Abstract
Stretches of cytosine-rich DNA are capable of adopting a dynamic secondary structure, the i-motif. When within promoter regions, the i-motif has the potential to act as a molecular switch for controlling gene expression. However, i-motif structures in genomic areas of repetitive nucleotide sequences may play a role in facilitating or hindering expansion of these DNA elements. Despite research on the i-motif trailing behind the complementary G-quadruplex structure, recent discoveries including the identification of a specific i-motif antibody are pushing this field forward. This perspective reviews initial and current work characterizing the i-motif and providing insight into the biological function of this DNA structure, with a focus on how the i-motif can serve as a molecular target for developing new therapeutic approaches to modulate gene expression and extension of repetitive DNA.
1. Introduction
In 1953, Watson and Crick first published the structure of the DNA double helix [1]. They described a DNA molecule as a right-handed, twisted coil composed of a purine and pyrimidine inner core held together by hydrogen bonds, with a sugar–phosphate backbone that extended from these paired bases. The x-ray diffraction photograph produced by Rosalind Franklin is one of the most readily identifiable images from scientific literature. Initially, two variations of the double helix were identified, the A- and B-forms [2]. These two conformations have distinct positions of the deoxyribose sugar that result in different spacing between nucleotide bases, with DNA most commonly found in the B-form [3]. In the 1970s, Z-DNA, the left-handed duplex, was observed under certain laboratory conditions [4]. This discovery gradually led to the realization that DNA can adopt different configurations. Since this early work uncovering the varied helices of duplex DNA, continued investigations demonstrated that nucleic acids also form sequence-dependent secondary structures, such as triplex DNA, hairpins, and cruciforms, as well as tetraplex structures: the guanine-quadruplex (G4) and the intercalated-motif (i-motif) (reviewed in [5]).
G4 and i-motif structures are formed from stacked tetrads of guanine or cytosine nucleotides, respectively, that can occur in both single strands of DNA and where two or four strands assemble together. G4s were first identified in 1962 and biologically relevant structures in human telomeres were first discovered in 1987 [6,7] to arise from sequences of contiguous guanines stabilized by cations such as K+ [8]. Extensive research has explored the formation of G4 structures, their role in transcriptional regulation and telomere maintenance, and therapeutic potential, particularly in the area of silencing oncogene expression in the treatment of cancer [9,10,11,12,13,14,15]. Nearly 30 years later in 1993, another quadruplex-based DNA structure, the i-motif, was shown in foundational studies using nuclear magnetic resonance (NMR), but unlike the G4, this structure is built from adjacent runs of cytosines [16]. Specifically, the i-motif consists of intercalated hemi-protonated cytosine (C-C+) base pairs with loop regions composed of various bases intervening between the cytosine runs (Figure 1). Similar to the guanines in the G4, base pairing of cytosines occurs via Hoogsteen hydrogen bonds; however, since the formation of i-motif structures also requires the protonation of cytosines, i-motifs form more readily at acidic pH. This pH-dependent formation of i-motifs and the initial lack of evidence that i-motifs could form in solutions at physiological pH led to skepticism within the DNA scientific community as to whether i-motifs existed in vivo. Contrary to the belief that these structures may not form in cellular nuclei, the prevalence of C-rich regions in telomeres and promoter regions suggest that the i-motif serves a biological function [17]. Shortly after the discovery of the i-motif, structures are now recognized to form from C-rich sequences within telomeres as well as those found in the promoters of primarily oncogenes, including c-MYC [17]. Beginning in 1997 with i-motif structure formation in the insulin-linked polymorphic region, studies showing that i-motifs have the ability to block DNA replication furthered the idea that i-motifs may regulate nuclear processes [18]. The continuing research pursuits to demonstrate their formation and role in physiological relevant contexts led to several breakthroughs in the i-motif field, particularly in the last few years, and posit these structures as the next likely DNA structure to target for therapeutic development.
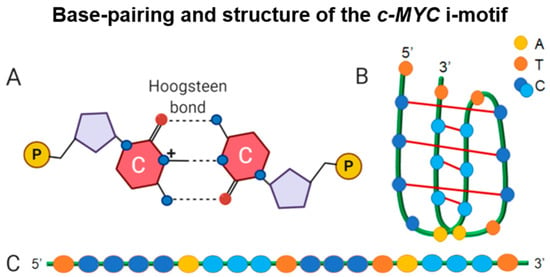
Figure 1.
Building blocks of the i-motif structure using c-MYC as a model. (A) A representation of Hoogsteen base pairing within an i-motif; (B) diagram of the c-MYC i-motif folded structure; and (C) the C-rich sequence that gives rise to an i-motif with the pairing pattern of cytosine tracts illustrated in two different shades of blue [24].
This review focuses on the proposed models for the biological role of i-motifs in the context of select diseases. We discuss how discovery of i-motif ligands serve as both molecular tools to tease out these nuclear functions and potential new therapeutics for drug development. We also highlight the different screening approaches used to identify i-motif interactive agents. For background, we include a brief discussion on the requirements for i-motif formation and the recent detection of these structures in the nuclei of cells, but direct readers to more comprehensive reviews for detailed descriptions of factors affecting i-motif structure and formation [19,20,21,22,23].
2. i-Motif Formation and Detection in Living Cells
Even though initial evidence supports the biological relevance of i-motifs, the study of these structures lags behind that of the well-characterized complementary G4 structures. The pH dependency of i-motifs raises the important question of whether these sequences fold into tetraplexes and persist in living cells. Rather than immediate research efforts focused on their biological function, i-motifs received attention as potential molecular switches with applications in nanotechnology, allowing for the development of technologies such as intracellular pH indicators and targeted drug delivery systems [25]. It would take 25 years from the discovery of the i-motif for confirmation of the existence of these structures inside the nuclei of living cells.
2.1. Role of Molecular Crowding, Nucleotide Sequence, and Epigenetic Modification
Demonstration that pH was not the only factor affecting the stability of i-motifs substantially progressed the field and indicated the real possibility these structures may form inside the nucleus. Many factors are now recognized to influence whether a nucleotide sequence is likely to form an i-motif, including the local, molecular environment, loop length and base composition, and epigenetic modification. When molecular crowding agents such as polyethylene glycol (PEG) are included in buffer solutions to mimic the crowded nuclear environment within a cell, the transitional pH for i-motif formation increases to approximately 6.8, substantially closer to physiological levels [26]. It is important to note that the degradation of PEG can lower the pH of a solution, which raises the question as to whether the stabilization effect of PEG is due to this potential change in pH or its molecular crowding effects [27]. Similarly, the torsional strain placed on duplex DNA within the nucleus induces negative superhelicity. When recapitulated in vitro by introducing i-motif sequences into supercoiled plasmids, this negative superhelicity also facilitates the preferential formation of i-motifs in highly C-rich elements over duplex DNA [28]. The specific nucleotide sequence of the i-motif-forming region also impacts the stability of the consequent structure, where, for example, the melting temperature of an i-motif increases with the number of cytosines in sequence [29,30]. Likewise, the length and composition of the loop regions are important and originally, longer lateral loops were thought to permit assembly of more stable structures. However, recent studies show that certain i-motif sequences with shorter loops exhibit increased thermal stability [31]. Longer lateral loops may still contribute to a greater stability due to interactions between specific bases contained within the loop regions. An increased number of cytosines within these regions results in stable structures while a high number of purine bases is associated with decreased stability [32,33]. Evidence also suggests that some longer loop regions form stable hairpins, creating a more stable hybrid i-motif/hairpin structure [34]. Additionally, modifications of the sugar backbone of nucleic acids can alter i-motif stability. Substitution of cytidines with an artificial nucleic acid containing an acyclic scaffold, threoninol, destabilizes i-motifs [35] and 2′-fluoroarabinonucleic acid modifications increase i-motif stability [36]. These modifications provide some control over i-motif stability in laboratory experiments and provide insight into the structure and folding process of i-motifs. Lastly, epigenetic modification of cytosines within i-motif sequences also appears to affect stability. The Waller laboratory examined a range of cytosine modifications and determined that i-motifs stable at physiological pH were more likely to consist of methylated cytosines [37]. The presence of i-motif sequences within CpG islands, which are well-known sites of methylation, supports this finding and indicates that methylation may contribute to i-motif formation within living cells [37,38,39].
2.2. Visualization of the i-Motif Using Fluorescent Antibodies and In-Cell NMR
In 2018, Christ et al. identified an antibody fragment, known as iMab, that binds specifically to formed i-motif structures [40]. Their experiments confirmed that the interaction between iMab and i-motifs differentiates between i-motifs and other secondary structures, including G4s. Using three human cancer cell lines, MCF7, U2OS, and HeLa, iMab enables the visualization of formed i-motif structures within telomeres and promoter regions within the nuclei of these living cells. More recently, an antibody against the transcription factor BmILF, which binds with high specificity to the i-motif structure within the promoter region of the gene BmPOUM2, further discussed in Section 3.2, detected folded i-motifs in cell nuclei from the testes of the invertebrate, Bombyx mori [41]. In support of an active role in gene transcription, the formation of these i-motifs within the testes were cell cycle dependent as indicated by enhanced staining during the G1 phase. In addition to the use of antibodies, cutting-edge in-cell NMR also demonstrates that stable, persistent i-motifs form within HeLa cells under physiological conditions [42]. Collectively, these findings confirm what i-motif researchers long suspected, that despite their in vitro dependence on low pH, DNA C-rich sequences can adopt i-motif conformations within the nucleus.
3. i-Motifs Function as Molecular Switches for Gene Regulation
Lending further credibility to i-motifs as biologically relevant structures, i-motif-forming sequences are evolutionarily conserved across species, in both prokaryotes and eukaryotes, within telomeres and gene promoter regions [43,44]. Similar to G4s, the close proximity of i-motif elements to transcription start sites infers a role in regulating gene expression. Thus far, data support this cis-regulatory function of the i-motif and indicate the potential to act as an activator and repressor of transcription depending on the specific promoter region, the unique i-motif sequence, and the associated transcription factors (Figure 2). In the human genome, genes encoding oncogenes, bone development proteins, sequence-specific DNA binding domain transcription factors, and molecules related to the positive regulation of transcription from the RNA polymerase II promoter have a high incidence of C-rich sequences capable of forming i-motif structures [45]. Here, we limit our discussion to published work involving three key oncogene i-motif structures as well as an i-motif recently identified from the genome of an insect that offer insight into how i-motifs function as regulators of gene expression. Additional genes are covered in later sections of the review.
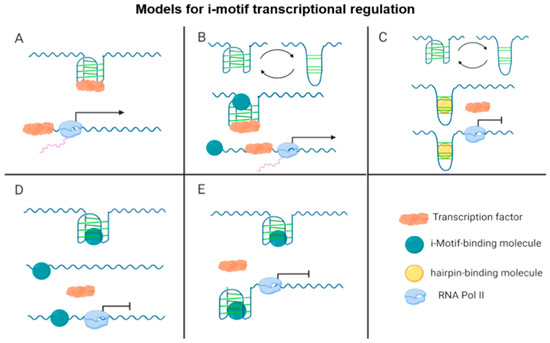
Figure 2.
Schematic of the different working models for i-motif function in transcription. (A) An i-motif serves as a scaffold for transcription factor binding that leads to i-motif unfolding and transcriptional activation and (B–E) depict potential effects of i-motif-binding molecules. A small molecule can: (B) stabilize the i-motif over a hairpin structure, allowing transcription factor recognition of binding sites on the lateral loops of the i-motif, leading to i-motif unfolding and transcriptional activation; (C) bind to a hairpin structure, preventing i-motif formation, resulting in loss of transcription factor binding, and transcriptional repression; (D) destabilize the i-motif, leading to i-motif unfolding, loss of transcription factor binding, and transcriptional repression; or (E) stabilize the i-motif blocking the transcription factor binding site, leading to transcriptional repression.
3.1. Transcriptional Activator
While early research often speculated about the biological relevance of i-motif structures, there was no direct evidence of i-motifs regulating transcription until 2014. Two foundational studies investigating the i-motif-forming sequence within the promoter region of the BCL2 oncogene demonstrate i-motif elements exhibit a dynamic equilibrium of structures recognized by small molecules analogous to RNA riboswitches [46,47]. BCL2 encodes an anti-apoptotic protein important in maintaining cell survival and its expression is dysregulated in many cancers, allowing cells to bypass cell cycle checkpoints and contribute to cancer initiation and progression [48]. By screening the NCI Diversity Set library of small molecules, two compounds were identified to interact with the BCL2 i-motif that either stabilized, IMC-48 (Figure 3), or destabilized the structure, IMC-76 (Figure 4) [46]. The observed destabilization was not as much as an unfolding of the i-motif, but rather a stabilization and trapping out of the hairpin species, thereby preventing i-motif assembly. In contrast, IMC-48 bound to the folded i-motif, presumably at the central loop, and increased stability of the structure. Utilizing these compounds as molecular tools to modulate BCL2 i-motif formation within B-cell lymphoma and breast cancer cell lines, BCL2 mRNA expression was substantially reduced, with less i-motif formation, while the presence of a stable i-motif increased mRNA levels. The ability for i-motif formation to elevate gene expression that was then mitigated by resolution of the structure indicates that, in the BCL2 promoter, the i-motif has the potential to activate transcription.
