
Improved Bioavailability of Poorly Water-Soluble Drug by Targeting Increased Absorption through Solubility Enhancement and Precipitation Inhibition

 , and
, and
Abstract

1. Introduction
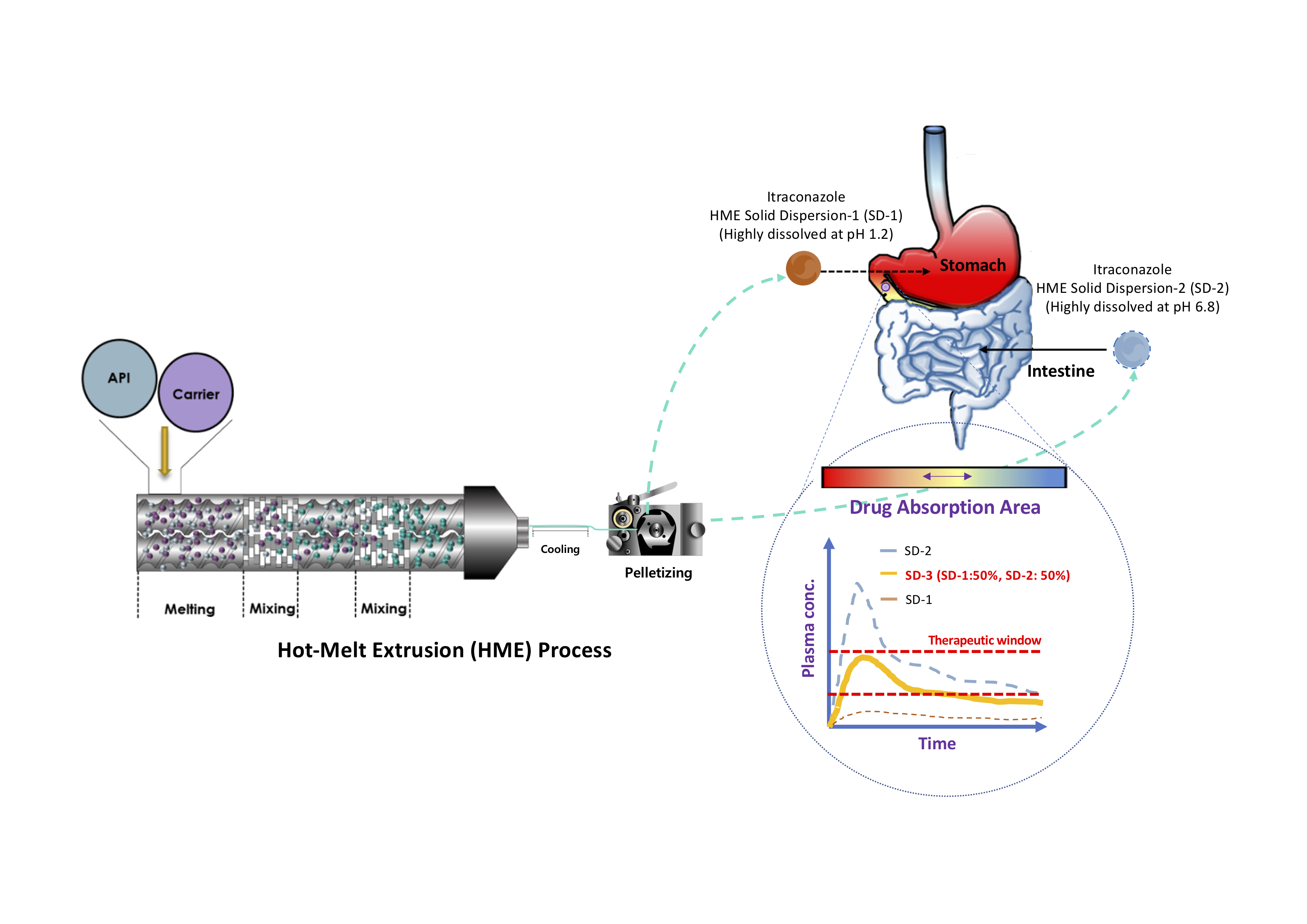
2. Results and Discussion
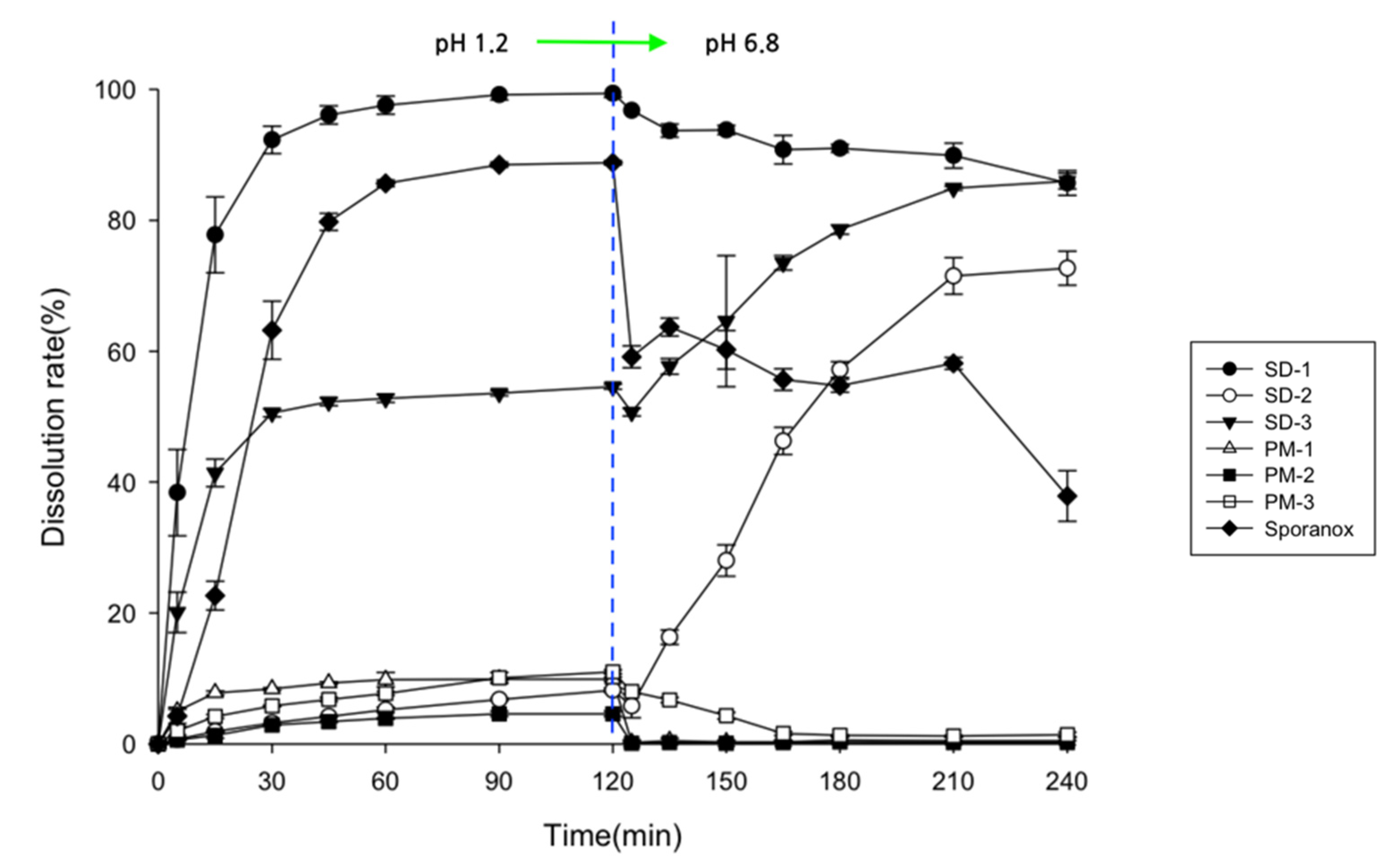
2.1. Assessment of Drug Contents, Release Behaviors, and Morphological Changes
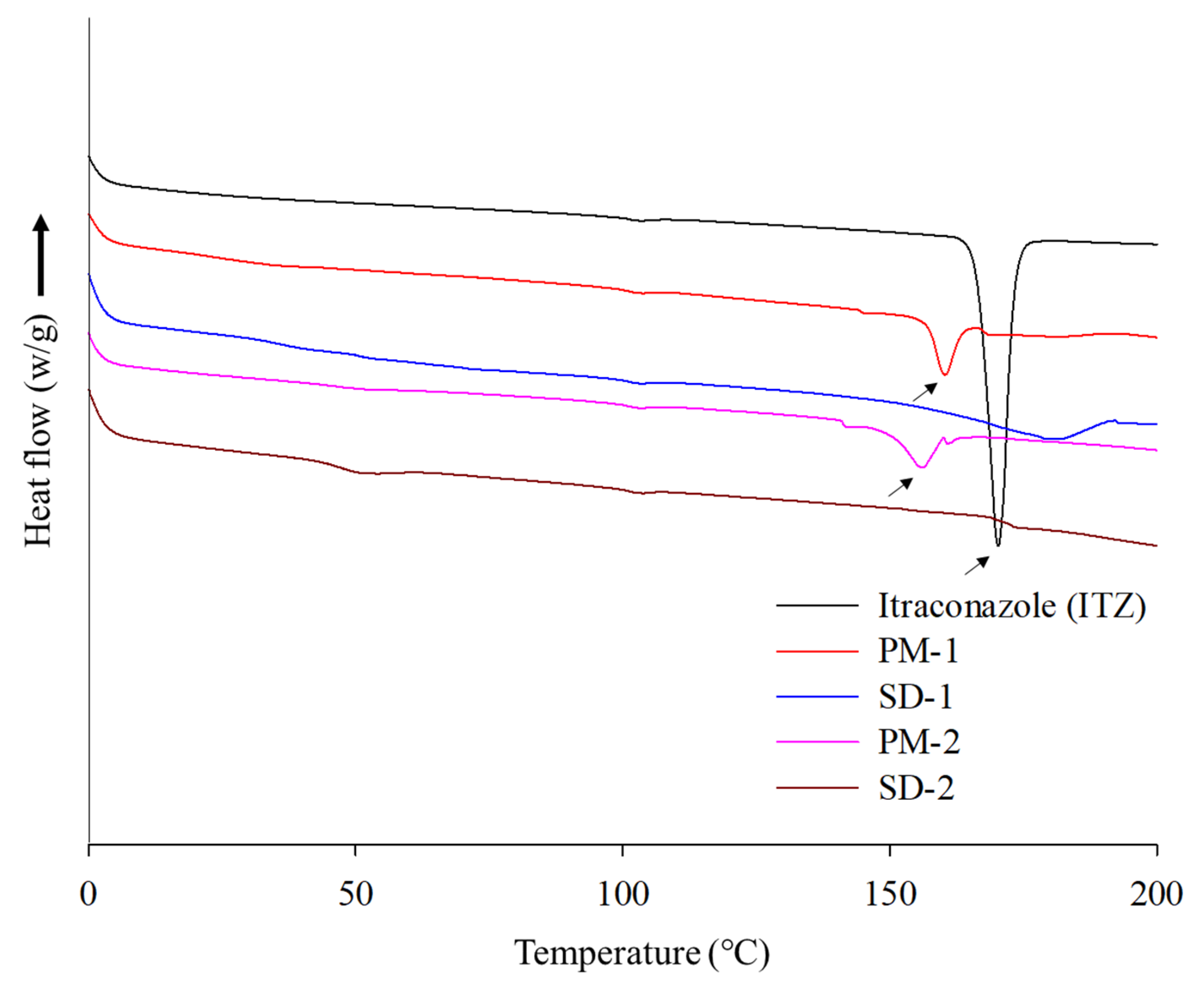
2.2. Physicochemical Properties of ITZ SD
2.3. Stability Test
2.4. In Vivo Studies
3. Materials and Methods
3.1. Materials
3.2. Preparation of SD Pellets Using HME Technology
3.3. Determination of Drug Content in SDs
3.4. Drug Release
3.5. Recrystallization Behavior of ITZ in pH 6.8 Medium
3.6. Physicochemical Properties of SDs
3.7. Stability Test
3.8. In Vivo Studies
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Davis, M.; Walker, G. Recent strategies in spray drying for the enhanced bioavailability of poorly water-soluble drugs. J. Control Release 2018, 269, 110–127. [Google Scholar] [CrossRef] [PubMed]
- Alonzo, D.E.; Zhang, G.G.; Zhou, D.; Gao, Y.; Taylor, L.S. Understanding the behavior of amorphous pharmaceutical systems during dissolution. Pharm. Res. 2010, 27, 608–618. [Google Scholar] [CrossRef] [PubMed]
- Bevernage, J.; Brouwers, J.; Clarysse, S.; Vertzoni, M.; Tack, J.; Annaert, P.; Augustijns, P. Drug supersaturation in simulated and human intestinal fluids representing different nutritional states. J. Pharm. Sci. 2010, 99, 4525–4534. [Google Scholar] [CrossRef]
- Tanaka, Y.; Kawakami, A.; Nanimatsu, A.; Horio, M.; Matsuoka, J.; Wada, T.; Kasaoka, S.; Yoshikawa, H. In vivo evaluation of supersaturation/precipitation/re-dissolution behavior of cinnarizine, a lipophilic weak base, in the gastrointestinal tract: The key process of oral absorption. Eur. J. Pharm. Sci. 2017, 96, 464–471. [Google Scholar] [CrossRef]
