
Preclinical Pharmacokinetics and Biodistribution of Anticancer Dinuclear Palladium(II)-Spermine Complex (Pd2Spm) in Mice

 ,
, 
 ,
,  , , ,
, , , 
Abstract
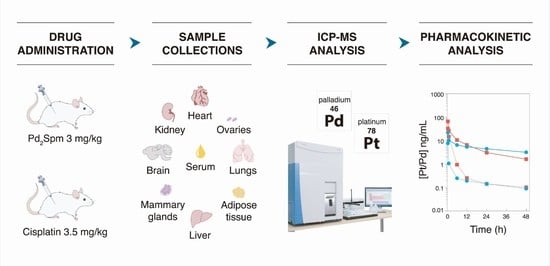
1. Introduction
2. Results
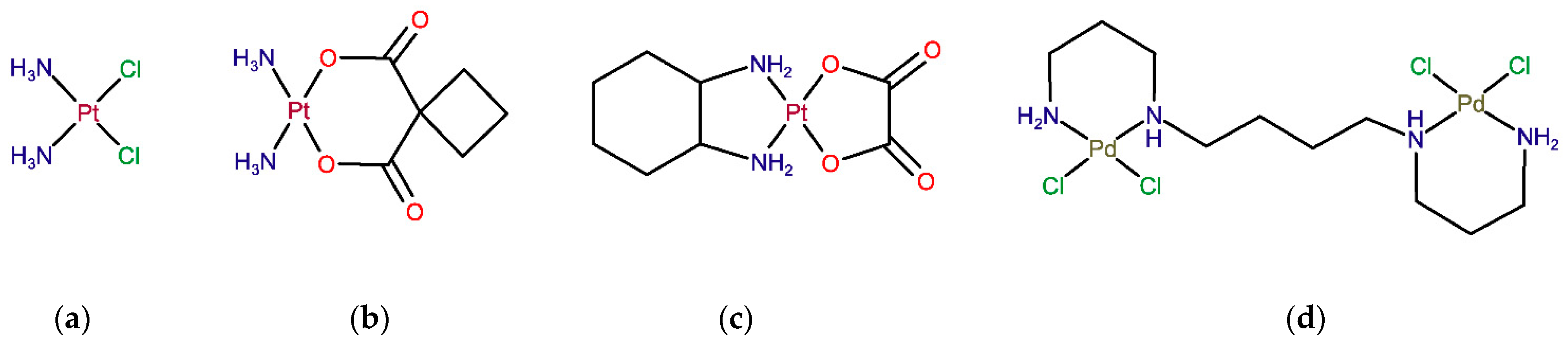
2.1. Drug Pharmacokinetics in Serum and Serum Ultrafiltrate
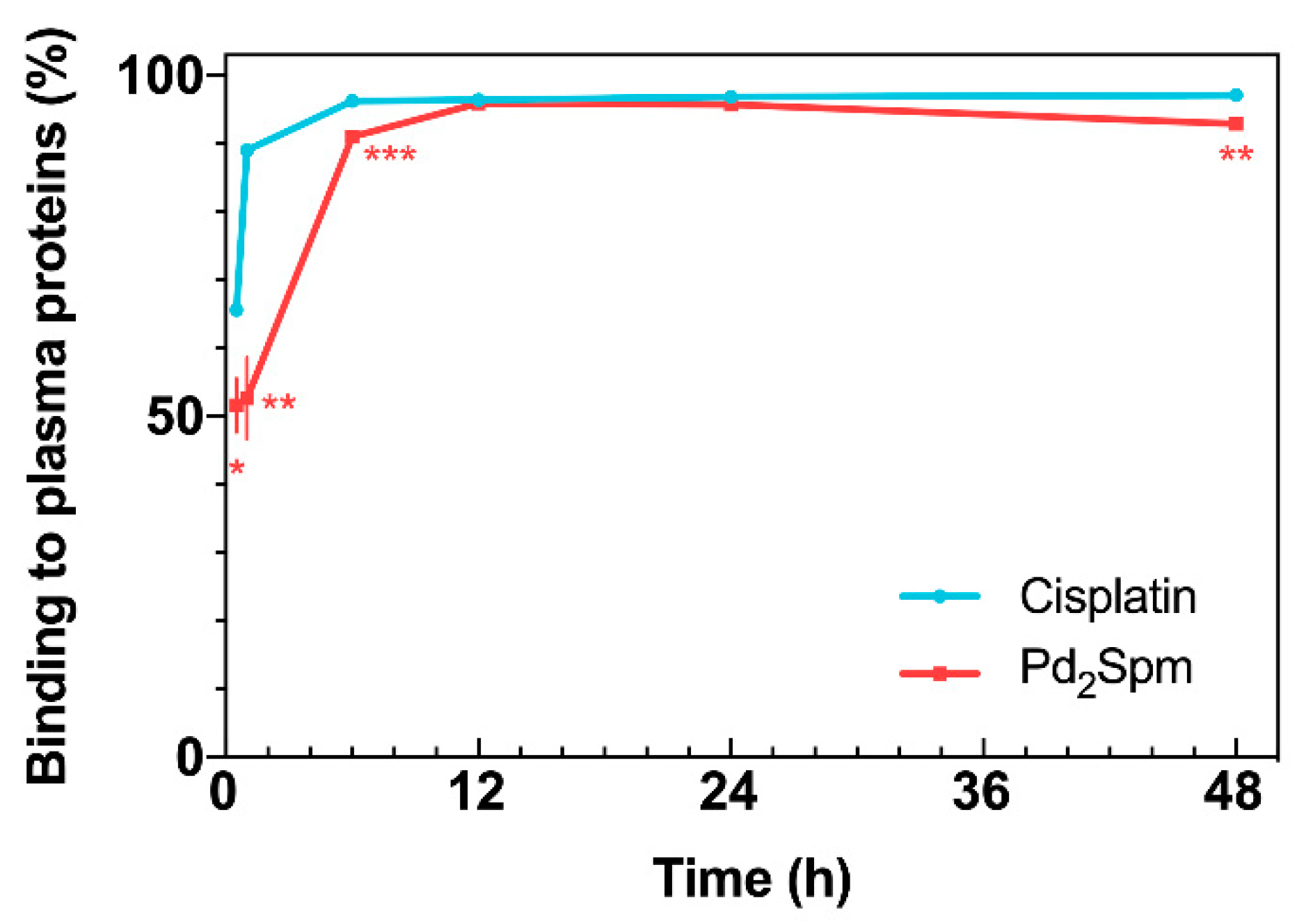
2.2. Drug Binding to Plasma Proteins
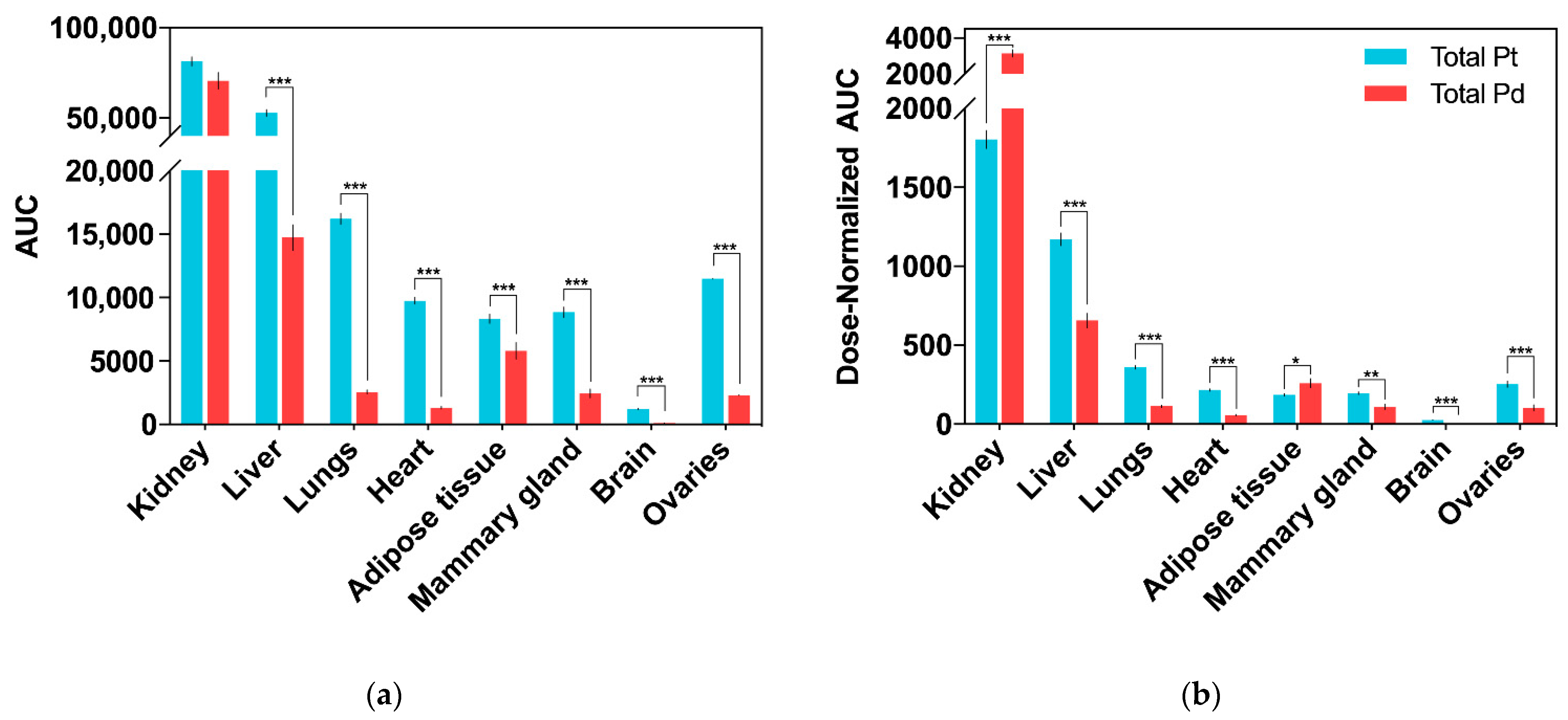
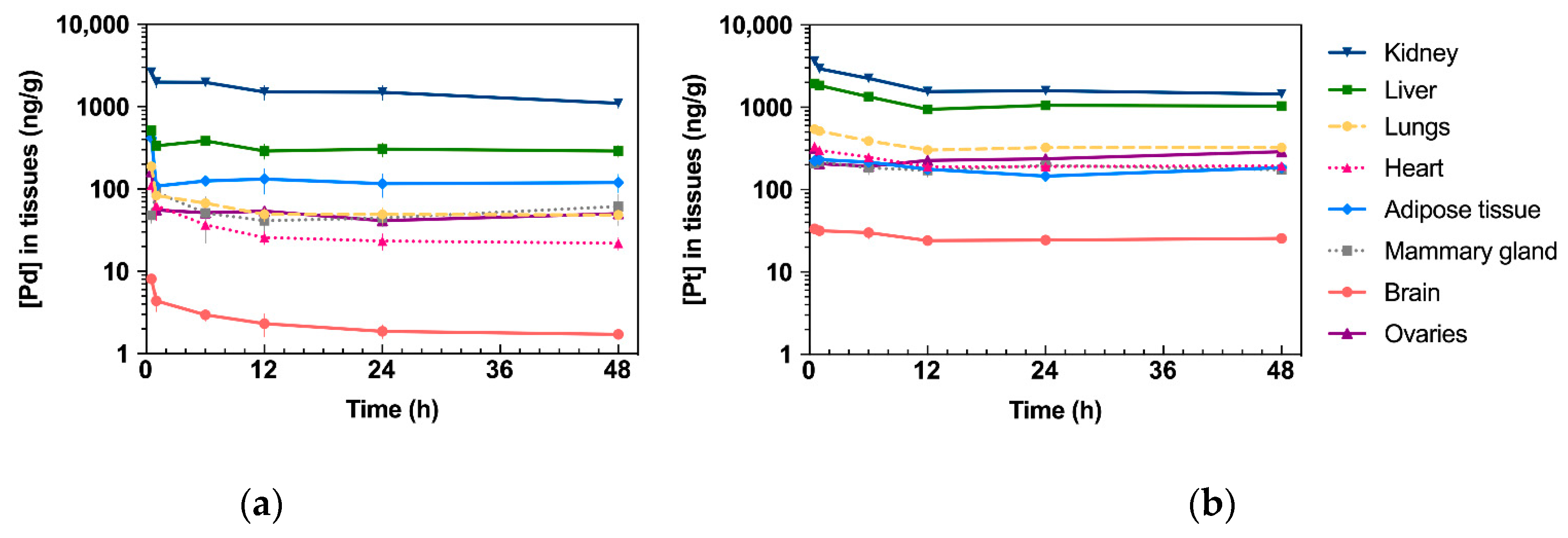
2.3. Drug Tissue Biodistribution
2.4. In Vitro Cellular Drug Uptake
3. Discussion
4. Materials and Methods
4.1. Reagents and Chemicals
4.2. Cell Culture
4.3. Ethical Considerations
4.4. Animals
4.5. Pd2Spm Synthesis and Formulation
4.6. Pharmacokinetic Study Design and Sample Collection
4.7. Sample Preparation and ICP-MS Analysis
4.8. Pharmacokinetic Analysis
4.9. Intracellular Accumulation of Pd2Spm or Cisplatin in Breast Cancer Cells
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Time (h) | Body Weight (g) | Kidney (mg) | Liver (mg) | Lungs (mg) | Heart (mg) | Brain (mg) | Adipose Tissue (mg) | Mammary Gland (mg) |
|---|---|---|---|---|---|---|---|---|---|
| Ciplatin | 0.5 | 20.6 ± 2.3 | 157.7 ± 19.4 | 962.3 ± 120.8 | 192.5 ± 31.5 | 110.9 ± 10.6 | 182.8 ± 28.0 | 101.8 ± 44.0 | 110.8 ± 31.1 |
| 1 | 20.0 ± 1.6 | 151.8 ± 23.4 | 765.8 ± 378.5 | 159.0 ± 32.9 | 99.5 ± 7.6 | 178.0 ± 29.4 | 95.0 ± 46.7 | 81.5 ± 37.1 | |
| 6 | 19.6 ± 0.9 | 140.4 ± 10.1 | 824.5 ± 144.3 | 197.1 ± 7.2 | 104.8 ± 7.5 | 188.9 ± 22.3 | 86.6 ± 30.9 | 100.3 ± 45.3 | |
| 12 | 20.0 ± 2.1 | 156.3 ± 20.2 | 878.3 ± 224.3 | 184.3 ± 23.5 | 114.4 ± 19.0 | 166.9 ± 34.9 | 93.8 ± 32.1 | 105.3 ± 44.4 | |
| 24 | 19.8 ± 1.5 | 153.6 ± 14.9 | 824.5 ± 155.8 | 173.0 ± 21.1 | 109.6 ± 5.6 | 183.3 ± 22.8 | 87.9 ± 28.5 | 95.0 ± 46.4 | |
| 48 | 19.8 ± 2.0 | 149.3 ± 20.2 | 939.7 ± 137.0 | 166.1 ± 17.2 | 107.6 ± 8.9 | 195.7 ± 24.6 | 79.3 ± 16.9 | 116.8 ± 21.2 | |
| Pd2Spm | 0.5 | 21.2 ± 1.3 | 160.9 ± 11.2 | 897.9 ± 199.2 | 188.8 ± 16.8 | 113.4 ± 8.1 | 200.7 ± 31.7 | 82.6 ± 12.9 | 119.0 ± 12.4 |
| 1 | 22.2 ± 1.1 | 161.2 ± 7.2 | 1034.4 ± 89.6 | 187.9 ± 27.8 | 117.0 ± 7.0 | 191.5 ± 20.7 | 87.7 ± 12.3 | 119.0 ± 18,6 | |
| 6 | 21.0 ± 1.2 | 153.3 ± 12.6 | 1056.8 ± 264.3 | 188.3 ± 18.9 | 112.7 ± 12.0 | 212.8 ± 23.4 | 86.8 ± 14.2 | 115.0 ± 23.3 | |
