
Design and Mechanism of Action of a New Prototype of Combi-Molecule “Programed” to Release Bioactive Species at a pH Range Akin to That of the Tumor Microenvironment


Abstract
1. Introduction
2. Results
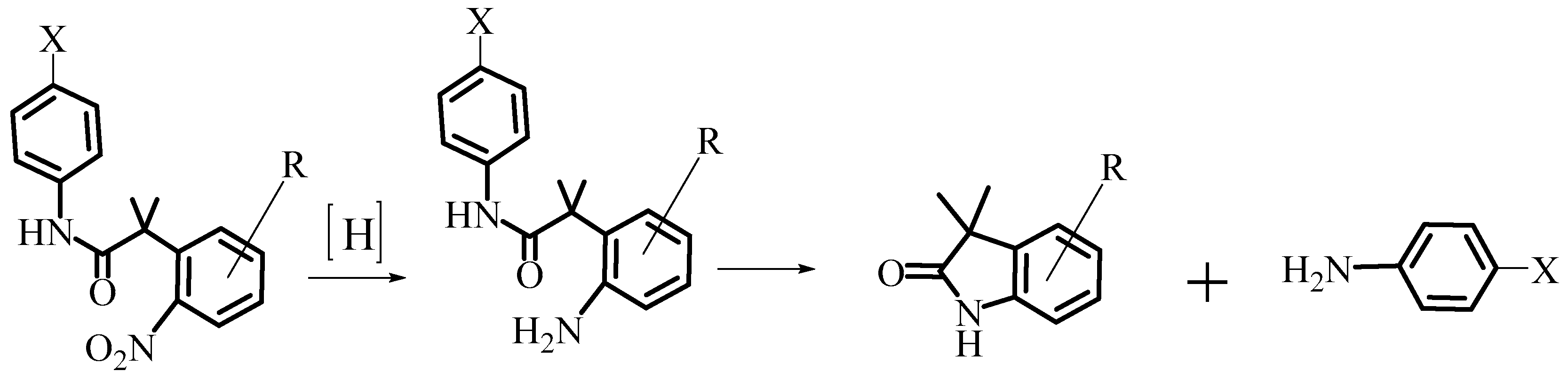
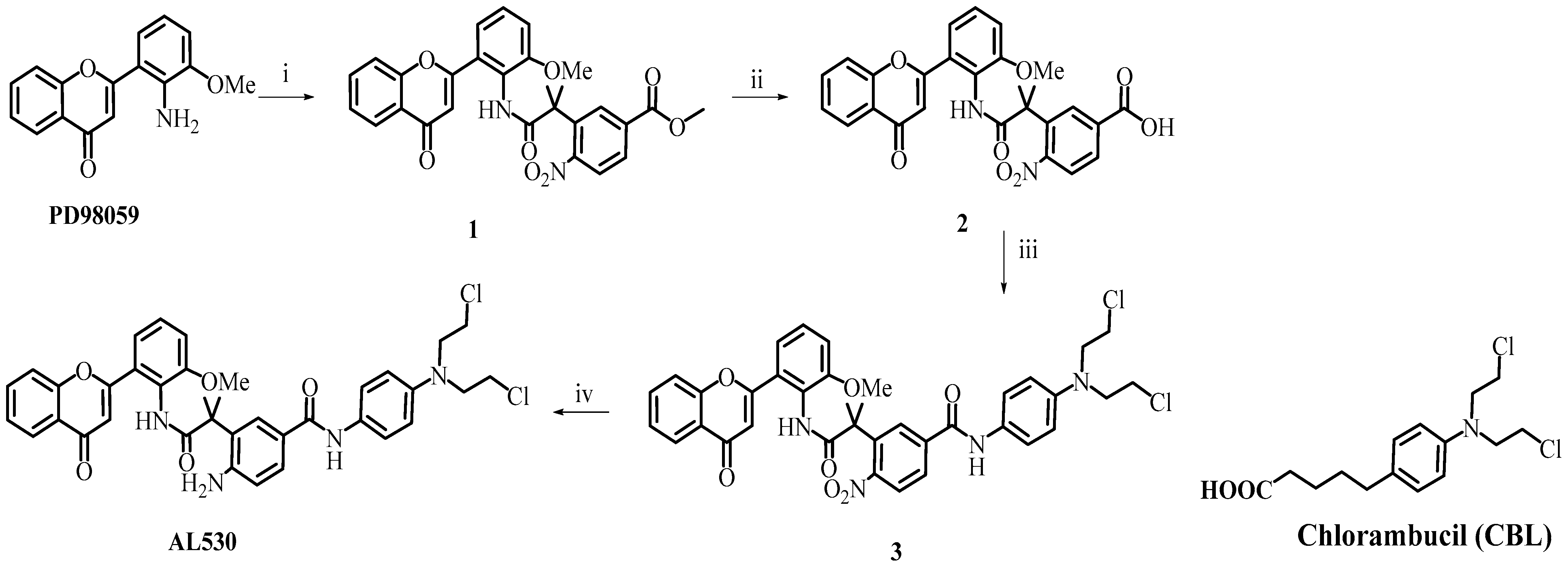
2.1. Design and Synthesis of AL530
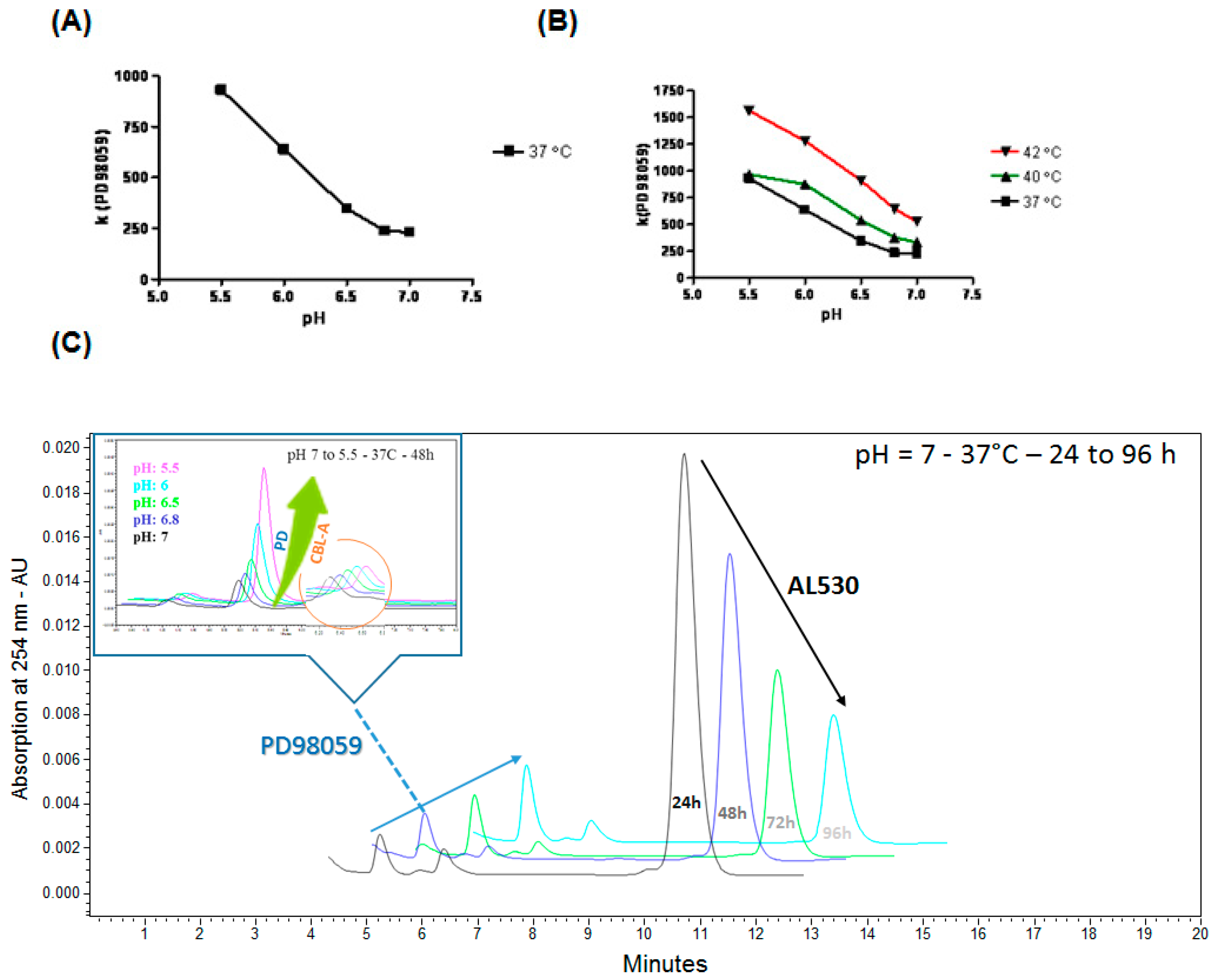
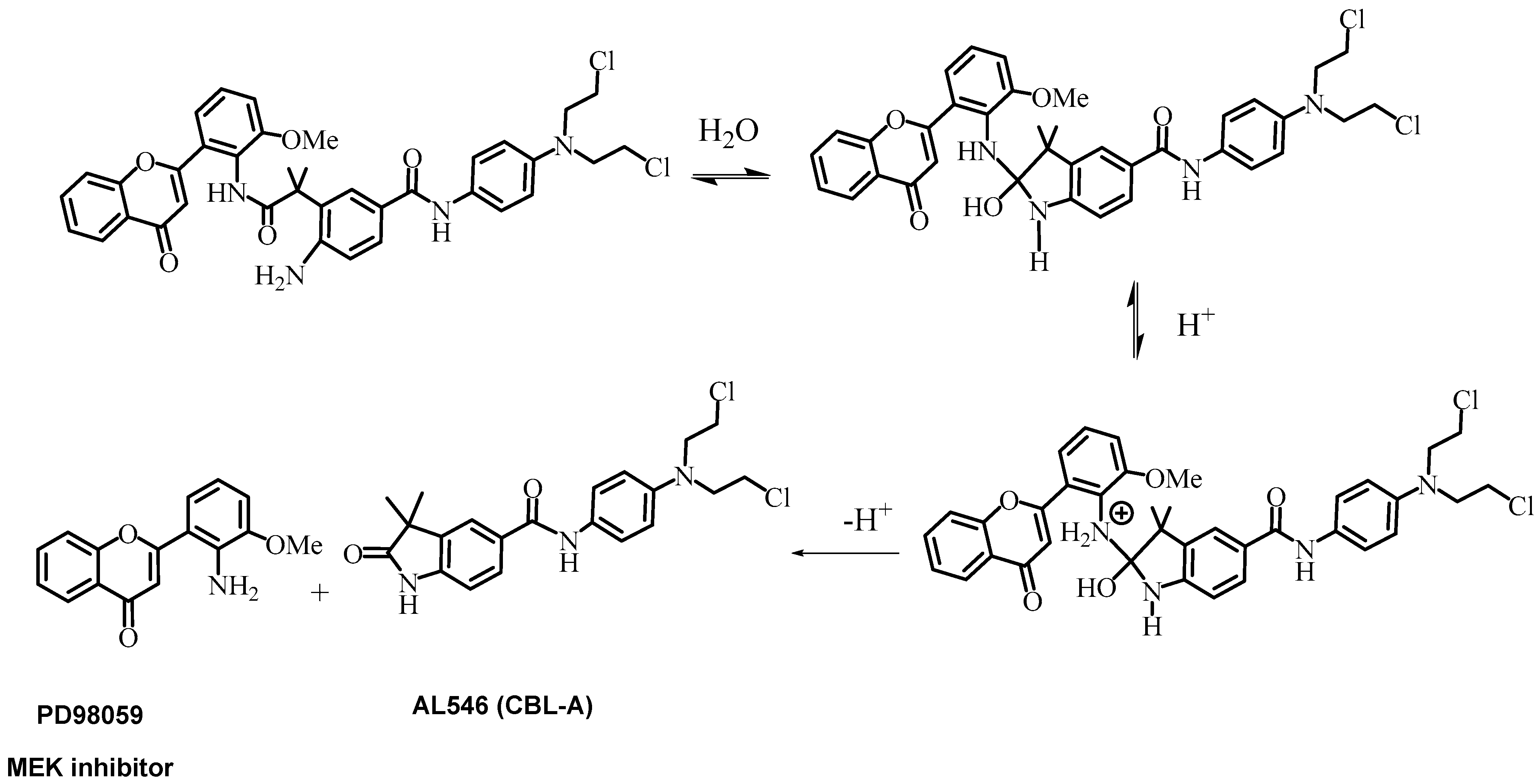
2.2. Kinetics of AL530 Hydrolysis at Variable pH and Temperature
2.3. Growth Inhibition of AL530
2.4. Erk Phosphorylation Inhibition by AL530
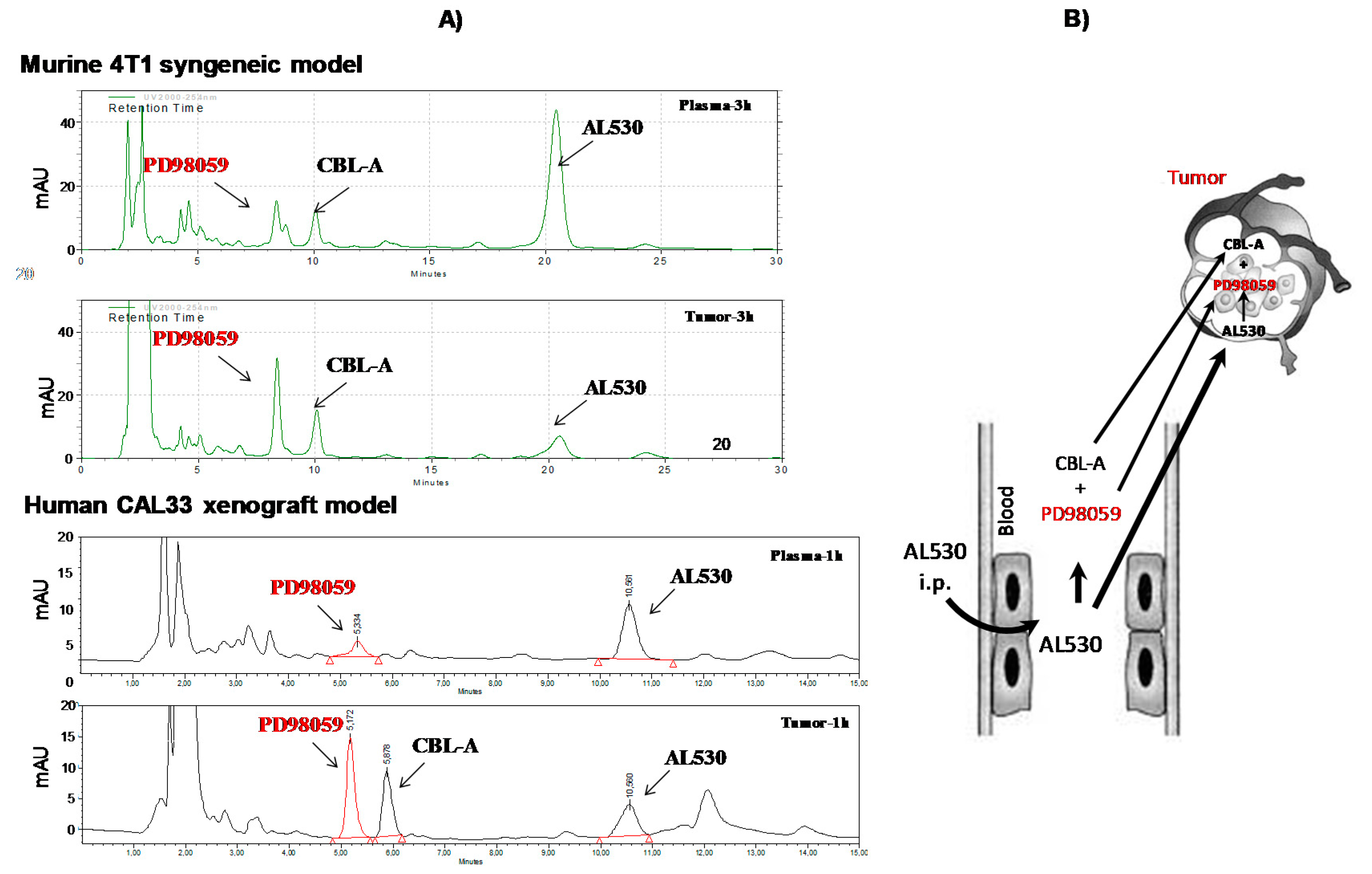
2.5. Pharmacokinetics and Pharmacodynamics of AL530
2.5.1. Study of the Release of PD98059 and CBL-A in Plasma and Tumor
2.5.2. Pharmacodynamics/Immunohistochemistry Analysis of AL530 Effect on Tumor Tissue
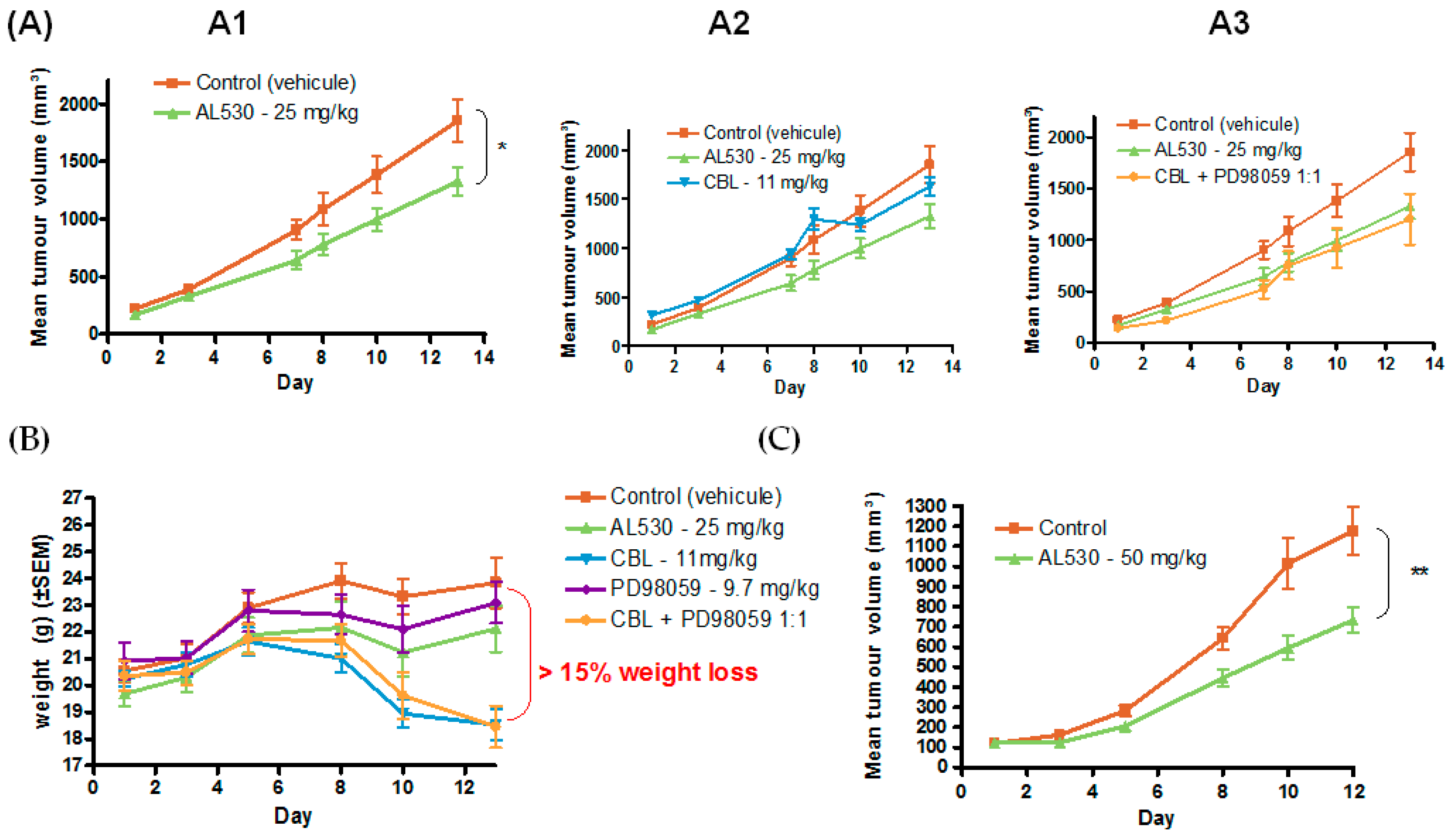
2.5.3. Efficacy of AL530 in Vivo
Dose Tolerance Efficacy
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.1.1. Compound 1
4.1.2. Compound 2
4.1.3. Compound 3
4.1.4. Compound AL530
4.2. Cell Lines
4.3. Cell Culture
4.4. In Vitro Drug Treatment
4.5. Growth Inhibition Assay and IC50 Determination
4.6. Temperature and pH Course Study of AL530 Hydrolysis
4.7. In Vivo Drug Treatment