- DiNunzio, J.C.; Brough, C.; Miller, D.A.; Williams, R.O., 3rd; McGinity, J.W. Fusion processing of itraconazole solid dispersions by kinetisol® dispersing: A comparative study to hot melt extrusion. J. Pharm. Sci. 2010, 99, 1239–1253. [Google Scholar] [CrossRef] [PubMed]
- Duret, C.; Wauthoz, N.; Sebti, T.; Vanderbist, F.; Amighi, K. Solid dispersions of itraconazole for inhalation with enhanced dissolution, solubility and dispersion properties. Int. J. Pharm. 2012, 428, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, S.L.; Trividic, A.; Davis, A.F.; Hadgraft, J. Crystallization of hydrocortisone acetate: Influence of polymers. Int. J. Pharm. 2001, 212, 213–221. [Google Scholar] [CrossRef]
- Konno, H.; Handa, T.; Alonzo, D.E.; Taylor, L.S. Effect of polymer type on the dissolution profile of amorphous solid dispersions containing felodipine. Eur. J. Pharm. Biopharm. 2008, 70, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Blaabjerg, L.I.; Grohganz, H.; Lindenberg, E.; Löbmann, K.; Müllertz, A.; Rades, T. The influence of polymers on the supersaturation potential of poor and good glass formers. Pharmaceutics 2018, 10, 164. [Google Scholar] [CrossRef] [PubMed]
- Costa, B.L.A.; Sauceau, M.; Del Confetto, S.; Sescousse, R.; Ré, M.I. Determination of drug-polymer solubility from supersaturated spray-dried amorphous solid dispersions: A case study with Efavirenz and Soluplus®. Eur. J. Pharm. Biopharm. 2019, 142, 300–306. [Google Scholar] [CrossRef]
- Hu, X.Y.; Lou, H.; Hageman, M.J. Preparation of lapatinib ditosylate solid dispersions using solvent rotary evaporation and hot melt extrusion for solubility and dissolution enhancement. Int. J. Pharm. 2018, 552, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Repka, M.A.; Battu, S.K.; Upadhye, S.B.; Thumma, S.; Crowley, M.M.; Zhang, F.; Martin, C.; McGinity, J.W. Pharmaceutical applications of hot-melt extrusion: Part II. Drug. Dev. Ind. Pharm. 2007, 33, 1043–1057. [Google Scholar] [CrossRef]
- Mendonsa, N.S.; Pradhan, A.; Sharma, P.; Prado, R.M.B.; Murthy, S.N.; Kundu, S.; Repka, M.A. A quality by design approach to develop topical creams via hot-melt extrusion technology. Eur. J. Pharm. Sci. 2019, 136, 104948. [Google Scholar] [CrossRef]
