, Z.; Li, X.-J. Stem Cell Models and Gene Targeting for Human Motor Neuron Diseases. Pharmaceuticals 2021, 14, 565. https://doi.org/10.3390/ph14060565
Karpe Y, Chen Z, Li X-J. Stem Cell Models and Gene Targeting for Human Motor Neuron Diseases. Pharmaceuticals. 2021; 14(6):565. https://doi.org/10.3390/ph14060565
Chicago/Turabian StyleKarpe, Yashashree, Zhenyu Chen, and Xue-Jun Li. 2021. "Stem Cell Models and Gene Targeting for Human Motor Neuron Diseases" Pharmaceuticals 14, no. 6: 565. https://doi.org/10.3390/ph14060565
APA StyleKarpe, Y., Chen, Z., & Li, X.-J. (2021). Stem Cell Models and Gene Targeting for Human Motor Neuron Diseases. Pharmaceuticals, 14(6), 565. https://doi.org/10.3390/ph14060565