4.7.1. (a) Syngeneic 4T1 Mouse Model for Tumor Distribution Analysis
4.7.2. (b) Xenograft Model
4.8. Pharmacokinetics
4.9. HPLC Sample Preparation
4.10. Western Blot Analysis
4.11. Immunohistochemistry
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Todorova, M.I.; Larroque, A.L. Subcellular distribution of a fluorescence-labeled combi-molecule designed to block epidermal growth factor receptor tyrosine kinase and damage DNA with a green fluorescent species. Mol. Cancer Ther. 2010, 9, 869–882. [Google Scholar] [CrossRef]
- Banerjee, R.; Huang, Y. The combi-targeting concept: Selective targeting of the epidermal growth factor receptor- and Her2-expressing cancer cells by the complex combi-molecule RB24. J. Pharmacol. Exp. Ther. 2010, 334, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, R.; Huang, Y. The combi-targeting concept: Mechanism of action of the pleiotropic combi-molecule RB24 and discovery of a novel cell signaling-based combination principle. Cell. Signal. 2011, 4, 630–640. [Google Scholar] [CrossRef]
- Banerjee, R.; Rachid, Z.J. Synthesis of a Prodrug Designed To Release Multiple Inhibitors of the Epidermal Growth Factor Receptor Tyrosine Kinase and an Alkylating Agent: A Novel Tumor Targeting Concept. J. Med. Chem. 2003, 46, 5546–5551. [Google Scholar] [CrossRef]