- Desai, P.M.; Hogan, R.C.; Brancazio, D.; Puri, V.; Jensen, K.D.; Chun, J.H.; Myerson, A.S.; Trout, B.L. Integrated hot-melt extrusion—Injection molding continuous tablet manufacturing platform: Effects of critical process parameters and formulation attributes on product robustness and dimensional stability. Int. J. Pharm. 2017, 531, 332–342. [Google Scholar] [CrossRef]
- Yang, Z.; Yang, H.; Gang, T.; Mingming, D.; Qi, L.; Xia, L. Development of ibuprofen dry suspensions by hot melt extrusion: Characterization, physical stability and pharmacokinetic studies. J. Drug Deliv. Sci. Technol. 2019, 54, 101313. [Google Scholar] [CrossRef]
- Zhao, Y.; Xie, X.; Zhao, Y.; Gao, Y.; Cai, C.; Zhang, Q.; Ding, Z.; Fan, Z.; Zhang, H.; Liu, M.; et al. Effect of plasticizers on manufacturing ritonavir/copovidone solid dispersions via hot-melt extrusion: Preformulation, physicochemical characterization, and pharmacokinetics in rats. Eur. J. Pharm. Sci. 2019, 127, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Simões, M.F.; Pinto, R.M.A.; Simões, S. Hot-melt extrusion in the pharmaceutical industry: Toward filing a new drug application. Drug Discov. Today. 2019, 24, 1749–1768. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Daintree, L.S.; Ding, S.; Ledger, D.M.; Wang, B.; Zhao, W.; Qi, J.; Wu, W.; Han, J. Itraconazole solid dispersion prepared by a supercritical fluid technique: Preparation, in vitro characterization, and bioavailability in beagle dogs. Drug Des. Devel. Ther. 2015, 9, 2801–2810. [Google Scholar]
- Vasconcelos, T.; Marques, S.; das Neves, J.; Sarmento, B. Amorphous solid dispersions: Rational selection of a manufacturing process. Adv. Drug Deliv. Rev. 2016, 100, 85–101. [Google Scholar] [CrossRef] [PubMed]
- Haser, A.; Huang, S.; Listro, T.; White, D.; Zhang, F. An approach for chemical stability during melt extrusion of a drug substance with a high melting point. Int. J. Pharm. 2017, 524, 55–64. [Google Scholar] [CrossRef]
