
The Protein-Binding Behavior of Platinum Anticancer Drugs in Blood Revealed by Mass Spectrometry


Abstract
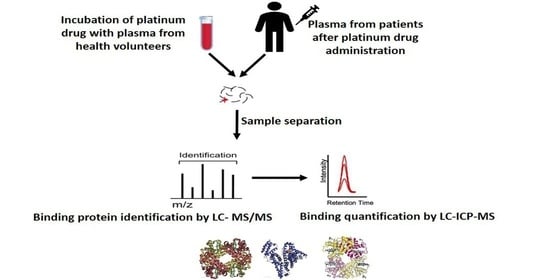
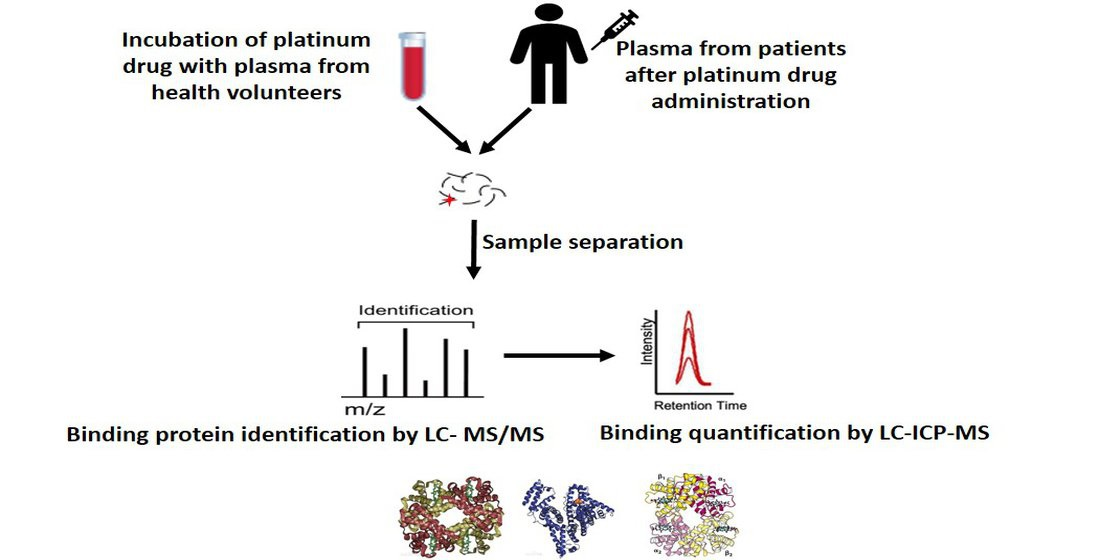
1. Introduction
2. Mass Spectrometry Techniques Used in Metallomics
2.1. Inductively Coupled Plasma Mass Spectrometry (ICP-MS)
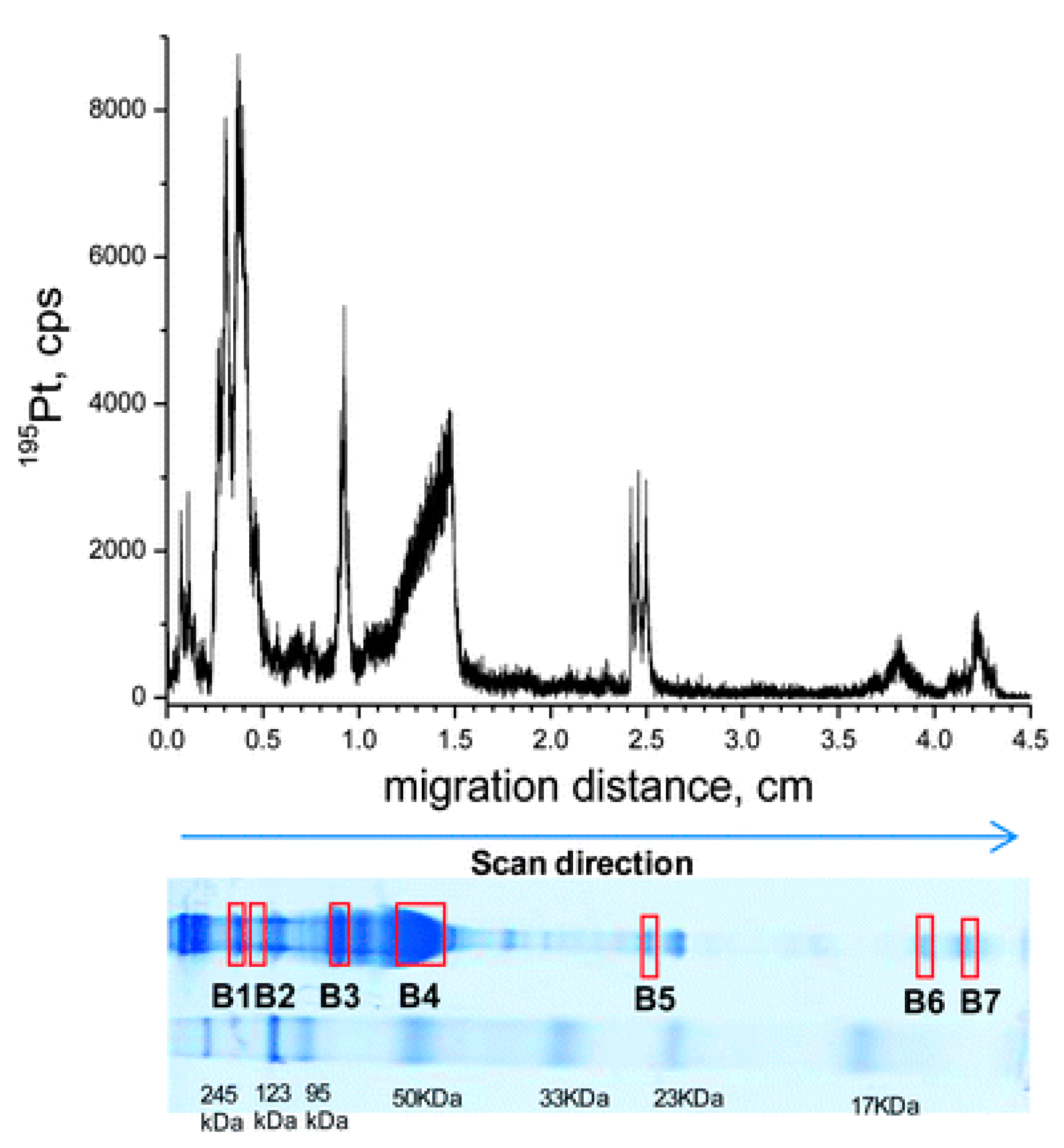
2.2. Laser Ablation (LA)–ICP-MS
2.3. Electrospray Ionization (ESI)-MS
3. Protein-Binding Behavior of Platinum Drugs in Blood Elucidated by Mass Spectrometry
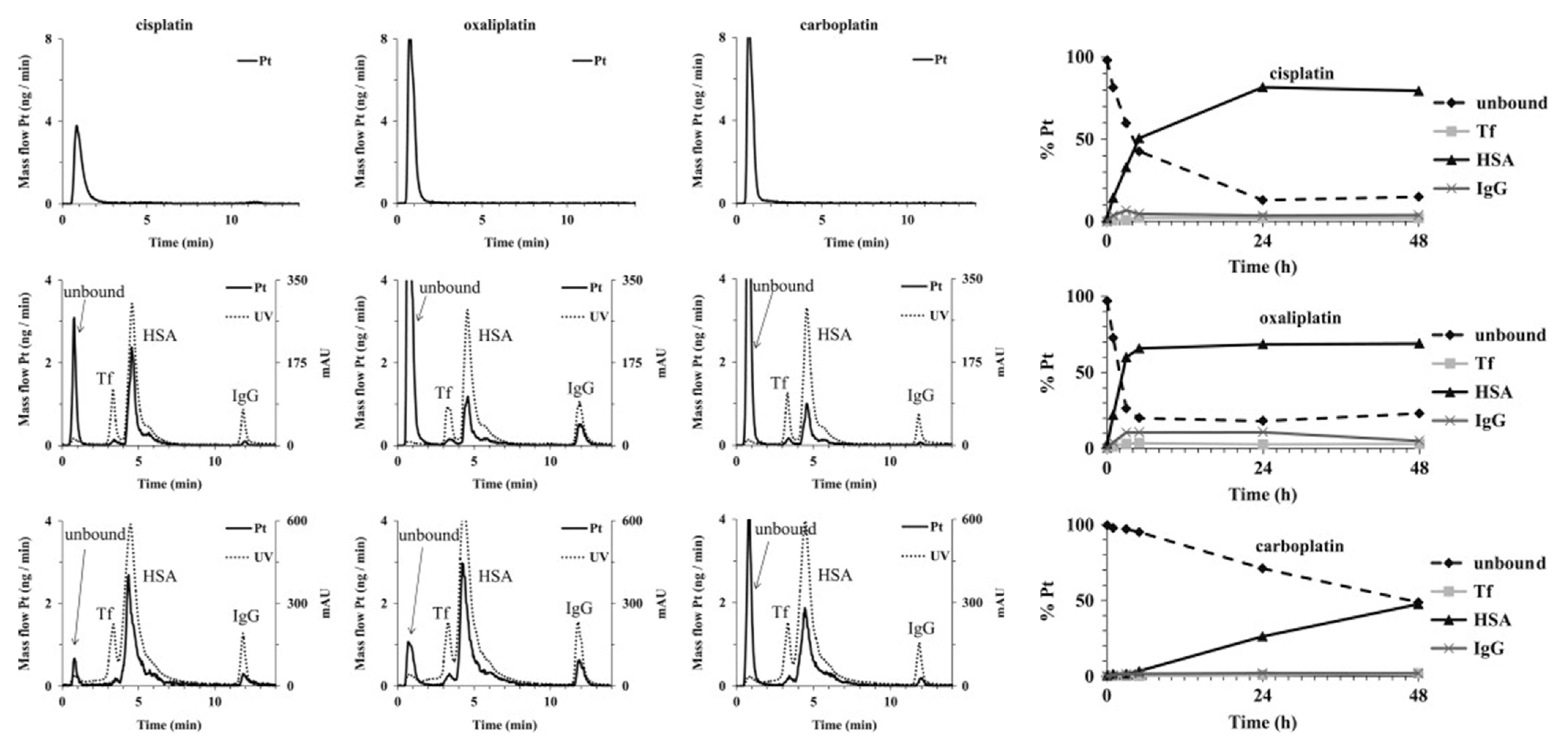
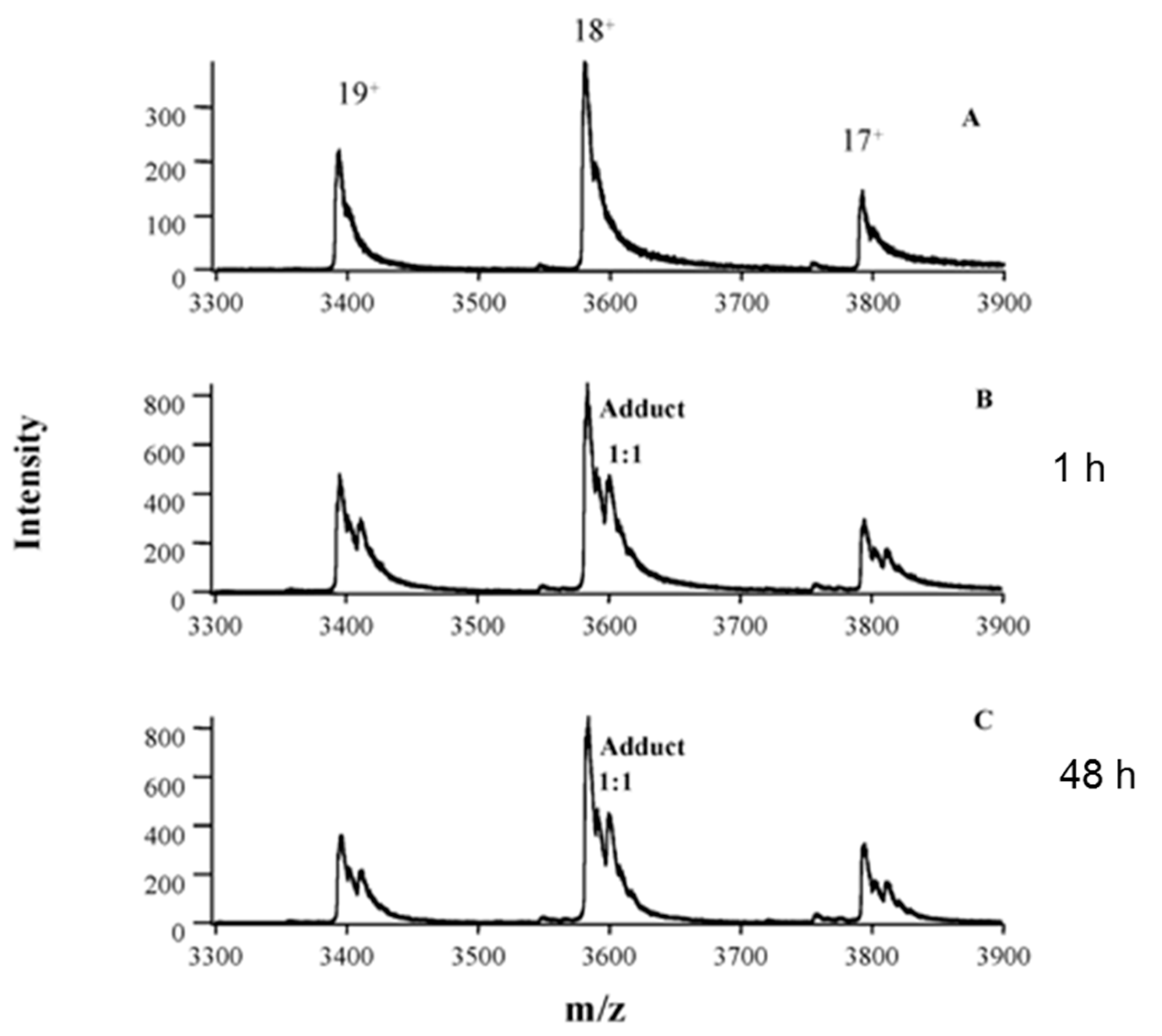
3.1. In Vitro Binding Analysis
3.2. In Vivo Binding Analysis
3.3. Software for the Identification of Proteins Binding with Platinum Drugs
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| A1at | α-2-antitrypsin |
| α2MG | α-2-macroglobulin |
| AES | atomic emission spectroscopy |
| Apoa1 | Apolipoprotein A-I |
| Apoa2 | Apolipoprotein A-II |
| APOC2 | apolipoprotein C-II |
| CBB | Coomassie brilliant blue |
| CD | circular dichroism |
| CE | capillary electrophoresis |
| CID | collision-induced dissociation |
| DEAE | diethylamino |
| ESI-MS/MS | electrospray ionization mass spectrometry |
| FASP | filter-aided sample preparation |
| HSA | human serum albumin, |
| Hb | hemoglobin |
| HPLC–ICP-MS | high-performance liquid chromatography/inductively coupled plasma mass spectrometry |
| IE | ion exchange |
| IEF | isoelectric focusing |
| IgG | immunoglobulin |
| LA-ICP-MS | laser ablation/inductively coupled plasma mass spectrometry |
| MudPIT | multidimensional protein identification technology |
| PBR | protein-binding rate |
| RBC | red blood cells |
| SCX | strong cation exchange |
| SEC | size-exclusion chromatography |
| SNAP | Smart Numerical Annotation Procedure |
| Tf | transferrin |
| Trfe | serotransferrin |
References
- Rosenberg, B.; Vancamp, L.; Krigas, T. Inhibition of Cell Division in Escherichia coli by Electrolysis Products from a Platinum Electrode. Nature 1965, 205, 698–699. [Google Scholar] [CrossRef] [PubMed]
- Wiltshaw, E. A review of clinical experience with cis-platinum diammine dichloride: 1972–1978. Biochimie 1978, 60, 925–929. [Google Scholar] [CrossRef]
- Becher, R.; Schütt, P.; Osieka, R.; Schmidt, C.G. Peripheral neuropathy and ophthalmologic toxicity after treatment with cis-dichlorodiaminoplatinum II. J. Cancer Res. Clin. Oncol. 1980, 96, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Hanif, M.; Hartinger, C.G. Anticancer metallodrugs: Where is the next cisplatin? Future Med. Chem. 2018, 10, 615–617. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Zhao, Y.; Luo, Q.; Zhang, Y.; Wu, K.; Wang, F. Rational design of multi-targeting ruthenium- and platinum-based anticancer complexes. Sci. China Chem. 2016, 59, 1240–1249. [Google Scholar] [CrossRef]
- Dilruba, S.; Kalayda, G.V. Platinum-based drugs: Past, present and future. Cancer Chemother. Pharmacol. 2016, 77, 1103–1124. [Google Scholar] [CrossRef]
- Go, R.S.; Adjei, A.A. Review of the comparative pharmacology and clinical activity of cisplatin and carboplatin. J. Clin. Oncol. 1999, 17, 409–422. [Google Scholar] [CrossRef]
- Wiltshaw, E. Ovarian trials at the Royal Marsden. Cancer Treat. Rev. 1985, 12 (Suppl. A), 67–71. [Google Scholar] [CrossRef]
- Grothey, A. Oxaliplatin-safety profile: Neurotoxicity. Semin. Oncol. 2003, 30, 5–13. [Google Scholar] [CrossRef]
- Rixe, O.; Ortuzar, W.; Alvarez, M.; Parker, R.; Reed, E.; Paull, K.; Fojo, T. Oxaliplatin, tetraplatin, cisplatin, and carboplatin: Spectrum of activity in drug-resistant cell lines and in the cell lines of the national cancer institute’s anticancer drug screen panel. Biochem. Pharmacol. 1996, 52, 1855–1865. [Google Scholar] [CrossRef]
