
Advances on Greener Asymmetric Synthesis of Antiviral Drugs via Organocatalysis




Abstract
:
1. Introduction
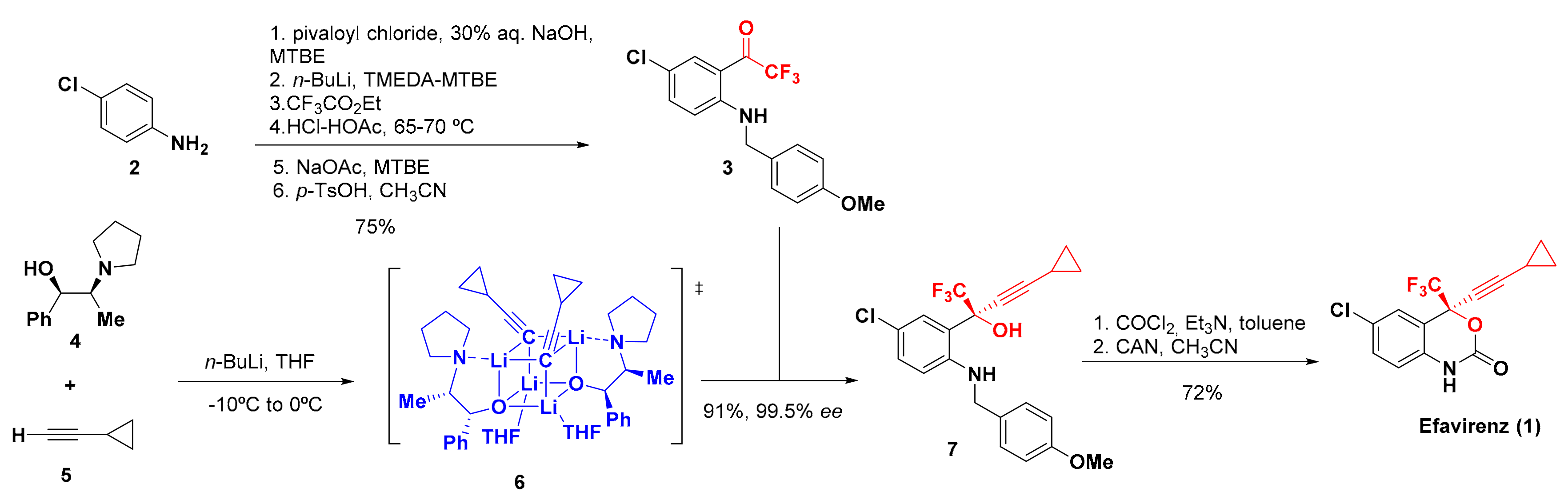
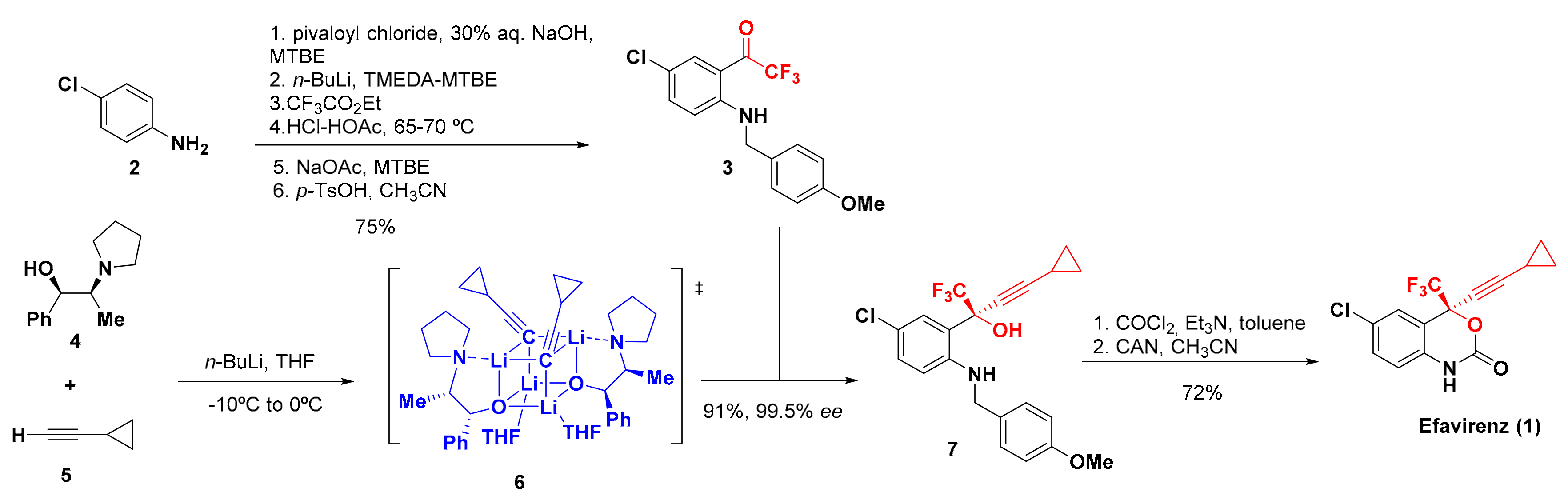
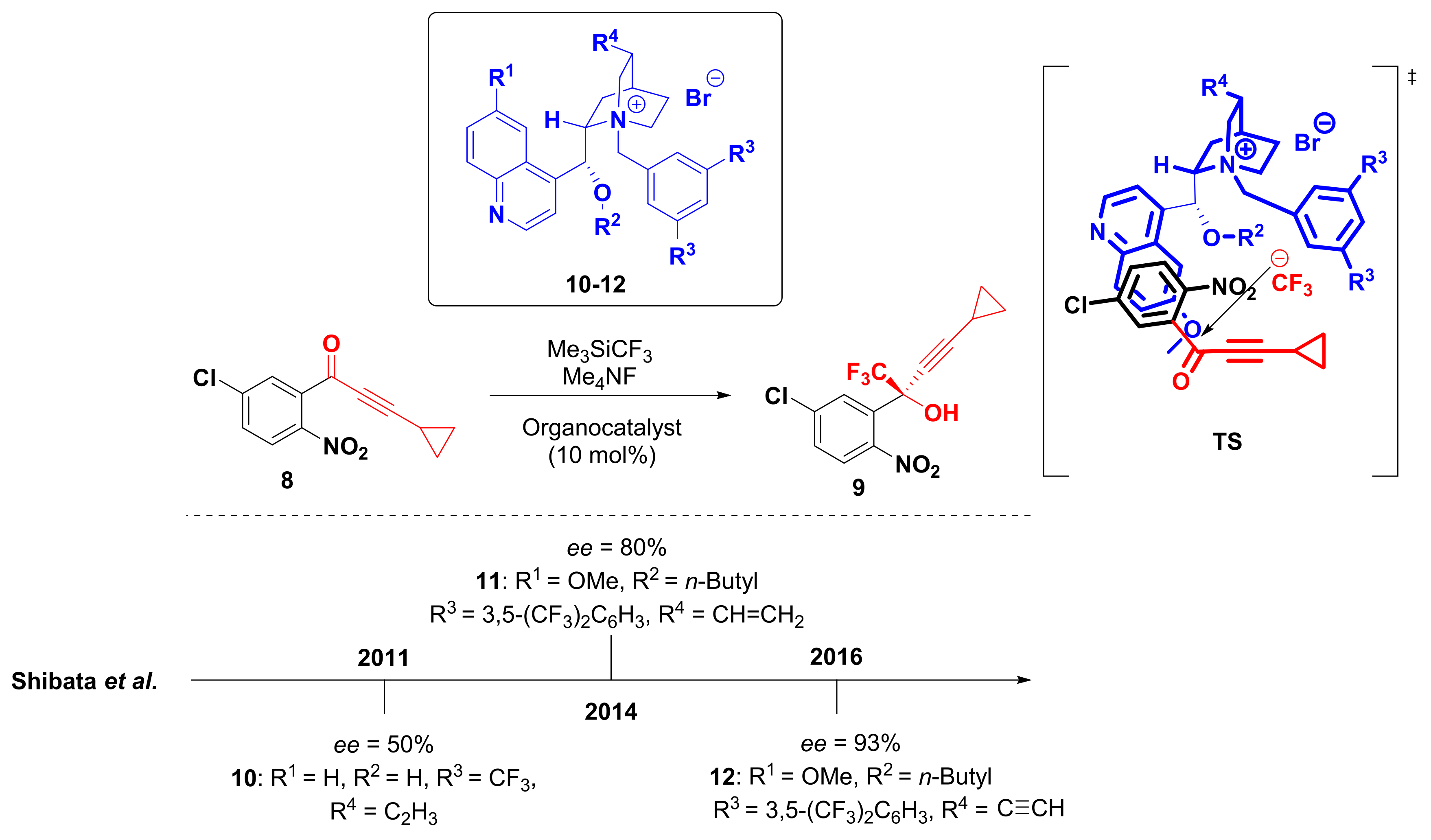