- Malaquias, L.F.B.; Sá-Barreto, L.C.L.; Daniel, O.F.; Izabel, C.R.S.; Kapish, K.; Thomas, D.; Eliana, M.L.; Ricardo, N.M.; Guilherme, M.G.; Tais, G.; et al. Taste masking and rheology improvement of drug complexed with beta-cyclodextrin and hydroxypropyl-beta-cyclodextrin by hot-melt extrusion. Carbohydr. Polym. 2018, 185, 19–26. [Google Scholar]
- Jung, J.-Y.; Yoo, S.D.; Lee, S.H.; Kim, K.H.; Yoon, D.S.; Lee, K.H. Enhanced solubility and dissolution rate of itraconazole by a solid dispersion technique. Int. J. Pharm. 1999, 187, 209–218. [Google Scholar] [CrossRef]
- Tao, T.; Zhao, Y.; Wu, J.; Zhou, B. Preparation and evaluation of itraconazole dihydrochloride for the solubility and dissolution rate enhancement. Int. J. Pharm. 2009, 367, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.K.; Poudel, B.K.; Marasini, N.; Yang, K.Y.; Kim, J.W.; Kim, J.O.; Choi, H.G.; Yong, C.S. Enhanced solubility and oral bioavailability of itraconazole by combining membrane emulsification and spray drying technique. Int. J. Pharm. 2012, 434, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Amit, C.; Atul, G.; Kartik, H.; Tejal, M. Exploring novel carrier for improving bioavailability of Itraconazole: Solid dispersion through hot-melt extrusion. J. Drug Deliv. Sci. Technol. 2021, 63, 102541. [Google Scholar]
- Thiry, J.; Kok, M.G.; Collard, L.; Frère, A.; Krier, F.; Fillet, M.; Evrard, B. Bioavailability enhancement of itraconazole-based solid dispersions produced by hot melt extrusion in the framework of the Three Rs rule. Eur. J. Pharm. Sci. 2017, 99, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Solanki, N.G.; Lam, K.; Tahsin, M.; Gumaste, S.G.; Shah, A.V.; Serajuddin, A.T.M. Effects of surfactants on itraconazole-HPMCAS solid dispersion prepared by hot-melt extrusion I: Miscibility and drug release. J. Pharm. Sci. 2019, 108, 1453–1465. [Google Scholar] [CrossRef]
