
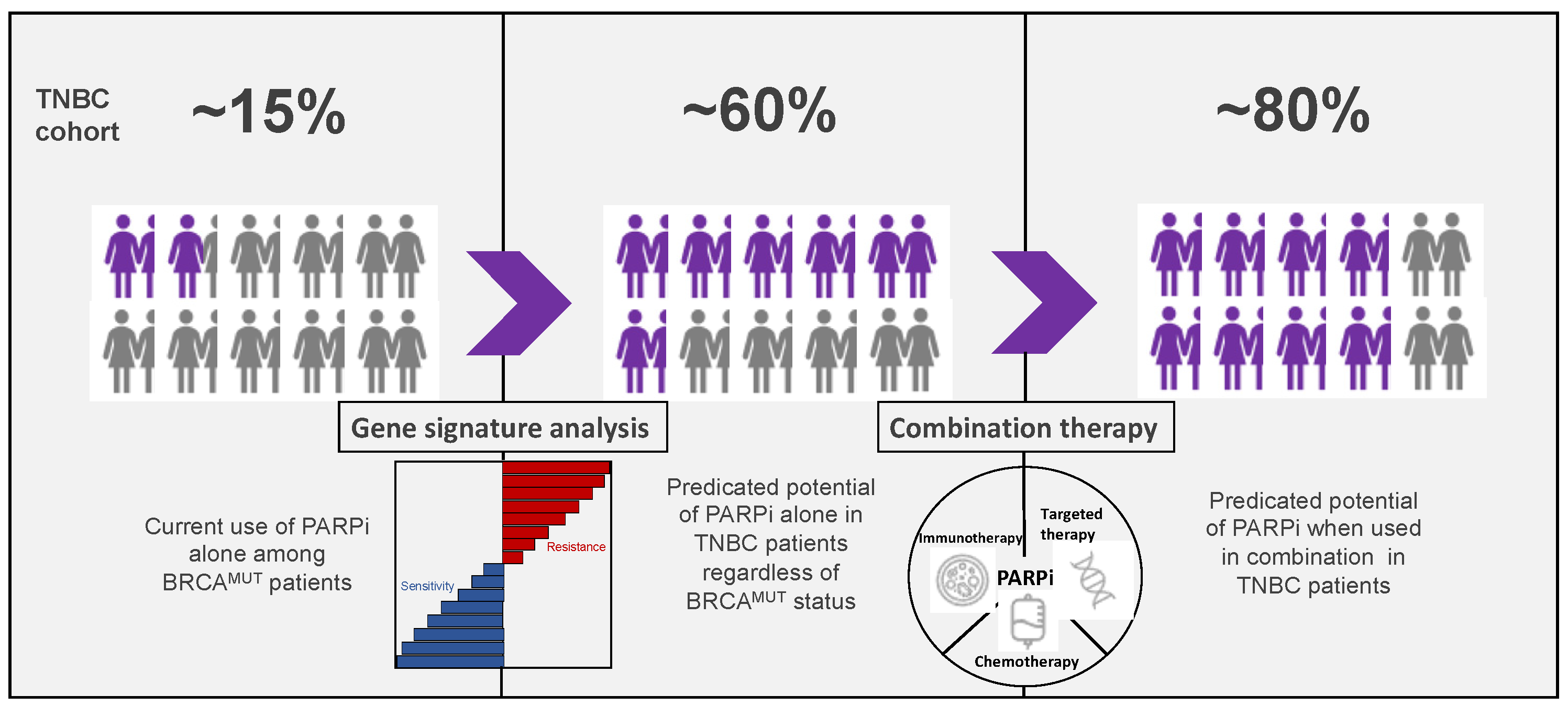
Expanding the Use of PARP Inhibitors as Monotherapy and in Combination in Triple-Negative Breast Cancer

Abstract
:
1. Introduction
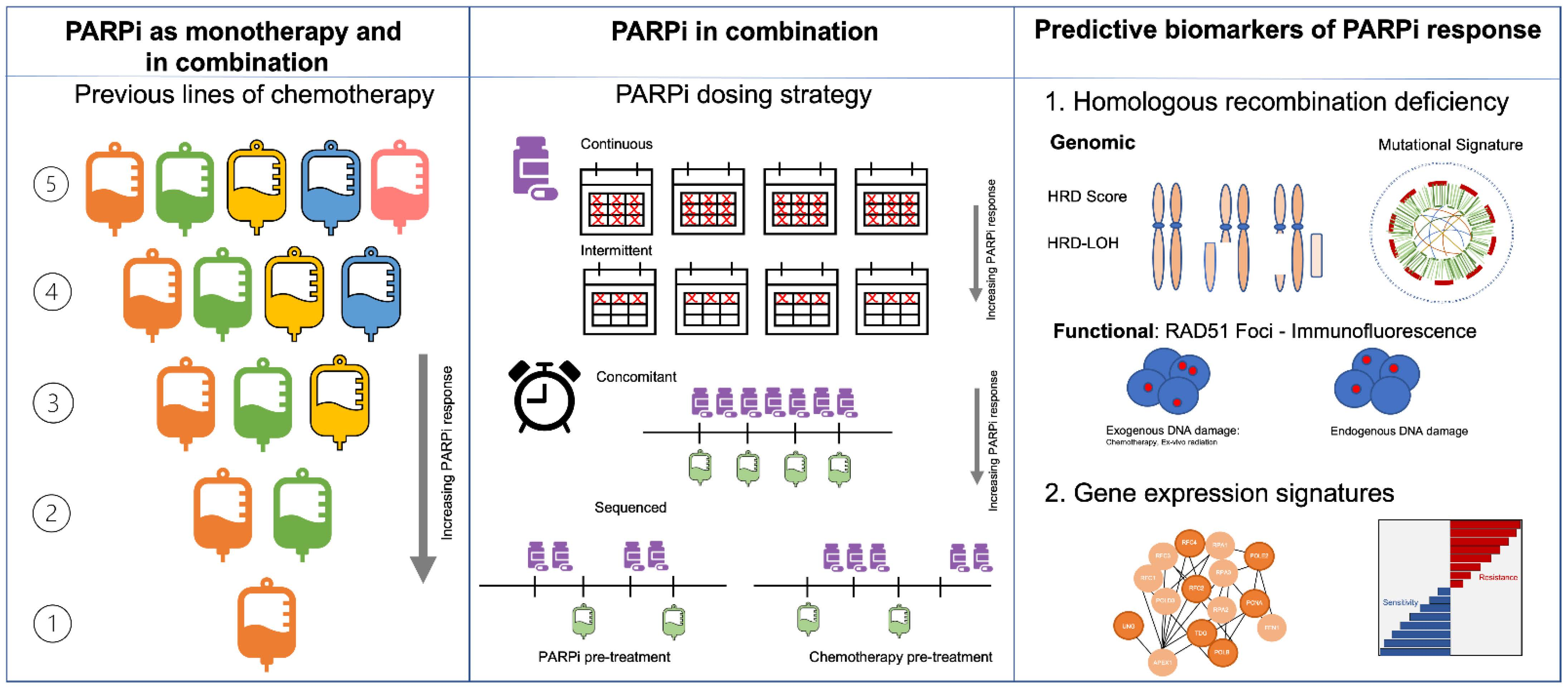
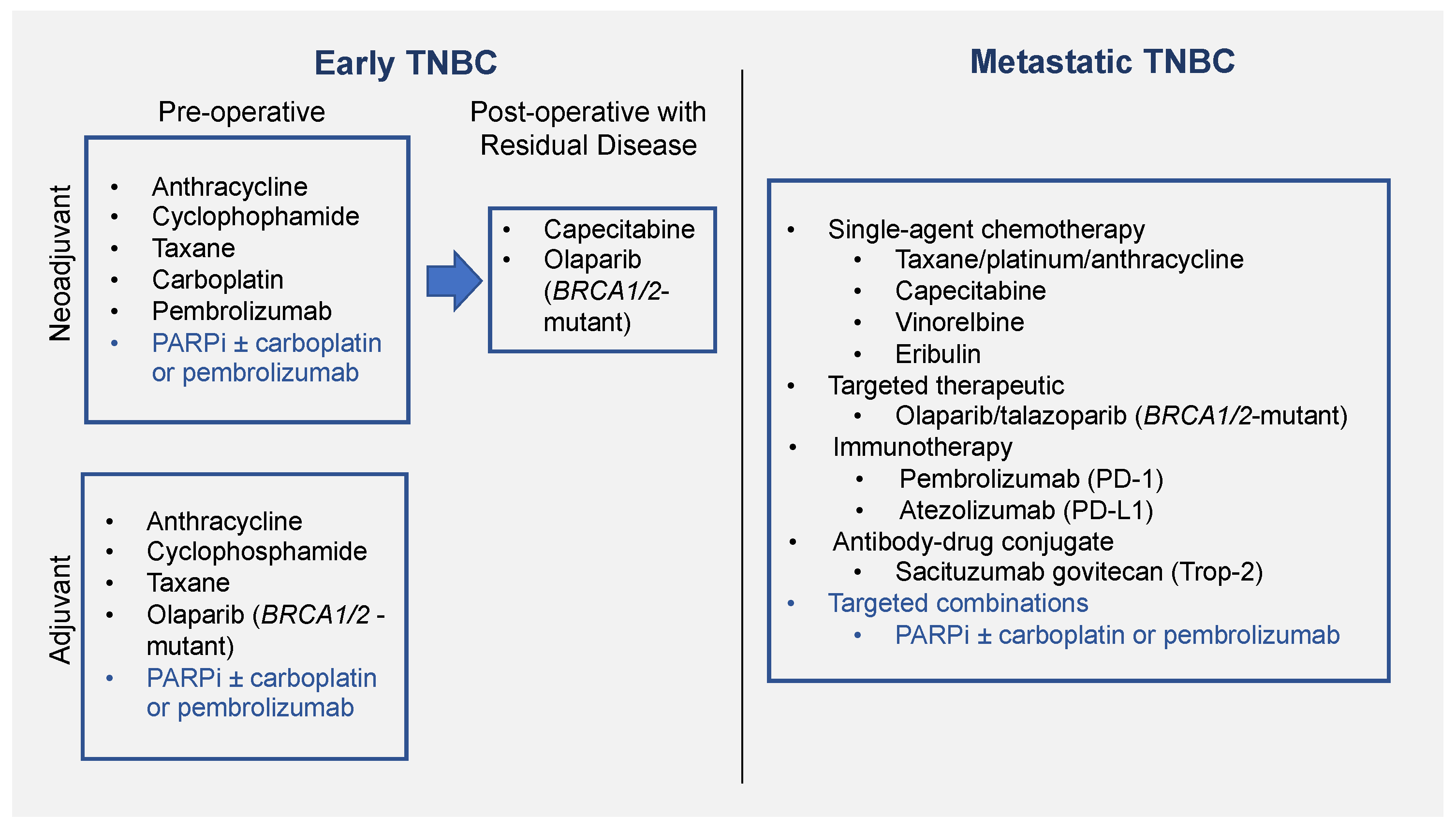
2. PARPi as Monotherapy
2.1. Use in Metastatic Setting

{kind=link}
{kind=link}
{kind=link}
| First Author Study Name | Year of Study | No. of Participants (BC Patients) | Type of Study | Median/Mean No. of Prior Chemotherapy Regimens (Range) | Comparative Arms | Patient Population | Outcome (Objective Response Rate, Progression Free Survival) | |
|---|---|---|---|---|---|---|---|---|
| PARPi | Comparative Agent/ Standard Chemotherapy | |||||||
| Fong, P.C. et al. [42] | 2009 | 60 (9 BC) | Phase I | 28% ≤ 2 lines * 18%—3 lines * 53% ≥ 4 lines * | Olaparib Cohort 1: 10 mg/day to 600 mg twice daily Cohort 2: 200 mg twice daily | None | BRCAMUT: N = 22 BRCAWT: N = 38 | ORR in all BRCAMUT: 47.4% No objective response in BRCAWT |
| de Bono, J. et al. [43] | 2017 | 110 (20 BC) | Phase I | 2.5 (0–13) | Talazoparib Part 1: 0.025 to 1.1 mg/day Part 2: 1.0 mg/day | None | Part 1: DNA repair deficiency; Part 2: gBRCAMUT | ORR in BRCAMUT in breast cancer: 50% PFS in breast cancer: 34.6 weeks |
| Puhalla S et al. [44] Pahuja S et al. [45] | 2014 | 98 (40 BC) | Phase I | gBRCAMUT: 6 (1–14) * BRCAWT: 4 (1–12) * | Veliparib 50–500 mg twice daily | None | gBRCAMUT: N = 70 BRCAWT: N = 28 | ORR in all BRCAMUT: 23%, breast BRCAMUT: 29% ORR in all BRCAWT: 4%, breast BRCAMUT: 5% |
| Tutt, A. et al. [46] | 2010 | 54 (54 BC) | Phase II, non-randomized sequential-cohort | Cohort 1: 3 (1–5) Cohort 2: 3 (2–4) | Olaparib Cohort 1: 400 mg twice daily Cohort 2: 100 mg twice daily | None | gBRCAMUT | ORR in cohort 1: 41%, cohort 2: 22% PFS in cohort 1: 5.7 months; PFS in cohort 2: 3.8 months |
| Gelmon, K.A. et al. [47] | 2011 | 91 (26 BC) | Phase II, non-randomized | 3 (1–7) | Olaparib capsule 400 mg twice daily | None | BRCAMUT: N = 27 BRCAWT: N = 63 | ORR in ovarian cancer: BRCAMUT 41%, BRCAWT 24%; breast cancer: BRCAMUT 0% BRCAWT 0% PFS in ovarian cancer: BRCAMUT 7.4 months, BRCAWT: 6.4 months; breast cancer: BRCAMUT 3.6 months, BRCAWT: 1.8 months |
| Kaufman, B. et al. [48] | 2015 | 298 (62 BC) | Phase II, single-arm, non-randomized | BC cohort: 4.6 (3–11) | Olaparib capsule 400 mg twice daily | None | gBRCAMUT | Response rate for all: 26.2%; breast cancer 12.9% PFS in breast cancer 3.7 months |
| Robson, M.E. et al. [26,49] OlympiAD | 2017 | 302 (302 BC) | Phase III, randomized | ≤2 lines | Olaparib tablet 300 mg twice daily | Capecitabine, eribulin, or vinorelbine | gBRCAMUT HER2-negative | ORR 59.9% vs. 28.8% (olaparib versus standard chemotherapy) PFS 7.0 months vs. 4.2 months (olaparib versus standard chemotherapy) |
| Litton J.K. et al. [27] EMBRACA | 2018 | 431 (431 BC) | Phase III, randomized | ≤3 lines | Talazoparib 1 mg once daily | Capecitabine, eribulin, gemcitabine, or vinorelbine | gBRCAMUT HER2-negative | ORR 62.2% vs. 27.2% (olaparib versus standard chemotherapy) PFS 8.6 months vs. 5.6 months (olaparib versus standard chemotherapy) |
| Tung N.M. et al. [50] TBCRC 048 | 2020 | 54 (54 BC) | Phase II, non-randomized | 1 (0–4) | Olaparib tablet 300 mg twice daily | None | Cohort 1: Germline mutation in HR-related gene (not gBRCA1/2) Cohort 2: Somatic mutations in same genes (including BRCA1/2) | ORR in all cohort 1, 33%; gPALB2MUT 82%; all cohort 2, 31%; sBRCAMUT 50% PFS for gPALB2MUT, 13.3 months; sBRCAMUT 6.3 months |
2.2. Use in Adjuvant Setting
2.3. Use in Neoadjuvant Setting
3. PARPi as Combination Therapy
3.1. PARPi in Combination with Chemotherapy
3.1.1. Use in Metastatic Setting
3.1.2. Use in Neoadjuvant Setting
3.2. PARPi in Combination with Immunotherapy
3.3. PARPi in Combination with Targeted Therapies
4. PARPi in Elderly Patients