2. Synthesis of Efavirenz
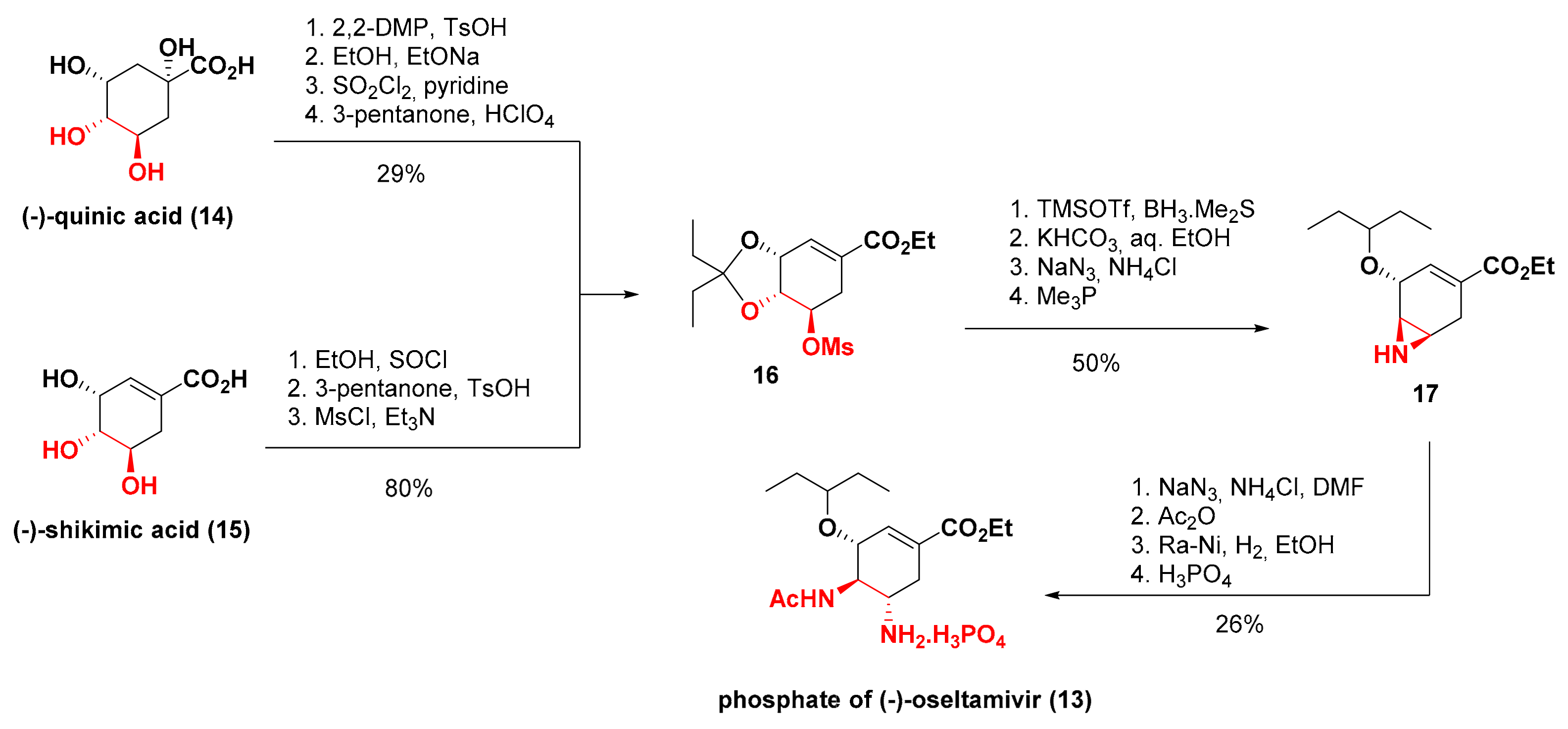
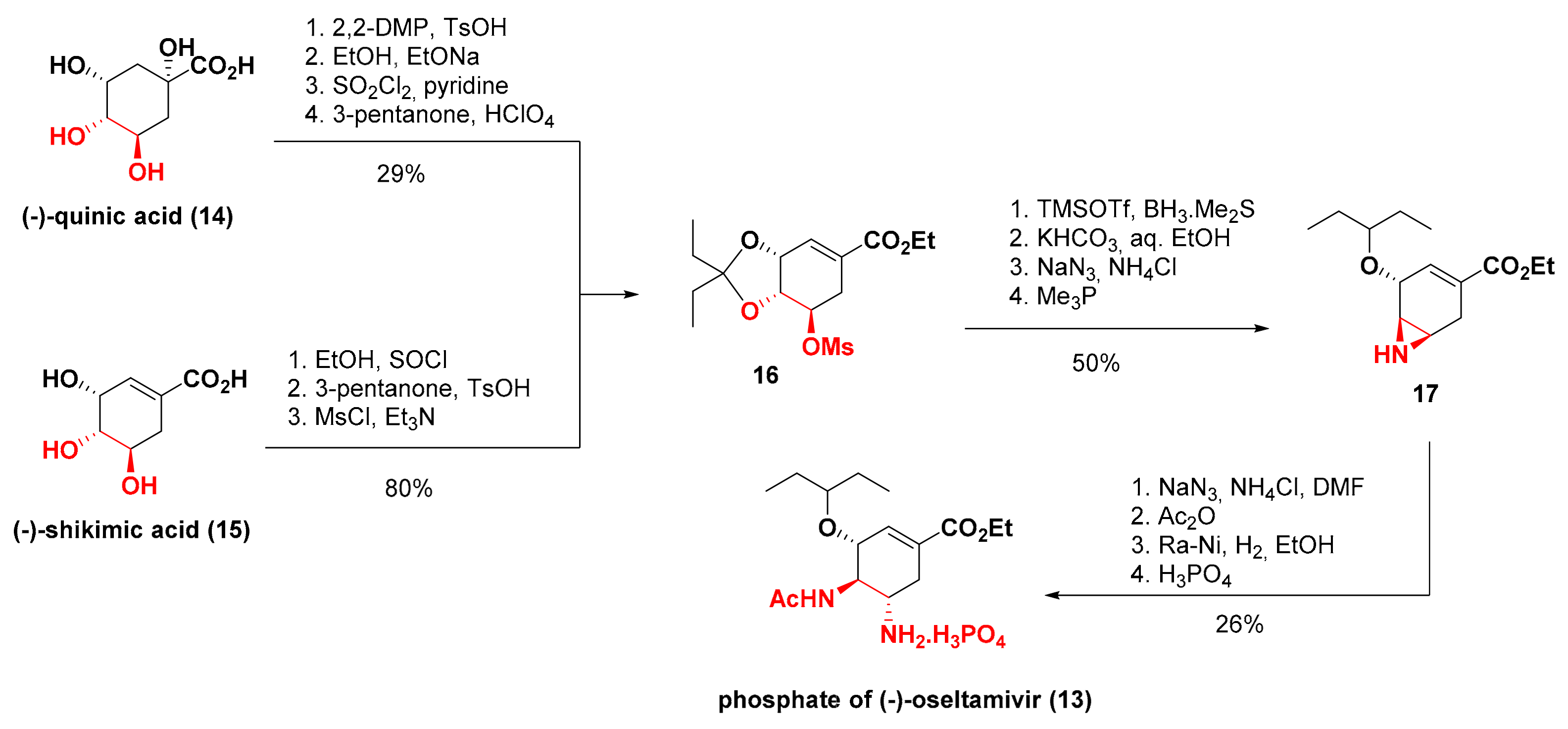