| 12 | 19.6 ± 2.2 | 148.9 ± 14.4 | 1010.1 ± 196.4 | 188.4 ± 11.9 | 105.3 ± 7.9 | 179.8 ± 18.7 | 89.3 ± 20.8 | 88.1 ± 21.7 | |
| 24 | 20.6 ± 1.1 | 168.4 ± 14.8 | 868.8 ± 76.0 | 190.6 ± 19.8 | 115.9 ± 0.4 | 190.1 ± 11.4 | 63.1 ± 17.3 | 83.0 ± 27.9 | |
| 48 | 20.6 ± 0.9 | 154.4 ± 6.3 | 983.9 ± 279.7 | 197.1 ± 21.0 | 105.5 ± 6.1 | 203.1 ± 24.6 | 68.6 ± 8.0 | 87.7 ± 26.3 |
Appendix B


References
- Dasari, S.; Bernard Tchounwou, P. Cisplatin in Cancer Therapy: Molecular Mechanisms of Action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Lippard, S.J. Cellular Processing of Platinum Anticancer Drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular Mechanisms of Cisplatin Resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef] [PubMed]
- Mohn, C.; Häcker, H.G.; Hilger, R.A.; Gütschow, M.; Jaehde, U. Defining the Role of MRP-Mediated Efflux and Glutathione in Detoxification of Oxaliplatin. Pharmazie 2013, 68, 622–627. [Google Scholar] [CrossRef] [PubMed]
- Schoch, S.; Gajewski, S.; Rothfuß, J.; Hartwig, A.; Köberle, B. Comparative Study of the Mode of Action of Clinically Approved Platinum-Based Chemotherapeutics. Int. J. Mol. Sci. 2020, 21, 6928. [Google Scholar] [CrossRef]
- Makovec, T. Cisplatin and beyond: Molecular Mechanisms of Action and Drug Resistance Development in Cancer Chemotherapy. Radiol. Oncol. 2019, 53, 148–158. [Google Scholar] [CrossRef]
- Marques, M.P.M.; Batista De Carvalho, A.L.M.; Sakai, V.G.; Hatter, L.; Batista De Carvalho, L.A.E. Intracellular Water-an Overlooked Drug Target? Cisplatin Impact in Cancer Cells Probed by Neutrons. Phys. Chem. Chem. Phys. 2017, 19, 2702–2713. [Google Scholar] [CrossRef]
- Eljack, N.D.; Ma, H.Y.M.; Drucker, J.; Shen, C.; Hambley, T.W.; New, E.J.; Friedrich, T.; Clarke, R.J. Mechanisms of Cell Uptake and Toxicity of the Anticancer Drug Cisplatin. Metallomics 2014, 6, 2126–2133. [Google Scholar] [CrossRef]
- Dilruba, S.; Kalayda, G.V. Platinum-Based Drugs: Past, Present and Future. Cancer Chemother. Pharmacol. 2016, 77, 1103–1124. [Google Scholar] [CrossRef] [PubMed]
- Frezza, M.; Hindo, S.; Chen, D.; Davenport, A.; Schmitt, S.; Tomco, D.; Dou, Q.P. Novel Metals and Metal Complexes as Platforms for Cancer Therapy. Curr. Pharm. Des. 2010, 16, 1813–1825. [Google Scholar] [CrossRef]
