
Bacteriophages as Therapeutic and Diagnostic Vehicles in Cancer



Abstract
1. Introduction
2. Bacteriophages: An Overview
3. Discovery of Cancer Biomarkers and Targeting Moieties by Phage Display
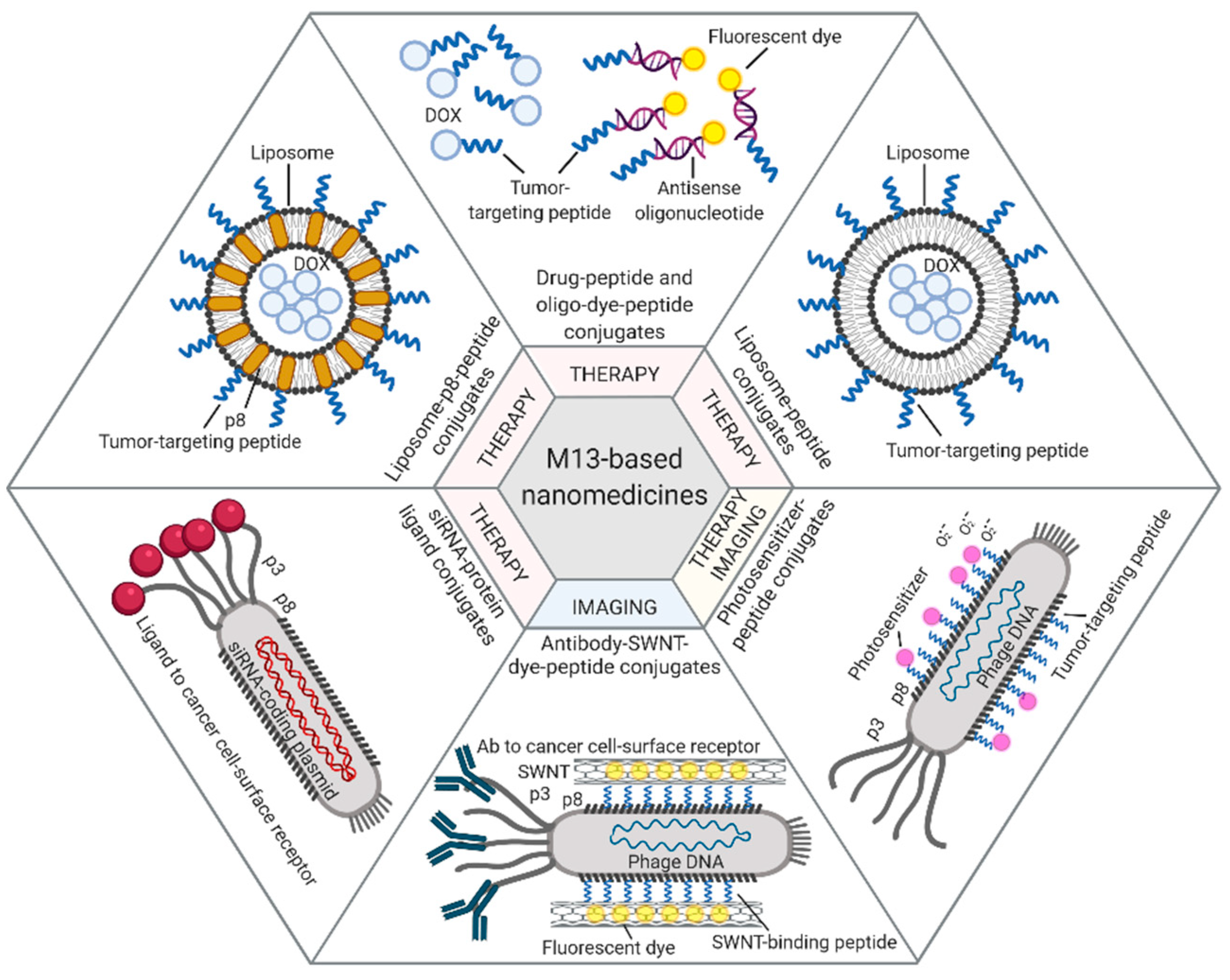
4. Bacteriophages and Their Derivatives for Drug Delivery and Targeted Imaging
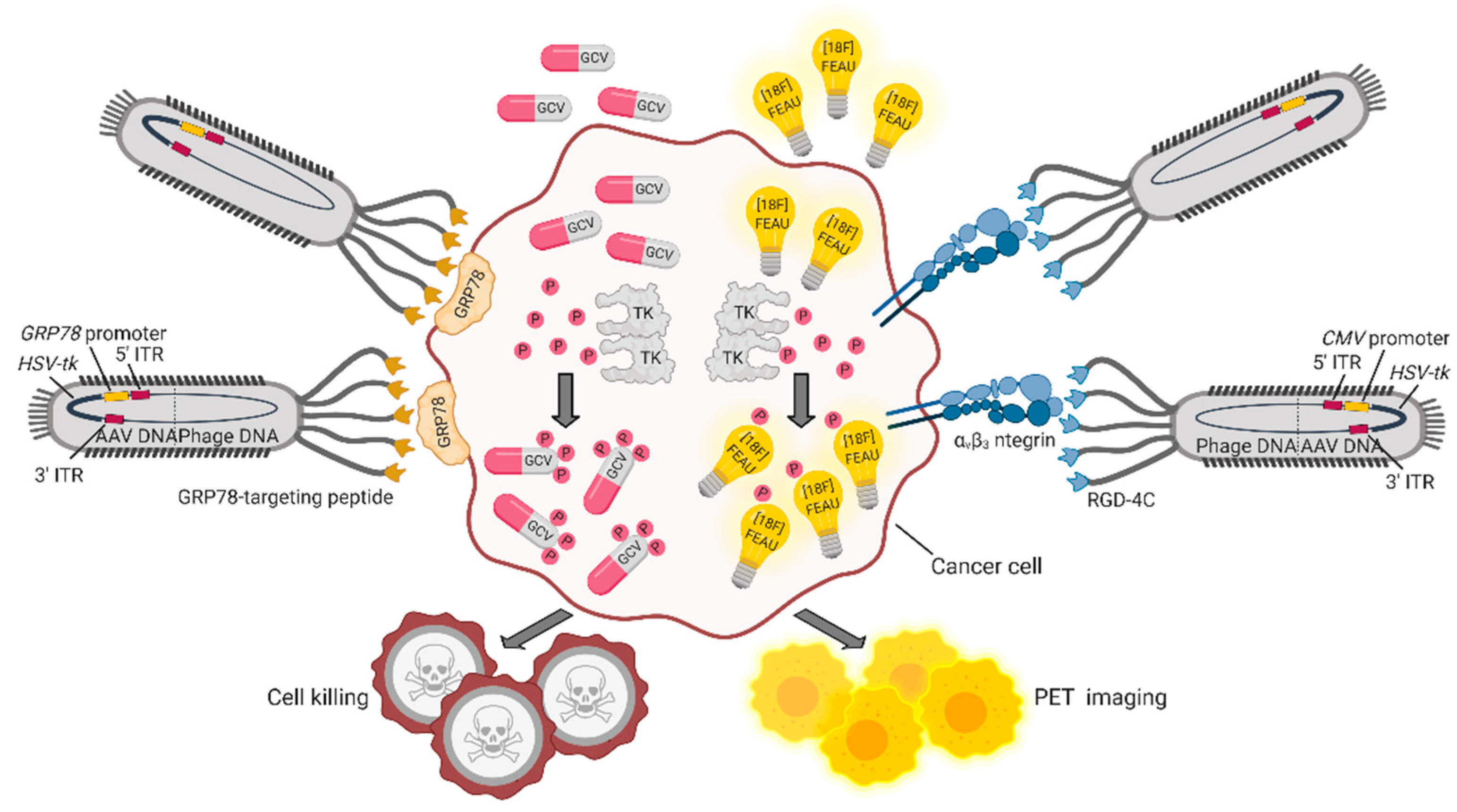
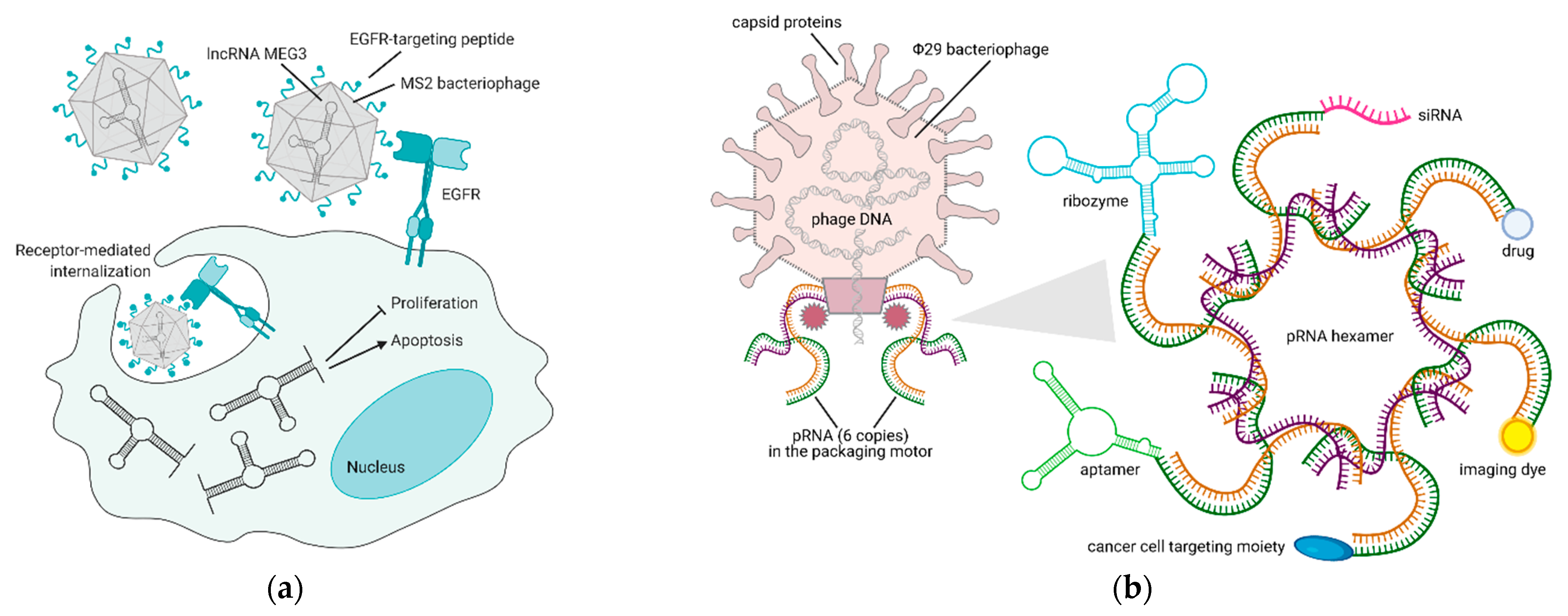
5. Bacteriophages and Their Derivatives for Gene Therapy
6. Bacteriophages as Carriers for Cancer Vaccines
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lammers, T.; Rizzo, L.Y.; Storm, G.; Kiessling, F. Personalized nanomedicine. Clin. Cancer Res. 2012, 18, 4889–4894. [Google Scholar] [CrossRef]
- Perik, P.J.; Lub-De Hooge, M.N.; Gietema, J.A.; van der Graaf, W.T.; de Korte, M.A.; Jonkman, S.; Kosterink, J.G.; van Veldhuisen, D.J.; Sleijfer, D.T.; Jager, P.L.; et al. Indium-111-labeled trastuzumab scintigraphy in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer. J. Clin. Oncol. 2006, 24, 2276–2282. [Google Scholar] [CrossRef] [PubMed]
- Amin, S.; Bathe, O.F. Response biomarkers: Re-envisioning the approach to tailoring drug therapy for cancer. BMC Cancer 2016, 16, 850. [Google Scholar] [CrossRef]
- Hait, W.N.; Hambley, T.W. Targeted cancer therapeutics. Cancer Res. 2009, 69, 1263–1267. [Google Scholar] [CrossRef] [PubMed]
- Ju, Z.; Sun, W. Drug delivery vectors based on filamentous bacteriophages and phage-mimetic nanoparticles. Drug Deliv. 2017, 24, 1898–1908. [Google Scholar] [CrossRef] [PubMed]
- Yao, V.J.; D’Angelo, S.; Butler, K.S.; Theron, C.; Smith, T.L.; Marchio, S.; Gelovani, J.G.; Sidman, R.L.; Dobroff, A.S.; Brinker, C.J.; et al. Ligand-targeted theranostic nanomedicines against cancer. J. Control Release 2016, 240, 267–286. [Google Scholar] [CrossRef]
- Awasthi, R.; Roseblade, A.; Hansbro, P.M.; Rathbone, M.J.; Dua, K.; Bebawy, M. Nanoparticles in cancer treatment: Opportunities and obstacles. Curr. Drug Targets 2018, 19, 1696–1709. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Wu, J.; Sawa, T.; Matsumura, Y.; Hori, K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: A review. J. Control Release 2000, 65, 271–284. [Google Scholar] [CrossRef]
- Thomas, O.S.; Weber, W. Overcoming physiological barriers to nanoparticle delivery-are we there yet? Front. Bioeng. Biotechnol. 2019, 7, 415. [Google Scholar] [CrossRef]
- Boraschi, D.; Italiani, P.; Palomba, R.; Decuzzi, P.; Duschl, A.; Fadeel, B.; Moghimi, S.M. Nanoparticles and innate immunity: New perspectives on host defence. Semin. Immunol. 2017, 34, 33–51. [Google Scholar] [CrossRef]
- Durymanov, M.O.; Rosenkranz, A.A.; Sobolev, A.S. Current approaches for improving intratumoral accumulation and distribution of nanomedicines. Theranostics 2015, 5, 1007–1020. [Google Scholar] [CrossRef]
- Karimi, M.; Mirshekari, H.; Moosavi Basri, S.M.; Bahrami, S.; Moghoofei, M.; Hamblin, M.R. Bacteriophages and phage-inspired nanocarriers for targeted delivery of therapeutic cargos. Adv. Drug Deliv. Rev. 2016, 106, 45–62. [Google Scholar] [CrossRef]