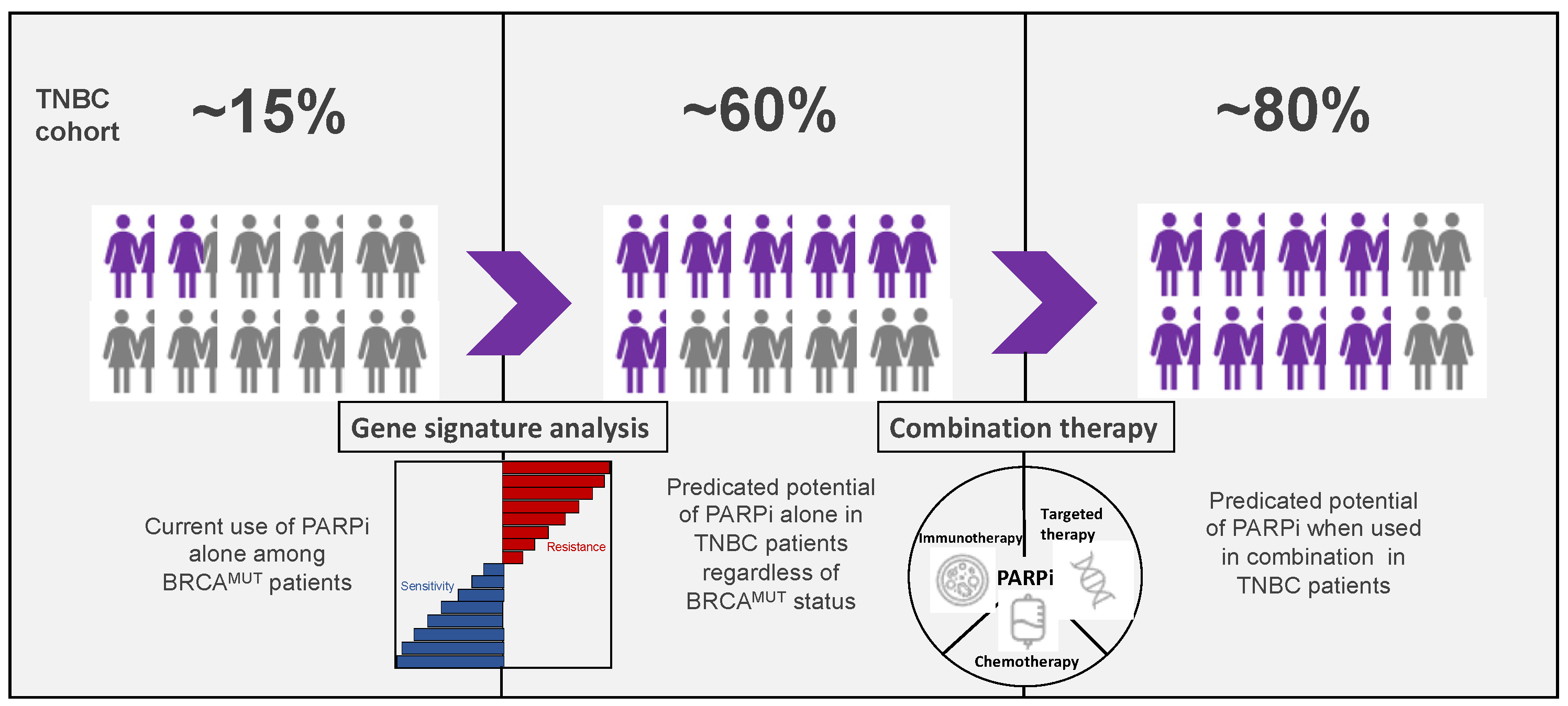
5. Predictive Biomarkers of Response to PARPi
5.1. BRCA1/2 Mutations
5.2. HRR Gene Mutations
5.3. Copy Number Based “Genomic Scar” Assays
5.4. Mutational Signatures
5.5. Functional Biomarkers of HR Deficiency
5.6. Gene Expression Signatures
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Papadimitriou, M.; Mountzios, G.; Papadimitriou, C.A. The role of PARP inhibition in triple-negative breast cancer: Unraveling the wide spectrum of synthetic lethality. Cancer Treat. Rev. 2018, 67, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Couch, F.J.; Hart, S.N.; Sharma, P.; Toland, A.E.; Wang, X.; Miron, P.; Olson, J.E.; Godwin, A.K.; Pankratz, V.S.; Olswold, C.; et al. Inherited Mutations in 17 Breast Cancer Susceptibility Genes Among a Large Triple-Negative Breast Cancer Cohort Unselected for Family History of Breast Cancer. J. Clin. Oncol. 2015, 33, 304–311. [Google Scholar] [CrossRef]
- Engel, C.; Rhiem, K.; Hahnen, E.; Loibl, S.; Weber, K.E.; Seiler, S.; Zachariae, S.; Hauke, J.; Wappenschmidt, B.; Waha, A.; et al. Prevalence of pathogenic BRCA1/2 germline mutations among 802 women with unilateral triple-negative breast cancer without family cancer history. BMC Cancer 2018, 18, 265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Angulo, A.M.; Timms, K.M.; Liu, S.; Chen, H.; Litton, J.K.; Potter, J.; Lanchbury, J.S.; Stemke-Hale, K.; Hennessy, B.T.; Arun, B.K.; et al. Incidence and Outcome of BRCA Mutations in Unselected Patients with Triple Receptor-Negative Breast Cancer. Clin. Cancer Res. 2011, 17, 1082–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Incorvaia, L.; Fanale, D.; Bono, M.; Calò, V.; Fiorino, A.; Brando, C.; Corsini, L.R.; Cutaia, S.; Cancelliere, D.; Pivetti, A.; et al. BRCA1/2 pathogenic variants in triple-negative versus luminal-like breast cancers: Genotype–phenotype correlation in a cohort of 531 patients. Ther. Adv. Med. Oncol. 2020, 12, 1758835920975326. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Tung, N.; Lin, N.U.; Kidd, J.; Allen, B.A.; Singh, N.; Wenstrup, R.J.; Hartman, A.-R.; Winer, E.P.; Garber, J.E. Frequency of Germline Mutations in 25 Cancer Susceptibility Genes in a Sequential Series of Patients with Breast Cancer. J. Clin. Oncol. 2016, 34, 1460–1468. [Google Scholar] [CrossRef] [Green Version]
- Winter, C.; Nilsson, M.P.; Olsson, E.; George, A.M.; Chen, Y.; Kvist, A.; Törngren, T.; Vallon-Christersson, J.; Hegardt, C.; Häkkinen, J.; et al. Targeted sequencing of BRCA1 and BRCA2 across a large unselected breast cancer cohort suggests that one-third of mutations are somatic. Ann. Oncol. 2016, 27, 1532–1538. [Google Scholar] [CrossRef]
- Kim, C.; Gao, R.; Sei, E.; Brandt, R.; Hartman, J.; Hatschek, T.; Crosetto, N.; Foukakis, T.; Navin, N.E. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell 2018, 173, 879–893.e13. [Google Scholar] [CrossRef] [Green Version]
- Cohen-Sharir, Y.; McFarland, J.M.; Abdusamad, M.; Marquis, C.; Bernhard, S.V.; Kazachkova, M.; Tang, H.; Ippolito, M.R.; Laue, K.; Zerbib, J.; et al. Aneuploidy renders cancer cells vulnerable to mitotic checkpoint inhibition. Nature 2021, 590, 486–491. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, C.K.; Carey, L.A. Biology, Metastatic Patterns, and Treatment of Patients with Triple-Negative Breast Cancer. Clin. Breast Cancer 2009, 9, S73–S81. [Google Scholar] [CrossRef] [PubMed]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-Negative Breast Cancer: Clinical Features and Patterns of Recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef]
- Yin, L.; Duan, J.-J.; Bian, X.-W.; Yu, S.-C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020, 22, 61. [Google Scholar] [CrossRef]