- Kostova, I. Platinum Complexes as Anticancer Agents. Recent Pat. Anticancer. Drug Discov. 2006, 1, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Vojtek, M.; Marques, M.P.M.; Ferreira, I.M.P.L.V.O.; Mota-Filipe, H.; Diniz, C. Anticancer Activity of Palladium-Based Complexes against Triple-Negative Breast Cancer. Drug Discov. Today 2019, 24, 1044–1058. [Google Scholar] [CrossRef]
- Ndagi, U.; Mhlongo, N.; Soliman, M.E. Metal Complexes in Cancer Therapy—An Update from Drug Design Perspective. Drug Des. Devel. Ther. 2017, 11, 599–616. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Kim, C.Y.; Nam, T.G. Ruthenium Complexes as Anticancer Agents: A Brief History and Perspectives. Drug Des. Devel. Ther. 2020, 14, 5375–5392. [Google Scholar] [CrossRef] [PubMed]
- Hussain, A.; AlAjmi, M.F.; Rehman, M.T.; Amir, S.; Husain, F.M.; Alsalme, A.; Siddiqui, M.A.; AlKhedhairy, A.A.; Khan, R.A. Copper(II) Complexes as Potential Anticancer and Nonsteroidal Anti-Inflammatory Agents: In Vitro and in Vivo Studies. Sci. Rep. 2019, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Lazarević, T.; Rilak, A.; Bugarčić, Ž.D. Platinum, Palladium, Gold and Ruthenium Complexes as Anticancer Agents: Current Clinical Uses, Cytotoxicity Studies and Future Perspectives. Eur. J. Med. Chem. 2017, 142, 8–31. [Google Scholar] [CrossRef]
- Marques, M.P.M. Platinum and Palladium Polyamine Complexes as Anticancer Agents: The Structural Factor. ISRN Spectrosc. 2013, 2013, 1–29. [Google Scholar] [CrossRef]
- Martins, A.S.; Batista de Carvalho, A.L.M.; Lamego, I.; Marques, M.P.M.; Gil, A.M. Cytotoxicity of Platinum and Palladium Chelates against Osteosarcoma. ChemistrySelect 2020, 5, 5993–6000. [Google Scholar] [CrossRef]
- Batista de Carvalho, A.L.M.; Medeiros, P.S.C.; Costa, F.M.; Ribeiro, V.P.; Sousa, J.B.; Diniz, C.; Marques, M.P.M. Anti-Invasive and Anti-Proliferative Synergism between Docetaxel and a Polynuclear Pd-Spermine Agent. PLoS ONE 2016, 11, e0167218. [Google Scholar] [CrossRef]
- Batista de Carvalho, A.L.M.; Pilling, M.; Gardner, P.; Doherty, J.; Cinque, G.; Wehbe, K.; Kelley, C.; Batista de Carvalho, L.A.E.; Marques, M.P.M. Chemotherapeutic Response to Cisplatin-like Drugs in Human Breast Cancer Cells Probed by Vibrational Microspectroscopy. Faraday Discuss. 2016, 187, 273–298. [Google Scholar] [CrossRef]