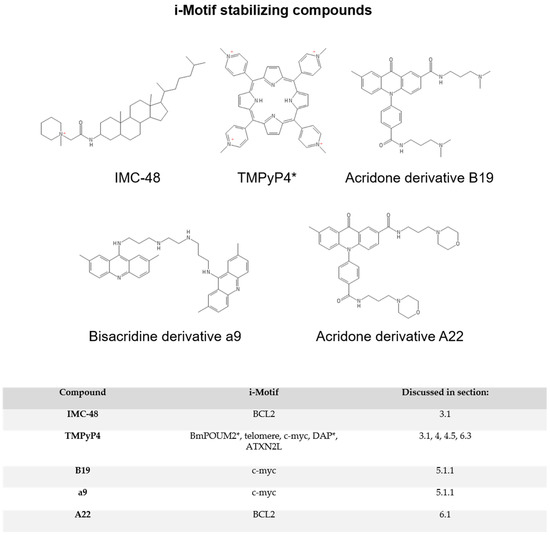
Figure 3.
i-Motif stabilizing compounds. * TMPyP4 can also be a destabilizer, depending on the i-motif in question. In the table below, asterisks denote i-motifs destabilized by TMPyP4.
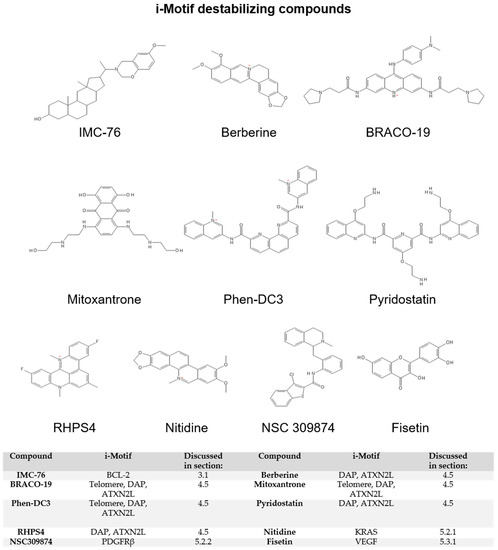
Figure 4.
i-Motif destabilizing compounds.
In the companion study, discovery of the transcription factor HNRNP LL binding to the BCL2 i-motif and enhancing mRNA expression further supported the role of the i-motif as a transcriptional activator and provided a potential mechanism [47]. HNRNP LL recognizes the lateral loops of the folded i-motif and initiates unfolding of the structure upon binding, consequently leading to increased transcription (Figure 2A,B). When the BCL2 i-motif is already in a partially unfolded or hairpin state, this interaction cannot occur and gene expression is downregulated (Figure 2C).
Of note, HNRNP LL is not the only HNRNP family member involved in i-motif driven mechanisms for regulating transcription. HNRNPK is known to recognize and bind i-motif structures, including in the KRAS promoter [49]. Similar to the interaction between the BCL2 i-motif and HNRNP LL, destabilization of the KRAS i-motif disrupts the interaction between the transcription factor and the structure, causing downregulation of KRAS expression (Figure 2D).
In addition to mammalian cells, the i-motif was shown to activate transcription in the invertebrate Bombyx mori, commonly known as the silkworm moth [50]. The transcription factor gene BmPOUM2, which coordinates a gene expression program critical for proper wing disc formation during metamorphosis, contains an i-motif-forming sequence within its promoter region. This i-motif element was characterized to form a stable structure and destabilization of the i-motif with either non-cytosine base substitutions, hybridization with an anti-sense oligonucleotide, or a small-molecule ligand decreased transcription of BmPOUM2. Treatment with anti-sense oligonucleotides against the complementary G4-forming sequence to prevent G4 formation confirmed that this effect was due to the formation of i-motifs rather than G4s. To further establish the regulatory role of the i-motif, a luciferase reporter assay showed that treatment with the porphyrin compound TMPyP4, which destabilizes the BmPOUM2 i-motif, significantly reduces BmPOUM2 transcription. This compound, detailed in Section 4, also interacts with G4 structures. While interaction with the BmPOUM2 G4 was not discussed so its effect cannot be attributed solely to i-motif destabilization, the results of this assay confirm that the secondary structures formed in this sequence regulate gene expression [50]. Consistent with mammalian counterparts, a nuclear protein, BmILF, was identified to bind to the BmPOUM2 i-motif and enhance promoter activity. The corroborating data across species are suggestive of a generalizable rule that i-motifs positively regulate gene expression. However, as with other cis-regulatory elements, inherent differences across promoters in the distance of the i-motif sequence from the transcription start site, composition of flanking nucleotide motifs, and the i-motif sequence itself, most likely confer promoter-specific variations on i-motif function.
3.2. Transcriptional Repressor
In the context of the c-MYC promoter, the relative position of the i-motif-forming sequence to a transcription factor binding site alters the protein–DNA interaction and we see how the i-motif structure can act to repress transcription. c-MYC is one of the most frequently perturbed oncogenes in human cancers, with over 40% of tumors showing overexpression at the protein level. The cancer-enabling properties of the c-MYC protein come from its activity as a “master regulator” to propagate expression of multiple pathways associated with hallmarks of cancer, such as cell cycle signaling, and glycolysis and other metabolic cascades [51,52,53]. The expression of c-MYC is required for normal cell growth, tissue development, and apoptosis, but levels must be downregulated when growth and proliferation are complete, allowing for appropriate differentiation and senescence [54]. Because c-MYC affects the expression of a myriad of genes involved in these processes, the expression of c-MYC must be tightly regulated to prevent the development of cancers. The presence of an i-motif-forming sequence within the c-MYC promoter caused researchers to immediately hypothesize about its potential role in transcriptional regulation and subsequent studies show that the c-MYC i-motif can modulate c-MYC expression.
In keeping with the theme of HNRNP proteins recognizing i-motif sequences as observed for BCL2 and KRAS, HNRNPK also interacts with the c-MYC i-motif for transcription control. Although a seminal study from 1996 established the HNRNPK transcription factor bound to the C-rich segment within the c-MYC promoter and this interaction is required for activating c-MYC expression, this sequence was not yet known to form an i-motif structure [55]. Surprisingly, in contrast to the BCL2 i-motif, formation of the c-MYC i-motif leads to lower transcriptional activity than when unfolded [56]. While i-motif formation is still necessary for initial HNRNPK recognition of cytosine tracts in the lateral loops, this interaction is not strong enough to fully activate transcription. Increasing superhelicity unwinds the i-motif and exposes additional cytosine tracts to HNRNPK. These additional elements increase the binding strength of HNRNPK sufficiently to activate high levels of transcription [56]. In this case, the i-motif plays a role in the initial binding site recognition. However, the persistence of the i-motif prevents access to the primary site for maximum transcriptional activation by HNRNPK and therefore has potential to also act as a repressor of transcription. This model of transcriptional regulation is further supported by the later use of small-molecule stabilizers [57], which are discussed below in Section 5.1.1 (Figure 2E).
4. Identification of i-Motif Interactive Small Molecules
Small molecules, including some of those discussed in Section 3.1, were critical molecular tools in defining the biological functions of i-motifs, particularly in the case of the BCL2 i-motif and compounds IMC-48 and IMC-76. While these i-motif interactive compounds were used to elucidate the function of i-motifs, the identification of such compounds also establishes the i-motif as a therapeutic target and will facilitate drug discovery. The porphyrin, TMPyP4 (Figure 3), a previously validated pan-G4-binding compound, was the first i-motif interactive small molecule identified in 2000 [58]. Initially, TMPyP4 was found to interact with i-motif-forming sequences from human telomeres and induce i-motif formation [59]. After further study, it became increasingly clear that similar to interactions with G4s, the effect of TMPyP4 on i-motif formation varies depending on the structure. For instance, TMPyP4 increases the stability of the c-MYC i-motif and prevents binding of the transcription factor HNRNPK [60], but decreases the stability of the BmPOUM2 i-motif and leads to downregulation of transcription [50]. Even though extensive structure–activity relationship analyses are lacking, i-motif destabilizing compounds (Figure 4) tend to consist of more complex, multicyclic ring systems relative to i-motif stabilizers (Figure 3). Beyond the chemical features of the ligands, what is striking about the i-motif destabilizers identified thus far is their ability to function dually as strong G4 stabilizers. These compounds are discussed in Section 4.5 below. Regardless of effect on the structure, i-motif interactive compounds tend to contain either a quaternary amine or a highly basic amine that most likely is ionized at physiological pH and presumably interacts with the negative DNA phosphate backbone to anchor the molecule in the DNA. In this section, we will discuss commonly used techniques as well as newer approaches that effectively discovered both i-motif stabilizing and destabilizing agents.
4.1. Fluorescence Resonance Energy Transfer (FRET)-Based Screen
Many i-motif-binding molecules are identified using a FRET-based screen, including the BCL2 i-motif interactive small molecules IMC-76 and IMC-48 [46]. In a FRET screening assay, the i-motif sequence of interest serves as the FRET probe, with a fluorescent donor molecule attached to the 5′ end and an acceptor, or quencher, molecule to the 3′end [61]. Probes are suspended in a buffer conducive to i-motif formation, typically sodium cacodylate as it maintains a constant pH despite variations in temperature [62], and then incubated with compound libraries. The probe–ligand mixtures are exposed to a light source at the excitation wavelength of the donor fluorophore. If the i-motif structure is folded, emission from the donor fluorophore is absorbed by the acceptor fluorophore due to the close proximity of the fluorophore-labeled ends of the probe and results in a loss of fluorescence at the donor emission wavelength. Conversely, if the i-motif is unfolded, the donor and acceptor/quencher fluorophores are spatially distant and the emission wavelength of the donor fluorophore is not transferred. Thus, a compound that yields a decrease in fluorescence indicates a stabilizing interaction while an increase in fluorescence at the emission wavelength of the donor indicates a destabilizing effect on the i-motif. A melting assay is sometimes applied to this FRET screen for i-motif interactive compounds [63]. This technique uses the same principle of donor and acceptor fluorophores, but measures fluorescence across a range of increasing temperatures to determine the thermal stability inferred by the melting temperature at which half the signal is lost. In this case, increased melting temperature indicates an increase in structure stability. When using FRET to screen for interactive compounds, it is important to use a secondary method to confirm interaction such as circular dichroism spectroscopy (CD) or UV spectroscopy as some molecules can interact with the fluorophores attached to the probes or the fluorophores themselves can alter the stability of i-motif structures [64].
4.2. Fluorescent Intercalator Displacement (FID) Assay
In recent years, other approaches successfully identified i-motif interacting compounds without the need for fluorescently labeled i-motif probes. One such method is the FID assay [65]. FID assays are routinely employed to discover compounds that bind to various DNA structures and recently the technique was adapted for the i-motif using the fluorescent probe thiazole orange [66]. Thiazole orange is not fluorescent until it binds to DNA, and when thiazole orange binds to an i-motif the compound exhibits detectable fluorescence. Thiazole orange is considered ideal for this assay because of the high affinity for i-motif DNA that is strong enough to create an observable fluorescent signal, but still allows for displacement by other i-motif interacting ligands to create a fluorescence detection window for stabilizers. Potential i-motif interacting compounds are screened by adding compounds to a solution containing thiazole orange already bound to the i-motif oligomer of interest and observing changes in fluorescence. The loss of thiazole orange fluorescence intensity identifies molecules that bind to the i-motif with higher affinity. Development of this assay for i-motif-binding ligands led to the discovery of compounds mitoxantrone, tobramycin, tilorone, Harmalol, and quinalizarin, which interact with telomeric and c-MYC i-motifs [66].
4.3. Electrospray Ionization Mass Spectrometry (ESI-MS)
Similar to most assays utilized to study the i-motif, a rapid ESI-MS screening assay was originally adapted to detect G4 interactive ligands [10]. This method is recognized as equally effective at screening small molecules for i-motif interaction [67]. The ESI-MS is combined with capillary electrophoresis to create a rapid, automated screening process that uses a minimum amount of sample. A low input is possible because, rather than evaluate molecules in solution, ESI-MS involves conversion of molecules from a liquid solution into a gas phase [68]. This process allows for a highly sensitive determination of i-motif formation and assessment of stability in addition to providing information about ligand binding interactions. ESI-MS coupled with capillary electrophoresis is a valuable tool for screening potential i-motif interactive compounds that provides more data regarding binding strength and stoichiometry in comparison to many other methods. The drawback to the ability of assaying minimal oligomer and ligand material is that ESI-MS cannot evaluate ligand binding in solution, which is necessary to determine the kinetics of the interaction and the folding/unfolding of the i-motif structure in response to ligand binding. Thus, while this assay serves as an efficient first-screening method to save on reagents and generate a short ‘hit’ list of compounds, follow-up assays, such as CD and NMR, are necessary to validate these compounds as i-motif interactive agents in solution.