- Pendyala, L.; Creaven, P.J. In Vitro Cytotoxicity, Protein Binding, Red Blood Cell Partitioning, and Biotransformation of Oxaliplatin. Cancer Res. 1993, 53, 5970–5976. [Google Scholar] [PubMed]
- Alberts, D.S.; Fanta, P.T.; Running, K.L.; Adair, L.P., Jr.; Garcia, D.J.; Liu-Stevens, R.; Salmon, S.E. In vitro phase II comparison of the cytotoxicity of a novel platinum analog, nedaplatin (254-S), with that of cisplatin and carboplatin against fresh, human ovarian cancers. Cancer Chemother. Pharmacol. 1997, 39, 493–497. [Google Scholar] [CrossRef] [PubMed]
- Akaza, H.; Togashi, M.; Nishio, Y.; Miki, T.; Kotake, T.; Matsumura, Y.; Yoshida, O.; Aso, Y. Phase II study of cis-diammine(glycolato)platinum, 254-S, in patients with advanced germ-cell testicular cancer, prostatic cancer, and transitional-cell carcinoma of the urinary tract. 254-S Urological Cancer Study Group. Cancer Chemother. Pharmacol. 1992, 31, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, M.; Shinkai, T.; Eguchi, K.; Sasaki, Y.; Tamura, T.; Ohe, Y.; Kojima, A.; Oshita, F.; Hara, K.; Saijo, N. Phase II study of (glycolate-O,O′) diammineplatinum(II), a novel platinum complex, in the treatment of non-small-cell lung cancer. Cancer Chemother. Pharmacol. 1990, 26, 393–396. [Google Scholar] [CrossRef] [PubMed]
- Inuyama, Y.; Miyake, H.; Horiuchi, M.; Hayasaki, K.; Komiyama, S.; Ota, K. A late phase II clinical study of cis-diammine glycolato platinum, 254-S, for head and neck cancers. Cancer Chemother. 1992, 19, 871–877. [Google Scholar]
- Sasaki, Y.; Tamura, T.; Eguchi, K.; Shinkai, T.; Fujiwara, Y.; Fukuda, M.; Ohe, Y.; Bungo, M.; Horichi, N.; Niimi, S.; et al. Pharmacokinetics of (glycolate-0,0′)-diammine platinum (II), a new platinum derivative, in comparison with cisplatin and carboplatin. Cancer Chemother. Pharmacol. 1989, 23, 243–246. [Google Scholar] [CrossRef]
- Zalba, S.; Garrido, M.J. Liposomes, a promising strategy for clinical application of platinum derivatives. Expert Opin. Drug Delivery. 2013, 10, 829–844. [Google Scholar] [CrossRef]
- Nicholas, P.F. Platinum Formulations as Anticancer Drugs Clinical and Pre-Clinical Studies. Curr. Trends Med. Chem. 2011, 11, 2623–2631. [Google Scholar]
- O’Dwyer, P.J.; Stevenson, J.P.; Johnson, S.W. Clinical pharmacokinetics and administration of established platinum drugs. Drugs. 2000, 59 (Suppl. 4), 19–27. [Google Scholar]
- Deconti, R.C.; Toftness, B.R.; Lange, R.C.; Creasey, W.A. Clinical and pharmacological studies with cis-diamminedichloroplatinum (II). Cancer Res. 1973, 33, 1310–1315. [Google Scholar]
- Kato, R.; Sato, T.; Iwamoto, A.; Yamazaki, T.; Nakashiro, S.; Yoshikai, S.; Fujimoto, A.; Imano, H.; Ijiri, Y.; Mino, Y.J.B.; et al. Interaction of Platinum Agents, Cisplatin, Carboplatin, and Oxaliplatin against Albumin in Vivo Rats and In Vitro Study Using Inductively Coupled Plasma-Mass Spectrometer. Biopharm. Drug Dispos. 2019, 40, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Gormley, P.E.; Bull, J.M.; LeRoy, A.F.; Cysyk, R. Kinetics of cis-dichlorodiammineplatinum. Clin. Pharmacol. Ther. 1979, 25, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Belt, R.J.; Himmelstein, K.J.; Patton, T.F.; Bannister, S.J.; Repta, A.J. Pharmacokinetics of non-protein-bound platinum species following administration of cis-dichlorodiammineplatinum (II). Cancer Treat. Rep. 1979, 63, 1515. [Google Scholar] [PubMed]
- Harland, S.J.; Newell, D.R.; Siddik, Z.H.; Chadwick, R.; Calvert, A.H.; Harrap, K.R. Pharmacokinetics of cis-diammine-1,1-cyclobutane dicarboxylate platinum (II) in patients with normal and impaired renal function. Cancer Res. 1984, 44, 1693–1697. [Google Scholar] [PubMed]