- Brahimi, F.; Matheson, S.L. Inhibition of epidermal growth factor receptor-mediated signaling by Combi-Triazene BJ2000, a new probe for Combi-Targeting postulates. J. Pharmacol. Exp. Ther. 2002, 303, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Q.; Dudouit, F. The combi-targeting concept: A novel 3,3-disubstituted nitrosourea with EGFR tyrosine kinase inhibitory properties. Cancer Chemother. Pharmacol. 2003, 51, 1–10. [Google Scholar] [CrossRef]
- Rachid, Z.; Brahimi, F. The Combi-Targeting Concept: Chemical Dissection of the Dual Targeting Properties of a Series of Combi-Triazenes. J. Med. Chem. 2003, 46, 4313–4321. [Google Scholar] [CrossRef]
- Rao, S.; Larroque-Lombard, A.-L. Target Modulation by a Kinase Inhibitor Engineered to Induce a Tandem Blockade of the Epidermal Growth Factor Receptor (EGFR) and c-Src: The Concept of Type III Combi-Targeting. PLoS ONE 2015, 10, e0117215. [Google Scholar] [CrossRef]
- Banerjee, R.; Rachid, Z. Sustained antiproliferative mechanisms by RB24, a targeted precursor of multiple inhibitors of epidermal growth factor receptor and a DNA alkylating agent in the A431 epidermal carcinoma of the vulva cell line. Br. J. Cancer 2004, 91, 1066–1073. [Google Scholar] [CrossRef][Green Version]