- Brough, C.; Miller, D.A.; Keen, J.M.; Kucera, S.A.; Lubda, D.; Williams, R.O., 3rd. Use of polyvinyl alcohol as a solubility-enhancing polymer for poorly water soluble drug delivery (part 1). AAPS Pharm. Sci. Tech. 2016, 17, 167–179. [Google Scholar] [CrossRef]
- Chan, S.-Y.; Chung, Y.-Y.; Cheah, X.-Z.; Tan, E.Y.-L.; Quah, J. The characterization and dissolution performances of spray dried solid dispersion of ketoprofen in hydrophilic carriers. Asian. J. Pharm. Sci. 2015, 10, 372–385. [Google Scholar] [CrossRef]
- Albadarin, A.B.; Potter, C.B.; Davis, M.T.; Iqbal, J.; Korde, S.; Pagire, S.; Paradkar, A.; Walker, G. Development of stability-enhanced ternary solid dispersions via combinations of HPMCP and Soluplus® processed by hot melt extrusion. Int. J. Pharm. 2017, 532, 603–611. [Google Scholar] [CrossRef]
- Berben, P.; Vink, M.J.A.; Domínguez-Vega, E.; Somsen, G.W.; Brouwers, J.; Augustijns, P. Linking the concentrations of itraconazole and 2-hydroxypropyl-beta-cyclodextrin in human intestinal fluids after oral intake of Sporanox®. Eur. J. Pharm. Biopharm. 2018, 132, 231–236. [Google Scholar] [CrossRef]
- Six, K.; Verreck, G.; Peeters, J.; Brewster, M.; Van Den Mooter, G. Increased physical stability and improved dissolution properties of itraconazole, a class II drug, by solid dispersions that combine fast- and slow-dissolving polymers. J. Pharm. Sci. 2004, 93, 124–131. [Google Scholar] [CrossRef]
- Janssens, S.; de Armas, H.N.; Remon, J.P.; Van den Mooter, G. The use of a new hydrophilic polymer, Kollicoat IR, in the formulation of solid dispersions of itraconazole. Eur. J. Pharm. Sci. 2007, 30, 288–294. [Google Scholar] [CrossRef]
- Thiry, J.; Lebrun, P.; Vinassa, C.; Adam, M.; Netchacovitch, L.; Ziemons, E.; Hubert, P.; Krier, F.; Evrard, B. Continuous production of itraconazole-based solid dispersions by hot melt extrusion: Preformulation, optimization and design space determination. Int. J. Pharm. 2016, 515, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Verreck, G.; Six, K.; Van den Mooter, G.; Baert, L.; Peeters, J.; Brewster, M.E. Characterization of solid dispersions of itraconazole and hydroxypropylmethylcellulose prepared by melt extrusion part I. Int. J. Pharm. 2002, 251, 165–174. [Google Scholar] [CrossRef]