- Loibl, S.; Sikov, W.; Huober, J.; Rugo, H.; Wolmark, N.; O’Shaughnessy, J.; Maag, D.; Untch, M.; Golshan, M.; Lorenzo, J.P.; et al. 119O Event-free survival (EFS), overall survival (OS), and safety of adding veliparib (V) plus carboplatin (Cb) or carboplatin alone to neoadjuvant chemotherapy in triple-negative breast cancer (TNBC) after ≥4 years of follow-up: BrighTNess, a randomized phase III trial. Ann. Oncol. 2021, 32, S408. [Google Scholar] [CrossRef]
- Pareja, F.; Geyer, F.C.; Marchiò, C.; Burke, K.A.; Weigelt, B.; Reis-Filho, J.S. Triple-negative breast cancer: The importance of molecular and histologic subtyping, and recognition of low-grade variants. npj Breast Cancer 2016, 2, 16036. [Google Scholar] [CrossRef]
- Denkert, C.; Liedtke, C.; Tutt, A.; von Minckwitz, G. Molecular alterations in triple-negative breast cancer—the road to new treatment strategies. Lancet 2016, 389, 2430–2442. [Google Scholar] [CrossRef] [Green Version]
- Masuda, N.; Lee, S.-J.; Ohtani, S.; Im, Y.-H.; Lee, E.-S.; Yokota, I.; Kuroi, K.; Im, S.-A.; Park, B.-W.; Kim, S.-B.; et al. Adjuvant Capecitabine for Breast Cancer after Preoperative Chemotherapy. N. Engl. J. Med. 2017, 376, 2147–2159. [Google Scholar] [CrossRef]
- Emens, L.; Adams, S.; Barrios, C.; Diéras, V.; Iwata, H.; Loi, S.; Rugo, H.; Schneeweiss, A.; Winer, E.; Patel, S.; et al. First-line atezolizumab plus nab-paclitaxel for unresectable, locally advanced, or metastatic triple-negative breast cancer: IMpassion130 final overall survival analysis. Ann. Oncol. 2021, 32, 983–993. [Google Scholar] [CrossRef]
- Schmid, P.; Cortes, J.; Dent, R.; Pusztai, L.; McArthur, H.; Kümmel, S.; Bergh, J.; Denkert, C.; Park, Y.; Hui, R.; et al. VP7-2021: KEYNOTE-522: Phase III study of neoadjuvant pembrolizumab + chemotherapy vs. placebo + chemotherapy, followed by adjuvant pembrolizumab vs. placebo for early-stage TNBC. Ann. Oncol. 2021, 32, 1198–1200. [Google Scholar] [CrossRef]
- Schmid, P.; Cortes, J.; Pusztai, L.; McArthur, H.; Kümmel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; Harbeck, N.; et al. Pembrolizumab for Early Triple-Negative Breast Cancer. N. Engl. J. Med. 2020, 382, 810–821. [Google Scholar] [CrossRef] [PubMed]
- Berger, E.; Park, T.; Saridakis, A.; Golshan, M.; Greenup, R.; Ahuja, N. Immunotherapy Treatment for Triple Negative Breast Cancer. Pharmaceuticals 2021, 14, 763. [Google Scholar] [CrossRef]
- Bardia, A.; Hurvitz, S.A.; Tolaney, S.M.; Loirat, D.; Punie, K.; Oliveira, M.; Brufsky, A.; Sardesai, S.D.; Kalinsky, K.; Zelnak, A.B.; et al. Sacituzumab Govitecan in Metastatic Triple-Negative Breast Cancer. N. Engl. J. Med. 2021, 384, 1529–1541. [Google Scholar] [CrossRef]
- Robson, M.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.-H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.-Y.N.; DAS, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [Green Version]
- Lord, C.; Ashworth, A. Mechanisms of resistance to therapies targeting BRCA-mutant cancers. Nat. Med. 2013, 19, 1381–1388. [Google Scholar] [CrossRef]
- Lord, C.J.; Tutt, A.N.; Ashworth, A. Synthetic Lethality and Cancer Therapy: Lessons Learned from the Development of PARP Inhibitors. Annu. Rev. Med. 2015, 66, 455–470. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.-Y.N.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific PARP Trapping by BMN 673 and Comparison with Olaparib and Rucaparib. Mol. Cancer Ther. 2013, 13, 433–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell Dev. Biol. 2020, 8, 564601. [Google Scholar] [CrossRef] [PubMed]
- Turk, A.A.; Wisinski, K.B. PARP inhibitors in breast cancer: Bringing synthetic lethality to the bedside. Cancer 2018, 124, 2498–2506. [Google Scholar] [CrossRef]
- Pommier, Y.; O’Connor, M.J.; de Bono, J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med. 2016, 8, 362. [Google Scholar] [CrossRef]
- FDA Approves Olaparib for Germline BRCA-Mutated Metastatic Breast Cancer. Available online: https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm592357.htm (accessed on 23 August 2018).