3. Synthesis of Oseltamivir
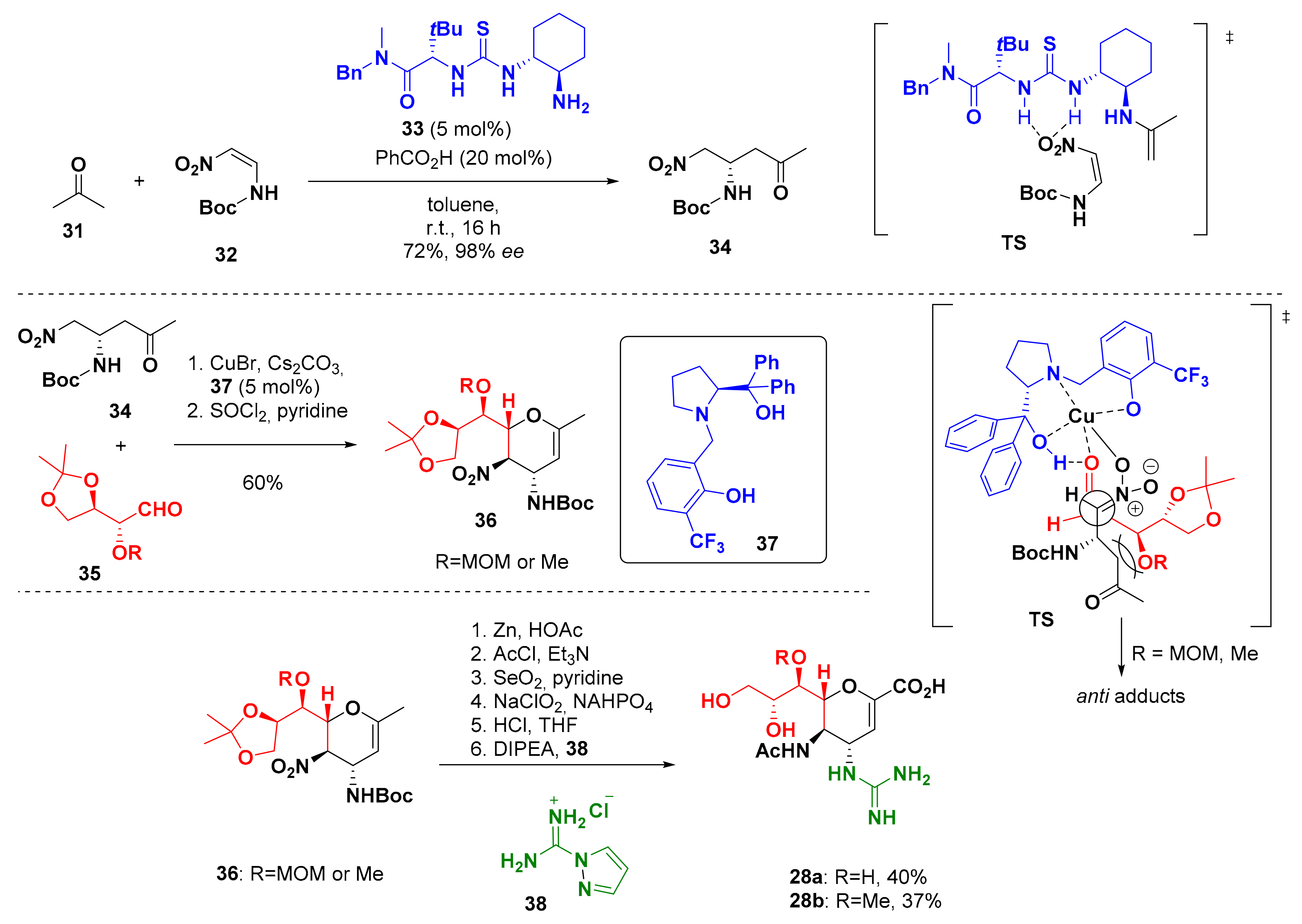
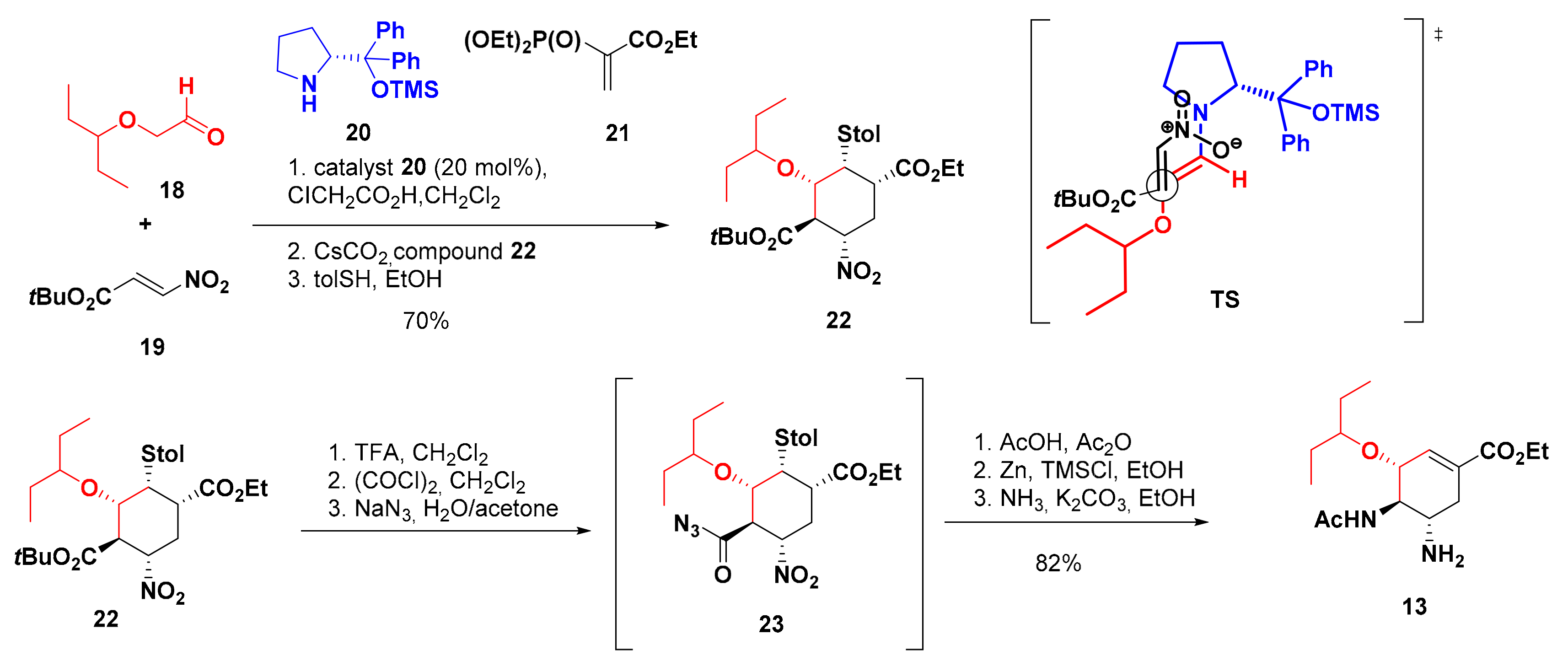