- Rachid, Z.; Brahimi, F. Synthesis of half-mustard combi-molecules with fluorescence properties: Correlation with EGFR status. Bioorg. Med. Chem. Lett. 2005, 15, 1135–1138. [Google Scholar] [CrossRef] [PubMed]
- Golabi, N.; Brahimi, F. A bioanalytical investigation on the exquisitely strong in vitro potency of the EGFR-DNA targeting type II combi-molecule ZR2003 and its mitigated in vivo antitumour activity. J. Pharmaceut. Biomed. Anal. 2011, 56, 592–599. [Google Scholar] [CrossRef]
- Sharifi, Z.; Abdulkarim, B. Mechanisms and Antitumor Activity of a Binary EGFR/DNA-Targeting Strategy Overcomes Resistance of Glioblastoma Stem Cells to Temozolomide. Clin. Cancer Res. 2019, 25, 7594–7608. [Google Scholar] [CrossRef]
- Yacoub, A.; McKinstry, R. Epidermal growth factor and ionizing radiation up-regulate the DNA repair genes XRCC1 and ERCC1 in DU145 and LNCaP prostate carcinoma through MAPK signaling. Radiat. Res. 2003, 159, 439–452. [Google Scholar] [CrossRef]
- Fang, J.S.; Gillies, R.D. Adaptation to hypoxia and acidosis in carcinogenesis and tumor progression. Semin. Cancer Biol. 2008, 18, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer. 2004, 4, 891–899. [Google Scholar] [CrossRef]
- Kato, Y.; Ozawa, S. Acidic extracellular microenvironment and cancer. Cancer Cell Int. 2013, 13, 1–8. [Google Scholar] [CrossRef]
- Atwell, G.J.; Sykes, B.M. Denny, Relationships between structure and kinetics of cyclization of 2-aminoaryl amides: Potential prodrugs of cyclization-activated aromatic mustards. J. Med. Chem. 1994, 37, 371–380. [Google Scholar] [CrossRef]
- Dudley, D.T.; Pang, L. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc. Natl. Acad. Sci. USA 1995, 92, 7686–7689. [Google Scholar] [CrossRef]
- Ullah Mughal, E.; Ayaz, M. Synthesis and antibacterial activity of substituted flavones, 4-thioflavones and 4-iminoflavones. Bioorg. Med. Chem. 2006, 14, 4704–4711. [Google Scholar] [CrossRef]
- Gesson-Paute, A.; Ferron, G. Pharmacokinetics of oxaliplatin during open versus laparoscopically assisted heated intraoperative intraperitoneal chemotherapy (HIPEC): An experimental study. Ann. Surg. Oncol. 2008, 15, 339–344. [Google Scholar] [CrossRef]