- Loh, B.; Kuhn, A.; Leptihn, S. The fascinating biology behind phage display: Filamentous phage assembly. Mol. Microbiol. 2019, 111, 1132–1138. [Google Scholar] [CrossRef]
- Rakonjac, J.; Bennett, N.J.; Spagnuolo, J.; Gagic, D.; Russel, M. Filamentous bacteriophage: Biology, phage display and nanotechnology applications. Curr. Issues Mol. Biol. 2011, 13, 51–76. [Google Scholar]
- Harada, L.K.; Silva, E.C.; Campos, W.F.; Del Fiol, F.S.; Vila, M.; Dabrowska, K.; Krylov, V.N.; Balcao, V.M. Biotechnological applications of bacteriophages: State of the art. Microbiol. Res. 2018, 212, 38–58. [Google Scholar] [CrossRef] [PubMed]
- Adhya, S.; Merril, C.R.; Biswas, B. Therapeutic and prophylactic applications of bacteriophage components in modern medicine. Cold Spring Harb. Perspect. Med. 2014, 4, a012518. [Google Scholar] [CrossRef]
- Gamkrelidze, M.; Dabrowska, K. T4 bacteriophage as a phage display platform. Arch. Microbiol. 2014, 196, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Lindner, T.; Kolmar, H.; Haberkorn, U.; Mier, W. DNA libraries for the construction of phage libraries: Statistical and structural requirements and synthetic methods. Molecules 2011, 16, 1625–1641. [Google Scholar] [CrossRef]
- Yacoby, I.; Benhar, I. Targeted filamentous bacteriophages as therapeutic agents. Expert. Opin. Drug Deliv. 2008, 5, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Mimmi, S.; Maisano, D.; Quinto, I.; Iaccino, E. Phage Display: An Overview in Context to Drug Discovery. Trends Pharmacol. Sci. 2019, 40, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Saw, P.E.; Song, E.W. Phage display screening of therapeutic peptide for cancer targeting and therapy. Protein Cell 2019, 10, 787–807. [Google Scholar] [CrossRef] [PubMed]
- Fagbohun, O.A.; Kazmierczak, R.A.; Petrenko, V.A.; Eisenstark, A. Metastatic prostate cancer cell-specific phage-like particles as a targeted gene-delivery system. J. Nanobiotechnol. 2013, 11, 31. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.; Riechmann, L. Antibody VH domains as small recognition units. Biotechnology 1995, 13, 475–479. [Google Scholar] [CrossRef]
- Peabody, D.S. Subunit fusion confers tolerance to peptide insertions in a virus coat protein. Arch. Biochem. Biophys. 1997, 347, 85–92. [Google Scholar] [CrossRef]
- Ren, Z.; Black, L.W. Phage T4 SOC and HOC display of biologically active, full-length proteins on the viral capsid. Gene 1998, 215, 439–444. [Google Scholar] [CrossRef]
- Houshmand, H.; Froman, G.; Magnusson, G. Use of bacteriophage T7 displayed peptides for determination of monoclonal antibody specificity and biosensor analysis of the binding reaction. Anal. Biochem. 1999, 268, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Casjens, S.R.; Hendrix, R.W. Locations and amounts of major structural proteins in bacteriophage lambda. J. Mol. Biol. 1974, 88, 535–545. [Google Scholar] [CrossRef]