- Newell, D.R.; Pearson, A.D.J.; Balmanno, K.; Price, L.; Stevens, M.C.G. Carboplatin pharmacokinetics in children: The development of a pediatric dosing formula. The United Kingdom Children’s Cancer Study Group. J. Clin. Oncol. 1993, 11, 2314–2323. [Google Scholar] [CrossRef]
- Jodrell, D.I.; Egorin, M.J.; Canetta, R.M.; Langenberg, P.; Goldbloom, E.P.; Burroughs, J.N.; Goodlow, J.L.; Tan, S.; Wiltshaw, E. Relationships between carboplatin exposure and tumor response and toxicity in patients with ovarian cancer. J. Clin. Oncol. 1992, 10, 520–528. [Google Scholar] [CrossRef]
- Calvert, A.H.; Newell, D.R.; Gumbrell, L.A.; O’Reilly, S.; Burnell, M.; Boxall, F.E.; Siddik, Z.H.; Judson, I.R.; Gore, M.E.; Wiltshaw, E. Carboplatin dosage: Prospective evaluation of a simple formula based on renal function. J. Clin. Oncol. 1989, 7, 1748–1756. [Google Scholar] [CrossRef]
- Graham, M.A.; Lockwood, G.F.; Greenslade, D.; Brienza, S.; Bayssas, M.; Gamelin, E. Clinical pharmacokinetics of oxaliplatin: A critical review. Clin. Cancer Res. 2000, 6, 1205–1218. [Google Scholar]
- Boulikas, T.; Vougiouka, M. Cisplatin and platinum drugs at the molecular level. (Review). Oncol. Rep. 2003, 10, 1663–1682. [Google Scholar] [CrossRef]
- Wang, D.; Lippard, S.J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320. [Google Scholar] [CrossRef]
- Akaboshi, M.; Kawai, K.; Maki, H.; Akuta, K.; Ujeno, Y.; Miyahara, T. The number of platinum atoms binding to DNA, RNA and protein molecules of HeLa cells treated with cisplatin at its mean lethal concentration. Jpn. J. Cancer Res. 1992, 83, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Akaboshi, M.; Kawai, K.; Ujeno, Y.; Takada, S.; Miyahara, T. Binding characteristics of (-)-(R)-2-Aminomethylpyrrolidine(1,1-cyclobutanedi-carboxylato)-2-platinum (II) to DNA, RNA and protein molecules in HeLa cells and its lethal effect: Comparison with cis- and trans-diamminedichloroplatinums (II). Jpn. J. Cancer Res. 1994, 85, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Cepeda, V.; Fuertes, M.A.; Castilla, J.; Alonso, C.; Quevedo, C.; Pérez, J.M. Biochemical mechanisms of cisplatin cytotoxicity. Anticancer Agents Med. Chem. 2007, 7, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, B.T.; Peterson, E.J.; Kabolizadeh, P.; Martínez, A.; Kipping, R.; Farrell, N.P. Effects of Noncovalent Platinum Drug–Protein Interactions on Drug Efficacy: Use of Fluorescent Conjugates as Probes for Drug Metabolism. Mol. Pharm. 2011, 8, 940–948. [Google Scholar] [CrossRef]
- Theiner, S.; Varbanov, H.P.; Galanski, M.; Egger, A.E.; Berger, W.; Heffeter, P.; Keppler, B.K. Comparative in vitro and in vivo pharmacological investigation of platinum (IV) complexes as novel anticancer drug candidates for oral application. J. Biol. Inorg. Chem. 2015, 20, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Messori, L.; Merlino, A. Cisplatin binding to proteins: A structural perspective. Coord. Chem. Rev. 2016, 315, 67–89. [Google Scholar] [CrossRef]
- Mayr, J.; Heffeter, P.; Groza, D.; Galvez, L.; Koellensperger, G.; Roller, A.; Alte, B.; Haider, M.; Berger, W.; Kowol, C.R. An albumin-based tumor-targeted oxaliplatin prodrug with distinctly improved anticancer activity in vivo. Chem. Sci. 2017, 8, 2241–2250. [Google Scholar] [CrossRef]
- Göschl, S.; Schreiber-Brynzak, E.; Pichler, V.; Cseh, K.; Heffeter, P.; Jungwirth, U.; Jakupec, M.A.; Berger, W.; Keppler, B.K. Comparative studies of oxaliplatin-based platinum (iv) complexes in different in vitro and in vivo tumor models. Metallomics. 2017, 9, 309–322. [Google Scholar] [CrossRef]