- Tummala, R.; Diegelman, P.; Fiuza, S.M.; Batista de Carvalho, L.A.E.; Marques, M.P.M.; Kramer, D.L.; Clark, K.; Vujcic, S.; Porter, C.W.; Pendyala, L. Characterization of Pt-, Pd-Spermine Complexes for Their Effect on Polyamine Pathway and Cisplatin Resistance in A2780 Ovarian Carcinoma Cells. Oncol. Rep. 2010, 24, 15–24. [Google Scholar] [CrossRef]
- Lamego, I.; Marques, M.P.M.; Duarte, I.F.; Martins, A.S.; Oliveira, H.; Gil, A.M. Impact of the Pd2Spermine Chelate on Osteosarcoma Metabolism: An NMR Metabolomics Study. J. Proteome Res. 2017, 16, 1773–1783. [Google Scholar] [CrossRef]
- Marques, M.P.M.; Batista de Carvalho, A.L.M.; Mamede, A.P.; Dopplapudi, A.; Rudić, S.; Tyagi, M.; Garcia Sakai, V.; Batista de Carvalho, L.A.E. A New Look into the Mode of Action of Metal-Based Anticancer Drugs. Molecules 2020, 25, 246. [Google Scholar] [CrossRef]
- Perše, M.; Veceric-Haler, Z. Cisplatin-Induced Rodent Model of Kidney Injury: Characteristics and Challenges. Biomed. Res. Int. 2018, 2018, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Jahromi, E.Z.; Divsalar, A.; Saboury, A.A.; Khaleghizadeh, S.; Mansouri-Torshizi, H.; Kostova, I. Palladium Complexes: New Candidates for Anti-Cancer Drugs. J. Iran. Chem. Soc. 2016, 13, 967–989. [Google Scholar] [CrossRef]
- Brun, P.H.; DeGroot, J.L.; Gudgin Dickson, E.F.; Farahani, M.; Pottier, R.H. Determination of the in Vivo Pharmacokinetics of Palladium-Bacteriopheophorbide (WST09) in EMT6 Tumour-Bearing Balb/c Mice Using Graphite Furnace Atomic Absorption Spectroscopy. Photochem. Photobiol. Sci. 2004, 3, 1006–1010. [Google Scholar] [CrossRef]
- Mazor, O.; Brandis, A.; Plaks, V.; Neumark, E.; Rosenbach-Belkin, V.; Salomon, Y.; Scherz, A. WST11, A Novel Water-Soluble Bacteriochlorophyll Derivative; Cellular Uptake, Pharmacokinetics, Biodistribution and Vascular-Targeted Photodynamic Activity Using Melanoma Tumors as a Model. Photochem. Photobiol. 2007, 81, 342–351. [Google Scholar] [CrossRef]
- Fiuza, S.M.; Holy, J.; Batista de Carvalho, L.A.E.; Marques, M.P.M. Biologic Activity of a Dinuclear Pd(II)-Spermine Complex Toward Human Breast Cancer. Chem. Biol. Drug Des. 2011, 77, 477–488. [Google Scholar] [CrossRef]