4.4. Small-Molecule Microarrays (SMMs)
The final screening technique we will discuss is the SMMs, which efficiently supports high-throughput screening of small compounds. SMMs were developed to screen for molecules that bind to various proteins and effectively adapted for nucleic acid applications [69]. In this assay, collections of thousands of small molecules are immobilized as spots on glass slides arrayed into rows that are subsequently probed with a fluorescently tagged i-motif-forming oligomer of interest [70]. Under conditions for inducing i-motif formation, slides are incubated with the labeled i-motif and then through a series of wash steps, non-specific, low-affinity interactions are rinsed away. A microarray scanner detects spots with fluorescent activity that correspond to compounds with potential to bind to the i-motif. This technique is highly successful for detection of G4 ligands and, subsequently, SMMs were used to identify candidate small molecules that interact with the i-motif within the HRAS oncogene promoter [71]. To confirm that the compounds interact with the folded i-motif rather than duplex or hairpin formations, the experiments were conducted at various pH levels and only compounds that demonstrated pH-dependent binding were considered for further study. While SMMs are not yet widely used to identify i-motif-binding ligands, the high-throughput capabilities of this method make it an attractive option for studying i-motifs as therapeutic targets.
4.5. G4 Interaction as an Important Consideration for Identifying i-Motif Ligands
Since i-motifs were discovered later than G4s and only recently pursued as potential targets for therapeutics, many reported G4 stabilizing compounds were not tested on the complementary i-motif structures. In 2018 and 2019, however, two studies were published examining the effects of G4 stabilizers on i-motifs. The 2018 study treated oligomers of the telomeric i-motif sequence with six known G4 stabilizers: berberine, BRACO-19, mitoxantrone, phen-DC3, Pyridostatin, and RHPS4 (Figure 3 and Figure 4) [64]. All six compounds proved to bind to the i-motif via FID assay and three of them (BRACO-19, Mitoxantrone, and Phen-DC3) caused destabilization of the i-motif structure as measured by changes in thermal stability. Although FID assays showed that the other compounds bound to the i-motif, subsequent experiments showed that these interactions had no effect on structure stability. In the 2019 paper, the same compounds along with TmPyP4 were tested on i-motif sequences from promoter regions that form structures at neutral pH, death-associated protein (DAP) and ataxin type 2-related protein (ATXN2L) [72]. Again, all compounds were shown to interact with these two promoter i-motif structures. In the case of the DAP i-motif sequence, the small molecules destabilized the structure. For the ATXN2L i-motif, only berberine, BRACO-19, phen-DC3, and RHPS4 lowered the stability of the structure, while mitoxantrone, pyridostatin, and TmPyP4 stabilized the i-motif as measured by an increase in the thermal stability. The authors hypothesized that the stabilization of the ATXN2L i-motif by these compounds was likely due to stabilization of an alternate secondary structure or a change in the unfolding kinetics, leading to the higher melting temperature. These studies not only emphasize the importance of considering the effects on i-motif structures when developing G4 ligands, but they also demonstrate the possibility of simultaneously targeting both G4s and i-motifs with a single ligand in order to modulate gene expression. This multi-targeted approach could be beneficial when designing therapeutics to treat drug-resistant cancers.
5. Therapeutic Potential of Targeting the i-Motif in Cancer
i-Motif sequences are prevalent in promoter regions, including those of oncogenes (Table 1). Given the evidence that i-motifs act as transcriptional regulators, these structures are ideal targets for shutting down oncogenic signaling. Oncogenes are normal genes (proto-oncogenes) that lead to the initiation and/or progression of cancer when aberrantly activated due to mutations or other genomic events. i-Motifs are also present in telomeres, where stable i-motifs are shown to block the progression of telomerase. Dysfunctional telomeres and abnormal telomerase activity prolong cell survival, a critical hallmark of cancer, and increase the likelihood of accumulating oncogenic mutations with the ability to bypass apoptotic cell death. Thus, pharmacological targeting telomeric i-motifs present another avenue for therapeutic intervention in cancer.

Table 1.
Oncogenes, tumor suppressors, and other cancer-related genes with promoter i-motif-forming sequences.
5.1. Stabilization of the c-MYC and Telomeric Structures Inhibits Tumor Growth
5.1.1. c-MYC
As discussed in Section 3.2, the c-MYC promoter contains an i-motif-forming sequence that plays a role in its transcriptional regulation. While c-MYC protein is important for normal cellular function in many cell types, when mutations eliminate the reliance of c-MYC activation on growth factor signaling, this oncogene helps drive cancer initiation and progression. Small-molecule stabilizers of the c-MYC i-motif that decrease expression of this key oncogene have immense utility as new agents for cancer treatment especially given that the c-MYC protein is considered undruggable. Recent efforts to develop therapeutics against this well-sought-after molecular target from researchers at Sun Yat-sen University in China led to the identification of promising compounds that stabilize the c-MYC i-motif [57,73]. In 2018, they identified an acridone derivative, 2-methylacridin-9(10H)-one, as a c-MYC i-motif stabilizer and generated analogs to increase selectivity [57]. While multiple compounds were found to increase i-motif stability in comparison to the parent compound, B19 stood out as a prime candidate (Figure 3). This molecule displayed strong binding affinity and considerable preference for the c-MYC promoter i-motif structure over G4 and duplex DNA. Compound B19 not only stabilized the i-motif, but induced folding of the structure from a linear conformation. In confirmatory assays, compound B19 significantly reduced transcriptional activation as demonstrated in a dual-luciferase reporter assay. B19 also lowered mRNA and protein expression, reduced the proliferation of cervical cancer cells through inducing apoptosis, and prevented metastasis in vitro [57].
Two years later, the same group discovered a bisacridine derivative, compound a9 (Figure 3), that targets both the c-MYC i-motif and G4 structures with higher selectivity than duplex and hairpin DNA [73]. They also showed that compound a9 preferentially stabilizes the c-MYC i-motif and G4 over secondary structures from promoter regions of other genes. Relative to compound B19, a9 exhibited high potency at nanomolar to low micromolar concentrations in effectively reducing c-MYC mRNA and protein levels. Compound a9 inhibited proliferation in multiple cancer cell lines, including those derived from cervical, lymphoma, lung, bone, colon, and liver cancers, and tumor growth in an in vivo model of cervical cancer. Notably, compound a9 was not toxic to the mice in terms of overall weight loss or changes in organ weight compared to cisplatin (a mainstay of cervical cancer therapy), which caused significant reductions in body weight and weight of the liver and spleen. Compounds that target both i-motifs and G4s are of particular interest because the development of drug resistance is the primary obstacle to achieving successful treatment of many human cancers. As stabilization of the c-MYC G4 is known to also block transcription [74], stabilizing both transcription regulators with a single drug could minimize drug resistance.
5.1.2. Telomeres
When a cell duplicates its DNA, the DNA polymerase fails to replicate to the very ends of chromosomes such that with each cellular replication cycle, chromosomes are shortened. To prevent premature shortening and protect the valuable, internal genetic information from this gradual erosion, the DNA at chromosome ends, referred to as the telomeres, consist of TTAGGG/AATCCC repeats that are elongated during DNA replication by the telomerase enzyme [75,76,77]. However, over the lifetime of a cell, telomeres reach a critical length and programmed cell death is initiated. In some cancers, this process is hijacked with higher than normal expression or activity of telomerase, permitting a continuous extension of telomeres, which contribute to the sustained proliferation and immortalization tumor phenotypes [77]. Accordingly, telomerase inhibition offers a reasonable targeted approach to hinder tumor growth. With the G/C-rich telomere sequences comprising some of the most well-studied G4 and i-motifs [78,79], the stabilization of these structures is an active area of drug development in the field. The first reports that stabilization of G4s with small molecules inhibited telomerase activity started in the late 1990s [58], while telomeric i-motif stabilization was not demonstrated until 2012.
The first compounds shown to selectively stabilize the telomeric i-motif and inhibit telomerase were carboxylated single-walled carbon nanotubes (SWNTs) [80]. SWNTs can enter cells through the lipid bilayer and accumulate in the nucleus, where they bind to the major groove of DNA and induce stable formation of i-motif structures. This effect is unique to i-motifs, with no effect observed on the formation or stability of G4s. In vitro assays showed SWNT-induced stabilization of telomeric i-motifs directly leads to the loss of telomerase activity, which was then confirmed in human cancer cells. It is important to note, SWNT treatment was not immediately cytotoxic. There was a two-week delay before observing a significant decline in cellular proliferation with an additional week until proliferation ceased altogether. i-Motif stabilization by SWNTs causes the uncapping of telomeres and induces a DNA damage response localized to the telomeres that eventually initiates cell cycle arrest, apoptosis, or senescence. Subsequent evidence suggests that the mechanism for SWNT stabilization of i-motifs involves the proton exchange between the carboxyl group on the SWNT and the i-motif cytosines, resulting in hemi-protonation of the i-motif-forming sequences and folding of a stable structure [81]. SWNTs have received attention as potential drug delivery devices and their ability to inhibit telomerase via i-motif stabilization and induce anti-proliferative effects hints at an additional intriguing potential strategy for future cancer therapeutics [80].
5.2. Destabilization of the KRAS and PDGFR-β i-Motifs Blocks Oncogenic Signaling
5.2.1. KRAS
In healthy cells, the protein KRAS, a member of the RAS family of GTPases, acts as an on/off switch for cell proliferation, survival, differentiation, and metabolism signaling pathways [82,83,84]. Mutations within KRAS are often seen in human cancers and account for approximately 86% of RAS mutations [82]. These mutations prevent the hydrolysis of GTP to GDP, which disables the “off switch”, constitutively activating KRAS regulated signaling pathways that drive tumor growth [85]. There is also evidence that KRAS mutations are involved in tumor cell evasion of host immune responses [86]. Similar to c-MYC, KRAS was considered impossible to target for many years. The active site of KRAS consists of small and transient allosteric areas that make designing inhibitory drugs difficult [87]. Although development of small molecules to inhibit KRAS has progressed, targeting KRAS at the gene level still remains a more attractive strategy [83].
Along with BCL2 and c-MYC, the i-motifs in the KRAS promoter are among the more thoroughly investigated. Three potential i-motifs have potential to form within the KRAS promoter with the most stable structure arising from the mid-region sequence [49]. The mid-region i-motif is also the structure to which the transcription factor HNRNPK binds, resulting in transcriptional activation. The same study discovered a benzophenanthridine alkaloid, nitidine (Figure 4), from a FRET screen that binds to and destabilizes the mid-region i-motif as well as prevents interaction between HNRNPK and the i-motif. Nitidine seems selective for the KRAS i-motif structure since there was no measurable effect on the stability of the BCL2 or c-MYC i-motif structures. The mid-region KRAS i-motif contains a long loop region that forms a short hairpin structure. It was proposed that this additional hairpin structure may be important for HNRNPK binding and that nitidine preferentially acts on the KRAS i-motif due to this hybrid structure. Thus, nitidine interaction with other i-motifs that form similar hairpins within their loops cannot be ruled out. Treatment with nitidine downregulated KRAS mRNA production in the pancreatic cancer cells; however, whether this persisted through to the protein level or altered cell proliferation is not known. While there are unanswered questions regarding the potential of nitidine as a lead compound for therapeutic development, this initial study provides evidence for targeting KRAS expression through modulating i-motif stability.
5.2.2. PDGFRβ
PDGFRβ is a tyrosine kinase receptor that activates Ras-MAPK, PI3K, and PLC-γ growth and calcium-dependent transduction pathways [88,89]. High levels of PDGFRβ expression and activity through different mechanisms including gene translocations are implicated in solid, epithelial-based cancers and vascular diseases [90,91]. Cells overexpressing the receptor can release PDGFRβ ligands, propagating an autocrine signal loop that leads to proliferation of these unstable cells and contributes to tumor development. These ligands are also known to recruit stromal cells to the tumor through paracrine signaling [92,93]. While there are therapies that target the PDGFR-β protein, including tyrosine kinase inhibitors such as imatinib and neutralizing antibodies against PDGFR-β [94], there is still a need for more effective drugs to inhibit aberrant signaling through this receptor [91].