- Le Brun, J.; Campion, L. Survival benefit of hyperthermic intraperitoneal chemotherapy for recurrent ovarian cancer: A multi-institutional case control study. Ann. Surg. Oncol. 2014, 21, 3621–3627. [Google Scholar] [CrossRef] [PubMed]
- Morgan, A.M.; Parsels, L.A. Improving the Efficacy of Chemoradiation with Targeted Agents. Cancer Discov. 2014, 4, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, H.; Nakashiro, K. Possible contribution of active MMP2 to lymph-node metastasis and secreted cathepsin L to bone invasion of newly established human oral-squamous-cancer cell lines. Int. J. Cancer 1997, 7, 120–127. [Google Scholar] [CrossRef]
- Gioanni, J.; Fischel, J.L. Two new human tumor cell lines derived from squamous cell carcinomas of the tongue: Establishment, characterization and response to cytotoxic treatment. Eur. J. Cancer Clin. Oncol. 1988, 24, 1445–1455. [Google Scholar] [CrossRef]
- Lo Nigro, C.; Maffi, M. Impact of erythropoietin on the effects of irradiation under hypoxia. J. Cancer Res. Clin. Oncol. 2009, 135, 1615–1623. [Google Scholar] [CrossRef] [PubMed]
- Skehan, P.; Storeng, R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell line | Tumor Type | AL530 IC50 (µM) |
|---|---|---|
| DU145 | prostate | 16.83 ± 1.10 |
| MDA-MB-468 | breast | 3.15 |
| A427 | lung | 3.33 ± 0.07 |