- FDA Approves Talazoparib for gBRCAm HER2-Negative Locally Advanced or Metastatic Breast Cancer. Available online: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-talazoparib-gbrcam-her2-negative-locally-advanced-or-metastatic-breast-cancer (accessed on 1 March 2020).
- Lynparza® (olaparib) Receives New Indication for BRCA-Mutated Metastatic Breast Cancer. Available online: https://www.astrazeneca.ca/en/media/press-releases/2018/lynparza---olaparib--receives-new-indication-for-brca-mutated-me.html (accessed on 12 September 2018).
- Summary Basis of Decision Talzenna Health Canada. Available online: https://hpr-rps.hres.ca/reg-content/summary-basis-decision-detailTwo.php?linkID=SBD00458 (accessed on 1 March 2020).
- Beniey, M.; Haque, T.; Hassan, S. Translating the role of PARP inhibitors in triple-negative breast cancer. Oncoscience 2018, 6, 287–288. [Google Scholar] [CrossRef]
- Sonnenblick, A.; de Azambuja, E.; Azim, H.A.; Piccart-Gebhart, M. An update on PARP inhibitors—moving to the adjuvant setting. Nat. Rev. Clin. Oncol. 2014, 12, 27–41. [Google Scholar] [CrossRef]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of Poly(ADP-Ribose) Polymerase in Tumors from BRCAMutation Carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [Green Version]
- de Bono, J.; Ramanathan, R.K.; Mina, L.; Chugh, R.; Glaspy, J.; Rafii, S.; Kaye, S.; Sachdev, J.; Heymach, J.; Smith, D.C.; et al. Phase I, Dose-Escalation, 2-Part Trial of Poly(ADP-Ribose) Polymerase Inhibitor Talazoparib in Patients with Advanced Germline BRCA1/2 Mutations and Selected Sporadic Cancers. Cancer Discov. 2017, 7, 620–629. [Google Scholar] [CrossRef] [Green Version]
- Puhalla, S.; Beumer, J.H.; Pahuja, S.; Appleman, L.J.; Tawbi, H.A.-H.; Stoller, R.G.; Lee, J.J.; Lin, Y.; Kiesel, B.; Yu, J.; et al. Final results of a phase 1 study of single-agent veliparib (V) in patients (pts) with either BRCA1/2-mutated cancer (BRCA+), platinum-refractory ovarian, or basal-like breast cancer (BRCA-wt). J. Clin. Oncol. 2014, 32, 2570. [Google Scholar] [CrossRef]
- Pahuja, S.; Beumer, J.H.; Appleman, L.J.; Tawbi, H.A.-H.; Stoller, R.G.; Lee, J.J.; Lin, Y.; Kiesel, B.; Yu, J.; Tan, A.R.; et al. Outcome of BRCA 1/2-mutated (BRCA+) and triple-negative, BRCA wild type (BRCA-wt) breast cancer patients in a phase I study of single-agent veliparib (V). J. Clin. Oncol. 2014, 32, 135. [Google Scholar] [CrossRef]
- Tutt, A.; Robson, M.; Garber, J.E.; Domchek, S.M.; Audeh, M.W.; Weitzel, J.N.; Friedlander, M.; Arun, B.; Loman, N.; Schmutzler, R.K.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: A proof-of-concept trial. Lancet 2010, 376, 235–244. [Google Scholar] [CrossRef]
- Gelmon, K.A.; Tischkowitz, M.; Mackay, H.; Swenerton, K.; Robidoux, A.; Tonkin, K.; Hirte, H.; Huntsman, D.; Clemons, M.; Gilks, B.; et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: A phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011, 12, 852–861. [Google Scholar] [CrossRef]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmaña, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib Monotherapy in Patients with Advanced Cancer and a Germline BRCA1/2 Mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Robson, M.; Tung, N.; Conte, P.; Im, S.-A.; Senkus, E.; Xu, B.; Masuda, N.; Delaloge, S.; Li, W.; Armstrong, A.; et al. OlympiAD final overall survival and tolerability results: Olaparib versus chemotherapy treatment of physician’s choice in patients with a germline BRCA mutation and HER2-negative metastatic breast cancer. Ann. Oncol. 2019, 30, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Tung, N.M.; Robson, M.E.; Ventz, S.; Santa-Maria, C.A.; Nanda, R.; Marcom, P.K.; Shah, P.D.; Ballinger, T.J.; Yang, E.S.; Vinayak, S.; et al. TBCRC 048: Phase II Study of Olaparib for Metastatic Breast Cancer and Mutations in Homologous Recombination-Related Genes. J. Clin. Oncol. 2020, 38, 4274–4282. [Google Scholar] [CrossRef] [PubMed]
- Poggio, F.; Bruzzone, M.; Ceppi, M.; Conte, B.; Martel, S.; Maurer, C.; Tagliamento, M.; Viglietti, G.; Del Mastro, L.; de Azambuja, E.; et al. Single-agent PARP inhibitors for the treatment of patients with BRCA-mutated HER2-negative metastatic breast cancer: A systematic review and meta-analysis. ESMO Open 2018, 3, e000361. [Google Scholar] [CrossRef] [Green Version]
- Tutt, A.N.; Garber, J.E.; Kaufman, B.; Viale, G.; Fumagalli, D.; Rastogi, P.; Gelber, R.D.; de Azambuja, E.; Fielding, A.; Balmaña, J.; et al. Adjuvant Olaparib for Patients with BRCA1- or BRCA2-Mutated Breast Cancer. N. Engl. J. Med. 2021, 384, 2394–2405. [Google Scholar] [CrossRef] [PubMed]
- Litton, J.K.; Scoggins, M.E.; Hess, K.R.; Adrada, B.E.; Murthy, R.K.; Damodaran, S.; DeSnyder, S.M.; Brewster, A.M.; Barcenas, C.H.; Valero, V.; et al. Neoadjuvant Talazoparib for Patients With Operable Breast Cancer With a Germline BRCA Pathogenic Variant. J. Clin. Oncol. 2020, 38, 388–394. [Google Scholar] [CrossRef]
- Litton, J.K.; Beck, J.T.; Jones, J.M.; Andersen, J.; Blum, J.L.; Mina, L.A.; Brig, R.; Danso, M.A.; Yuan, Y.; Abbattista, A.; et al. Neoadjuvant talazoparib in patients with germline BRCA1/2 (gBRCA1/2) mutation-positive, early HER2-negative breast cancer (BC): Results of a phase 2 study. J. Clin. Oncol. 2021, 39, 505. [Google Scholar] [CrossRef]
- Eikesdal, H.; Yndestad, S.; Elzawahry, A.; Llop-Guevara, A.; Gilje, B.; Blix, E.; Espelid, H.; Lundgren, S.; Geisler, J.; Vagstad, G.; et al. Olaparib monotherapy as primary treatment in unselected triple negative breast cancer. Ann. Oncol. 2020, 32, 240–249. [Google Scholar] [CrossRef]
- Chopra, N.; Tovey, H.; Pearson, A.; Cutts, R.; Toms, C.; Proszek, P.; Hubank, M.; Dowsett, M.; Dodson, A.; Daley, F.; et al. Homologous recombination DNA repair deficiency and PARP inhibition activity in primary triple negative breast cancer. Nat. Commun. 2020, 11, 2662. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-M.; Hays, J.L.; Annunziata, C.M.; Noonan, A.M.; Minasian, L.; Zujewski, J.A.; Yu, M.; Gordon, N.; Ji, J.; Sissung, T.M.; et al. Phase I/Ib Study of Olaparib and Carboplatin in BRCA1 or BRCA2 Mutation-Associated Breast or Ovarian Cancer with Biomarker Analyses. J. Natl. Cancer Inst. 2014, 106, dju089. [Google Scholar] [CrossRef]
- Lee, J.-M.; Peer, C.J.; Yu, M.; Amable, L.; Gordon, N.; Annunziata, C.M.; Houston, N.; Goey, A.K.; Sissung, T.M.; Parker, B.; et al. Sequence-Specific Pharmacokinetic and Pharmacodynamic Phase I/Ib Study of Olaparib Tablets and Carboplatin in Women’s Cancer. Clin. Cancer Res. 2016, 23, 1397–1406. [Google Scholar] [CrossRef] [Green Version]
- Dhawan, M.S.; Bartelink, I.H.; Aggarwal, R.R.; Leng, J.; Zhang, J.Z.; Pawlowska, N.; Terranova-Barberio, M.; Grabowsky, J.A.; Gewitz, A.; Chien, A.J.; et al. Differential Toxicity in Patients with and without DNA Repair Mutations: Phase I Study of Carboplatin and Talazoparib in Advanced Solid Tumors. Clin. Cancer Res. 2017, 23, 6400–6410. [Google Scholar] [CrossRef] [Green Version]