- Gong, C.; Qian, L.; Yang, H.; Ji, L.-L.; Wei, H.; Zhou, W.-B.; Qi, C.; Wang, C.-H. Hepatotoxicity and Pharmacokinetics of Cisplatin in Combination Therapy with a Traditional Chinese Medicine Compound of Zengmian Yiliu Granules in ICR Mice and SKOV-3-Bearing Nude Mice. BMC Complement. Altern. Med. 2015, 15, 1–12. [Google Scholar] [CrossRef]
- Chen, Y.; Brott, D.; Luo, W.; Gangl, E.; Kamendi, H.; Barthlow, H.; Lengel, D.; Fikes, J.; Kinter, L.; Valentin, J.P.; et al. Assessment of Cisplatin-Induced Kidney Injury Using an Integrated Rodent Platform. Toxicol. Appl. Pharmacol 2013, 268, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.-H.; Gong, C.; Yang, H.; Wei, H.; Zhou, W.-B.; Qi, C.; Wang, C.-H. Pharmacokinetics of Cisplatin in the Absence or Presence of Zengmian Yiliu Granules (a Traditional Chinese Medicine Compound) in Rats Determined via ICP-MS: An Investigation on Drug-Herb Interactions. Pharm. Biol. 2015, 53, 159–166. [Google Scholar] [CrossRef]
- Hanada, K.; Ninomiya, K.; Ogata, H. Pharmacokinetics and Toxicodynamics of Cisplatin and Its Metabolites in Rats: Relationship between Renal Handling and Nephrotoxicity of Cisplatin. J. Pharm. Pharmacol. 2000, 52, 1345–1353. [Google Scholar] [CrossRef]
- Bohnert, T.; Gan, L.S. Plasma Protein Binding: From Discovery to Development. J. Pharm. Sci. 2013, 102, 2953–2994. [Google Scholar] [CrossRef] [PubMed]
- Ueda, T.; Yasumasu, T.; Uozumi, J. Experimental Studies on the Pharmacokinetics and Nephrotoxicity of Carboplatin (Cis-Diammine-1, 1-Cyclobutane Dicarboxylate Platinum II) in Rats. J. Toxicol. Sci. 1991, 16, 101–109. [Google Scholar] [CrossRef][Green Version]
- Kato, R.; Sato, T.; Iwamoto, A.; Yamazaki, T.; Nakashiro, S.; Yoshikai, S.; Fujimoto, A.; Imano, H.; Ijiri, Y.; Mino, Y.; et al. Interaction of Platinum Agents, Cisplatin, Carboplatin and Oxaliplatin against Albumin in Vivo Rats and in Vitro Study Using Inductively Coupled Plasma-Mass Spectrometory. Biopharm. Drug Dispos. 2019, 40, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Yasumasu, T.; Ueda, T.; Uozumi, J.; Mihara, Y.; Kumazawa, J. Comparative Study of Cisplatin and Carboplatin on Pharmacokinetics, Nephrotoxicity and Effect on Renal Nuclear DNA Synthesis in Rats. Pharmacol. Toxicol. 1992, 70, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Will, J.; Wolters, D.A.; Sheldrick, W.S. Characterisation of Cisplatin Binding Sites in Human Serum Proteins Using Hyphenated Multidimensional Liquid Chromatography and ESI Tandem Mass Spectrometry. ChemMedChem 2008, 3, 1696–1707. [Google Scholar] [CrossRef]