- Garg, A.K.; Sachdeva, R.K.; Kapoor, G. Comparison of crystalline and amorphous carriers to improve the dissolution profile of water insoluble drug itraconazole. Int. J. Pharma Bio. Sci. 2013, 4, 934–948. [Google Scholar]
- Darji, M.A.; Lalge, R.M.; Marathe, S.P.; Mulay, T.D.; Fatima, T.; Alshammari, A.; Lee, H.K.; Repka, M.A.; Murthy, S.N. Excipient stability in oral solid dosage forms: A review. AAPS Pharm. Sci. Tech. 2018, 19, 12–26. [Google Scholar] [CrossRef] [PubMed]
- Mendiratta, C.; Kadam, V.; Pokharkar, V. Lansoprazole solid dispersion using a novel amphiphillic polymer Soluplus®. J. Chem. Pharm. Res. 2011, 3, 536–543. [Google Scholar]
- Karandikar, H.; Ambardekar, R.; Kelly, A.; Gough, T.; Paradkar, A. Systematic identification of thermal degradation products of HPMCP during hot melt extrusion process. Int. J. Pharm. 2015, 486, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.T.; Potter, C.B.; Mohammadpour, M.; Albadarin, A.B.; Walker, G.M. Design of spray dried ternary solid dispersions comprising itraconazole, soluplus and HPMCP: Effect of constituent compositions. Int. J. Pharm. 2017, 519, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.; Peng, T.; Huang, Z.; Singh, V.; Shi, Y.; Wen, T.; Lu, M.; Quan, G.; Pan, X.; Wu, C. Polymer(-)surfactant system based amorphous solid dispersion: Precipitation inhibition and bioavailability enhancement of itraconazole. Pharmaceutics 2018, 10, 53. [Google Scholar] [CrossRef]
- Sagawa, K.; Li, F.; Liese, R.; Sutton, S.C. Fed and fasted gastric pH and gastric residence time in conscious beagle dogs. J. Pharm. Sci. 2009, 98, 2494–2500. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.D.; Lee, S.H.; Kang, E.; Jun, H.; Jung, J.Y.; Park, J.W.; Lee, K.H. Bioavailability of itraconazole in rats and rabbits after administration of tablets containing solid dispersion particles. Drug Dev. Ind. Pharm. 2000, 26, 27–34. [Google Scholar] [CrossRef]