- Somlo, G.; Frankel, P.H.; Arun, B.K.; Ma, C.X.; Garcia, A.A.; Cigler, T.; Cream, L.V.; Harvey, H.A.; Sparano, J.A.; Nanda, R.; et al. Efficacy of the PARP Inhibitor Veliparib with Carboplatin or as a Single Agent in Patients with Germline BRCA1- or BRCA2-Associated Metastatic Breast Cancer: California Cancer Consortium Trial NCT01149083. Clin. Cancer Res. 2017, 23, 4066–4076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appleman, L.J.; Beumer, J.H.; Jiang, Y.; Lin, Y.; Ding, F.; Puhalla, S.; Swartz, L.; Owonikoko, T.K.; Harvey, R.D.; Stoller, R.; et al. Phase 1 study of veliparib (ABT-888), a poly (ADP-ribose) polymerase inhibitor, with carboplatin and paclitaxel in advanced solid malignancies. Cancer Chemother. Pharmacol. 2019, 84, 1289–1301. [Google Scholar] [CrossRef]
- Han, H.S.; Diéras, V.; Robson, M.; Palácová, M.; Marcom, P.K.; Jager, A.; Bondarenko, I.; Citrin, D.; Campone, M.; Telli, M.L.; et al. Veliparib with temozolomide or carboplatin/paclitaxel versus placebo with carboplatin/paclitaxel in patients with BRCA1/2 locally recurrent/metastatic breast cancer: Randomized phase II study. Ann. Oncol. 2018, 29, 154–161. [Google Scholar] [CrossRef]
- Diéras, V.; Han, H.S.; Kaufman, B.; Wildiers, H.; Friedlander, M.; Ayoub, J.-P.; Puhalla, S.L.; Bondarenko, I.; Campone, M.; Jakobsen, E.H.; et al. Veliparib with carboplatin and paclitaxel in BRCA-mutated advanced breast cancer (BROCADE3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020, 21, 1269–1282. [Google Scholar] [CrossRef]
- Arun, B.K.; Han, H.S.; Kaufman, B.; Wildiers, H.; Friedlander, M.; Ayoub, J.-P.; Puhalla, S.L.; Bell-McGuinn, K.M.; Bach, B.A.; Kundu, M.G.; et al. Efficacy and safety of first-line veliparib and carboplatin–paclitaxel in patients with HER2− advanced germline BRCA+ breast cancer: Subgroup analysis of a randomised clinical trial. Eur. J. Cancer 2021, 154, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Rugo, H.S.; Olopade, O.I.; DeMichele, A.; Yau, C.; Van’t Veer, L.J.; Buxton, M.B.; Hogarth, M.; Hylton, N.M.; Paoloni, M.; Perlmutter, J.; et al. Adaptive Randomization of Veliparib–Carboplatin Treatment in Breast Cancer. N. Engl. J. Med. 2016, 375, 23–34. [Google Scholar] [CrossRef]
- Loibl, S.; O’Shaughnessy, J.; Untch, M.; Sikov, W.M.; Rugo, H.S.; McKee, M.D.; Huober, J.; Golshan, M.; von Minckwitz, G.; Maag, D.; et al. Addition of the PARP inhibitor veliparib plus carboplatin or carboplatin alone to standard neoadjuvant chemotherapy in triple-negative breast cancer (BrighTNess): A randomised, phase 3 trial. Lancet Oncol. 2018, 19, 497–509. [Google Scholar] [CrossRef]
- Kuznar, W. ESMO 2021 Immunotherapy but not PARP Inhibitor Improves Outcomes when Added to Chemotherapy in Early TNBC. Available online: https://oncoxchange.org/view/16972/esmo-2021-immunotherapy-but-not-parp-inhibitor-improves-outcomes-when-added-to-chemotherapy-in-early-tnbc?utm_source=Cyberimpact&utm_medium=email&utm_campaign=Precision-Medicine-Breast-cancer-News---October-2021 (accessed on 6 November 2021).
- Fasching, P.; Link, T.; Hauke, J.; Seither, F.; Jackisch, C.; Klare, P.; Schmatloch, S.; Hanusch, C.; Huober, J.; Stefek, A.; et al. Neoadjuvant paclitaxel/olaparib in comparison to paclitaxel/carboplatinum in patients with HER2-negative breast cancer and homologous recombination deficiency (GeparOLA study). Ann. Oncol. 2020, 32, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, A.; Bertucci, A.; Bertucci, F. PARP Inhibitors in the Treatment of Early Breast Cancer: The Step Beyond? Cancers 2020, 12, 1378. [Google Scholar] [CrossRef]
- Alba, K.P.; McMurtry, E.; Vallier, A.-L.; Grybowicz, L.; Copson, E.; Armstrong, A.; Roylance, R.; Qian, W.; Demiris, N.; Thomas, S.; et al. Abstract P3-10-05: Preliminary safety data from stage 1 and 2 of the phase II/III PARTNER trial: Addition of olaparib to platinum-based neoadjuvant chemotherapy in triple negative and/or germline BRCA mutated breast cancer patients. Cancer Res. 2020, 80, P3-10-05. [Google Scholar] [CrossRef]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.-K.; Hsu, J.-M.; Hsu, J.L.; Yu, W.-H.; Du, Y.; Lee, H.-H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef] [Green Version]
- Vinayak, S.; Tolaney, S.M.; Schwartzberg, L.; Mita, M.; McCann, G.; Tan, A.R.; Wahner-Hendrickson, A.E.; Forero, A.; Anders, C.; Wulf, G.M.; et al. Open-label Clinical Trial of Niraparib Combined with Pembrolizumab for Treatment of Advanced or Metastatic Triple-Negative Breast Cancer. JAMA Oncol. 2019, 5, 1132–1140. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Kim, H.-J.; Wang, Q.; Kearns, M.; Jiang, T.; Ohlson, C.E.; Li, B.B.; Xie, S.; Liu, J.F.; Stover, E.H.; et al. PARP Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1-Deficient Ovarian Cancer. Cell Rep. 2018, 25, 2972–2980.e5. [Google Scholar] [CrossRef] [Green Version]
- Pantelidou, C.; Sonzogni, O.; De Oliveria Taveira, M.; Mehta, A.K.; Kothari, A.; Wang, D.; Visal, T.; Li, M.K.; Pinto, J.; Castrillon, J.A.; et al. PARP Inhibitor Efficacy Depends on CD8+ T-cell Recruitment via Intratumoral STING Pathway Activation in BRCA-Deficient Models of Triple-Negative Breast Cancer. Cancer Discov. 2019, 9, 722–737. [Google Scholar] [CrossRef] [Green Version]
- Mehta, A.K.; Cheney, E.M.; Hartl, C.A.; Pantelidou, C.; Oliwa, M.; Castrillon, J.A.; Lin, J.-R.; Hurst, K.E.; Taveira, M.D.O.; Johnson, N.T.; et al. Targeting immunosuppressive macrophages overcomes PARP inhibitor resistance in BRCA1-associated triple-negative breast cancer. Nat. Rev. Cancer 2020, 2, 66–82. [Google Scholar] [CrossRef] [PubMed]
- Domchek, S.M.; Postel-Vinay, S.; Im, S.-A.; Park, Y.H.; Delord, J.-P.; Italiano, A.; Alexandre, J.; You, B.; Bastian, S.; Krebs, M.G.; et al. Olaparib and durvalumab in patients with germline BRCA-mutated metastatic breast cancer (MEDIOLA): An open-label, multicentre, phase 1/2, basket study. Lancet Oncol. 2020, 21, 1155–1164. [Google Scholar] [CrossRef]
- Domchek, S.; Postel-Vinay, S.; Im, S.-A.; Park, Y.; Delord, J.-P.; Italiano, A.; Alexandre, J.; You, B.; Bastian, S.; Krebs, M.; et al. Phase II study of olaparib (O) and durvalumab (D) (MEDIOLA): Updated results in patients (pts) with germline BRCA-mutated (gBRCAm) metastatic breast cancer (MBC). Ann. Oncol. 2019, 30, v477. [Google Scholar] [CrossRef]
- Wang, X.; Shi, Y.; Huang, D.; Guan, X. Emerging therapeutic modalities of PARP inhibitors in breast cancer. Cancer Treat. Rev. 2018, 68, 62–68. [Google Scholar] [CrossRef]
- Moiseeva, T.N.; Qian, C.; Sugitani, N.; Osmanbeyoglu, H.; Bakkenist, C.J. WEE1 kinase inhibitor AZD1775 induces CDK1 kinase-dependent origin firing in unperturbed G1- and S-phase cells. Proc. Natl. Acad. Sci. USA 2019, 116, 23891–23893. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; McGrail, D.J.; Sun, C.; Labrie, M.; Chen, X.; Zhang, D.; Ju, Z.; Vellano, C.P.; Lu, Y.; Li, Y.; et al. Sequential Therapy with PARP and WEE1 Inhibitors Minimizes Toxicity while Maintaining Efficacy. Cancer Cell 2019, 35, 851–867.e7. [Google Scholar] [CrossRef] [PubMed]
- Mak, J.P.; Ma, H.T.; Poon, R.Y. Synergism between ATM and PARP1 Inhibition Involves DNA Damage and Abrogating the G2 DNA Damage Checkpoint. Mol. Cancer Ther. 2019, 19, 123–134. [Google Scholar] [CrossRef] [Green Version]
- Nieto-Jimenez, C.; Alcaraz-Sanabria, A.; Martinez-Canales, S.; Corrales-Sanchez, V.; Montero, J.C.; Burgos, M.; Nuncia-Cantarero, M.; Pandiella, A.; Galan-Moya, E.M.; Ocaña, A. Checkpoint Kinase 1 Pharmacological Inhibition Synergizes with DNA-Damaging Agents and Overcomes Platinum Resistance in Basal-Like Breast Cancer. Int. J. Mol. Sci. 2020, 21, 9034. [Google Scholar] [CrossRef]