| HN | head and neck | 4.14 ± 0.18 |
| CAL166 | head and neck | 20.31 ± 1.37 |
| CAL33 | head and neck | 11.50 ± 1.32 |
| Drug | HN (µM) | CAL166 (µM) | CAL33 (µM) |
|---|---|---|---|
| CBL | 21.34 ± 5.18 | 61.00 ± 8.92 | 57.4 ± 2.30 |
| PD98059 | nd | nd | nd |
| CBL + fixed PD (20 µM) | 4.63 ± 0.70 | nd | 33.7 ± 4.40 |
| CBL + PD (1:1) | 12.3 ± 0.30 | nd | 24.0 ± 2.90 |
| AL530 | 4.14 ± 0.18 | 20.31 ± 1.37 | 11.50 ± 1.32 |
| (A) | AUC0–24h mg/µL·h | AUC0–24h (mg/g·h) | T1/2 (h) | Ke (min−1) | Cmax mg/µL for Plasma Cmax mg/g for Tumor | tmax (h) | |
|---|---|---|---|---|---|---|---|
| Plasma (P) | AL530 | 3984.8 | 3,984,750 | 1.6 | 7.17 | 51.02 | 2 |
| PD98059 released | 339.8 | 339,816.7 | 11.71 | 0.0009 | 3.61 | 2 | |
| Tumor (T) | AL530 | 6,393,616.7 | 3.93 | 2.94 | 32,345.62 | 2.5 | |
| PD98059 released | 1,658,845 | 6.77 | 0.0017 | 7054.31 | 2.5 |
| (B) | Ratio AUC0–24h_Tumor/AUC0–24h_Plasma |
|---|---|
| AL530 | 1.6 |
| PD98059 | 4.88 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Larroque-Lombard, A.-L.; Chatelut, E.; Delord, J.-P.; Imbs, D.-C.; Rochaix, P.; Jean-Claude, B.; Allal, B. Design and Mechanism of Action of a New Prototype of Combi-Molecule “Programed” to Release Bioactive Species at a pH Range Akin to That of the Tumor Microenvironment. Pharmaceuticals 2021, 14, 160. https://doi.org/10.3390/ph14020160