- Seflova, J.; Cechova, P.; Stenclova, T.; Sebela, M.; Kubala, M. Identification of cisplatin-binding sites on the large cytoplasmic loop of the Na+/K+-ATPase. J. Enzym. Inhib. Med. Chem. 2018, 33, 701–706. [Google Scholar] [CrossRef]
- Massai, L.; Pratesi, A.; Gailer, J.; Marzo, T.; Messori, L. The Cisplatin/Serum Albumin System: A Reappraisal. Inorg. Chim. Acta 2019, 495, 118983. [Google Scholar] [CrossRef]
- Makovec, T. Cisplatin and beyond: Molecular mechanisms of action and drug resistance development in cancer chemotherapy. Radiol. Oncol. 2019, 53, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Park, C.R.; Kim, H.Y.; Song, M.G.; Lee, Y.-S.; Youn, H.; Chung, J.-K.; Cheon, G.J.; Kang, K.W. Efficacy and Safety of Human Serum Albumin-Cisplatin Complex in U87MG Xenograft Mouse Models. Int. J. Mol. Sci. 2020, 21, 7932. [Google Scholar] [CrossRef] [PubMed]
- Jun, P.; Rupasri, M.; Michael, S.; Xing-Fang, L.J.C.C. Characterization of Intact Hemoglobin and Oxaliplatin Interaction by Nanoelectrospray Ionization Tandem Mass Spectrometry. Clin. Chem. 2005, 51, 2274–2281. [Google Scholar]
- Hartinger, C.G.; Groessl, M.; Meier, S.M.; Casini, A.; Dyson, P.J. Application of mass spectrometric techniques to delineate the modes-of-action of anticancer metallodrugs. Chem. Soc. Rev. 2013, 42, 6186–6199. [Google Scholar] [CrossRef] [PubMed]
- Pinato, O.; Musetti, C.; Sissi, C. Pt-based drugs: The spotlight will be on proteins. Metallomics 2014, 6, 380–395. [Google Scholar] [CrossRef] [PubMed]
- Michelucci, E.; Pieraccini, G.; Moneti, G.; Gabbiani, C.; Pratesi, A.; Messori, L. Mass spectrometry and metallomics: A general protocol to assess stability of metallodrug-protein adducts in bottom-up MS experiments. Talanta 2017, 167, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Luo, Q.; Zhao, Y.; Zheng, W.; Wang, F. Investigations of molecular mechanism of action of metal-based anticancer complexes by mass spectrometry. Sci. Sin. Chim. 2017, 47, 233–248. [Google Scholar]
- Holtkamp, H.U.; Hartinger, C.G. Advanced metallomics methods in anticancer metallodrug mode of action studies. Trac Trends Anal. Chem. 2018, 104, 110–117. [Google Scholar] [CrossRef]
- Harper, B.W.J.; Morris, T.T.; Gailer, J.; Aldrich-Wright, J.R. Probing the interaction of bisintercalating (2,2′:6′,2″-terpyridine)platinum (II) complexes with glutathione and rabbit plasma. J. Inorg. Biochem. 2016, 163, 95–102. [Google Scholar] [CrossRef]
- Dolman, R.C.; Deacon, G.B.; Hambley, T.W. Studies of the binding of a series of platinum (IV) complexes to plasma proteins. J. Inorg. Biochem. 2002, 88, 260–267. [Google Scholar] [CrossRef]
- Chen, C.K.J.; Gui, X.; Kappen, P.; Renfrew, A.K.; Hambley, T.W. The effect of charge on the uptake and resistance to reduction of platinum(iv) complexes in human serum and whole blood models. Metallomics 2020, 12, 1599–1615. [Google Scholar] [CrossRef] [PubMed]
- Brouwers, E.E.; Tibben, M.; Rosing, H.; Schellens, J.H.; Beijnen, J.H. The application of inductively coupled plasma mass spectrometry in clinical pharmacological oncology research. Mass Spectrom. Rev. 2008, 27, 67–100. [Google Scholar] [CrossRef] [PubMed]
- Lobinski, R.; Schaumloffel, D.; Szpunar, J. Mass spectrometry in bioinorganic analytical chemistry. Mass Spectrom. Rev. 2006, 25, 255–289. [Google Scholar] [CrossRef] [PubMed]
- Klose, M.H.M.; Schoberl, A.; Heffeter, P.; Berger, W.; Hartinger, C.G.; Koellensperger, G.; Meier-Menches, S.M.; Keppler, B.K. Serum-binding properties of isosteric ruthenium and osmium anticancer agents elucidated by SEC-ICP-MS. Mon. Chem. 2018, 149, 1719–1726. [Google Scholar] [CrossRef] [PubMed]