- Do, K.T.; Kochupurakkal, B.S.; Kelland, S.; de Jonge, A.; Hedglin, J.; Powers, A.; Quinn, N.; Gannon, C.; Vuong, L.; Parmar, K.; et al. Phase 1 Combination Study of the CHK1 Inhibitor Prexasertib and the PARP Inhibitor Olaparib in High-grade Serous Ovarian Cancer and Other Solid Tumors. Clin. Cancer Res. 2021, 27, 4710–4716. [Google Scholar] [CrossRef] [PubMed]
- Liposits, G.; Loh, K.P.; Soto-Perez-De-Celis, E.; Dumas, L.; Battisti, N.M.L.; Kadambi, S.; Baldini, C.; Banerjee, S.; Lichtman, S.M. PARP inhibitors in older patients with ovarian and breast cancer: Young International Society of Geriatric Oncology review paper. J. Geriatr. Oncol. 2018, 10, 337–345. [Google Scholar] [CrossRef]
- Crozier, J.; Pezzi, T.; Hodge, C.; Janeva, S.; Lesnikoski, B.-A.; Samiian, L.; Devereaux, A.; Hammond, W.; Audisio, R.; Pezzi, C.M. Addition of chemotherapy to local therapy in women aged 70 years or older with triple-negative breast cancer: A propensity-matched analysis. Lancet Oncol. 2020, 21, 1611–1619. [Google Scholar] [CrossRef]
- Dockery, L.E.; Tew, W.P.; Ding, K.; Moore, K.N. Tolerance and toxicity of the PARP inhibitor olaparib in older women with epithelial ovarian cancer. Gynecol. Oncol. 2017, 147, 509–513. [Google Scholar] [CrossRef]
- Hassan, S.; Esch, A.; Liby, T.; Gray, J.W.; Heiser, L.M. Pathway-Enriched Gene Signature Associated with 53BP1 Response to PARP Inhibition in Triple-Negative Breast Cancer. Mol. Cancer Ther. 2017, 16, 2892–2901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daemen, A.; Wolf, D.M.; Korkola, J.E.; Griffith, O.L.; Frankum, J.R.; Brough, R.; Jakkula, L.R.; Wang, N.J.; Natrajan, R.; Reis-Filho, J.S.; et al. Cross-platform pathway-based analysis identifies markers of response to the PARP inhibitor olaparib. Breast Cancer Res. Treat. 2012, 135, 505–517. [Google Scholar] [CrossRef] [Green Version]
- Evans, K.W.; Yuca, E.; Akcakanat, A.; Scott, S.M.; Arango, N.P.; Zheng, X.; Chen, K.; Tapia, C.; Tarco, E.; Eterovic, A.K.; et al. A Population of Heterogeneous Breast Cancer Patient-Derived Xenografts Demonstrate Broad Activity of PARP Inhibitor in BRCA1/2 Wild-Type Tumors. Clin. Cancer Res. 2017, 23, 6468–6477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.L.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014, 15, 852–861. [Google Scholar] [CrossRef]
- Mohyuddin, G.R.; Aziz, M.; Britt, A.; Wade, L.; Sun, W.; Baranda, J.; Al-Rajabi, R.; Saeed, A.; Kasi, A. Similar response rates and survival with PARP inhibitors for patients with solid tumors harboring somatic versus Germline BRCA mutations: A Meta-analysis and systematic review. BMC Cancer 2020, 20, 507. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, B.T.; Timms, K.M.; Carey, M.S.; Gutin, A.; Meyer, L.A.; Flake, D.D.; Abkevich, V.; Potter, J.; Pruss, D.; Glenn, P.; et al. Somatic Mutations in BRCA1 and BRCA2 Could Expand the Number of Patients That Benefit from Poly (ADP Ribose) Polymerase Inhibitors in Ovarian Cancer. J. Clin. Oncol. 2010, 28, 3570–3576. [Google Scholar] [CrossRef] [Green Version]
- Pascual, T.; Gonzalez-Farre, B.; Teixidó, C.; Oleaga, L.; Oses, G.; Ganau, S.; Chic, N.; Riu, G.; Adamo, B.; Galván, P.; et al. Significant Clinical Activity of Olaparib in a Somatic BRCA1-Mutated Triple-Negative Breast Cancer with Brain Metastasis. JCO Precis. Oncol. 2019, 3, 1–6. [Google Scholar] [CrossRef]
- Li, S.; Tao, L.; Dai, H.; Gong, X.; Zhuo, Y.; Xiang, H.; Zhao, Y.; Gao, Q.; Deng, L. BRCA1 Versus BRCA2 and PARP Inhibitors Efficacy in Solid Tumors: A Meta-Analysis of Randomized Controlled Trials. Front. Oncol. 2021, 11, 718871. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.K.; Harrell, M.I.; Oza, A.; Oaknin, A.; Ray-Coquard, I.; Tinker, A.V.; Helman, E.; Radke, M.R.; Say, C.; Vo, L.-T.; et al. BRCA Reversion Mutations in Circulating Tumor DNA Predict Primary and Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2018, 9, 210–219. [Google Scholar] [CrossRef] [Green Version]
- Carneiro, B.A.; Collier, K.A.; Nagy, R.J.; Pamarthy, S.; Sagar, V.; Fairclough, S.; Odegaard, J.; Lanman, R.B.; Costa, R.; Taxter, T.; et al. Acquired Resistance to Poly (ADP-ribose) Polymerase Inhibitor Olaparib in BRCA2-Associated Prostate Cancer Resulting From Biallelic BRCA2 Reversion Mutations Restores Both Germline and Somatic Loss-of-Function Mutations. JCO Precis. Oncol. 2018, 2, PO.17.00176. [Google Scholar] [CrossRef] [PubMed]
- Goodall, J.; Mateo, J.; Yuan, W.; Mossop, H.; Porta, N.; Miranda, S.; Perez-Lopez, R.; Dolling, D.; Robinson, D.R.; Sandhu, S.; et al. Circulating Cell-Free DNA to Guide Prostate Cancer Treatment with PARP Inhibition. Cancer Discov. 2017, 7, 1006–1017. [Google Scholar] [CrossRef] [Green Version]
- Ganesan, S.; Garber, J. Poly (ADP-Ribose) Polymerase Inhibitor Activity in Prostate Cancers Harboring Mutations in DNA Repair Genes: Who Benefits? JCO Precis. Oncol. 2020. [Google Scholar] [CrossRef]
- Sorrells, S.; McKinnon, K.E.; McBratney, A.; Sumey, C. Longitudinal and multi-tissue molecular diagnostics track somatic BRCA2 reversion mutations that correct the open reading frame of germline alteration upon clinical relapse. Npj Genom. Med. 2021, 6, 17. [Google Scholar] [CrossRef] [PubMed]
- Weigelt, B.; Comino-Mendez, I.; de Bruijn, I.; Tian, L.; Meisel, J.L.; Garcia-Murillas, I.; Fribbens, C.; Cutts, R.; Martelotto, L.G.; Ng, C.K.Y.; et al. Diverse BRCA1 and BRCA2 Reversion Mutations in Circulating Cell-Free DNA of Therapy-Resistant Breast or Ovarian Cancer. Clin. Cancer Res. 2017, 23, 6708–6720. [Google Scholar] [CrossRef] [Green Version]
- Waks, A.; Cohen, O.; Kochupurakkal, B.; Kim, D.; Dunn, C.; Buendia, J.B.; Wander, S.; Helvie, K.; Lloyd, M.; Marini, L.; et al. Reversion and non-reversion mechanisms of resistance to PARP inhibitor or platinum chemotherapy in BRCA1/2-mutant metastatic breast cancer. Ann. Oncol. 2020, 31, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Puhalla, S.L.; Diéras, V.; Arun, B.K.; Kaufman, B.; Wildiers, H.; Han, H.S.; Ayoub, J.-P.; Stearns, V.; Yuan, Y.; Helsten, T.; et al. Relevance of Platinum-free Interval and BRCA Reversion Mutations for Veliparib Monotherapy after Progression on Carboplatin/Paclitaxel for gBRCA Advanced Breast Cancer (BROCADE3 Crossover). Clin. Cancer Res. 2021, 27, 4983–4993. [Google Scholar] [CrossRef]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Rodrigues, D.N.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef]
- Mateo, J.; Porta, N.; Bianchini, D.; McGovern, U.; Elliott, T.; Jones, R.; Syndikus, I.; Ralph, C.; Jain, S.; Varughese, M.; et al. Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): A multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 2019, 21, 162–174. [Google Scholar] [CrossRef]
- Swisher, E.M.; Kwan, T.T.; Oza, A.M.; Tinker, A.V.; Ray-Coquard, I.; Oaknin, A.; Coleman, R.L.; Aghajanian, C.; Konecny, G.E.; O’Malley, D.M.; et al. Molecular and clinical determinants of response and resistance to rucaparib for recurrent ovarian cancer treatment in ARIEL2 (Parts 1 and 2). Nat. Commun. 2021, 12, 2487. [Google Scholar] [CrossRef]