- Kojo, Y.; Kobayashi, K.; Matsunaga, S.; Suzuki, H.; Seto, Y.; Sato, H.; Onoue, S. Avoidance of food effect on oral absorption profile of itraconazole by self-micellizing solid dispersion approach. Drug Metab Pharmacokinet 2017, 32, 273–276. [Google Scholar] [CrossRef]
- Hong, J.Y.; Kim, J.K.; Song, Y.K.; Park, J.S.; Kim, C.K. A new self-emulsifying formulation of itraconazole with improved dissolution and oral absorption. J. Control. Release 2006, 110, 332–338. [Google Scholar] [CrossRef]
- Brouwers, J.; Brewster, M.E.; Augustijns, P. Supersaturating drug delivery systems: The answer to solubility-limited oral bioavailability. J. Pharm. Sci. 2009, 98, 2549–2572. [Google Scholar] [CrossRef]
- Tsinman, O.; Tsinman, K.; Ali, S. Excipient update—Soluplus®: An understanding of supersaturation from amorphous solid dispersions. Drug Dev. Deliv. 2015, 15, 1–11. [Google Scholar]
- Park, J.-B.; Lee, B.-J.; Kang, C.Y.; Repka, M.A. Process analytical quality control of tailored drug release formulation prepared via hot-melt extrusion technology. J. Drug Deliv. Sci. Technol. 2017, 38, 51–58. [Google Scholar] [CrossRef]
- Kim, E.J.; Kim, J.H.; Kim, M.S.; Jeong, S.H.; Choi, D.H. Process Analytical Technology Tools for Monitoring Pharmaceutical Unit Operations: A Control Strategy for Continuous Process Verification. Pharmaceutics 2021, 13, 919. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time and Stability Testing Conditions | SD-1 | SD-2 |
|---|---|---|
| Initial | 99.20 ± 2.51 | 98.43 ± 3.11 |
| Long-term 3 months | 97.30 ± 1.71 | 95.41 ± 3.66 |
| Long-term 6 months | 98.83 ± 0.93 | 96.71 ± 2.21 |
| Accelerated 2 months | 97.15 ± 2.00 | 98.55 ± 1.15 |
| Accelerated 4 months | 97.15 ± 2.71 | 98.31 ± 1.05 |
| Accelerated 6 months | 97.97 ± 1.39 | 97.90 ± 1.91 |