- Koning, R.I.; Gomez-Blanco, J.; Akopjana, I.; Vargas, J.; Kazaks, A.; Tars, K.; Carazo, J.M.; Koster, A.J. Asymmetric cryo-EM reconstruction of phage MS2 reveals genome structure in situ. Nat. Commun. 2016, 7, 12524. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Abu-Shilbayeh, L.; Rao, V.B. Display of a PorA peptide from Neisseria meningitidis on the bacteriophage T4 capsid surface. Infect. Immun. 1997, 65, 4770–4777. [Google Scholar] [CrossRef]
- Ionel, A.; Velazquez-Muriel, J.A.; Luque, D.; Cuervo, A.; Caston, J.R.; Valpuesta, J.M.; Martin-Benito, J.; Carrascosa, J.L. Molecular rearrangements involved in the capsid shell maturation of bacteriophage T7. J. Biol. Chem. 2011, 286, 234–242. [Google Scholar] [CrossRef]
- Poul, M.A.; Becerril, B.; Nielsen, U.B.; Morisson, P.; Marks, J.D. Selection of tumor-specific internalizing human antibodies from phage libraries. J. Mol. Biol. 2000, 301, 1149–1161. [Google Scholar] [CrossRef]
- Tordsson, J.; Lavasani, S.; Ohlsson, L.; Karlstrom, P.; Svedberg, H.; Abrahmsen, L.; Brodin, T. A3—A novel colon and pancreatic cancer reactive antibody from a primate phage library selected using intact tumour cells. Int. J. Cancer 2000, 87, 559–568. [Google Scholar] [CrossRef]
- Pavoni, E.; Vaccaro, P.; Pucci, A.; Monteriu, G.; Beghetto, E.; Barca, S.; Dupuis, M.L.; De Pasquale Ceratti, A.; Lugini, A.; Cianfriglia, M.; et al. Identification of a panel of tumor-associated antigens from breast carcinoma cell lines, solid tumors and testis cDNA libraries displayed on lambda phage. BMC Cancer 2004, 4, 78. [Google Scholar] [CrossRef]
- Larsen, S.A.; Meldgaard, T.; Fridriksdottir, A.J.; Lykkemark, S.; Poulsen, P.C.; Overgaard, L.F.; Petersen, H.B.; Petersen, O.W.; Kristensen, P. Raising an antibody specific to breast cancer subpopulations using phage display on tissue sections. Cancer Genom. Proteom. 2016, 13, 21–30. [Google Scholar]
- Mandrup, O.A.; Friis, N.A.; Lykkemark, S.; Just, J.; Kristensen, P. A novel heavy domain antibody library with functionally optimized complementarity determining regions. PLoS ONE 2013, 8, e76834. [Google Scholar] [CrossRef] [PubMed]
- Mueller, J.; Gaertner, F.C.; Blechert, B.; Janssen, K.P.; Essler, M. Targeting of tumor blood vessels: A phage-displayed tumor-homing peptide specifically binds to matrix metalloproteinase-2-processed collagen IV and blocks angiogenesis in vivo. Mol. Cancer Res. 2009, 7, 1078–1085. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Jia, H.; Li, W.; Hou, Y.; Lu, S.; He, S. Screening and identification of a phage display derived peptide that specifically binds to the CD44 protein region encoded by variable exons. J. Biomol. Screen. 2016, 21, 44–53. [Google Scholar] [CrossRef][Green Version]
- Zuo, S.; Dai, G.; Wang, L.; Wen, Y.; Huang, Z.; Yang, W.; Ma, W.; Ren, X. Suppression of angiogenesis and tumor growth by recombinant T4 phages displaying extracellular domain of vascular endothelial growth factor receptor 2. Arch. Virol. 2019, 164, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Peng, X.; Leal, J.; Mohanty, R. Peptides as drug delivery vehicles across biological barriers. J. Pharm. Investig. 2018, 48, 89–111. [Google Scholar] [CrossRef]