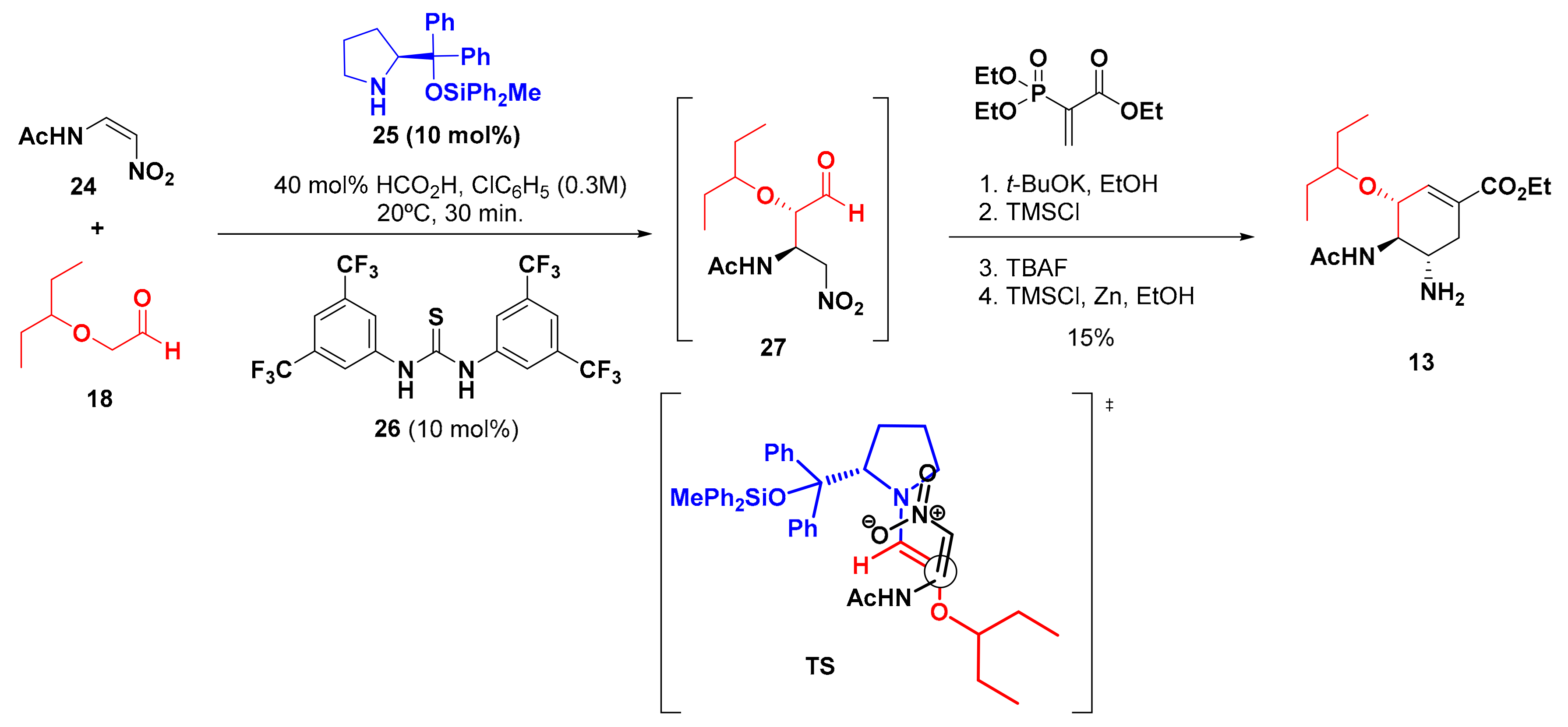
4. Synthesis of Zanamivir
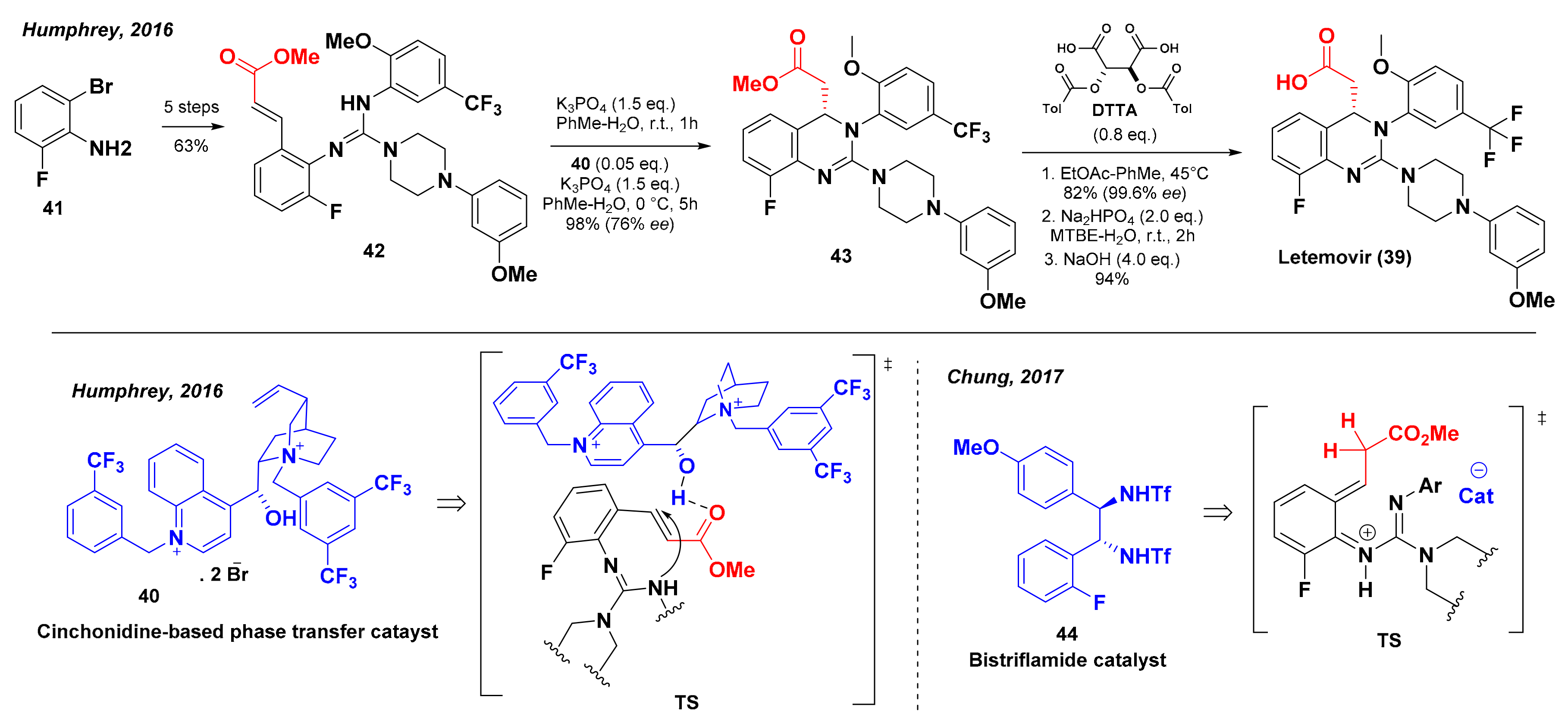
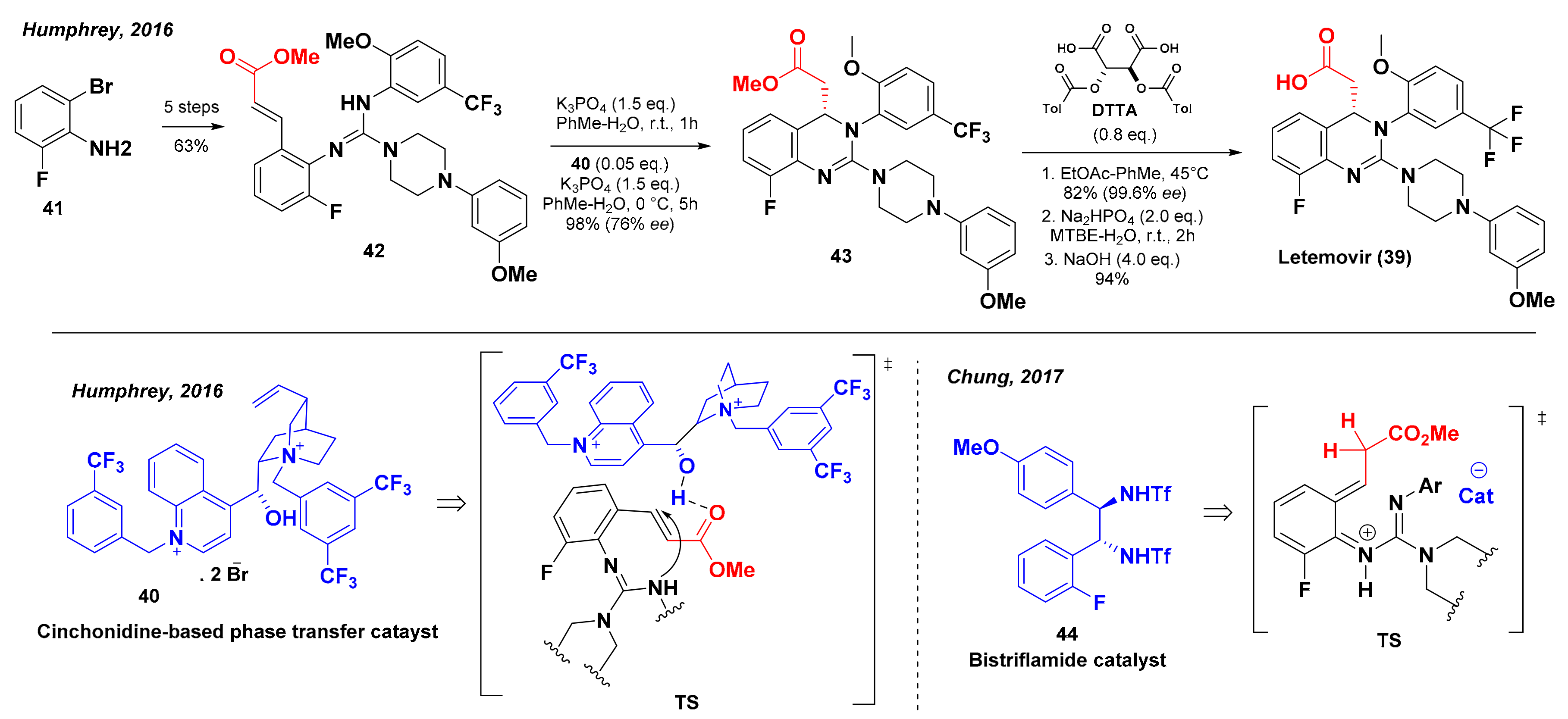