- Galvez, L.; Theiner, S.; Grabarics, M.; Kowol, C.R.; Keppler, B.K.; Hann, S.; Koellensperger, G. Critical assessment of different methods for quantitative measurement of metallodrug-protein associations. Anal. Bioanal. Chem. 2018, 410, 7211–7220. [Google Scholar] [CrossRef]
- Esteban-Fernández, D.; Moreno-Gordaliza, E.; Caas, B.; Palacios, M.A.; Gómez-Gómez, M.M. Analytical methodologies for metallomics studies of antitumor Pt-containing drugs. Metallomics 2010, 2, 19–38. [Google Scholar] [CrossRef] [PubMed]
- Galvez, L.; Rusz, M.; Jakupec, M.A.; Koellensperger, G. Heart-cut 2DSEC-RP-LC-ICP-MS as a screening tool in metal-based anticancer research Electronic supplementary information (ESI) available. J. Anal. Atomic Spectrom. 2019, 34, 1279–1286. [Google Scholar] [CrossRef]
- Bytzek, A.K.; Hartinger, C.G. Capillary electrophoretic methods in the development of metal-based therapeutics and diagnostics: New methodology and applications. Electrophoresis 2012, 33, 622–634. [Google Scholar] [CrossRef]
- Meermann, B.; Sperling, M. Hyphenated techniques as tools for speciation analysis of metal-based pharmaceuticals: Developments and applications. Anal. Bioanal. Chem. 2012, 403, 1501–1522. [Google Scholar] [CrossRef]
- Polec-Pawlak, K.; Abramski, J.K.; Semenova, O.; Hartinger, C.G.; Timerbaev, A.R.; Keppler, B.K.; Jarosz, M. Platinum group metallodrug-protein binding studies by capillary electrophoresis—Inductively coupled plasma-mass spectrometry: A further insight into the reactivity of a novel antitumor ruthenium(III) complex toward human serum proteins. Electrophoresis 2006, 27, 1128–1135. [Google Scholar] [CrossRef]
- Konz, I.; Fernández, B.; Fernández, M.L.; Pereiro, R.; Sanz-Medel, A. Laser ablation ICP-MS for quantitative biomedical applications. Anal. Bioanal. Chem. 2012, 403, 2113–2125. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Gordaliza, E.; Giesen, C.; Lazaro, A.; Esteban-Fernandez, D.; Humanes, B.; Canas, B.; Panne, U.; Tejedor, A.; Jakubowski, N.; Gomez-Gomez, M.M. Elemental bioimaging in kidney by LA-ICP-MS as a tool to study nephrotoxicity and renal protective strategies in cisplatin therapies. Anal. Chem. 2011, 83, 7933–7940. [Google Scholar] [CrossRef] [PubMed]
- Will, J.; Kyas, A.; Sheldrick, W.S.; Wolters, D. Identification of (eta6-arene)ruthenium(II) protein binding sites in E. coli cells by combined multidimensional liquid chromatography and ESI tandem mass spectrometry: Specific binding of [(eta6-p-cymene)RuCl2 (DMSO)] to stress-regulated proteins and to helicases. J. Biol. Inorg. Chem. 2007, 12, 883–894. [Google Scholar] [PubMed]
- Will, J.; Sheldrick, W.S.; Wolters, D. Characterisation of cisplatin coordination sites in cellular Escherichia coli DNA-binding proteins by combined biphasic liquid chromatography and ESI tandem mass spectrometry. J. Biol. Inorg. Chem. 2008, 13, 421–434. [Google Scholar] [CrossRef] [PubMed]
- Will, J.; Wolters, D.A.; Sheldrick, W.S. Characterisation of Cisplatin Binding Sites in Human Serum Proteins Using Hyphenated Multidimensional Liquid Chromatography and ESI Tandem Mass Spectrometry. Chem. Med. Chem. 2010, 3, 1696–1707. [Google Scholar] [CrossRef]
- Wiglusz, K.; Trynda-Lemiesz, L. Platinum drugs binding to human serum albumin: Effect of non-steroidal anti-inflammatory drugs. J. Photochem. Photobiol. A 2014, 289, 1–6. [Google Scholar] [CrossRef]
- Shafaei, Z.; Abazari, O.; Divsalar, A.; Ghalandari, B.; Poursoleiman, A.; Saboury, A.A.; Ahmad, F. Effect of a Synthesized Amyl-Glycine1, 10-Phenanthroline Platinum Nitrate on Structure and Stability of Human Blood Carrier Protein, Albumin: Spectroscopic and Modeling Approaches. J. Fluoresc. 2017, 27, 1829–1838. [Google Scholar] [CrossRef]