- Shadidi, M.; Sioud, M. Identification of novel carrier peptides for the specific delivery of therapeutics into cancer cells. FASEB J. 2003, 17, 256–258. [Google Scholar] [CrossRef] [PubMed]
- Bolhassani, A. Potential efficacy of cell-penetrating peptides for nucleic acid and drug delivery in cancer. Biochim. Biophys. Acta 2011, 1816, 232–246. [Google Scholar] [CrossRef]
- Du, B.; Han, H.; Wang, Z.; Kuang, L.; Wang, L.; Yu, L.; Wu, M.; Zhou, Z.; Qian, M. Targeted drug delivery to hepatocarcinoma in vivo by phage-displayed specific binding peptide. Mol. Cancer Res. 2010, 8, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Fukuta, T.; Asai, T.; Kiyokawa, Y.; Nakada, T.; Bessyo-Hirashima, K.; Fukaya, N.; Hyodo, K.; Takase, K.; Kikuchi, H.; Oku, N. Targeted delivery of anticancer drugs to tumor vessels by use of liposomes modified with a peptide identified by phage biopanning with human endothelial progenitor cells. Int. J. Pharm. 2017, 524, 364–372. [Google Scholar] [CrossRef]
- Wang, T.; D’Souza, G.G.; Bedi, D.; Fagbohun, O.A.; Potturi, L.P.; Papahadjopoulos-Sternberg, B.; Petrenko, V.A.; Torchilin, V.P. Enhanced binding and killing of target tumor cells by drug-loaded liposomes modified with tumor-specific phage fusion coat protein. Nanomedicine 2010, 5, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Hartner, W.C.; Gillespie, J.W.; Praveen, K.P.; Yang, S.; Mei, L.A.; Petrenko, V.A.; Torchilin, V.P. Enhanced tumor delivery and antitumor activity in vivo of liposomal doxorubicin modified with MCF-7-specific phage fusion protein. Nanomedicine 2014, 10, 421–430. [Google Scholar] [CrossRef]
- Cai, X.M.; Xie, H.L.; Liu, M.Z.; Zha, X.L. Inhibition of cell growth and invasion by epidermal growth factor-targeted phagemid particles carrying siRNA against focal adhesion kinase in the presence of hydroxycamptothecin. BMC Biotechnol. 2008, 8, 74. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.K.; Steinmetz, N.F.; Fu, C.Y.; Manchester, M.; Johnson, J.E. Transferrin-mediated targeting of bacteriophage HK97 nanoparticles into tumor cells. Nanomedicine 2011, 6, 55–68. [Google Scholar] [CrossRef]
- Yi, H.; Ghosh, D.; Ham, M.H.; Qi, J.; Barone, P.W.; Strano, M.S.; Belcher, A.M. M13 phage-functionalized single-walled carbon nanotubes as nanoprobes for second near-infrared window fluorescence imaging of targeted tumors. Nano Lett. 2012, 12, 1176–1183. [Google Scholar] [CrossRef] [PubMed]
- Cohen, B.A.; Bergkvist, M. Targeted in vitro photodynamic therapy via aptamer-labeled, porphyrin-loaded virus capsids. J. Photochem. Photobiol. B 2013, 121, 67–74. [Google Scholar] [CrossRef]
- Gandra, N.; Abbineni, G.; Qu, X.; Huai, Y.; Wang, L.; Mao, C. Bacteriophage bionanowire as a carrier for both cancer-targeting peptides and photosensitizers and its use in selective cancer cell killing by photodynamic therapy. Small 2013, 9, 215–221. [Google Scholar] [CrossRef]
- Sánchez-Sánchez, L.; Tapia-Moreno, A.; Juarez-Moreno, K.; Patterson, D.P.; Cadena-Nava, R.D.; Douglas, T.; Vazquez-Duhalt, R. Design of a VLP-nanovehicle for CYP450 enzymatic activity delivery. J. Nanobiotechnol. 2015, 13, 66. [Google Scholar] [CrossRef]