In 2017, Brown et al. published a study targeting transcription of PDGFRβ through G4s within the complex nuclease hypersensitive element in the promoter region in order to prevent its expression [95]. This sequence can form four different G4s, with the 3′ end G4 structure responsible for repression of transcription. To define this repressor effect, they created oligomers with three different point mutations within the 3′ end G4 sequence. Mutation of the 3′ adenine to a guanine resulted in a drastic increase in G4 stability. Paradoxically, the stabilization of the repressor G4 increased transcription. The counterintuitive result incited attention on the i-motif sequence and the corresponding thymine to cytosine transition. The additional cytosine within the i-motif-forming sequence stabilized the i-motif and attributed to the transcriptional activation. Comparison of the wild-type i-motif sequence, which is capable of forming at least two i-motifs in equilibrium, led to the discovery that the mutant sequence is locked into forming a single, highly stable structure [95]. Screening the NCI Diversity Set library identified benothiophene-2-carboxamide (NSC309874; Figure 4) as a destabilizer of both the wild-type and mutant i-motifs. NSC309874, which did not significantly interact with the PDGFRβ G4 structures, repressed PDGFRβ transcription in an aggressive neuroblastoma cell line indicating the potential of this compound as a new PDGFRβ inhibitor.
5.3. Potential of Targeting i-Motifs in Other Cancer-Related Genes
In addition to the cancer-enabling genes with defined i-motifs discussed above, there are several other strong candidates as future drug targets worth mentioning (Table 1). The precise function of the i-motifs shown to form in the genes listed below is not fully teased out, but considering the growing body of evidence that i-motifs are modulators of gene expression, it is likely the structures found in these genes also regulate transcription.
5.3.1. i-Motifs in Oncogenes
While the characterization of the VEGF i-motif [96] was a part of the early cohort of oncogenes, i-motifs in the BRAF [97], c-MYB [98], and HRAS [99] oncogenes were only detected in the last five years. Both the BRAF kinase and the HRAS GTP hydrolase promote RAF/MEK/ERK signaling and activating mutations in either protein causes constitutive signaling and subsequent uncontrolled growth and proliferation [100,101]. Such mutations occur in many cancers, but are especially common in melanoma for BRAF [102] and head and neck, bladder, and thyroid cancers for HRAS [103]. c-MYB is a transcription factor that controls expression of genes involved in hematopoiesis and colonic crypt formation, and may also play a role in tumor immune surveillance in natural killer cells [104,105]. Oncogenic mutations within c-MYB are associated with multiple cancer types including pancreatic, colon, prostate, and liver cancers [106]. The VEGF oncogene is actively pursued as a cancer target with already FDA-approved inhibitors (sorafenib, sunitinib, and bevacizumab) that shut down this primary driver of angiogenesis [107,108]. The flavonol fisetin (Figure 4) destabilizes the VEGF i-motif, potentially binding preferentially to the alternative hairpin structure, but the biological role of this i-motif and the effects of its destabilization on VEGF transcription remain unclear [109].
5.3.2. i-Motifs in Tumor Suppressor Genes
The enrichment of G4 and i-motif-forming sequences within oncogene promoter regions [11,12,45] naturally steers drug discovery efforts towards oncogenes. Furthermore, in light of research characterizing G4 structures as transcription repressors, the ability to turn off gene expression is seemingly more attainable from a drug development perspective than turning on expression, which is needed in the case of restoring tumor suppressors. Based on data supporting a switch-like function of i-motifs for modulating transcription [46], targeting these structures in tumor suppressor promoters is a feasible approach to reactivate the lost cellular guardians. Thus far, DAP [45,71], RAD17 [110,111], Rb [112], and SMARCA4 [113] tumor suppressor genes are reported to contain i-motif-forming elements within their promoters. Reduced expression of DAP promotes cell migration and tumor growth and is correlated with the pathology of breast and colon cancers [114,115]. Loss of RAD17 is implicated in colorectal [116], non-small-cell lung [117], and head and neck cancers [118]. RAD17 normally suppresses genomic instability by maintaining the integrity of DNA repair and cell cycle checkpoint prior to DNA replication in the S phase [119,120]. Similarly, Rb is a negative regulator of cell cycle progression. Mutations and deletions of Rb are found in many cancers [121,122,123,124]. Interestingly, SMARCA4 acts primarily as a tumor suppressor, as its loss leads to inactivation of the Rb pathway in ovarian and non-small-cell lung cancers, but is also considered an oncogene in other contexts [125,126,127,128]. Overexpression of SMARCA4 in breast and late-stage pancreatic drives tumor cell proliferation and is associated with progressive disease and poor prognosis [129,130,131]. The i-motif upstream of the SMARCA4 promoter is moderately stabilized by TMPyP4 [113], but to date no studies seek to determine the effect of i-motif stabilization on SMARCA4 expression.
5.3.3. i-Motifs in Other Cancer-Related Genes
Additional genes related to propagating cancer cell phenotypes that do not fit into either the oncogene or tumor suppressor categories were recently discovered to harbor i-motif-forming sequences capable of adopting structures. Acetyl CoA Carboxylase 1 (ACC1) is the control enzyme for fatty acid metabolism [132]. Mutations in the oncoprotein BRCA1 can activate ACC1 and perturb regulatory mechanisms, allowing cancer cells to synthesize fatty acids more quickly [133]. Many cancer types exhibit ACC1 overexpression enhancing migration and invasion in breast cancer [134] and proliferation of cancer stem cells [135]. An i-motif structure that forms from the C-rich ACC1 promoter sequence inhibited transcription in a luciferase assay, whereas a mutant sequence that prevented i-motif assembly increased transcription [136]. While this work nicely demonstrates formation of a novel i-motif, a follow-up study is needed to assess whether the effects on gene expression were due to loss of the i-motif or changes to the complementary G4 [136].
ATXN2L, which was also recently discovered to contain an i-motif-forming sequence, encodes a key component of stress granules. During times of cellular stress, granules containing RNA-protein complexes are formed in the cytoplasm in response to impaired translational initiation [137]. Overexpression of ATXN2L leads to excessive formation of stress granules and reduced expression prevents their formation [138]. Dysregulation of ATXN2L-dependent stress granule formation is seen in gastric cancer and T-cell lymphomas and can promote cell survival, proliferation, and metastasis [139,140].
6. i-Motif-Targeting Compounds as Potential Therapies for Non-Cancer Human Diseases
Numerous human diseases are linked to a particular defect in the cell genotype. Besides genes integral to the pathology of cancer, i-motif sequences are found in areas of the genome associated with other diseases (Figure 5). Certain genetic diseases, such as fragile X syndrome, stem from expanded guanine and cytosine-rich (G/C) repeats in 5′ untranslated regions capable of folding into i-motif structures. This section examines the current literature on i-motifs that form within key genes in liver disease and select neurological disorders, although we acknowledge there are other illness such as diabetes with recognized connections to i-motif-containing genes.
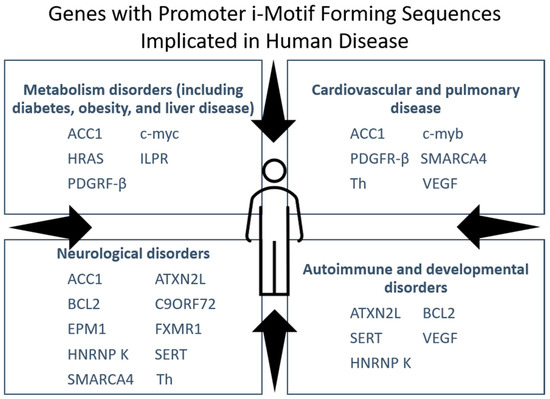
Figure 5.
i-Motif sequences are found in the promoters of genes implicated in many human diseases besides cancer.
6.1. Liver Diseases
Non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH) are both characterized by liver injury and inflammation, which are driven in part by excessive apoptosis of hepatocytes due to low BCL2 expression [141,142]. Previous studies establishing the i-motif within the BCL2 promoter as a transcriptional activator [46,47] support the premise that stabilization of the BCL2 i-motif in NAFLD and NASH is an effective approach to increase BCL2 levels and prevent hepatocyte apoptosis. Ding Li’s laboratory identified an acridone derivative, A22, as a high-affinity stabilizer of the BCL2 i-motif (Figure 3) [143]. In order to demonstrate preferential selectivity, they tested the compound against the BCL2 G4, duplex DNA, and several other i-motif sequences. Moreover, treatment of hepatocytes, grown in palmitic acid oil to mimic the lipid-induced apoptotic environment of NASH, with A22 resulted in a robust increase in cell viability and a decrease in activation of the apoptotic cell death pathway. Since NAFLD and NASH are tightly linked to metabolic syndrome, the authors also examined the effect of A22 treatment on glucose uptake and demonstrated that A22 was significantly more efficacious in stimulating the influx of glucose into hepatocytes compared to the metformin, the frequently prescribed treatment for insulin resistance [143]. Compound A22 also performed well in a NAFLD/NASH mouse model where treatment led to elevated BCL2 levels, less apoptosis, and decreased markers of hepatocyte injury and metabolic syndrome (e.g., weight gain not associated with food intake, insulin resistance, and elevated serum cholesterol and triglycerides). In view of these data, compound A22 and increasing BCL2 expression is a promising therapeutic strategy for diseases where cell survival is compromised, including NAFLD/NASH. Caution is warranted, of course, for approaches that aim to upregulate proto-oncogenes. Treatment with A22 also elicited a dose-dependent increase in BCL2 mRNA and protein in a hepatocellular carcinoma cell line [143], emphasizing the need for further work to fine tune dosage and define the appropriate patient subsets with a favorable risk–benefit ratio to avoid development of therapy-induced malignancies.
6.2. Genetic Neurological Disorders Linked to Nucleotide Repeat Expansions
Nucleotide repeats, or microsatellites, are short, simple repeats that occur frequently in the genome, typically in non-coding regions, and are evolutionarily conserved [144]. Expansion of these nucleotide repeats are a type of mutation that results from amplification of these short repeats. When nucleotide repeat expansions occur in repeats with high G/C content, the elongated sequences have potential for G4 or i-motif structure formation [145]. i-Motifs and G4s facilitate DNA polymerase stalling during replication and cause the polymerase to dissociate from the DNA [146]. When the DNA polymerase initiates replication after this dissociation, incorrect realignment along the highly repetitive DNA often occurs and results in expansion of the segment. Depending on the location, the extra stretches of repeats disrupt the reading frame for proper gene expression and increase the likelihood for secondary structure formation, which can further perturb gene replication and transcription. Nucleotide repeat expansions with repeated G/C sequences are implicated in several human diseases including fragile X syndrome (CGG), progressive myoclonus epilepsy (CCCCGCCCCGCG), and amyotrophic lateral sclerosis/frontotemporal dementia (GGGGCC) [147,148,149].
In fragile X syndrome, the loss of fragile X mental retardation 1 gene (FRM1) expression confers a defect in neuronal synaptic transmission and subsequent intellectual disabilities. FRM1 is susceptible to expanded C-repeats, which are known to form i-motif structures and thought to play a role in suppressing expression of this gene [29]. A recent study revealed that the addition of a metal complex CoII(Chromomycin)2 causes the i-motif structures to transition into duplex DNA [147]. CoII(Chromomycin)2 complex preferentially binds to the duplex conformation of DNA and the authors suggest that this metal complex may prevent i-motif formation by forcing the DNA to remain in double-stranded form and, in turn, minimize polymerase slippage and inhibit further repeat expansion [147].
i-Motif formation is also observed in the relevant genomic loci of two other DNA repeat expansion-associated diseases, progressive myoclonus epilepsy [148] and amyotrophic lateral sclerosis [149]. In agreement with the BCL2 i-motif, the i-motif-forming sequences in the genes that underlie each disease exist in a dynamic equilibrium with a hairpin [149,150]. As well, for amyotrophic lateral sclerosis, the i-motif and hairpin structures in C9ORF72 are capable of forming even when in a duplex DNA environment, where the complimentary G-rich strand is present [150]. This preliminary work demonstrates the structural flexibility of these C-repeat regions and indicates the possibility of targeting the structures to develop new therapeutic approaches. Further research is required, however, to determine the role of i-motif formation in both the progressive myoclonus epilepsy 1 and C9ORF72 genes in maintaining nucleotide expansion and/or transcription.
6.3. Other Neurological Disorders
A recent discovery highlights a role for i-motifs in the neurodegenerative disorder Parkinson’s disease. A 2020 study examined the activity of α-synuclein (α-syn), a protein that forms oligomers and amyloid fibrils linked to cell death of neurons [151]. While α-syn was discovered to bind telomeric G4s and i-motifs, the neuronal protein did not alter the stability of G4 structures, but promoted the folding and stability of i-motifs. Although human telomeric i-motif sequences were used as a model, the findings provide a proof of concept that α-syn may exert its cellular function through interactions with i-motifs [151]. As the mechanisms driving Parkinson’s disease are not fully defined, uncovering the interplay between α-syn and i-motifs in regulatory regions of genes critical in this disease will provide insight into the pathological defects and identify new targets for treatment.