- Sooriyaarachchi, M.; Narendran, A.; Gailer, J. Comparative Hydrolysis and Plasma Protein Binding of Cis-Platin and Carboplatin in Human Plasma in Vitro. Metallomics 2011, 3, 49–55. [Google Scholar] [CrossRef]
- Li, Y.; Meng, F.; Chen, Z.; Han, F.; He, D.; Hao, Y.; Gao, A.; Jiang, J.; Wang, Z.; Liu, W.; et al. Pharmacokinetics and Tissue Distribution in Rats of a Novel Anticancer Platinum Compound LLC-1903. Xenobiotica 2020, 50, 980–987. [Google Scholar] [CrossRef]
- Zhao, J.; Wen, Y.; Zhang, W.; Zhao, D.; Fan, A.; Zhang, Y.; Deng, S.; Wang, X.; Liu, Q.; Lu, Y.; et al. Investigation on Pharmacokinetics, Tissue Distribution and Excretion of a Novel Platinum Anticancer Agent in Rats by Inductively Coupled Plasma Mass Spectrometry (ICP-MS). Xenobiotica 2014, 44, 757–762. [Google Scholar] [CrossRef]
- Navolotskii, D.V.; Ivanenko, N.B.; Solovyev, N.D.; Fedoros, E.I.; Panchenko, A.V. Pharmacokinetics and Tissue Distribution of Novel Platinum Containing Anticancer Agent BP-C1 Studied in Rabbits Using Sector Field Inductively Coupled Plasma Mass Spectrometry. Drug Test. Anal. 2015, 7, 737–744. [Google Scholar] [CrossRef]
- Esteban-Fernández, D.; Verdaguer, J.M.; Ramírez-Camacho, R.; Palacios, M.A.; Gómez-Gómez, M.M. Accumulation, Fractionation, and Analysis of Platinum in Toxicologically Affected Tissues after Cisplatin, Oxaliplatin, and Carboplatin Administration. J. Anal. Toxicol. 2008, 32, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Au-Yeung, S.C.F.; Ho, Y.P. Pharmacokinetics and Tissue Distribution of Novel Traditional Chinese Medicine-Platinum Anticancer Agents in Rats. J. Inorg. Biochem. 2007, 101, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Divsalar, A.; Saboury, A.A.; Ahadi, L.; Zemanatiyar, E.; Mansouri-Torshizi, H.; Ajloo, D.; Sarma, R.H. Biological Evaluation and Interaction of a Newly Designed Anti-Cancer Pd(II) Complex and Human Serum Albumin. J. Biomol. Struct. Dyn. 2011, 29, 283–296. [Google Scholar] [CrossRef]