Larroque-Lombard A-L, Chatelut E, Delord J-P, Imbs D-C, Rochaix P, Jean-Claude B, Allal B. Design and Mechanism of Action of a New Prototype of Combi-Molecule “Programed” to Release Bioactive Species at a pH Range Akin to That of the Tumor Microenvironment. Pharmaceuticals. 2021; 14(2):160. https://doi.org/10.3390/ph14020160
Chicago/Turabian StyleLarroque-Lombard, Anne-Laure, Etienne Chatelut, Jean-Pierre Delord, Diane-Charlotte Imbs, Philippe Rochaix, Bertrand Jean-Claude, and Ben Allal. 2021. "Design and Mechanism of Action of a New Prototype of Combi-Molecule “Programed” to Release Bioactive Species at a pH Range Akin to That of the Tumor Microenvironment" Pharmaceuticals 14, no. 2: 160. https://doi.org/10.3390/ph14020160
APA StyleLarroque-Lombard, A.-L., Chatelut, E., Delord, J.-P., Imbs, D.-C., Rochaix, P., Jean-Claude, B., & Allal, B. (2021). Design and Mechanism of Action of a New Prototype of Combi-Molecule “Programed” to Release Bioactive Species at a pH Range Akin to That of the Tumor Microenvironment. Pharmaceuticals, 14(2), 160. https://doi.org/10.3390/ph14020160