- Hajitou, A.; Trepel, M.; Lilley, C.E.; Soghomonyan, S.; Alauddin, M.M.; Marini, F.C., III; Restel, B.H.; Ozawa, M.G.; Moya, C.A.; Rangel, R.; et al. A hybrid vector for ligand-directed tumor targeting and molecular imaging. Cell 2006, 125, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Hajitou, A.; Lev, D.C.; Hannay, J.A.; Korchin, B.; Staquicini, F.I.; Soghomonyan, S.; Alauddin, M.M.; Benjamin, R.S.; Pollock, R.E.; Gelovani, J.G.; et al. A preclinical model for predicting drug response in soft-tissue sarcoma with targeted AAVP molecular imaging. Proc. Natl. Acad. Sci. USA 2008, 105, 4471–4476. [Google Scholar] [CrossRef] [PubMed]
- Kia, A.; Przystal, J.M.; Nianiaris, N.; Mazarakis, N.D.; Mintz, P.J.; Hajitou, A. Dual systemic tumor targeting with ligand-directed phage and Grp78 promoter induces tumor regression. Mol. Cancer Ther. 2012, 11, 2566–2577. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Peng, G.L.; Liu, Q.C.; Li, F.L.; Zou, X.S.; He, J.X. Selective killing of lung cancer cells using carcinoembryonic antigen promoter and double suicide genes, thymidine kinase and cytosine deaminase (pCEA-TK/CD). Cancer Lett. 2012, 316, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Rama, A.R.; Hernandez, R.; Perazzoli, G.; Burgos, M.; Melguizo, C.; Velez, C.; Prados, J. Specific colon cancer cell cytotoxicity induced by bacteriophage E gene expression under transcriptional control of carcinoembryonic antigen promoter. Int. J. Mol. Sci. 2015, 16, 12601–12615. [Google Scholar] [CrossRef]
- Danda, R.; Krishnan, G.; Ganapathy, K.; Krishnan, U.M.; Vikas, K.; Elchuri, S.; Chatterjee, N.; Krishnakumar, S. Targeted expression of suicide gene by tissue-specific promoter and microRNA regulation for cancer gene therapy. PLoS ONE 2013, 8, e83398. [Google Scholar] [CrossRef]
- Higashi, K.; Hazama, S.; Araki, A.; Yoshimura, K.; Iizuka, N.; Yoshino, S.; Noma, T.; Oka, M. A novel cancer vaccine strategy with combined IL-18 and HSV-TK gene therapy driven by the hTERT promoter in a murine colorectal cancer model. Int. J. Oncol. 2014, 45, 1412–1420. [Google Scholar] [CrossRef]
- Dobroff, A.S.; D’Angelo, S.; Eckhardt, B.L.; Ferrara, F.; Staquicini, D.I.; Cardo-Vila, M.; Staquicini, F.I.; Nunes, D.N.; Kim, K.; Driessen, W.H.P.; et al. Towards a transcriptome-based theranostic platform for unfavorable breast cancer phenotypes. Proc. Natl. Acad. Sci. USA 2016, 113, 12780–12785. [Google Scholar] [CrossRef]
- Smith, T.L.; Yuan, Z.; Cardo-Vila, M.; Sanchez Claros, C.; Adem, A.; Cui, M.H.; Branch, C.A.; Gelovani, J.G.; Libutti, S.K.; Sidman, R.L.; et al. AAVP displaying octreotide for ligand-directed therapeutic transgene delivery in neuroendocrine tumors of the pancreas. Proc. Natl. Acad. Sci. USA 2016, 113, 2466–2471. [Google Scholar] [CrossRef]
- Przystal, J.M.; Umukoro, E.; Stoneham, C.A.; Yata, T.; O’Neill, K.; Syed, N.; Hajitou, A. Proteasome inhibition in cancer is associated with enhanced tumor targeting by the adeno-associated virus/phage. Mol. Oncol. 2013, 7, 55–66. [Google Scholar] [CrossRef]