- Heeke, A.L.; Pishvaian, M.J.; Lynce, F.; Xiu, J.; Brody, J.R.; Chen, W.-J.; Baker, T.M.; Marshall, J.L.; Isaacs, C. Prevalence of Homologous Recombination–Related Gene Mutations Across Multiple Cancer Types. JCO Precis. Oncol. 2018, 2018, PO.17.00286. [Google Scholar] [CrossRef]
- Hoppe, M.M.; Sundar, R.; Tan, D.S.P.; Jeyasekharan, A.D. Biomarkers for Homologous Recombination Deficiency in Cancer. J. Natl. Cancer Inst. 2018, 110, 704–713. [Google Scholar] [CrossRef] [Green Version]
- Riaz, N.; Blecua, P.; Lim, R.S.; Shen, R.; Higginson, D.; Weinhold, N.; Norton, L.; Weigelt, B.; Powell, S.N.; Reis-Filho, J.S. Pan-cancer analysis of bi-allelic alterations in homologous recombination DNA repair genes. Nat. Commun. 2017, 8, 857. [Google Scholar] [CrossRef]
- Marquard, A.M.; Eklund, A.C.; Joshi, T.; Krzystanek, M.; Favero, F.; Wang, Z.C.; Richardson, A.L.; Silver, D.P.; Szallasi, Z.; Birkbak, N.J. Pan-cancer analysis of genomic scar signatures associated with homologous recombination deficiency suggests novel indications for existing cancer drugs. Biomark. Res. 2015, 3, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, R.; Leary, A.; Scott, C.; Serra, V.; Lord, C.; Bowtell, D.; Chang, D.; Garsed, D.; Jonkers, J.; Ledermann, J.; et al. ESMO recommendations on predictive biomarker testing for homologous recombination deficiency and PARP inhibitor benefit in ovarian cancer. Ann. Oncol. 2020, 31, 1606–1622. [Google Scholar] [CrossRef] [PubMed]
- Abkevich, V.; Timms, K.M.; Hennessy, B.T.; Potter, J.; Carey, M.S.; Meyer, L.A.; Smith-McCune, K.; Broaddus, R.; Lu, K.H.; Chen, J.; et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br. J. Cancer 2012, 107, 1776–1782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Telli, M.L.; Timms, K.M.; Reid, J.; Hennessy, B.; Mills, G.B.; Jensen, K.C.; Szallasi, Z.; Barry, W.T.; Winer, E.P.; Tung, N.M.; et al. Homologous Recombination Deficiency (HRD) Score Predicts Response to Platinum-Containing Neoadjuvant Chemotherapy in Patients with Triple-Negative Breast Cancer. Clin. Cancer Res. 2016, 22, 3764–3773. [Google Scholar] [CrossRef] [Green Version]
- Loibl, S.; Weber, K.E.; Timms, K.M.; Elkin, E.P.; Hahnen, E.; Fasching, P.A.; Lederer, B.; Denkert, C.; Schneeweiss, A.; Braun, S.; et al. Survival analysis of carboplatin added to an anthracycline/taxane-based neoadjuvant chemotherapy and HRD score as predictor of response—final results from GeparSixto. Ann. Oncol. 2018, 29, 2341–2347. [Google Scholar] [CrossRef] [PubMed]
- Telli, M.L.; Metzger, O.; Timms, K.; Evans, B.; Vogel, D.; Wei, H.; Jones, J.T.; Wenstrup, R.J.; McKee, M.D.; Sullivan, D.M.; et al. Evaluation of homologous recombination deficiency (HRD) status with pathological response to carboplatin +/- veliparib in BrighTNess, a randomized phase 3 study in early stage TNBC. J. Clin. Oncol. 2018, 36, 519. [Google Scholar] [CrossRef]
- Mayer, E.L.; Abramson, V.; Jankowitz, R.; Falkson, C.; Marcom, P.K.; Traina, T.; Carey, L.; Rimawi, M.; Specht, J.; Miller, K.; et al. TBCRC 030: A phase II study of preoperative cisplatin versus paclitaxel in triple-negative breast cancer: Evaluating the homologous recombination deficiency (HRD) biomarker. Ann. Oncol. 2020, 31, 1518–1525. [Google Scholar] [CrossRef]
- Tutt, A.; Tovey, H.; Cheang, M.C.U.; Kernaghan, S.; Kilburn, L.; Gazinska, P.; Owen, J.; Abraham, J.; Barrett, S.; Barrett-Lee, P.; et al. Carboplatin in BRCA1/2-mutated and triple-negative breast cancer BRCAness subgroups: The TNT Trial. Nat. Med. 2018, 24, 628–637. [Google Scholar] [CrossRef] [Green Version]
- Hodgson, D.; Lai, Z.; Dearden, S.; Barrett, J.; Harrington, E.; Timms, K.; Lanchbury, J.; Wu, W.; Allen, A.; Senkus, E.; et al. Analysis of mutation status and homologous recombination deficiency in tumors of patients with germline BRCA1 or BRCA2 mutations and metastatic breast cancer: OlympiAD. Ann. Oncol. 2021, 32, 1582–1589. [Google Scholar] [CrossRef]
- Telli, M.L.; Jensen, K.C.; Vinayak, S.; Kurian, A.W.; Lipson, J.A.; Flaherty, P.J.; Timms, K.; Abkevich, V.; Schackmann, E.A.; Wapnir, I.L.; et al. Phase II Study of Gemcitabine, Carboplatin, and Iniparib As Neoadjuvant Therapy for Triple-Negative and BRCA1/2 Mutation–Associated Breast Cancer With Assessment of a Tumor-Based Measure of Genomic Instability: PrECOG 0105. J. Clin. Oncol. 2015, 33, 1895–1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isakoff, S.J.; Mayer, E.L.; He, L.; Traina, T.A.; Carey, L.A.; Krag, K.J.; Rugo, H.S.; Liu, M.C.; Stearns, V.; Come, S.E.; et al. TBCRC009: A Multicenter Phase II Clinical Trial of Platinum Monotherapy With Biomarker Assessment in Metastatic Triple-Negative Breast Cancer. J. Clin. Oncol. 2015, 33, 1902–1909. [Google Scholar] [CrossRef] [PubMed]
- Swisher, E.M.; Lin, K.K.; Oza, A.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, E.G.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef] [Green Version]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef] [Green Version]
- Funingana, I.-G.; Reinius, M.A.V.; Petrillo, A.; Ang, J.E.; Brenton, J.D. Can integrative biomarker approaches improve prediction of platinum and PARP inhibitor response in ovarian cancer? Semin. Cancer Biol. 2021, 77, 67–82. [Google Scholar] [CrossRef]
- Davies, H.; Glodzik, D.; Morganella, S.; Yates, L.R.; Staaf, J.; Zou, X.; Ramakrishna, M.; Martin, S.; Boyault, S.; Sieuwerts, A.M.; et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med. 2017, 23, 517–525. [Google Scholar] [CrossRef]
- Staaf, J.; Glodzik, D.; Bosch, A.; Vallon-Christersson, J.; Reuterswärd, C.; Häkkinen, J.; Degasperi, A.; Amarante, T.D.; Saal, L.H.; Hegardt, C.; et al. Whole-genome sequencing of triple-negative breast cancers in a population-based clinical study. Nat. Med. 2019, 25, 1526–1533. [Google Scholar] [CrossRef]
- Zhao, E.Y.; Shen, Y.; Pleasance, E.; Kasaian, K.; Leelakumari, S.; Jones, M.; Bose, P.; Ch’ng, C.; Reisle, C.; Eirew, P.; et al. Homologous Recombination Deficiency and Platinum-Based Therapy Outcomes in Advanced Breast Cancer. Clin. Cancer Res. 2017, 23, 7521–7530. [Google Scholar] [CrossRef] [Green Version]
- Willers, H.; Gheorghiu, L.; Liu, Q.; Efstathiou, J.A.; Wirth, L.J.; Krause, M.; von Neubeck, C. DNA Damage Response Assessments in Human Tumor Samples Provide Functional Biomarkers of Radiosensitivity. Semin. Radiat. Oncol. 2015, 25, 237–250. [Google Scholar] [CrossRef] [Green Version]
- Graeser, M.; McCarthy, A.; Lord, C.; Savage, K.; Hills, M.; Salter, J.; Orr, N.; Parton, M.; Smith, I.E.; Reis-Filho, J.S.; et al. A Marker of Homologous Recombination Predicts Pathologic Complete Response to Neoadjuvant Chemotherapy in Primary Breast Cancer. Clin. Cancer Res. 2010, 16, 6159–6168. [Google Scholar] [CrossRef] [Green Version]
- Naipal, K.A.; Verkaik, N.S.; Ameziane, N.; Van Deurzen, C.H.; Ter Brugge, P.; Meijers, M.; Sieuwerts, A.M.; Martens, J.W.; O’Connor, M.J.; Vrieling, H.; et al. Functional Ex Vivo Assay to Select Homologous Recombination–Deficient Breast Tumors for PARP Inhibitor Treatment. Clin. Cancer Res. 2014, 20, 4816–4826. [Google Scholar] [CrossRef] [Green Version]
- Cruz, C.; Castroviejo-Bermejo, M.; Gutiérrez-Enríquez, S.; Llop-Guevara, A.; Ibrahim, Y.; Oliver, A.G.; Bonache, S.; Morancho, B.; Bruna, A.; Rueda, O.; et al. RAD51 foci as a functional biomarker of homologous recombination repair and PARP inhibitor resistance in germline BRCA-mutated breast cancer. Ann. Oncol. 2018, 29, 1203–1210. [Google Scholar] [CrossRef]