A key pathway implicated in Parkinson’s disease and a range of other neurological conditions including bipolar disorder, schizophrenia, and addition and movement disorders is the synthesis pathway responsible for the production of dopamine, epinephrine, and norepinephrine [152]. Tyrosine hydroxylase (TH) is the rate-limiting enzyme of this pathway and contains a GC-rich element conserved across species within the TH promoter region capable of forming G4 and i-motif structures [153]. Interestingly, HNRNPK, the same transcription factor known to recognize and interact with the c-MYC i-motif sequence, also interacts with the C-rich region in the TH promoter in concert with cAMP Response Element-binding proteins. Consistent with promoter DNA secondary structure control of gene expression, TMPyP4 treatment of viable slices of mouse brain tissues resulted in a reduction in TH transcription. In further support of G4 and/or i-motif regulation of TH expression, a second group confirmed the ability of TMPyP4 to block transcriptional activity of the TH promoter [154]. This later study focused on the G4 structure and neglected the possibility of TMPyP4 perturbation of the i-motif structure, thus the predominant structure responsible for triggering the loss of transcription requires further resolution.
In addition to the TH promoter, the serotonin transporter-linked polymorphic region, which regulates serotonin transporter (SERT) gene expression, another important factor in neurological diseases and psychiatric disorders, also contains an i-motif-forming sequence [155]. This i-motif structure forms at neutral pH under molecular crowding conditions and could serve as an alternative target for SERT inhibition. Selective serotonin reuptake inhibitors are commonly prescribed anti-depression and anti-psychotic medications, but cause severe adverse effects in certain patient populations [156]. As further potential for targeting the i-motif in various neurological disease states, epigenetic modifications that favor i-motif formation are hypothesized to contribute to synaptic plasticity and memory formation [157].
Lastly, there is recognition that the unknown mechanism of action of current lithium-based therapies for neurological disorders [158,159,160,161] may involve i-motif regulation as lithium cations were shown to destabilize the i-motif of telomeric C-rich oligomers [162]. Given that i-motif folding and unfolding is a relatively long process, in order to obtain robust folding kinetics by adequately capturing multiple transition states, Kim et al. developed a modified single-molecule FRET technique with extended fluorescent reads. Instead of traditional FRET, where a single fluorescent end point is analyzed, multiple fluorescence signatures are captured during the entire read period and using bioinformatics aligned together to provide an overall picture of the kinetics throughout the structural changes of the i-motif [162]. This adapted technique revealed that lithium cations destabilized the i-motif structure by enhancing the unfolding of the structure, rather than preventing the folding. Furthermore, this handshaking repeated permutation (HaRP) kinetic analysis may offer future applications to characterize interactions of i-motif ligands for therapeutic development.
7. Conclusions
Over the last decade, major technological advances to detect and evaluate i-motif formation have contributed to the understanding of i-motif biology and addressed the long-standing question of whether DNA adopts such structures within cells. Whether mutual exclusive formation of the i-motif and G4 structure is a strict rule or context dependent requires further resolution. Occasionally, due to their different formation requirements or the offset position of the respective C- and G-rich sequences, both structures can form simultaneously [163]. In support of mutual exclusivity, a recent study from the Smith group demonstrates that the formation of one structure destabilizes the structure on the complementary strand [164]. Since G4s and i-motifs appear to have distinct roles in transcriptional regulation, it is reasonable that their formation within the same promoter is interdependent. From applying and modifying methods previously utilized for G4 characterization to the development of an i-motif-specific antibody that detects formed i-motifs within chromatin, molecular tools are in hand to further tease out the nuclear functions of the i-motif structure. Moving forward, additional genome-wide studies in different cellular contexts aimed at defining how i-motif formation alters the chromatin architecture and the localization of these events will aid in identifying new opportunities for therapeutic targeting. Considering the promiscuous nature of some G4 ligands to recognize the i-motif structure is also central for developing selective i-motif interactive compounds. However, perhaps achieving high levels of structure discrimination may not be necessary in genetically complex diseases such as cancer where multi-targeted approaches may provide enhanced therapeutic benefit. As research continues to delve deeper into the mechanisms behind i-motif control of nuclear processes and the structural requirements of formation within the genome, we can leverage this knowledge to design and develop a new class of targeted therapies with great potential to impact a diverse set of human diseases.
Author Contributions
Literature review, S.L.B.; writing—original draft preparation, S.L.B.; writing—review and editing, S.K. All authors have read and agreed to the published version of the manuscript.
Funding
The research activity of S.K. is funded by the National Institutes of Health, grant number P20GM121293.
Acknowledgments
We thank Brendan Frett for providing medicinal chemistry insights into i-motif stabilizing and destabilizing compounds. Figures created using Biorender.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Watson, J.D.; Crick, F.H. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature 1953, 171, 737–738. [Google Scholar] [CrossRef] [PubMed]
- Shakked, Z.; Rabinovich, D.; Kennard, O.; Cruse, W.B.; Salisbury, S.A.; Viswamitra, M.A. Sequence-dependent conformation of an A-DNA double helix. The crystal structure of the octamer d(G-G-T-A-T-A-C-C). J. Mol. Biol. 1983, 166, 183–201. [Google Scholar] [CrossRef]
- Dickerson, R.E.; Drew, H.R.; Conner, B.N.; Wing, R.M.; Fratini, A.V.; Kopka, M.L. The anatomy of A-, B-, and Z-DNA. Science 1982, 216, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Mitsui, Y.; Langridge, R.; Shortle, B.E.; Cantor, C.R.; Grant, R.C.; Kodama, M.; Wells, R.D. Physical and enzymatic studies on poly d(I-C)-poly d(I-C), an unusual double-helical DNA. Nature 1970, 228, 1166–1169. [Google Scholar] [CrossRef] [PubMed]
- Bochman, M.L.; Paeschke, K.; Zakian, V.A. DNA secondary structures: Stability and function of G-quadruplex structures. Nat. Rev. Genet. 2012, 13, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Gellert, M.; Lipsett, M.N.; Davies, D.R. Helix formation by guanylic acid. Proc. Natl. Acad. Sci. USA 1962, 48, 2013–2018. [Google Scholar] [CrossRef] [PubMed]
- Henderson, E.; Hardin, C.C.; Walk, S.K.; Tinoco, I.; Blackburn, E.H. Telomeric DNA oligonucleotides form novel intramolecular structures containing guanine-guanine base pairs. Cell 1987, 51, 899–908. [Google Scholar] [CrossRef]
- Bhattacharyya, D.; Mirihana Arachchilage, G.; Basu, S. Metal Cations in G-Quadruplex Folding and Stability. Front. Chem. 2016, 4, 38. [Google Scholar] [CrossRef]
- Sannohe, Y.; Sugiyama, H. Overview of formation of G-quadruplex structures. Curr. Protoc. Nucleic Acid Chem. 2010, 40, 17.2.1–17.2.17. [Google Scholar] [CrossRef]
- Yuan, G.; Zhang, Q.; Zhou, J.; Li, H. Mass spectrometry of G-quadruplex DNA: Formation, recognition, property, conversion, and conformation. Mass Spectrom. Rev. 2011, 30, 1121–1142. [Google Scholar] [CrossRef]
- Rhodes, D.; Lipps, H.J. G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res. 2015, 43, 8627–8637. [Google Scholar] [CrossRef] [PubMed]
- Hänsel-Hertsch, R.; Beraldi, D.; Lensing, S.V.; Marsico, G.; Zyner, K.; Parry, A.; Di Antonio, M.; Pike, J.; Kimura, H.; Narita, M.; et al. G-quadruplex structures mark human regulatory chromatin. Nat. Genet. 2016, 48, 1267–1272. [Google Scholar] [CrossRef] [PubMed]
- Huppert, J.L. Four-stranded nucleic acids: Structure, function and targeting of G-quadruplexes. Chem. Soc. Rev. 2008, 37, 1375–1384. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, J.; Mergny, J.L.; Salgado, G.F.; Queiroz, J.A.; Cruz, C. G-quadruplex, Friend or Foe: The Role of the G-quartet in Anticancer Strategies. Trends Mol. Med. 2020, 26, 848–861. [Google Scholar] [CrossRef] [PubMed]
- Sanij, E.; Hannan, K.M.; Xuan, J.; Yan, S.; Ahern, J.E.; Trigos, A.S.; Brajanovski, N.; Son, J.; Chan, K.T.; Kondrashova, O.; et al. CX-5461 activates the DNA damage response and demonstrates therapeutic efficacy in high-grade serous ovarian cancer. Nat. Commun. 2020, 11, 2641. [Google Scholar] [CrossRef] [PubMed]
- Gehring, K.; Leroy, J.L.; Guéron, M. A tetrameric DNA structure with protonated cytosine.cytosine base pairs. Nature 1993, 363, 561–565. [Google Scholar] [CrossRef]
- Manzini, G.; Yathindra, N.; Xodo, L.E. Evidence for intramolecularly folded i-DNA structures in biologically relevant CCC-repeat sequences. Nucleic Acids Res. 1994, 22, 4634–4640. [Google Scholar] [CrossRef]
- Catasti, P.; Chen, X.; Deaven, L.L.; Moyzis, R.K.; Bradbury, E.M.; Gupta, G. Cystosine-rich strands of the insulin minisatellite adopt hairpins with intercalated cytosine+.cytosine pairs. J. Mol. Biol. 1997, 272, 369–382. [Google Scholar] [CrossRef]
- Abou Assi, H.; Garavís, M.; González, C.; Damha, M.J. i-Motif DNA: Structural features and significance to cell biology. Nucleic Acids Res. 2018, 46, 8038–8056. [Google Scholar] [CrossRef]
- Kaushik, M.; Kaushik, S.; Roy, K.; Singh, A.; Mahendru, S.; Kumar, M.; Chaudhary, S.; Ahmed, S.; Kukreti, S. A bouquet of DNA structures: Emerging diversity. Biochem. Biophys. Rep. 2016, 5, 388–395. [Google Scholar] [CrossRef][Green Version]
- Oh, K.I.; Kim, J.; Park, C.J.; Lee, J.H. Dynamics Studies of DNA with Non-canonical Structure Using NMR Spectroscopy. Int. J. Mol. Sci. 2020, 21, 2673. [Google Scholar] [CrossRef] [PubMed]
- Day, H.A.; Pavlou, P.; Waller, Z.A. i-Motif DNA: Structure, stability and targeting with ligands. Bioorg. Med. Chem. 2014, 22, 4407–4418. [Google Scholar] [CrossRef] [PubMed]
- Benabou, S.; Aviñó, A.; Eritja, R.; González, C.; Gargallo, R. Fundamental aspects of the nucleic acid i-motif structures. RSC Adv. 2014, 4, 26956–26980. [Google Scholar] [CrossRef]