| Parameter | ITZ | Sporanox® | SD-3 |
|---|---|---|---|
| Cmax (ng·mL−1) | None | 167.8 ± 53.4 | 469.8 ± 108.0 |
| Tmax (h) | None | 2.8 ± 1.0 | 2.0 ± 0.0 |
| AUC0–48h (ng·h·mL−1) | None | 1073.9 ± 314.7 | 2969.7 ± 720.6 |
| Parameter | SD-1 | SD-2 | SD-3 |
|---|---|---|---|
| Cmax (ng·mL−1) | 0.26 ± 0.18 | 0.77 ± 0.58 | 0.55 ± 0.18 |
| Tmax (h) | 3.0 ± 2.8 | 3.0 ± 0.0 | 2.5 ± 0.7 |
| AUC0–24h (ng·h·mL−1) | 3.37 ± 3.28 | 7.50 ± 4.50 | 6.05 ± 1.71 |
| Formulation and Condition | SD-1 | SD-2 |
|---|---|---|
| Itraconazole (ITZ) | 20% | 20% |
| Parteck® MXP | 80% | 0% |
| Soluplus® | 0% | 60% |
| HPMCP-HP55 | 0% | 20% |
| Processing parameter | SD-1 | SD-2 |
| Screw speed (RPM) | 50 | 30 |
| Processing temperature (°C) | 200 | 170 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.-H.; Park, C.; Weon, K.-Y.; Kang, C.-Y.; Lee, B.-J.; Park, J.-B. Improved Bioavailability of Poorly Water-Soluble Drug by Targeting Increased Absorption through Solubility Enhancement and Precipitation Inhibition. Pharmaceuticals 2021, 14, 1255. https://doi.org/10.3390/ph14121255
Lee J-H, Park C, Weon K-Y, Kang C-Y, Lee B-J, Park J-B. Improved Bioavailability of Poorly Water-Soluble Drug by Targeting Increased Absorption through Solubility Enhancement and Precipitation Inhibition. Pharmaceuticals. 2021; 14(12):1255. https://doi.org/10.3390/ph14121255
Chicago/Turabian StyleLee, Ju-Hyun, Chulhun Park, Kwon-Yeon Weon, Chin-Yang Kang, Beom-Jin Lee, and Jun-Bom Park. 2021. "Improved Bioavailability of Poorly Water-Soluble Drug by Targeting Increased Absorption through Solubility Enhancement and Precipitation Inhibition" Pharmaceuticals 14, no. 12: 1255. https://doi.org/10.3390/ph14121255
APA StyleLee, J.-H., Park, C., Weon, K.-Y., Kang, C.-Y., Lee, B.-J., & Park, J.-B. (2021). Improved Bioavailability of Poorly Water-Soluble Drug by Targeting Increased Absorption through Solubility Enhancement and Precipitation Inhibition. Pharmaceuticals, 14(12), 1255. https://doi.org/10.3390/ph14121255