- Elizalde, V.S.; Llop-Guevara, A.; Pearson, A.; Cruz, C.; Castroviejo-Bermejo, M.; Chopra, N.; Tovey, H.; Toms, C.; Kriplani, D.; Gevensleben, H.; et al. 1O Detection of homologous recombination repair deficiency (HRD) in treatment-naive early triple-negative breast cancer (TNBC) by RAD51 foci and comparison with DNA-based tests. Ann. Oncol. 2021, 32, S21–S22. [Google Scholar] [CrossRef]
- Llop-Guevara, A.; Loibl, S.; Villacampa, G.; Vladimirova, V.; Schneeweiss, A.; Karn, T.; Zahm, D.M.; Herencia-Ropero, A.; Jank, P.; van Mackelenbergh, M.; et al. Association of RAD51 with homologous recombination deficiency (HRD) and clinical outcomes in untreated triple-negative breast cancer (TNBC): Analysis of the GeparSixto randomized clinical trial. Ann. Oncol. 2021, 32, 1590–1596. [Google Scholar] [CrossRef]
- Lord, C.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef] [PubMed]
- McGrail, D.J.; Lin, C.C.-J.; Garnett, J.; Liu, Q.; Mo, W.; Dai, H.; Lu, Y.; Yu, Q.; Ju, Z.; Yin, J.; et al. Improved prediction of PARP inhibitor response and identification of synergizing agents through use of a novel gene expression signature generation algorithm. Syst. Biol. Appl. 2017, 3, 8. [Google Scholar] [CrossRef] [Green Version]
- Severson, T.M.; Wolf, D.M.; Yau, C.; Peeters, J.; Wehkam, D.; Schouten, P.C.; Chin, S.-F.; Majewski, I.J.; Michaut, M.; Bosma, A.; et al. The BRCA1ness signature is associated significantly with response to PARP inhibitor treatment versus control in the I-SPY 2 randomized neoadjuvant setting. Breast Cancer Res. 2017, 19, 99. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2012, 41, D955–D961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, D.M.; Yau, C.; Sanil, A.; Glas, A.; Petricoin, E.; Wulfkuhle, J.; Severson, T.M.; Linn, S.; Brown-Swigart, L.; Hirst, G.; et al. DNA repair deficiency biomarkers and the 70-gene ultra-high risk signature as predictors of veliparib/carboplatin response in the I-SPY 2 breast cancer trial. npj Breast Cancer 2017, 3, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortes, J.; Cescon, D.W.; Rugo, H.S.; Nowecki, Z.; Im, S.-A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Holgado, E.; et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): A randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet 2020, 396, 1817–1828. [Google Scholar] [CrossRef]
- Staniszewska, A.D.; Jwt, J.W.Y.; Pike, A.; Fazenbaker, C.; Cook, K.; Bosco, E.; Smith, A.; Wilson, J.; Leo, E. Abstract 1270: The novel PARP1-selective inhibitor, AZD5305, is efficacious as monotherapy and in combination with standard of care chemotherapy in the in vivo preclinical models. Cancer Res. 2021, 81, 1270. [Google Scholar] [CrossRef]
| First Author Study Name | Year of Study | No. of Participants (BC Patients) | Type of Study | Median/Mean No. of Prior Chemotherapy Regimens (Range) | Therapeutic Agents | Patient Population | Outcome (Objective Response Rate (ORR), Progression Free Survival (PFS)) | ||
|---|---|---|---|---|---|---|---|---|---|
| PARPi | Combination Agent | Comparative Agent/ Standard Chemotherapy | |||||||
| Lee, J.M. et al. [57] | 2014 | 45 (8 BC) | Phase I/Ib | 5 (2–11) | Olaparib capsule, 100 mg twice daily continuous or Olaparib capsule, 200–400 mg twice daily days 1–7 | Carboplatin AUC 3–5 every 21 days | None | gBRCAMUT | ORR in all 52.4%, ORR in breast cancer 87.5% |
| Lee, J.M. et al. [58] | 2017 | 77 (14 BC) | Phase I/Ib | 4 (1–10) | Dose escalation: Olaparib tablet: 100–200 mg twice daily, days 1–7 300 mg twice daily maintenance after carboplatin Expansion cohort: Olaparib: Cohort A: Days 1–7 cycle 1, and days 2–8 for cycle 2; Cohort B: Days 2–8 cycle 1, and 1–7 cycle 2. Both cohorts: Days 1–7 cycle 3 up to 8; olaparib maintenance | Dose escalation: Carboplatin AUC4–5 every 21 days, up to 8 cycles Expansion cohort: Carboplatin: Cohort A: Day 8 cycle 1, day 1 cycle 2; Cohort B: Day 1 cycle 1, day 8 cycle 2 Both cohorts: Day 1 cycle 3, up to 8 | None | Recurrent or refractory gynecologic cancers or metastatic or inoperable breast cancer | ORR in all 46%, gBRCAMUT 68% |
| Dhawan, M.S. et al. [59] | 2017 | 24 (11 BC) | Phase I | 24% ≤ 2 lines * 12%—3 lines * 63% ≥ 4 lines* | Talazoparib 0.75 and 1 mg daily | Carboplatin AUC 1 and 1.5 every 2–3 weeks | None | Advanced solid tumors | ORR in all 14% |
| Somlo, G. et al. [60] | 2017 | 77 (77 BC) | Phase I/II | Phase I: 1 (0–5) Phase II: 1 (0–5) | Phase 1: Veliparib, 50–200 mg twice daily Phase 2: Veliparib, 400 mg twice daily and upon progression 150 mg twice daily in combination | Phase 1: Carboplatin AUC 5/6 every 21 days Phase 2: Carboplatin AUC 5 every 21 days in combination | None | gBRCAMUT breast cancer | Response rate in phase I, 56%; phase II—BRCA1MUT, 14%; BRCA2MUT, 36%, PFS in phase I, 8.7 months; phase II—on veliparib, 5.2 months; after combination therapy, 1.8 months |
| Appleman, L.J. et al. [61] | 2019 | 73 (16 BC) | Phase I | ≤3 lines | Veliparib 10–120 mg twice daily Days 1–7, starting cycle 2 | Carboplatin: AUC 6 Paclitaxel: 150–200 mg/m2 Day 1 of 21-day cycle 1, Day 3 of cycle 2 onwards | None | Advanced solid tumors | ORR in all 40%, ORR in breast cancer 69% |
| Han, H.S. et al. [62] BROCADE | 2018 | 294 (294 BC) | Phase II randomized controlled trial | ≤2 lines | Veliparib (V) 120 mg twice daily Days 1–7, 21-day cycles | Carboplatin (C): AUC 6 Paclitaxel (P): 75 mg/m2 Day 3 | PCP (placebo, carboplatin, paclitaxel) vs. V plus temozolomide (T) | gBRCAMUT breast cancer | ORR in VCP, 77.8%, PCP, 61.3%; VT, 28.6% PFS in VCP, 14.1 months; CP 12.3 months, V plus T, 7.4 months |
| Diéras, V. et al. [63] Arun, B.K. et al. [64] BROCADE3 | 2020 | 513 (513 BC) | Phase III Double-blinded, randomized controlled trial | ≤2 lines | Veliparib, 120 mg twice daily Days −2 to 5 If combination discontinued prior to progression, could continue with veliparib up to 400 mg twice daily | Carboplatin (C) AUC 6 Day 1 of 21-day cycle Paclitaxel (P) 80 mg/m2 Day 1, 8, 15 of 21-day cycle | PCP (placebo, carboplatin, paclitaxel) | gBRCAMUT HER2-negative breast cancer | All ORR in VCP 75.8%, PCP 74.1% PFS in VCP 14.5 months, PCP 12.6 months No previous chemotherapy ORR in VCP 79.7%, PCP 76.3% PFS in VCP 16.6 months, PCP 13.1 months |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yordanova, M.; Hubert, A.; Hassan, S. Expanding the Use of PARP Inhibitors as Monotherapy and in Combination in Triple-Negative Breast Cancer. Pharmaceuticals 2021, 14, 1270. https://doi.org/10.3390/ph14121270
Yordanova M, Hubert A, Hassan S. Expanding the Use of PARP Inhibitors as Monotherapy and in Combination in Triple-Negative Breast Cancer. Pharmaceuticals. 2021; 14(12):1270. https://doi.org/10.3390/ph14121270
Chicago/Turabian StyleYordanova, Mariya, Audrey Hubert, and Saima Hassan. 2021. "Expanding the Use of PARP Inhibitors as Monotherapy and in Combination in Triple-Negative Breast Cancer" Pharmaceuticals 14, no. 12: 1270. https://doi.org/10.3390/ph14121270
APA StyleYordanova, M., Hubert, A., & Hassan, S. (2021). Expanding the Use of PARP Inhibitors as Monotherapy and in Combination in Triple-Negative Breast Cancer. Pharmaceuticals, 14(12), 1270. https://doi.org/10.3390/ph14121270