- Dai, J.; Hatzakis, E.; Hurley, L.H.; Yang, D. I-motif structures formed in the human c-MYC promoter are highly dynamic--insights into sequence redundancy and I-motif stability. PLoS ONE 2010, 5, e11647. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Yang, Z.; Liu, D. DNA nanotechnology based on i-motif structures. Accounts Chem. Res. 2014, 47, 1853–1860. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Waltman, P.; Le, V.H.; Lewis, E.A. The effect of molecular crowding on the stability of human c-MYC promoter sequence I-motif at neutral pH. Molecules 2013, 18, 12751–12767. [Google Scholar] [CrossRef] [PubMed]
- Hemenway, J.N.; Carvalho, T.C.; Rao, V.M.; Wu, Y.; Levons, J.K.; Narang, A.S.; Paruchuri, S.R.; Stamato, H.J.; Varia, S.A. Formation of reactive impurities in aqueous and neat polyethylene glycol 400 and effects of antioxidants and oxidation inducers. J. Pharm. Sci. 2012, 101, 3305–3318. [Google Scholar] [CrossRef]
- Sun, D.; Hurley, L.H. The importance of negative superhelicity in inducing the formation of G-quadruplex and i-motif structures in the c-Myc promoter: Implications for drug targeting and control of gene expression. J. Med. Chem. 2009, 52, 2863–2874. [Google Scholar] [CrossRef]
- Fojtík, P.; Vorlícková, M. The fragile X chromosome (GCC) repeat folds into a DNA tetraplex at neutral pH. Nucleic Acids Res. 2001, 29, 4684–4690. [Google Scholar] [CrossRef]
- Fleming, A.M.; Ding, Y.; Rogers, R.A.; Zhu, J.; Burton, A.D.; Carlisle, C.B.; Burrows, C.J. 4n-1 Is a “Sweet Spot” in DNA i-Motif Folding of 2’-Deoxycytidine Homopolymers. J. Am. Chem. Soc. 2017, 139, 4682–4689. [Google Scholar] [CrossRef]
- Gurung, S.P.; Schwarz, C.; Hall, J.P.; Cardin, C.J.; Brazier, J.A. The importance of loop length on the stability of i-motif structures. Chem. Commun. 2015, 51, 5630–5632. [Google Scholar] [CrossRef] [PubMed]
- Fujii, T.; Sugimoto, N. Loop nucleotides impact the stability of intrastrand i-motif structures at neutral pH. Phys. Chem. Chem. Phys. 2015, 17, 16719–16722. [Google Scholar] [CrossRef] [PubMed]
- Benabou, S.; Garavís, M.; Lyonnais, S.; Eritja, R.; González, C.; Gargallo, R. Understanding the effect of the nature of the nucleobase in the loops on the stability of the i-motif structure. Phys. Chem. Chem. Phys. 2016, 18, 7997–8004. [Google Scholar] [CrossRef] [PubMed]
- Benabou, S.; Ferreira, R.; Aviñó, A.; González, C.; Lyonnais, S.; Solà, M.; Eritja, R.; Jaumot, J.; Gargallo, R. Solution equilibria of cytosine- and guanine-rich sequences near the promoter region of the n-myc gene that contain stable hairpins within lateral loops. Biochim. Biophys. Acta 2014, 1840, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Rentero, S.; Gargallo, R.; González, C.; Eritja, R. Modulation of the stability of i-motif structures using an acyclic threoninol cytidine derivative. RSC Adv. 2015, 5, 63278–63281. [Google Scholar] [CrossRef]
- Abou Assi, H.; El-Khoury, R.; González, C.; Damha, M.J. 2′-Fluoroarabinonucleic acid modification traps G-quadruplex and i-motif structures in human telomeric DNA. Nucleic Acids Res. 2017, 45, 11535–11546. [Google Scholar] [CrossRef]
- Wright, E.P.; Abdelhamid, M.A.S.; Ehiabor, M.O.; Grigg, M.C.; Irving, K.; Smith, N.M.; Waller, Z.A.E. Epigenetic modification of cytosines fine tunes the stability of i-motif DNA. Nucleic Acids Res. 2020, 48, 55–62. [Google Scholar] [CrossRef]
- Catasti, P.; Chen, X.; Mariappan, S.V.; Bradbury, E.M.; Gupta, G. DNA repeats in the human genome. Genetica 1999, 106, 15–36. [Google Scholar] [CrossRef] [PubMed]
- Bhavsar-Jog, Y.P.; Van Dornshuld, E.; Brooks, T.A.; Tschumper, G.S.; Wadkins, R.M. Co-Localization of DNA i-Motif-Forming Sequences and 5-Hydroxymethyl-cytosines in Human Embryonic Stem Cells. Molecules 2019, 24, 3619. [Google Scholar] [CrossRef] [PubMed]
- Zeraati, M.; Langley, D.B.; Schofield, P.; Moye, A.L.; Rouet, R.; Hughes, W.E.; Bryan, T.M.; Dinger, M.E.; Christ, D. I-motif DNA structures are formed in the nuclei of human cells. Nat. Chem. 2018, 10, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Niu, K.; Yu, G.; Jin, Y.; Zhang, X.; Peng, Y.; Chen, S.; Deng, H.; Li, S.; Wang, J.; et al. In vivo visualization of the i-motif DNA secondary structure in the Bombyx mori testis. Epigenetics Chromatin 2020, 13, 12. [Google Scholar] [CrossRef] [PubMed]
- Dzatko, S.; Krafcikova, M.; Hänsel-Hertsch, R.; Fessl, T.; Fiala, R.; Loja, T.; Krafcik, D.; Mergny, J.L.; Foldynova-Trantirkova, S.; Trantirek, L. Evaluation of the Stability of DNA i-Motifs in the Nuclei of Living Mammalian Cells. Angew. Chem. Int. Ed. 2018, 57, 2165–2169. [Google Scholar] [CrossRef] [PubMed]
- Garavís, M.; González, C.; Villasante, A. On the origin of the eukaryotic chromosome: The role of noncanonical DNA structures in telomere evolution. Genome Biol. Evol. 2013, 5, 1142–1150. [Google Scholar] [CrossRef] [PubMed]
- Školáková, P.; Badri, Z.; Foldynová-Trantírková, S.; Ryneš, J.; Šponer, J.; Fojtová, M.; Fajkus, J.; Marek, R.; Vorlíčková, M.; Mergny, J.L.; et al. Composite 5-methylations of cytosines modulate i-motif stability in a sequence-specific manner: Implications for DNA nanotechnology and epigenetic regulation of plant telomeric DNA. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129651. [Google Scholar] [CrossRef] [PubMed]
- Wright, E.P.; Huppert, J.L.; Waller, Z.A.E. Identification of multiple genomic DNA sequences which form i-motif structures at neutral pH. Nucleic Acids Res. 2017, 45, 2951–2959. [Google Scholar] [CrossRef] [PubMed]
- Kendrick, S.; Kang, H.J.; Alam, M.P.; Madathil, M.M.; Agrawal, P.; Gokhale, V.; Yang, D.; Hecht, S.M.; Hurley, L.H. The dynamic character of the BCL2 promoter i-motif provides a mechanism for modulation of gene expression by compounds that bind selectively to the alternative DNA hairpin structure. J. Am. Chem. Soc. 2014, 136, 4161–4171. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.J.; Kendrick, S.; Hecht, S.M.; Hurley, L.H. The transcriptional complex between the BCL2 i-motif and hnRNP LL is a molecular switch for control of gene expression that can be modulated by small molecules. J. Am. Chem. Soc. 2014, 136, 4172–4185. [Google Scholar] [CrossRef] [PubMed]
- Delbridge, A.R.; Grabow, S.; Strasser, A.; Vaux, D.L. Thirty years of BCL-2: Translating cell death discoveries into novel cancer therapies. Nat. Rev. Cancer 2016, 16, 99–109. [Google Scholar] [CrossRef]
- Kaiser, C.E.; Van Ert, N.A.; Agrawal, P.; Chawla, R.; Yang, D.; Hurley, L.H. Insight into the Complexity of the i-Motif and G-Quadruplex DNA Structures Formed in the KRAS Promoter and Subsequent Drug-Induced Gene Repression. J. Am. Chem. Soc. 2017, 139, 8522–8536. [Google Scholar] [CrossRef]
- Niu, K.; Zhang, X.; Deng, H.; Wu, F.; Ren, Y.; Xiang, H.; Zheng, S.; Liu, L.; Huang, L.; Zeng, B.; et al. BmILF and i-motif structure are involved in transcriptional regulation of BmPOUM2 in Bombyx mori. Nucleic Acids Res. 2018, 46, 1710–1723. [Google Scholar] [CrossRef]
- Miller, D.M.; Thomas, S.D.; Islam, A.; Muench, D.; Sedoris, K. c-Myc and cancer metabolism. Clin. Cancer Res. 2012, 18, 5546–5553. [Google Scholar] [CrossRef] [PubMed]
- Levens, D. You Don’t Muck with MYC. Genes Cancer 2010, 1, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V.; Le, A.; Gao, P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin. Cancer Res. 2009, 15, 6479–6483. [Google Scholar] [CrossRef] [PubMed]
- Levens, D. How the c-myc promoter works and why it sometimes does not. J. Natl. Cancer Inst. Monogr. 2008, 2008, 41–43. [Google Scholar] [CrossRef] [PubMed]
- Michelotti, E.F.; Michelotti, G.A.; Aronsohn, A.I.; Levens, D. Heterogeneous nuclear ribonucleoprotein K is a transcription factor. Mol. Cell Biol. 1996, 16, 2350–2360. [Google Scholar] [CrossRef]
- Sutherland, C.; Cui, Y.; Mao, H.; Hurley, L.H. A Mechanosensor Mechanism Controls the G-Quadruplex/i-Motif Molecular Switch in the MYC Promoter NHE III. J. Am. Chem. Soc. 2016, 138, 14138–14151. [Google Scholar] [CrossRef] [PubMed]
- Shu, B.; Cao, J.; Kuang, G.; Qiu, J.; Zhang, M.; Zhang, Y.; Wang, M.; Li, X.; Kang, S.; Ou, T.M.; et al. Syntheses and evaluation of new acridone derivatives for selective binding of oncogene c-myc promoter i-motifs in gene transcriptional regulation. Chem. Commun. 2018, 54, 2036–2039. [Google Scholar] [CrossRef]
- Sun, D.; Thompson, B.; Cathers, B.E.; Salazar, M.; Kerwin, S.M.; Trent, J.O.; Jenkins, T.C.; Neidle, S.; Hurley, L.H. Inhibition of human telomerase by a G-quadruplex-interactive compound. J. Med. Chem. 1997, 40, 2113–2116. [Google Scholar] [CrossRef]
- Fedoroff, O.Y.; Rangan, A.; Chemeris, V.V.; Hurley, L.H. Cationic porphyrins promote the formation of i-motif DNA and bind peripherally by a nonintercalative mechanism. Biochemistry 2000, 39, 15083–15090. [Google Scholar] [CrossRef]
- Bialis, T.; Dexheimer, T.; Gleason-Guzman, M.; Yang, D.; Hurley, L. Transcriptional Consequences of Targeting the i-Motif Structure of the c-Myc Promoter with TMPyP4. Cancer Res. 2007, 67, 3169. [Google Scholar]
- Wu, L.; Huang, C.; Emery, B.P.; Sedgwick, A.C.; Bull, S.D.; He, X.P.; Tian, H.; Yoon, J.; Sessler, J.L.; James, T.D. Förster resonance energy transfer (FRET)-based small-molecule sensors and imaging agents. Chem. Soc. Rev. 2020, 49, 5110–5139. [Google Scholar] [CrossRef] [PubMed]
- Day, H.A.; Huguin, C.; Waller, Z.A. Silver cations fold i-motif at neutral pH. Chem. Commun. 2013, 49, 7696–7698. [Google Scholar] [CrossRef] [PubMed]
- De Cian, A.; Guittat, L.; Kaiser, M.; Saccà, B.; Amrane, S.; Bourdoncle, A.; Alberti, P.; Teulade-Fichou, M.P.; Lacroix, L.; Mergny, J.L. Fluorescence-based melting assays for studying quadruplex ligands. Methods 2007, 42, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Pagano, A.; Iaccarino, N.; Abdelhamid, M.A.S.; Brancaccio, D.; Garzarella, E.U.; Di Porzio, A.; Novellino, E.; Waller, Z.A.E.; Pagano, B.; Amato, J.; et al. Common G-Quadruplex Binding Agents Found to Interact With i-Motif-Forming DNA: Unexpected Multi-Target-Directed Compounds. Front. Chem. 2018, 6, 281. [Google Scholar] [CrossRef] [PubMed]
- Tse, W.C.; Boger, D.L. A fluorescent intercalator displacement assay for establishing DNA binding selectivity and affinity. Accounts Chem. Res. 2004, 37, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Q.; Neaverson, J.C.; Mahmoud, T.; Stevenson, C.E.M.; Matthews, S.E.; Waller, Z.A.E. Identification of new DNA i-motif binding ligands through a fluorescent intercalator displacement assay. Org. Biomol. Chem. 2017, 15, 5669–5673. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Fu, H.; Qian, C.; Li, H.; Chen, D.D.Y. Characterization of interaction between Bcl-2 oncogene promoter I-Motif DNA and flavonoids using electrospray ionization mass spectrometry and pressure-assisted capillary electrophoresis frontal analysis. Talanta 2020, 215, 120885. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.S.; Lam, C.W.; Chan, M.H.; Cheung, R.C.; Law, L.K.; Lit, L.C.; Ng, K.F.; Suen, M.W.; Tai, H.L. Electrospray ionisation mass spectrometry: Principles and clinical applications. Clin. Biochem. Rev. 2003, 24, 3–12. [Google Scholar] [PubMed]