5. Synthesis of Letermovir
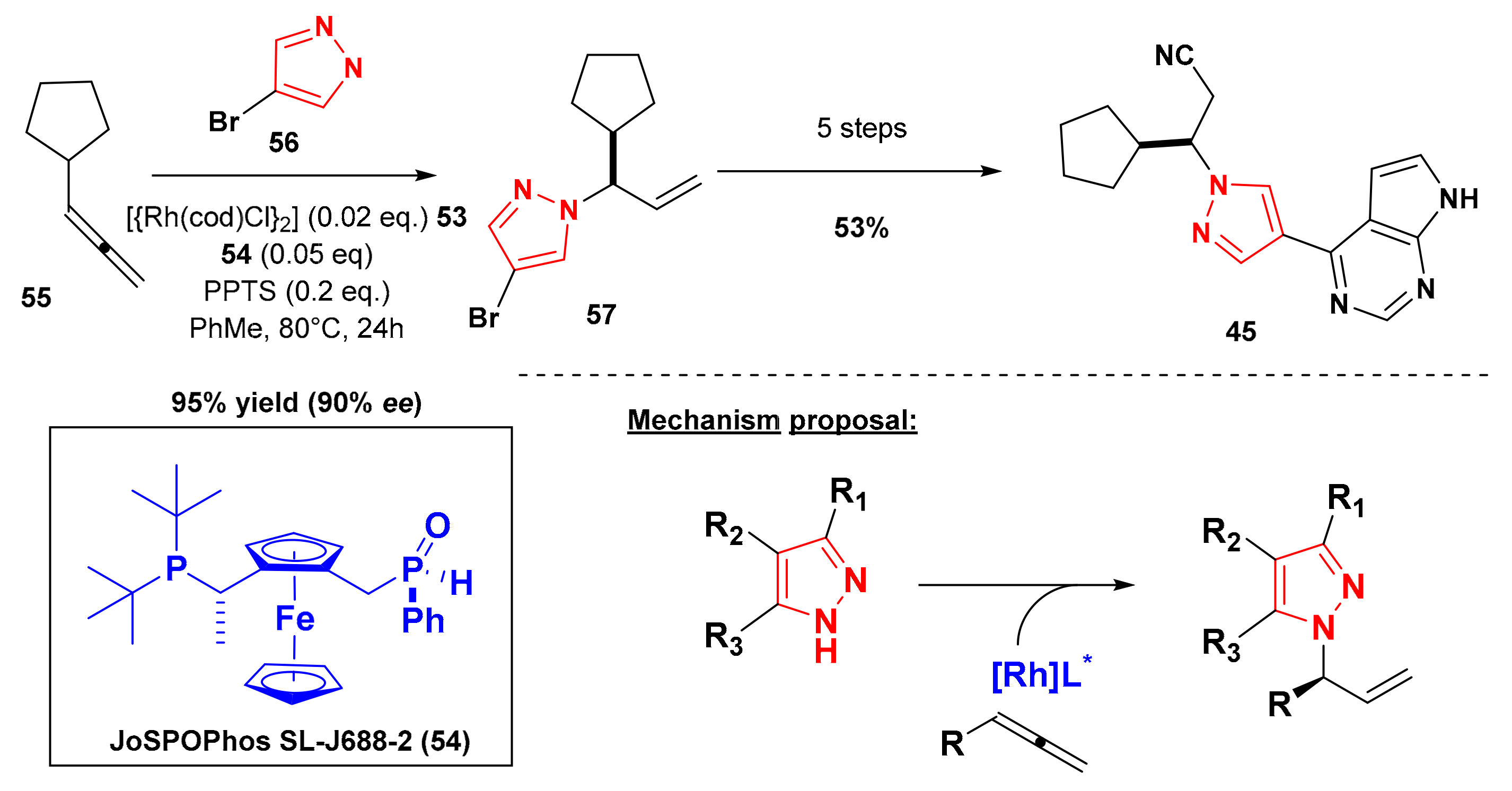
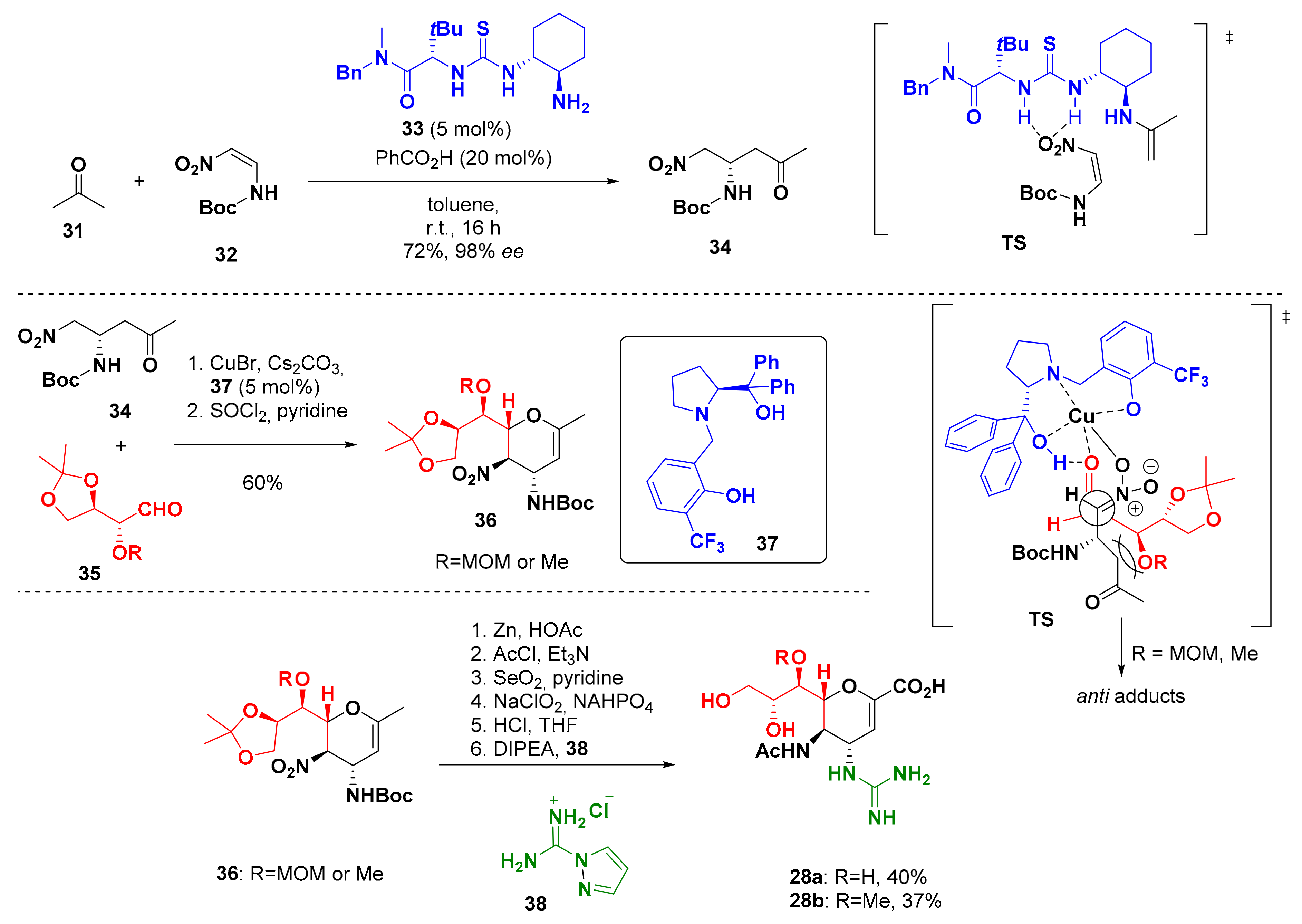