- Yata, T.; Lee, E.L.; Suwan, K.; Syed, N.; Asavarut, P.; Hajitou, A. Modulation of extracellular matrix in cancer is associated with enhanced tumor cell targeting by bacteriophage vectors. Mol. Cancer 2015, 14, 110. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Wang, G.; Jia, T.; Zhang, L.; Li, Y.; Han, Y.; Zhang, K.; Lin, G.; Zhang, R.; Li, J.; et al. Armored long non-coding RNA MEG3 targeting EGFR based on recombinant MS2 bacteriophage virus-like particles against hepatocellular carcinoma. Oncotarget 2016, 7, 23988–24004. [Google Scholar] [CrossRef]
- Shu, Y.; Haque, F.; Shu, D.; Li, W.; Zhu, Z.; Kotb, M.; Lyubchenko, Y.; Guo, P. Fabrication of 14 different RNA nanoparticles for specific tumor targeting without accumulation in normal organs. RNA 2013, 19, 767–777. [Google Scholar] [CrossRef]
- Zhang, Y.; Leonard, M.; Shu, Y.; Yang, Y.; Shu, D.; Guo, P.; Zhang, X. Overcoming tamoxifen resistance of human breast cancer by targeted gene silencing using multifunctional pRNA nanoparticles. ACS Nano 2017, 11, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Tschammer, N.; Mohammed, S.; Guo, P. Specific delivery of therapeutic RNAs to cancer cells via the dimerization mechanism of phi29 motor pRNA. Hum. Gene Ther. 2005, 16, 1097–1109. [Google Scholar] [CrossRef]
- Tarapore, P.; Shu, Y.; Guo, P.; Ho, S.M. Application of phi29 motor pRNA for targeted therapeutic delivery of siRNA silencing metallothionein-IIA and survivin in ovarian cancers. Mol. Ther. 2011, 19, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Sartorius, R.; Bettua, C.; D’Apice, L.; Caivano, A.; Trovato, M.; Russo, D.; Zanoni, I.; Granucci, F.; Mascolo, D.; Barba, P.; et al. Vaccination with filamentous bacteriophages targeting DEC-205 induces DC maturation and potent anti-tumor T-cell responses in the absence of adjuvants. Eur. J. Immunol. 2011, 41, 2573–2584. [Google Scholar] [CrossRef] [PubMed]
- Sartorius, R.; D’Apice, L.; Prisco, A.; De Berardinis, P. Arming filamentous bacteriophage, a nature-made nanoparticle, for new vaccine and immunotherapeutic strategies. Pharmaceutics 2019, 11, 437. [Google Scholar] [CrossRef]
- Krut, O.; Bekeredjian-Ding, I. Contribution of the iImmune response to phage therapy. J. Immunol. 2018, 200, 3037–3044. [Google Scholar] [CrossRef]
- Gaubin, M.; Fanutti, C.; Mishal, Z.; Durrbach, A.; De Berardinis, P.; Sartorius, R.; Del Pozzo, G.; Guardiola, J.; Perham, R.N.; Piatier-Tonneau, D. Processing of filamentous bacteriophage virions in antigen-presenting cells targets both HLA class I and class II peptide loading compartments. DNA Cell Biol. 2003, 22, 11–18. [Google Scholar] [CrossRef]
- Sartorius, R.; Pisu, P.; D’Apice, L.; Pizzella, L.; Romano, C.; Cortese, G.; Giorgini, A.; Santoni, A.; Velotti, F.; De Berardinis, P. The use of filamentous bacteriophage fd to deliver MAGE-A10 or MAGE-A3 HLA-A2-restricted peptides and to induce strong antitumor CTL responses. J. Immunol. 2008, 180, 3719–3728. [Google Scholar] [CrossRef]
- Van Houten, N.E.; Henry, K.A.; Smith, G.P.; Scott, J.K. Engineering filamentous phage carriers to improve focusing of antibody responses against peptides. Vaccine 2010, 28, 2174–2185. [Google Scholar] [CrossRef] [PubMed]