- Wang, Z.; Fang, L.; Zhao, J.; Gou, S. Insight into the antitumor actions of sterically hindered platinum(ii) complexes by a combination of STD NMR and LCMS techniques. Metallomics 2020, 12, 427–434. [Google Scholar] [CrossRef]
- Timerbaev, A.R.; Hartinger, C.G.; Aleksenko, S.S.; Keppler, B.K. Interactions of Antitumor Metallodrugs with Serum Proteins: Advances in Characterization Using Modern Analytical Methodology. Chem. Rev. 2006, 106, 2224–2248. [Google Scholar] [CrossRef]
- Chen, C.K.J.; Kappen, P.; Hambley, T.W. The reduction of cis-platinum(iv) complexes by ascorbate and in whole human blood models using H-1 NMR and XANES spectroscopy. Metallomics 2019, 11, 686–695. [Google Scholar] [CrossRef]
- Morris, T.T.; Ruan, Y.; Lewis, V.A.; Narendran, A.; Gailer, J. Fortification of blood plasma from cancer patients with human serum albumin decreases the concentration of cisplatin-derived toxic hydrolysis products in vitro. Metallomics 2014, 6, 2034–2041. [Google Scholar] [CrossRef] [PubMed]
- Theiner, S.; Grabarics, M.; Galvez, L.; Varbanov, H.P.; Sommerfeld, N.S.; Galanski, M.; Keppler, B.K.; Koellensperger, G. The impact of whole human blood on the kinetic inertness of platinum (IV) prodrugs—An HPLC-ICP-MS study. Dalton Trans. 2018, 47, 5252–5258. [Google Scholar] [CrossRef] [PubMed]
- Esteban-Fernández, D.; Montes-Bayón, M.; González, E.B.; Gómez-Gómez, M.M.; Palacios, M.A.; Sanz-Medel, A. Atomic (HPLC-ICP-MS) and molecular mass spectrometry (ESI-Q-TOF) to study cis-platin interactions with serum proteins. J. Anal. Atomic Spectrom. 2008, 23, 378. [Google Scholar] [CrossRef]
- Moraleja, I.; Moreno-Gordaliza, E.; Esteban-Fernández, D.; Mena, M.L.; Linscheid, M.W.; Gómez-Gómez, M.M.J.A.; Chemistry, B. A shotgun approach for the identification of platinum–protein complexes. Anal. Bioanal. Chem. 2015, 407, 2393–2403. [Google Scholar] [CrossRef] [PubMed]
- Allain, P.; Heudi, O.; Cailleux, A.; Bouil, A.L.; Gamelin, E.J.D.M. Disposition. Early Biotransformations of Oxaliplatin after Its Intravenous Administration to Cancer Patients. Drug Metab. Dispos. 2000, 28, 65–72. [Google Scholar]
- Xie, R.; Johnson, W.; Rodriguez, L.; Gounder, M.; Hall, G.S.; Buckley, B. A study of the interactions between carboplatin and blood plasma proteins using size exclusion chromatography coupled to inductively coupled plasma mass spectrometry. Trop. Med. Parasitol. 1990, 41, 419–421. [Google Scholar] [CrossRef]
- Moreno-Gordaliza, E.; Esteban-Fernandez, D.; Giesen, C.; Lehmann, K.; Lazaro, A.; Tejedor, A.; Scheler, C.; Canas, B.; Jakubowski, N.; Linscheid, M.W.; et al. LA-ICP-MS and nHPLC-ESI-LTQ-FT-MS/MS for the analysis of cisplatin–protein complexes separated by two dimensional gel electrophoresis in biological samples. J. Anal. Atomic Spectrom. 2012, 27, 1474–1483. [Google Scholar] [CrossRef]
- Mena, M.L.; Moreno-Gordaliza, E.; Moraleja, I.; Cañas, B.; Gómez-Gómez, M.M. OFFGEL isoelectric focusing and polyacrylamide gel electrophoresis separation of platinum-binding proteins. J. Chromatogr. A 2011, 1218, 1281–1290. [Google Scholar] [CrossRef]
- Martinčič, A.; Cemazar, M.; Sersa, G.; Kovač, V.; Milačič, R.; Ščančar, J. A novel method for speciation of Pt in human serum incubated with cisplatin, oxaliplatin and carboplatin by conjoint liquid chromatography on monolithic disks with UV and ICP-MS detection. Talanta 2013, 116, 141–148. [Google Scholar] [CrossRef]
- Larios, R.; Busto, M.E.D.C.; Garcia-Sar, D.; Ward-Deitrich, C.; Goenaga-Infante, H. Accurate quantification of carboplatin adducts with serum proteins by monolithic chromatography coupled to ICPMS with isotope dilution analysis. J. Anal. Atomic Spectrom. 2019, 34, 729–740. [Google Scholar] [CrossRef]