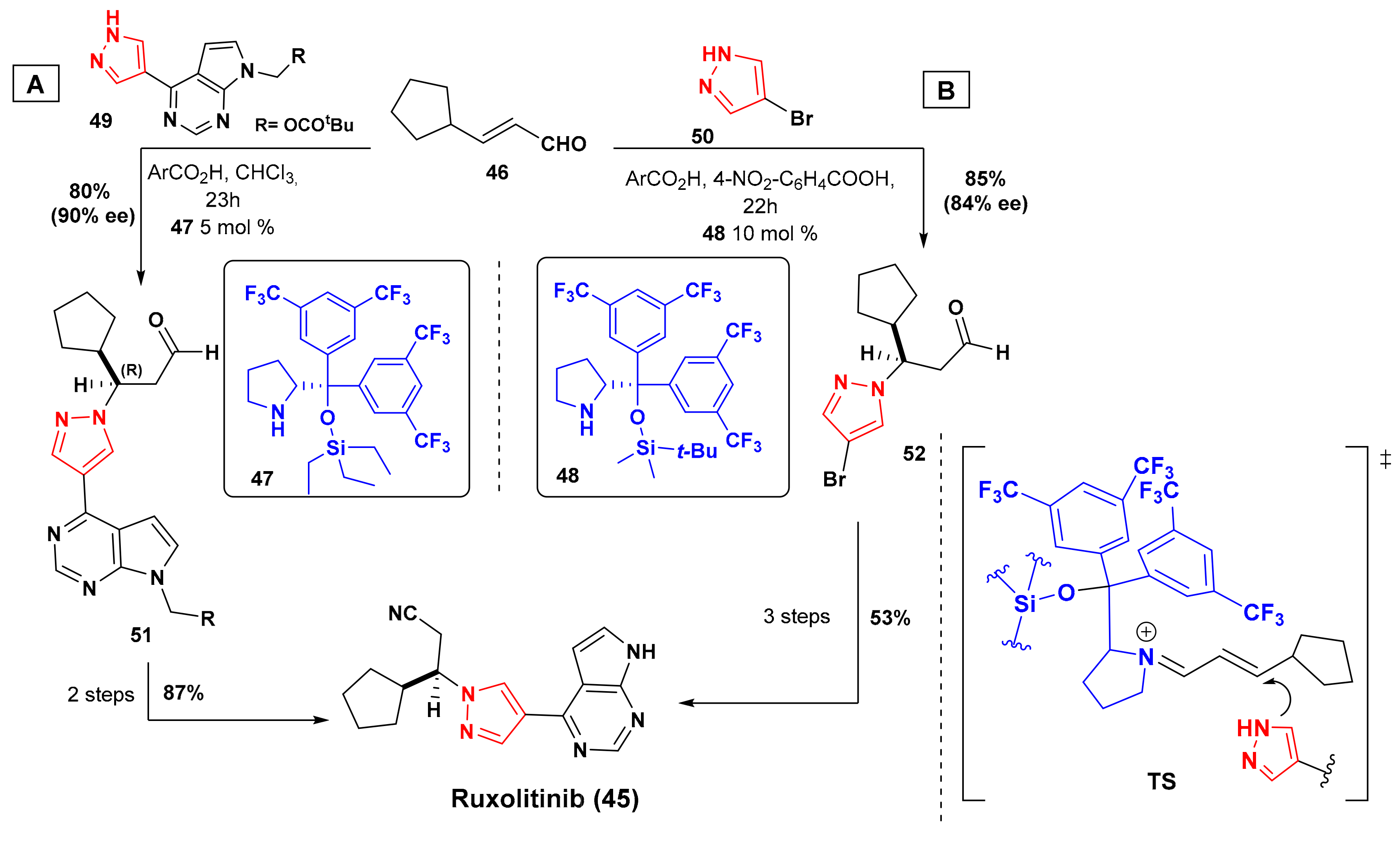
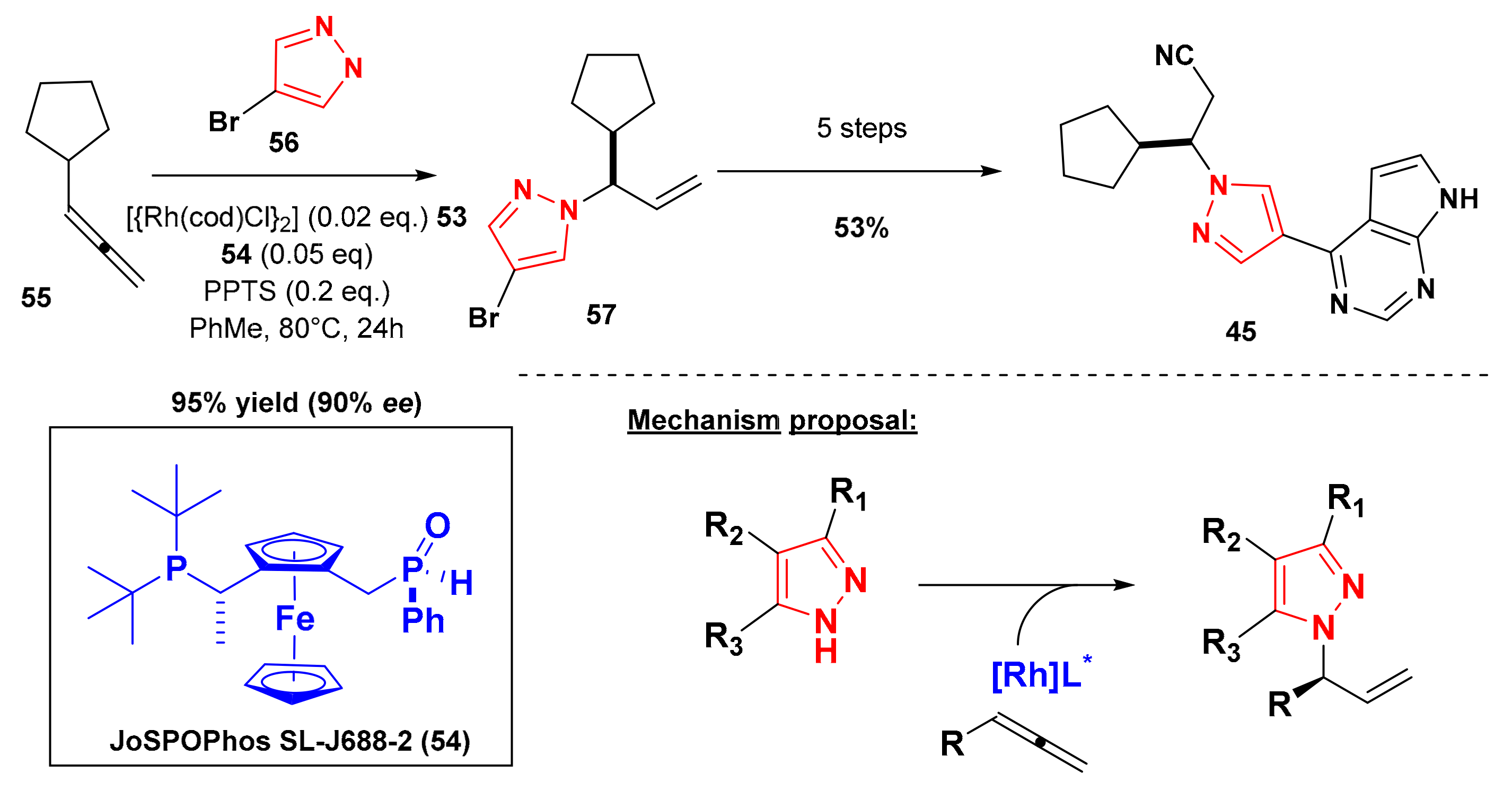
6. Synthesis of Ruxolitinib