- Carneiro, T.J.; Araújo, R.; Vojtek, M.; Gonçalves-Monteiro, S.; Diniz, C.; Batista de Carvalho, A.L.M.; Marques, M.P.M.; Gil, A.M. Multi-Organ NMR Metabolomics to Assess In Vivo Overall Metabolic Impact of Cisplatin in Mice. Metabolites 2019, 9, 279. [Google Scholar] [CrossRef]
- Carneiro, T.J.; Araújo, R.; Vojtek, M.; Gonçalves-Monteiro, S.; Diniz, C.; Batista de Carvalho, A.L.M.; Marques, M.P.M.; Gil, A.M. Novel Insights into Mice Multi-Organ Metabolism upon Exposure to a Potential Anticancer Pd(II)-Agent. Metabolites 2021, 11, 114. [Google Scholar] [CrossRef]
- He, M.; Guo, S.; Li, Z. In Situ Characterizing Membrane Lipid Phenotype of Breast Cancer Cells Using Mass Spectrometry Profiling. Sci. Rep. 2015, 5, 1–13. [Google Scholar] [CrossRef]
- Percie du Sert, N.; Hurst, V.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; et al. The ARRIVE Guidelines 2.0: Updated Guidelines for Reporting Animal Research. PLoS Biol. 2020, 18, e3000410. [Google Scholar] [CrossRef]
- Codina, G.; Caubet, A.; López, C.; Moreno, V.; Molins, E. Palladium(II) and Platinum(II) Polyamine Complexes: X-Ray Crystal Structures of (SP-4-2)-Chloro{N-[(3-amino-κN)propyl]propane-1,3-diamine-κN,κN’palladium(1+) Tetrachloropalladate (2–) (2 : 1) and (R,S)-Tetrachloro[μ-(spermine)]dipalladium(II) (={μ-{N,N’-Bis[(3-amino-κN)propyl]butane-1,4-diamine-κN:κN’}}tetrachlorodipalladium). Helv Chim. Acta 1999, 82, 1025–1037. [Google Scholar] [CrossRef]
- Fiuza, S.M.; Amado, A.M.; Parker, S.F.; Marques, M.P.M.; Batista de Carvalho, L.A.E. Conformational Insights and Vibrational Study of a Promising Anticancer Agent: The Role of the Ligand in Pd(II)–Amine Complexes. New J. Chem. 2015, 39, 6274–6283. [Google Scholar] [CrossRef]
- Batista de Carvalho, L.A.E.; Marques, M.P.M.; Martin, C.; Parker, S.F.; Tomkinson, J. Inelastic Neutron Scattering Study of Pt II Complexes Displaying Anticancer Properties. ChemPhysChem 2011, 12, 1334–1341. [Google Scholar] [CrossRef] [PubMed]
- Vojtek, M.; Pinto, E.; Gonçalves-Monteiro, S.; Almeida, A.; Marques, M.P.M.; Mota-Filipe, H.; Ferreira, I.M.P.L.V.O.; Diniz, C. Fast and Reliable ICP-MS Quantification of Palladium and Platinum-Based Drugs in Animal Pharmacokinetic and Biodistribution Studies. Anal. Methods 2020, 12. [Google Scholar] [CrossRef] [PubMed]





| Pharmacokinetic Parameter (Unit) | Pd2Spm | Cisplatin | ||
|---|---|---|---|---|