- Brodbelt, J.S.; Russell, D.H. Focus on the 20-year anniversary of SEQUEST. J. Am. Soc. Mass Spectrom. 2015, 26, 1797–1798. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.-Q.; Zeng, W.-F.; Fang, P.; Cao, W.-Q.; Liu, C.; Yan, G.-Q.; Zhang, Y.; Peng, C.; Wu, J.-Q.; Zhang, X.-J.; et al. pGlyco 2.0 enables precision N-glycoproteomics with comprehensive quality control and one-step mass spectrometry for intact glycopeptide identification. Nat. Commun. 2017, 8, 438. [Google Scholar] [CrossRef] [PubMed]
- Chi, H.; Liu, C.; Yang, H.; Zeng, W.-F.; Wu, L.; Zhou, W.-J.; Wang, R.-M.; Niu, X.-N.; Ding, Y.-H.; Zhang, Y.; et al. Comprehensive identification of peptides in tandem mass spectra using an efficient open search engine. Nat. Biotechnol. 2018, 36, 1059. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.F.S.; Menin, L.; Patiny, L.; Ortiz, D.; Dyson, P.J. Versatile Tool for the Analysis of Metal-Protein Interactions Reveals the Promiscuity of Metallodrug-Protein Interactions. Anal. Chem. 2017, 89, 11985–11989. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, D.; Gasilova, N.; Sepulveda, F.; Patiny, L.; Dyson, P.J.; Menin, L. Aom2S: A new web-based application for DNA/RNA tandem mass spectrometry data interpretation. Rapid Commun. Mass Spectrom. 2020, 34, e8927. [Google Scholar] [CrossRef]
- Wootton, C.A.; Lam, Y.P.Y.; Willetts, M.; van Agthoven, M.A.; Barrow, M.P.; Sadler, P.J.; O’Connor, P.B. Automatic assignment of metal-containing peptides in proteomic LC-MS and MS/MS data sets. Analyst 2017, 142, 2029–2037. [Google Scholar] [CrossRef]
- Wootton, C.A.; Millett, A.J.; Lopez-Clavijo, A.F.; Chiu, C.K.C.; Barrow, M.P.; Clarkson, G.J.; Sadler, P.J.; O’Connor, P.B. Structural analysis of peptides modified with organo-iridium complexes, opportunities from multi-mode fragmentation. Analyst 2019, 144, 1575–1581. [Google Scholar] [CrossRef]
- Chiu, C.K.C.; Lam, P.Y.Y.; Wootton, C.A.; Barrow, M.P.; O’Connor, P.B. Metallocomplex–Peptide Interactions Studied by Ultrahigh Resolution Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2020, 31, 594–601. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cisplatin | Carboplatin | Oxaliplatin | |
|---|---|---|---|
| t1/2α (min) | |||
| Total Platinum | 14–49 | 12–98 | 26 |
| Ultrafiltrate | 9–30 | 8–87 | 21 |
| t1/2β (h) | |||
| Total Platinum | 0.7–4.6 | 1.3–1.7 | |
| Ultrafiltrate | 0.7–0.8 | 1.7–5.9 | |
| t1/2γ (h) | |||
| Total Platinum | 24–127 | 8.2–40 | 38–47 |
| Ultrafiltrate | 24–27 | ||
| Protein Binding (%) | >90 | 24–50 | 85 |
| Urinary Excretion (%) | 23–50 | 54–82 | >50 |
| Drugs | Methods | Sample Type | Binding Proteins in Blood | Search Engine | Advantages | Disadvantages |
|---|---|---|---|---|---|---|
| Cisplatin | HPLC–ICP-MS and ESI-Q-TOF | Human serum (in vitro) | Tf, HSA [73] | - | Reliable identification | Requires pure protein; favorable for high-abundance proteins |
| Cisplatin | MudPIT | Human serum (in vitro) | HSA, Trfe, A2mg, A1at, Apoa1, Apoa2 [65] | SEQUEST | Platination site can be found | Unfavorable for low-abundance proteins |
| Cisplatin | SEC–ICP-MS and nLC–ESI-LTQ-Orbitrap-MS/MS | Human serum (in vitro) | HSA [74] | - | Reliable identification | Requires pure protein |
| Oxaliplatin | SEC–ICP-MS | Human plasma (in vivo) | γ-globulins, Hb, albumin [75] | - | Quantitative information for the binding can be obtained | Unfavorable for low-abundance proteins; platination site cannot be found |
| Carboplatin | SEC–ICP-MS | Human plasma (in vivo) | HSA, γ-globulins [76] | - | Quantitative information for the binding can be obtained | Unfavorable for low-abundance proteins; platination site cannot be found |