- Vegas, A.J.; Fuller, J.H.; Koehler, A.N. Small-molecule microarrays as tools in ligand discovery. Chem. Soc. Rev. 2008, 37, 1385–1394. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, K.M.; Saunders, L.B.; Simmons, J.K.; Leon, E.; Calabrese, D.R.; Zhang, S.; Michalowski, A.; Gareiss, P.; Mock, B.A.; Schneekloth, J.S. Small Molecule Microarrays Enable the Identification of a Selective, Quadruplex-Binding Inhibitor of MYC Expression. ACS Chem. Biol. 2016, 11, 139–148. [Google Scholar] [CrossRef]
- Journey, S.N.; Alden, S.L.; Hewitt, W.M.; Peach, M.L.; Nicklaus, M.C.; Schneekloth, J.S. Probing the hras-1Y i-motif with small molecules. Medchemcomm 2018, 9, 2000–2007. [Google Scholar] [CrossRef] [PubMed]
- Abdelhamid, M.A.S.; Gates, A.J.; Waller, Z.A.E. Destabilization of i-Motif DNA at Neutral pH by G-Quadruplex Ligands. Biochemistry 2019, 58, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Kuang, G.; Zhang, M.; Kang, S.; Hu, D.; Li, X.; Wei, Z.; Gong, X.; An, L.K.; Huang, Z.S.; Shu, B.; et al. Syntheses and Evaluation of New Bisacridine Derivatives for Dual Binding of G-Quadruplex and i-Motif in Regulating Oncogene. J. Med. Chem. 2020, 63, 9136–9153. [Google Scholar] [CrossRef] [PubMed]
- Ou, T.M.; Lin, J.; Lu, Y.J.; Hou, J.Q.; Tan, J.H.; Chen, S.H.; Li, Z.; Li, Y.P.; Li, D.; Gu, L.Q.; et al. Inhibition of cell proliferation by quindoline derivative (SYUIQ-05) through its preferential interaction with c-myc promoter G-quadruplex. J. Med. Chem. 2011, 54, 5671–5679. [Google Scholar] [CrossRef] [PubMed]
- Trybek, T.; Kowalik, A.; Góźdź, S.; Kowalska, A. Telomeres and telomerase in oncogenesis. Oncol. Lett. 2020, 20, 1015–1027. [Google Scholar] [CrossRef] [PubMed]
- Maciejowski, J.; de Lange, T. Telomeres in cancer: Tumour suppression and genome instability. Nat. Rev. Mol. Cell Biol. 2017, 18, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Meyne, J.; Ratliff, R.L.; Moyzis, R.K. Conservation of the human telomere sequence (TTAGGG)n among vertebrates. Proc. Natl. Acad. Sci. USA 1989, 86, 7049–7053. [Google Scholar] [CrossRef]
- Bryan, T.M. G-Quadruplexes at Telomeres: Friend or Foe? Molecules 2020, 25, 3686. [Google Scholar] [CrossRef]
- Abdelhamid, M.A.S.; Waller, Z.A.E. Tricky Topology: Persistence of Folded Human Telomeric i-Motif DNA at Ambient Temperature and Neutral pH. Front. Chem. 2020, 8, 40. [Google Scholar] [CrossRef]
- Chen, Y.; Qu, K.; Zhao, C.; Wu, L.; Ren, J.; Wang, J.; Qu, X. Insights into the biomedical effects of carboxylated single-wall carbon nanotubes on telomerase and telomeres. Nat. Commun. 2012, 3, 1074. [Google Scholar] [CrossRef]
- Wolski, P.; Wojton, P.; Nieszporek, K.; Panczyk, T. Interaction of Human Telomeric i-Motif DNA with Single-Walled Carbon Nanotubes: Insights from Molecular Dynamics Simulations. J. Phys. Chem. B 2019, 123, 10343–10353. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Wang, Y.; Li, X. Targeting the untargetable KRAS in cancer therapy. Acta Pharm. Sin. B 2019, 9, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Gillson, J.; Ramaswamy, Y.; Singh, G.; Gorfe, A.A.; Pavlakis, N.; Samra, J.; Mittal, A.; Sahni, S. Small Molecule KRAS Inhibitors: The Future for Targeted Pancreatic Cancer Therapy? Cancers 2020, 12, 1341. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.E.; Bar-Sagi, D.; Nassar, N. The structural basis for the transition from Ras-GTP to Ras-GDP. Proc. Natl. Acad. Sci. USA 2002, 99, 12138–12142. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Maldonado, C.; Zimmer, Y.; Medová, M. A Comparative Analysis of Individual RAS Mutations in Cancer Biology. Front. Oncol. 2019, 9, 1088. [Google Scholar] [CrossRef] [PubMed]
- Hamarsheh, S.; Groß, O.; Brummer, T.; Zeiser, R. Immune modulatory effects of oncogenic KRAS in cancer. Nat. Commun. 2020, 11, 5439. [Google Scholar] [CrossRef]
- Gorfe, A.A.; Cho, K.J. Approaches to inhibiting oncogenic K-Ras. Small GTPases 2019, 1–10. [Google Scholar] [CrossRef]
- Heldin, C.H.; Westermark, B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol. Rev. 1999, 79, 1283–1316. [Google Scholar] [CrossRef]
- Wu, E.; Palmer, N.; Tian, Z.; Moseman, A.P.; Galdzicki, M.; Wang, X.; Berger, B.; Zhang, H.; Kohane, I.S. Comprehensive dissection of PDGF-PDGFR signaling pathways in PDGFR genetically defined cells. PLoS ONE 2008, 3, e3794. [Google Scholar] [CrossRef] [PubMed]
- Hellström, M.; Kalén, M.; Lindahl, P.; Abramsson, A.; Betsholtz, C. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development 1999, 126, 3047–3055. [Google Scholar]
- Andrae, J.; Gallini, R.; Betsholtz, C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008, 22, 1276–1312. [Google Scholar] [CrossRef] [PubMed]
- Hermanson, M.; Funa, K.; Hartman, M.; Claesson-Welsh, L.; Heldin, C.H.; Westermark, B.; Nistér, M. Platelet-derived growth factor and its receptors in human glioma tissue: Expression of messenger RNA and protein suggests the presence of autocrine and paracrine loops. Cancer Res. 1992, 52, 3213–3219. [Google Scholar] [PubMed]
- Baxter, E.J.; Kulkarni, S.; Vizmanos, J.L.; Jaju, R.; Martinelli, G.; Testoni, N.; Hughes, G.; Salamanchuk, Z.; Calasanz, M.J.; Lahortiga, I.; et al. Novel translocations that disrupt the platelet-derived growth factor receptor beta (PDGFRB) gene in BCR-ABL-negative chronic myeloproliferative disorders. Br. J. Haematol. 2003, 120, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Raica, M.; Cimpean, A.M. Platelet-Derived Growth Factor (PDGF)/PDGF Receptors (PDGFR) Axis as Target for Antitumor and Antiangiogenic Therapy. Pharmaceuticals 2010, 3, 572–599. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.V.; Wang, T.; Chappeta, V.R.; Wu, G.; Onel, B.; Chawla, R.; Quijada, H.; Camp, S.M.; Chiang, E.T.; Lassiter, Q.R.; et al. The Consequences of Overlapping G-Quadruplexes and i-Motifs in the Platelet-Derived Growth Factor Receptor β Core Promoter Nuclease Hypersensitive Element Can Explain the Unexpected Effects of Mutations and Provide Opportunities for Selective Targeting of Both Structures by Small Molecules to Downregulate Gene Expression. J. Am. Chem. Soc. 2017, 139, 7456–7475. [Google Scholar] [CrossRef]
- Guo, K.; Gokhale, V.; Hurley, L.H.; Sun, D. Intramolecularly folded G-quadruplex and i-motif structures in the proximal promoter of the vascular endothelial growth factor gene. Nucleic Acids Res. 2008, 36, 4598–4608. [Google Scholar] [CrossRef]
- Greco, M.L.; Folini, M.; Sissi, C. Double stranded promoter region of BRAF undergoes to structural rearrangement in nearly physiological conditions. FEBS Lett. 2015, 589, 2117–2123. [Google Scholar] [CrossRef]
- Li, H.; Hai, J.; Zhou, J.; Yuan, G. The formation and characteristics of the i-motif structure within the promoter of the c-myb proto-oncogene. J. Photochem. Photobiol. B 2016, 162, 625–632. [Google Scholar] [CrossRef]
- Miglietta, G.; Cogoi, S.; Pedersen, E.B.; Xodo, L.E. GC-elements controlling HRAS transcription form i-motif structures unfolded by heterogeneous ribonucleoprotein particle A1. Sci. Rep. 2015, 5, 18097. [Google Scholar] [CrossRef]
- Buscà, R.; Abbe, P.; Mantoux, F.; Aberdam, E.; Peyssonnaux, C.; Eychène, A.; Ortonne, J.P.; Ballotti, R. Ras mediates the cAMP-dependent activation of extracellular signal-regulated kinases (ERKs) in melanocytes. EMBO J. 2000, 19, 2900–2910. [Google Scholar] [CrossRef]
- Jung, J.; Cho, K.J.; Naji, A.K.; Clemons, K.N.; Wong, C.O.; Villanueva, M.; Gregory, S.; Karagas, N.E.; Tan, L.; Liang, H.; et al. HRAS-driven cancer cells are vulnerable to TRPML1 inhibition. EMBO Rep. 2019, 20, e46685. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, R.G.; Gonda, T.J. MYB function in normal and cancer cells. Nat. Rev. Cancer 2008, 8, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.W.; Lee, Y.J.; Kim, J. Role of c-Myb in the regulation of natural killer cell activity. Biochem. Biophys. Res. Commun. 2018, 503, 2807–2813. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.K.; Bhardwaj, A.; Arora, S.; Singh, S.; Azim, S.; Tyagi, N.; Carter, J.E.; Wang, B.; Singh, A.P. MYB is a novel regulator of pancreatic tumour growth and metastasis. Br. J. Cancer 2015, 113, 1694–1703. [Google Scholar] [CrossRef]
- Karaman, S.; Leppänen, V.M.; Alitalo, K. Vascular endothelial growth factor signaling in development and disease. Development 2018, 145, 151019. [Google Scholar] [CrossRef]
- Estrada, C.C.; Maldonado, A.; Mallipattu, S.K. Therapeutic Inhibition of VEGF Signaling and Associated Nephrotoxicities. J. Am. Soc. Nephrol. 2019, 30, 187–200. [Google Scholar] [CrossRef]
- Takahashi, S.; Bhattacharjee, S.; Ghosh, S.; Sugimoto, N.; Bhowmik, S. Preferential targeting cancer-related i-motif DNAs by the plant flavonol fisetin for theranostics applications. Sci. Rep. 2020, 10, 2504. [Google Scholar] [CrossRef]
- Rogers, R.A.; Fleming, A.M.; Burrows, C.J. Unusual Isothermal Hysteresis in DNA i-Motif pH Transitions: A Study of the RAD17 Promoter Sequence. Biophys. J. 2018, 114, 1804–1815. [Google Scholar] [CrossRef]
- Rogers, R.A.; Fleming, A.M.; Burrows, C.J. Rapid Screen of Potential i-Motif Forming Sequences in DNA Repair Gene Promoters. ACS Omega 2018, 3, 9630–9635. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Sugiyama, H. Formation of the G-quadruplex and i-motif structures in retinoblastoma susceptibility genes (Rb). Nucleic Acids Res. 2006, 34, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Benabou, S.; Aviñó, A.; Lyonnais, S.; González, C.; Eritja, R.; De Juan, A.; Gargallo, R. i-motif structures in long cytosine-rich sequences found upstream of the promoter region of the SMARCA4 gene. Biochimie 2017, 140, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Levy-Strumpf, N.; Kimchi, A. Death associated proteins (DAPs): From gene identification to the analysis of their apoptotic and tumor suppressive functions. Oncogene 1998, 17, 3331–3340. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Ye, L.; Ji, K.; Toms, A.M.; Davies, M.L.; Ruge, F.; Ji, J.; Hargest, R.; Jiang, W.G. Death associated protein 1 is correlated with the clinical outcome of patients with colorectal cancer and has a role in the regulation of cell death. Oncol. Rep. 2014, 31, 175–182. [Google Scholar] [CrossRef]
- Yasuda, Y.; Sakai, A.; Ito, S.; Sasai, K.; Yamamoto, H.; Matsubara, N.; Ouchida, M.; Katayama, H.; Shimizu, K. Human RAD 17 Polymorphism at Codon 546 Is Associated with the Risk of Colorectal Cancer. Acta Med. Okayama 2017, 71, 59–68. [Google Scholar] [CrossRef]
- Sasaki, H.; Chen, L.B.; Auclair, D.; Moriyama, S.; Kaji, M.; Fukai, I.; Kiriyama, M.; Yamakawa, Y.; Fujii, Y. Overexpression of Hrad17 gene in non-small cell lung cancers correlated with lymph node metastasis. Lung Cancer 2001, 34, 47–52. [Google Scholar] [CrossRef]
- Valenti, F.; Sacconi, A.; Ganci, F.; Grasso, G.; Strano, S.; Blandino, G.; Di Agostino, S. The miR-205-5p/BRCA1/RAD17 Axis Promotes Genomic Instability in Head and Neck Squamous Cell Carcinomas. Cancers 2019, 11, 1347. [Google Scholar] [CrossRef]
- Cheng, J.; Demeulemeester, J.; Wedge, D.C.; Vollan, H.K.M.; Pitt, J.J.; Russnes, H.G.; Pandey, B.P.; Nilsen, G.; Nord, S.; Bignell, G.R.; et al. Pan-cancer analysis of homozygous deletions in primary tumours uncovers rare tumour suppressors. Nat. Commun. 2017, 8, 1221. [Google Scholar] [CrossRef]