| Total Pd in Serum | Free Pd in Ultrafiltrate | Total Pt in Serum | Free Pt in Ultrafiltrate | |
| Half-life (h) | 20.7 | 35.5 | 43.3 | 31.5 |
| λz (1/h) | 0.034 | 0.019 | 0.016 | 0.022 |
| Tmax (h) | 0.5 | 0.5 | 0.5 | 0.5 |
| Cmax (ng/mL) | 1499.4 ± 38.5 | 749.0 ± 120.6 | 1009.6 ± 64.2 | 350.7 ± 16.8 |
| Cmax/Dose (ng/mL/µg) | 65.9 ± 4.2 | 32.7 ± 5.3 | 21.9 ± 1.9 | 7.7 ± 0.4 |
| AUC0–48 h (h × ng/mL) | 6490.2 | 1219.7 | 10867.5 | 567.2 |
| AUC0–48 h/Dose (h × ng/mL/µg) | 285.0 | 53.2 | 240.5 | 12.5 |
| AUC0–∞ (h × ng/mL) | 7523.9 | 1339.1 | 19,758.5 | 751.5 |
| Vz/F (mL/kg) | 4555.0 | 43,992.0 | 7202.8 | 143,864.7 |
| CL/F (mL/h/kg) | 152.8 | 858.8 | 115.4 | 2989.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vojtek, M.; Gonçalves-Monteiro, S.; Pinto, E.; Kalivodová, S.; Almeida, A.; Marques, M.P.M.; Batista de Carvalho, A.L.M.; Martins, C.B.; Mota-Filipe, H.; Ferreira, I.M.P.L.V.O.; et al. Preclinical Pharmacokinetics and Biodistribution of Anticancer Dinuclear Palladium(II)-Spermine Complex (Pd2Spm) in Mice. Pharmaceuticals 2021, 14, 173. https://doi.org/10.3390/ph14020173
Vojtek M, Gonçalves-Monteiro S, Pinto E, Kalivodová S, Almeida A, Marques MPM, Batista de Carvalho ALM, Martins CB, Mota-Filipe H, Ferreira IMPLVO, et al. Preclinical Pharmacokinetics and Biodistribution of Anticancer Dinuclear Palladium(II)-Spermine Complex (Pd2Spm) in Mice. Pharmaceuticals. 2021; 14(2):173. https://doi.org/10.3390/ph14020173
Chicago/Turabian StyleVojtek, Martin, Salomé Gonçalves-Monteiro, Edgar Pinto, Sára Kalivodová, Agostinho Almeida, Maria P. M. Marques, Ana L. M. Batista de Carvalho, Clara B. Martins, Helder Mota-Filipe, Isabel M. P. L. V. O. Ferreira, and et al. 2021. "Preclinical Pharmacokinetics and Biodistribution of Anticancer Dinuclear Palladium(II)-Spermine Complex (Pd2Spm) in Mice" Pharmaceuticals 14, no. 2: 173. https://doi.org/10.3390/ph14020173
APA StyleVojtek, M., Gonçalves-Monteiro, S., Pinto, E., Kalivodová, S., Almeida, A., Marques, M. P. M., Batista de Carvalho, A. L. M., Martins, C. B., Mota-Filipe, H., Ferreira, I. M. P. L. V. O., & Diniz, C. (2021). Preclinical Pharmacokinetics and Biodistribution of Anticancer Dinuclear Palladium(II)-Spermine Complex (Pd2Spm) in Mice. Pharmaceuticals, 14(2), 173. https://doi.org/10.3390/ph14020173