- Van Houten, N.E.; Zwick, M.B.; Menendez, A.; Scott, J.K. Filamentous phage as an immunogenic carrier to elicit focused antibody responses against a synthetic peptide. Vaccine 2006, 24, 4188–4200. [Google Scholar] [CrossRef] [PubMed]
- Cuesta, A.M.; Suarez, E.; Larsen, M.; Jensen, K.B.; Sanz, L.; Compte, M.; Kristensen, P.; Alvarez-Vallina, L. Enhancement of DNA vaccine potency through linkage of antigen to filamentous bacteriophage coat protein III domain I. Immunology 2006, 117, 502–506. [Google Scholar] [CrossRef]
- Bartolacci, C.; Andreani, C.; Curcio, C.; Occhipinti, S.; Massaccesi, L.; Giovarelli, M.; Galeazzi, R.; Iezzi, M.; Tilio, M.; Gambini, V.; et al. Phage-based anti-HER2 vaccination can circumvent immune tolerance against breast cancer. Cancer Immunol. Res. 2018, 6, 1486–1498. [Google Scholar] [CrossRef]
- Roehnisch, T.; Then, C.; Nagel, W.; Blumenthal, C.; Braciak, T.; Donzeau, M.; Bohm, T.; Bourquin, C.; Oduncu, F. Chemically linked phage idiotype vaccination in the murine B cell lymphoma 1 model. J. Transl. Med. 2013, 11, 267. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.R.; March, J.B. Bacteriophage-mediated nucleic acid immunisation. FEMS Immunol. Med. Microbiol. 2004, 40, 21–26. [Google Scholar] [CrossRef]
- March, J.B.; Clark, J.R.; Jepson, C.D. Genetic immunisation against hepatitis B using whole bacteriophage lambda particles. Vaccine 2004, 22, 1666–1671. [Google Scholar] [CrossRef]
- Ghaemi, A.; Soleimanjahi, H.; Gill, P.; Hassan, Z.; Jahromi, S.R.; Roohvand, F. Recombinant lambda-phage nanobioparticles for tumor therapy in mice models. Genet. Vaccines Ther. 2010, 8, 3. [Google Scholar] [CrossRef]
- Ghaemi, A.; Soleimanjahi, H.; Gill, P.; Hassan, Z.M.; Razeghi, S.; Fazeli, M.; Razavinikoo, S.M. Protection of mice by a lambda-based therapeutic vaccine against cancer associated with human papillomavirus type 16. Intervirology 2011, 54, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Thomas, B.S.; Nishikawa, S.; Ito, K.; Chopra, P.; Sharma, N.; Evans, D.H.; Tyrrell, D.L.; Bathe, O.F.; Rancourt, D.E. Peptide vaccination is superior to genetic vaccination using a recombineered bacteriophage lambda subunit vaccine. Vaccine 2012, 30, 998–1008. [Google Scholar] [CrossRef]
- Arab, A.; Nicastro, J.; Slavcev, R.; Razazan, A.; Barati, N.; Nikpoor, A.R.; Brojeni, A.A.M.; Mosaffa, F.; Badiee, A.; Jaafari, M.R.; et al. Lambda phage nanoparticles displaying HER2-derived E75 peptide induce effective E75-CD8(+) T response. Immunol. Res. 2018, 66, 200–206. [Google Scholar] [CrossRef]
- Barati, N.; Razazan, A.; Nicastro, J.; Slavcev, R.; Arab, A.; Mosaffa, F.; Nikpoor, A.R.; Badiee, A.; Jaafari, M.R.; Behravan, J. Immunogenicity and antitumor activity of the superlytic lambdaF7 phage nanoparticles displaying a HER2/neu-derived peptide AE37 in a tumor model of BALB/c mice. Cancer Lett. 2018, 424, 109–116. [Google Scholar] [CrossRef]