- Wang, X.; Zou, L.; Zheng, H.; Wei, Q.; Elledge, S.J.; Li, L. Genomic instability and endoreduplication triggered by RAD17 deletion. Genes Dev. 2003, 17, 965–970. [Google Scholar] [CrossRef]
- Walter, D.M.; Yates, T.J.; Ruiz-Torres, M.; Kim-Kiselak, C.; Gudiel, A.A.; Deshpande, C.; Wang, W.Z.; Cicchini, M.; Stokes, K.L.; Tobias, J.W.; et al. RB constrains lineage fidelity and multiple stages of tumour progression and metastasis. Nature 2019, 569, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, S.; Takahashi, C. Intersection of retinoblastoma tumor suppressor function, stem cells, metabolism, and inflammation. Cancer Sci. 2017, 108, 1726–1731. [Google Scholar] [CrossRef] [PubMed]
- Labrecque, M.P.; Takhar, M.K.; Nason, R.; Santacruz, S.; Tam, K.J.; Massah, S.; Haegert, A.; Bell, R.H.; Altamirano-Dimas, M.; Collins, C.C.; et al. The retinoblastoma protein regulates hypoxia-inducible genetic programs, tumor cell invasiveness and neuroendocrine differentiation in prostate cancer cells. Oncotarget 2016, 7, 24284–24302. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Yeow, W.S.; Ertel, A.; Coleman, I.; Clegg, N.; Thangavel, C.; Morrissey, C.; Zhang, X.; Comstock, C.E.; Witkiewicz, A.K.; et al. The retinoblastoma tumor suppressor controls androgen signaling and human prostate cancer progression. J. Clin. Investig. 2010, 120, 4478–4492. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, J.C.; Zhang, L.; Evoli, S.; Schnicker, N.J.; Nunez-Hernandez, M.; Yu, L.; Wereszczynski, J.; Pufall, M.A.; Musselman, C.A. The molecular basis of selective DNA binding by the BRG1 AT-hook and bromodomain. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194566. [Google Scholar] [CrossRef] [PubMed]
- Güneş, C.; Paszkowski-Rogacz, M.; Rahmig, S.; Khattak, S.; Camgöz, A.; Wermke, M.; Dahl, A.; Bornhäuser, M.; Waskow, C.; Buchholz, F. Comparative RNAi Screens in Isogenic Human Stem Cells Reveal SMARCA4 as a Differential Regulator. Stem Cell Rep. 2019, 12, 1084–1098. [Google Scholar] [CrossRef] [PubMed]
- Kadoch, C.; Crabtree, G.R. Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Sci. Adv. 2015, 1, e1500447. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Meehan, B.; Fu, Z.; Wang, X.Q.D.; Fiset, P.O.; Rieker, R.; Levins, C.; Kong, T.; Zhu, X.; Morin, G.; et al. SMARCA4 loss is synthetic lethal with CDK4/6 inhibition in non-small cell lung cancer. Nat. Commun. 2019, 10, 557. [Google Scholar] [CrossRef]
- Numata, M.; Morinaga, S.; Watanabe, T.; Tamagawa, H.; Yamamoto, N.; Shiozawa, M.; Nakamura, Y.; Kameda, Y.; Okawa, S.; Rino, Y.; et al. The clinical significance of SWI/SNF complex in pancreatic cancer. Int. J. Oncol. 2013, 42, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Roy, N.; Malik, S.; Villanueva, K.E.; Urano, A.; Lu, X.; Von Figura, G.; Seeley, E.S.; Dawson, D.W.; Collisson, E.A.; Hebrok, M. Brg1 promotes both tumor-suppressive and oncogenic activities at distinct stages of pancreatic cancer formation. Genes Dev. 2015, 29, 658–671. [Google Scholar] [CrossRef]
- Wu, Q.; Madany, P.; Dobson, J.R.; Schnabl, J.M.; Sharma, S.; Smith, T.C.; van Wijnen, A.J.; Stein, J.L.; Lian, J.B.; Stein, G.S.; et al. The BRG1 chromatin remodeling enzyme links cancer cell metabolism and proliferation. Oncotarget 2016, 7, 38270–38281. [Google Scholar] [CrossRef] [PubMed]
- Abramson, H.N. The lipogenesis pathway as a cancer target. J. Med. Chem. 2011, 54, 5615–5638. [Google Scholar] [CrossRef] [PubMed]
- Moreau, K.; Dizin, E.; Ray, H.; Luquain, C.; Lefai, E.; Foufelle, F.; Billaud, M.; Lenoir, G.M.; Venezia, N.D. BRCA1 affects lipid synthesis through its interaction with acetyl-CoA carboxylase. J. Biol. Chem. 2006, 281, 3172–3181. [Google Scholar] [CrossRef] [PubMed]
- Rios Garcia, M.; Steinbauer, B.; Srivastava, K.; Singhal, M.; Mattijssen, F.; Maida, A.; Christian, S.; Hess-Stumpp, H.; Augustin, H.G.; Müller-Decker, K.; et al. Acetyl-CoA Carboxylase 1-Dependent Protein Acetylation Controls Breast Cancer Metastasis and Recurrence. Cell Metab. 2017, 26, 842–855.e5. [Google Scholar] [CrossRef] [PubMed]
- Corominas-Faja, B.; Cuyàs, E.; Gumuzio, J.; Bosch-Barrera, J.; Leis, O.; Martin, Á.; Menendez, J.A. Chemical inhibition of acetyl-CoA carboxylase suppresses self-renewal growth of cancer stem cells. Oncotarget 2014, 5, 8306–8316. [Google Scholar] [CrossRef]
- Kaulage, M.H.; Bhattacharya, S.; Muniyappa, K. Structural Characterization of i-Motif Structure in the Human Acetyl-CoA Carboxylase1 Gene Promoters and Their Role in the Regulation of Gene Expression. Chembiochem 2018, 19, 1078–1087. [Google Scholar] [CrossRef] [PubMed]
- Buchan, J.R.; Parker, R. Eukaryotic stress granules: The ins and outs of translation. Mol. Cell 2009, 36, 932–941. [Google Scholar] [CrossRef]
- Kaehler, C.; Isensee, J.; Nonhoff, U.; Terrey, M.; Hucho, T.; Lehrach, H.; Krobitsch, S. Ataxin-2-like is a regulator of stress granules and processing bodies. PLoS ONE 2012, 7, e50134. [Google Scholar] [CrossRef]
- Mahboubi, H.; Stochaj, U. Cytoplasmic stress granules: Dynamic modulators of cell signaling and disease. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 884–895. [Google Scholar] [CrossRef]
- Lin, L.; Li, X.; Pan, C.; Lin, W.; Shao, R.; Liu, Y.; Zhang, J.; Luo, Y.; Qian, K.; Shi, M.; et al. ATXN2L upregulated by epidermal growth factor promotes gastric cancer cell invasiveness and oxaliplatin resistance. Cell Death Dis. 2019, 10, 173. [Google Scholar] [CrossRef]
- Kanda, T.; Matsuoka, S.; Yamazaki, M.; Shibata, T.; Nirei, K.; Takahashi, H.; Kaneko, T.; Fujisawa, M.; Higuchi, T.; Nakamura, H.; et al. Apoptosis and non-alcoholic fatty liver diseases. World J. Gastroenterol. 2018, 24, 2661–2672. [Google Scholar] [CrossRef] [PubMed]
- Ramalho, R.M.; Cortez-Pinto, H.; Castro, R.E.; Solá, S.; Costa, A.; Moura, M.C.; Camilo, M.E.; Rodrigues, C.M. Apoptosis and Bcl-2 expression in the livers of patients with steatohepatitis. Eur. J. Gastroenterol. Hepatol. 2006, 18, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, J.; Gong, X.; Zhang, M.; Kang, S.; Shu, B.; Wei, Z.; Huang, Z.S.; Li, D. Upregulation of BCL-2 by acridone derivative through gene promoter i-motif for alleviating liver damage of NAFLD/NASH. Nucleic Acids Res. 2020, 48, 8255–8268. [Google Scholar] [CrossRef] [PubMed]
- Ellegren, H. Microsatellites: Simple sequences with complex evolution. Nat. Rev. Genet. 2004, 5, 435–445. [Google Scholar] [CrossRef] [PubMed]
- McMurray, C.T. DNA secondary structure: A common and causative factor for expansion in human disease. Proc. Natl. Acad. Sci. USA 1999, 96, 1823–1825. [Google Scholar] [CrossRef] [PubMed]
- Murat, P.; Guilbaud, G.; Sale, J.E. DNA polymerase stalling at structured DNA constrains the expansion of short tandem repeats. Genome Biol. 2020, 21, 209. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-W.; Satange, R.; Wu, P.-C.; Jhan, C.-R.; Chang, C.; Chung, K.-R.; Waring, M.J.; Lin, S.-W.; Hsieh, L.-C.; Hou, M.-H. CoII(Chromomycin)2 Complex Induces a Conformational Change of CCG Repeats from i-Motif to Base-Extruded DNA Duplex. Int. J. Mol. Sci. 2018, 19, 2796. [Google Scholar] [CrossRef] [PubMed]
- Pataskar, S.S.; Dash, D.; Brahmachari, S.K. Intramolecular i-motif structure at acidic pH for progressive myoclonus epilepsy (EPM1) repeat d(CCCCGCCCCGCG)n. J. Biomol. Struct. Dyn. 2001, 19, 307–313. [Google Scholar] [CrossRef]
- Kovanda, A.; Zalar, M.; Šket, P.; Plavec, J.; Rogelj, B. Anti-sense DNA d(GGCCCC)n expansions in C9ORF72 form i-motifs and protonated hairpins. Sci. Rep. 2015, 5, 17944. [Google Scholar] [CrossRef]
- Pataskar, S.S.; Dash, D.; Brahmachari, S.K. Progressive myoclonus epilepsy [EPM1] repeat d(CCCCGCCCCGCG)n forms folded hairpin structures at physiological pH. J. Biomol. Struct. Dyn. 2001, 19, 293–305. [Google Scholar] [CrossRef]
- Knop, J.M.; Mukherjee, S.K.; Oliva, R.; Möbitz, S.; Winter, R. Remodeling of the Conformational Dynamics of Noncanonical DNA Structures by Monomeric and Aggregated α-Synuclein. J. Am. Chem. Soc. 2020, 142, 18299–18303. [Google Scholar] [CrossRef] [PubMed]
- Daubner, S.C.; Le, T.; Wang, S. Tyrosine hydroxylase and regulation of dopamine synthesis. Arch. Biochem. Biophys. 2011, 508, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, K.; Wang, M.; Cai, E.; Fujiwara, N.; Baker, H.; Cave, J.W. Regulation of tyrosine hydroxylase transcription by hnRNP K and DNA secondary structure. Nat. Commun. 2014, 5, 5769. [Google Scholar] [CrossRef] [PubMed]
- Farhath, M.M.; Thompson, M.; Ray, S.; Sewell, A.; Balci, H.; Basu, S. G-Quadruplex-Enabling Sequence within the Human Tyrosine Hydroxylase Promoter Differentially Regulates Transcription. Biochemistry 2015, 54, 5533–5545. [Google Scholar] [CrossRef] [PubMed]
- Thorne, B.N.; Ellenbroek, B.A.; Day, D.J. Evaluation of i-Motif Formation in the Serotonin Transporter-Linked Polymorphic Region. Chembiochem 2020, 22, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Crawford, A.A.; Lewis, G.; Lewis, S.J.; Munafò, M.R. Systematic review and meta-analysis of serotonin transporter genotype and discontinuation from antidepressant treatment. Eur. Neuropsychopharmacol. 2013, 23, 1143–1150. [Google Scholar] [CrossRef] [PubMed]
- Todorov, G.; Cunha, C. Hypothesis: Regulation of neuroplasticity may involve I-motif and G-quadruplex DNA formation modulated by epigenetic mechanisms. Med. Hypotheses 2019, 127, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Machado-Vieira, R.; Manji, H.K.; Zarate, C.A. The role of lithium in the treatment of bipolar disorder: Convergent evidence for neurotrophic effects as a unifying hypothesis. Bipolar Disord. 2009, 11 (Suppl. 2), 92–109. [Google Scholar] [CrossRef]
- Bschor, T. Lithium in the treatment of major depressive disorder. Drugs 2014, 74, 855–862. [Google Scholar] [CrossRef]
- Nutt, D.J. Relationship of neurotransmitters to the symptoms of major depressive disorder. J. Clin. Psychiatry 2008, 69 (Suppl. E1), 4–7. [Google Scholar]
- Ashok, A.H.; Marques, T.R.; Jauhar, S.; Nour, M.M.; Goodwin, G.M.; Young, A.H.; Howes, O.D. The dopamine hypothesis of bipolar affective disorder: The state of the art and implications for treatment. Mol. Psychiatry 2017, 22, 666–679. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.E.; Lee, I.B.; Hyeon, C.; Hong, S.C. Destabilization of i-motif by submolar concentrations of a monovalent cation. J. Phys. Chem. B 2014, 118, 4753–4760. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Kong, D.; Ghimire, C.; Xu, C.; Mao, H. Mutually Exclusive Formation of G-Quadruplex and i-Motif Is a General Phenomenon Governed by Steric Hindrance in Duplex DNA. Biochemistry 2016, 55, 2291–2299. [Google Scholar] [CrossRef] [PubMed]
- King, J.J.; Irving, K.L.; Evans, C.W.; Chikhale, R.V.; Becker, R.; Morris, C.J.; Peña Martinez, C.D.; Schofield, P.; Christ, D.; Hurley, L.H.; et al. DNA G-Quadruplex and i-Motif Structure Formation Is Interdependent in Human Cells. J. Am. Chem. Soc. 2020, 142, 20600–20604. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).