| Oxaliplatin | nanoESI-QTOF-MS/MS | Human red blood cells (in vivo) | Hb [43] | - | Semiquantitative information for the binding can be obtained | Platination site cannot be found |
| Cisplatin | LA–ICP-MS and nLC–ESI-LTQ-FT-MS/MS | Wistar rat serum (in vivo) | A2mg, Tf, HSA, Hb, APOC2 [77] | Mascot | Semiquantitative information for the binding can be obtained | Platination site cannot be found due to sample preparation; unfavorable for low-abundance proteins |
| Cisplatin, carboplatin, oxaliplatin | ICP-MS | Wistar/ST rat plasma (in vivo) | Albumin [21] | - | Quantitative information for the binding can be obtained | Platination site cannot be found |
| Peptide Sequence [a] | Pt Fragment Mass [b] | Sample No. | Charge | SEQUEST Xcorr | Parameters ΔCn | Ions [c] | Other Possible Binding Site(s) [d] |
|---|---|---|---|---|---|---|---|
| 1. Serotransferrin (Trfe): Residues Y314/E385 | |||||||
| (a) K·313MOXY@LGYEYVTAIR324·N | 210 | 2 | 2 | 2.86 | 0.10 | 15/22 | |
| (b) K·381IM@NGEADAMSLDGGFVYIAGK401·C | 193 | 1 | 2 | 4.49 | 0.48 | 22/40 | E385 (0.11), D387 (0.31) |
| (c) K·381IM@NGEADAMSLDGGFVYIAGK401·C | 227 | 2 | 2 | 5.14 | 0.50 | 19/40 | E385 (0.18), D387 (0.33) |
| (d) M·383NGE@ADAMOXSLDGGFVYIAGK401·C | 263 | 2 | 2 | 4.57 | 0.64 | 21/36 | D387 (0.18) |
| 2. α-2-Macroglobulin (A2mg): Residues E300/S532 | |||||||
| (a) K·297LHTE@AQIQEEGTVVELTGR315·Q | 227 | 1 | 3 | 3.71 | 0.29 | 24/72 | T299 (0.05), H298 (0.05) |
| (b) R·517LLIYAVLPTGDVIGDS@532·A | 227 | 2 | 2 | 2.88 | 0.33 | 22/30 | D531 (0.08) |
| 3. α-1-Antitrypsin (A1at): Residues D107/K368 | |||||||
| (a) L·104NQPD@SQLQLTTGNGLFLSEGLK125·L | 210 | 1 | 2 | 3.12 | 0.28 | 15/42 | S108 (0.09) |
| (b) L·04NQPD@SQLQLTTGNGLFLSEGLK125·L | 210 | 2 | 2 | 3.44 | 0.34 | 16/42 | S108 (0.09) |
| (c) R·102TLNQPDS@QLQLTTGNGLFLSEGLK125·L | 227 | 1 | 3 | 4.12 | 0.30 | 30/92 | D107 (0.11) |
| (d) K·366FNK@PFVFLMIEQNTK380·S | 193 | 1 | 3 | 4.67 | 0.16 | 24/56 | |
| (e) N·368K@PFVFLMoxIEQNTK380 | 263 | 2 | 2 | 4.41 | 0.23 | 20/24 | |
| 4. Apolipoprotein AI (Apoa1): Residues D48/D73/S228 | |||||||
| (a) K·46LLD@NWDSVTSTFSK59·L | 210 | 2 | 2 | 3.50 | 0.15 | 16/26 | D51 (0.11) |
| (b) K·60LREQLGPVTQEFWD@N74·L | 227 | 1 | 2 | 3.34 | 0.22 | 16/28 | |
| (c) K·227VS@FLSALEEYT337·K | 263 | 1 | 2 | 3.08 | 0.29 | 15/20 | |
| 5. Apolipoprotein AII (Apoa2): Residues C6/E8 | |||||||
| (a) P·6C@VESLVSQYFQTVTDYGK23·D | 193 | 2 | 2 | 4.54 | 0.49 | 22/34 | E8 (0.07), S9 (0.08) |
| (b) E·5PC@VESLVSQYFQTVTDEYGK23·D | 210 | 1 | 2 | 3.94 | 0.26 | 17/36 | E8 (0.03), S9 (0.14) |
| (c) V·8E@SLVSQYFQTVTDYGK23·D | 227 | 1 | 2 | 5.46 | 0.52 | 22/30 | S9 (0.04) |
| (d) P·6C@VESLVSQYFQYFQTVTDYGK23·D | 263 | 1 | 2 | 3.65 | 0.25 | 21/34 | E8 (0.01), S9 (0.06) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Tao, J.; Jia, S.; Wang, M.; Jiang, H.; Du, Z. The Protein-Binding Behavior of Platinum Anticancer Drugs in Blood Revealed by Mass Spectrometry. Pharmaceuticals 2021, 14, 104. https://doi.org/10.3390/ph14020104
Wang J, Tao J, Jia S, Wang M, Jiang H, Du Z. The Protein-Binding Behavior of Platinum Anticancer Drugs in Blood Revealed by Mass Spectrometry. Pharmaceuticals. 2021; 14(2):104. https://doi.org/10.3390/ph14020104
Chicago/Turabian StyleWang, Jingchen, Jianmei Tao, Shuailong Jia, Meiqin Wang, Hongliang Jiang, and Zhifeng Du. 2021. "The Protein-Binding Behavior of Platinum Anticancer Drugs in Blood Revealed by Mass Spectrometry" Pharmaceuticals 14, no. 2: 104. https://doi.org/10.3390/ph14020104
APA StyleWang, J., Tao, J., Jia, S., Wang, M., Jiang, H., & Du, Z. (2021). The Protein-Binding Behavior of Platinum Anticancer Drugs in Blood Revealed by Mass Spectrometry. Pharmaceuticals, 14(2), 104. https://doi.org/10.3390/ph14020104