- Razazan, A.; Nicastro, J.; Slavcev, R.; Barati, N.; Arab, A.; Mosaffa, F.; Jaafari, M.R.; Behravan, J. Lambda bacteriophage nanoparticles displaying GP2, a HER2/neu derived peptide, induce prophylactic and therapeutic activities against TUBO tumor model in mice. Sci. Rep. 2019, 9, 2221. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.X.; Ren, Z.J.; Zhao, M.Y.; Wang, X.B.; Zuo, S.G.; Yu, F. Antitumor activity of endogenous mFlt4 displayed on a T4 phage nanoparticle surface. Acta Pharmacol. Sin. 2009, 30, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; FengYu; Zuo, S.; Zhao, M.; Wang, X.; Wang, X.; Chen, Y.; Wu, Z.; Ren, Z. Inhibition of tumor angiogenesis in lung cancer by T4 phage surface displaying mVEGFR2 vaccine. Vaccine 2011, 29, 5802–5811. [Google Scholar] [CrossRef]
- Pouyanfard, S.; Bamdad, T.; Hashemi, H.; Bandehpour, M.; Kazemi, B. Induction of protective anti-CTL epitope responses against HER-2-positive breast cancer based on multivalent T7 phage nanoparticles. PLoS ONE 2012, 7, e49539. [Google Scholar] [CrossRef]
- Shukla, G.S.; Sun, Y.J.; Pero, S.C.; Sholler, G.S.; Krag, D.N. Immunization with tumor neoantigens displayed on T7 phage nanoparticles elicits plasma antibody and vaccine-draining lymph node B cell responses. J. Immunol. Methods. 2018, 460, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Bakhshinejad, B.; Karimi, M.; Khalaj-Kondori, M. Phage display: Development of nanocarriers for targeted drug delivery to the brain. Neural. Regen. Res. 2015, 10, 862–865. [Google Scholar] [CrossRef]
- Samoylova, T.I.; Norris, M.D.; Samoylov, A.M.; Cochran, A.M.; Wolfe, K.G.; Petrenko, V.A.; Cox, N.R. Infective and inactivated filamentous phage as carriers for immunogenic peptides. J. Virol. Methods 2012, 183, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Brigati, J.R.; Petrenko, V.A. Thermostability of landscape phage probes. Anal. Bioanal. Chem. 2005, 382, 1346–1350. [Google Scholar] [CrossRef]
- Jepson, C.D.; March, J.B. Bacteriophage lambda is a highly stable DNA vaccine delivery vehicle. Vaccine 2004, 22, 2413–2419. [Google Scholar] [CrossRef] [PubMed]
- Dufour, N.; Delattre, R.; Debarbieux, L. In Vivo Bacteriophage Biodistribution. Methods Mol. Biol. 2018, 1693, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Rehman, S.; Ali, Z.; Khan, M.; Bostan, N.; Naseem, S. The dawn of phage therapy. Rev. Med. Virol. 2019, 29, e2041. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Foglizzo, V.; Marchiò, S. Bacteriophages as Therapeutic and Diagnostic Vehicles in Cancer. Pharmaceuticals 2021, 14, 161. https://doi.org/10.3390/ph14020161
Foglizzo V, Marchiò S. Bacteriophages as Therapeutic and Diagnostic Vehicles in Cancer. Pharmaceuticals. 2021; 14(2):161. https://doi.org/10.3390/ph14020161
Chicago/Turabian StyleFoglizzo, Valentina, and Serena Marchiò. 2021. "Bacteriophages as Therapeutic and Diagnostic Vehicles in Cancer" Pharmaceuticals 14, no. 2: 161. https://doi.org/10.3390/ph14020161
APA StyleFoglizzo, V., & Marchiò, S. (2021). Bacteriophages as Therapeutic and Diagnostic Vehicles in Cancer. Pharmaceuticals, 14(2), 161. https://doi.org/10.3390/ph14020161