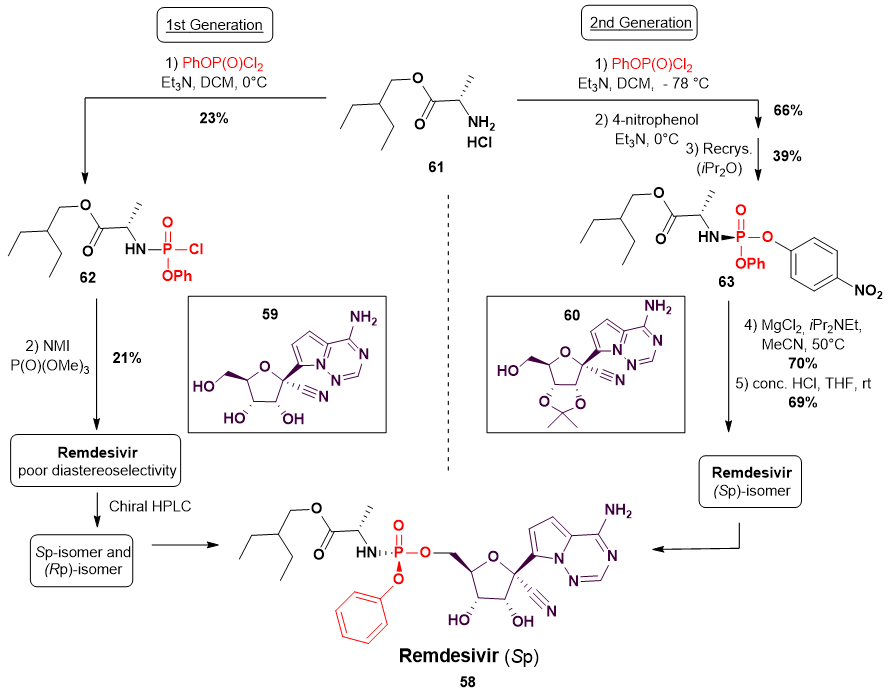
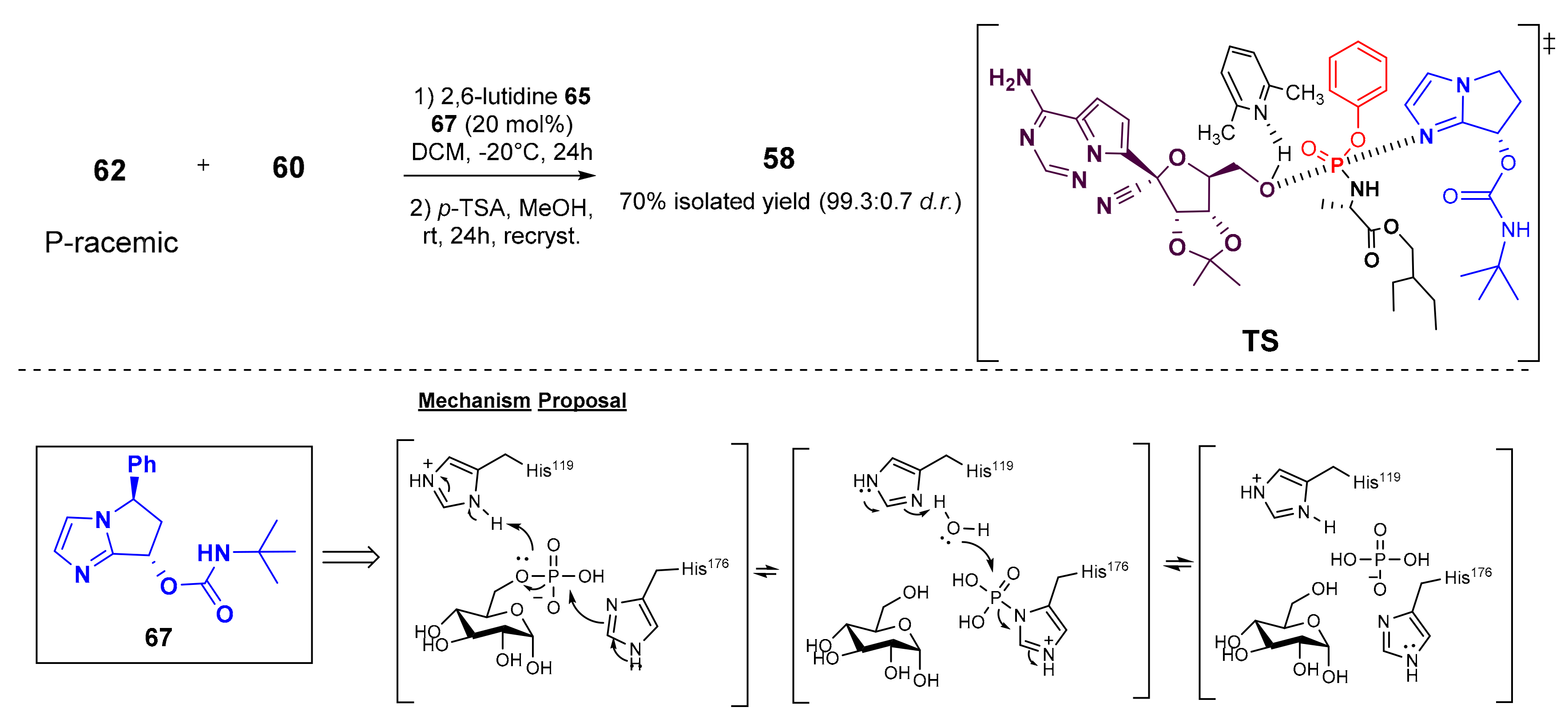
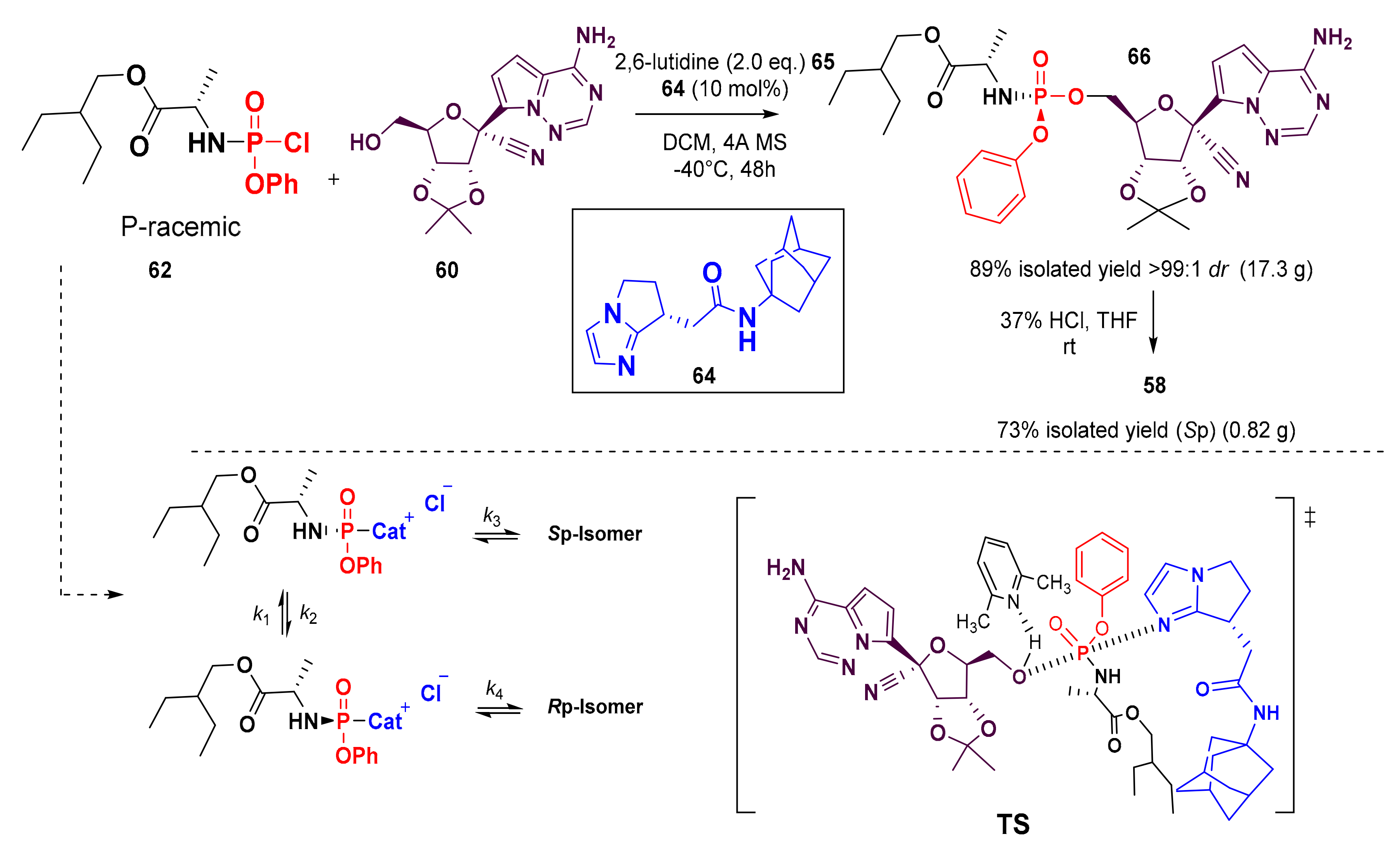
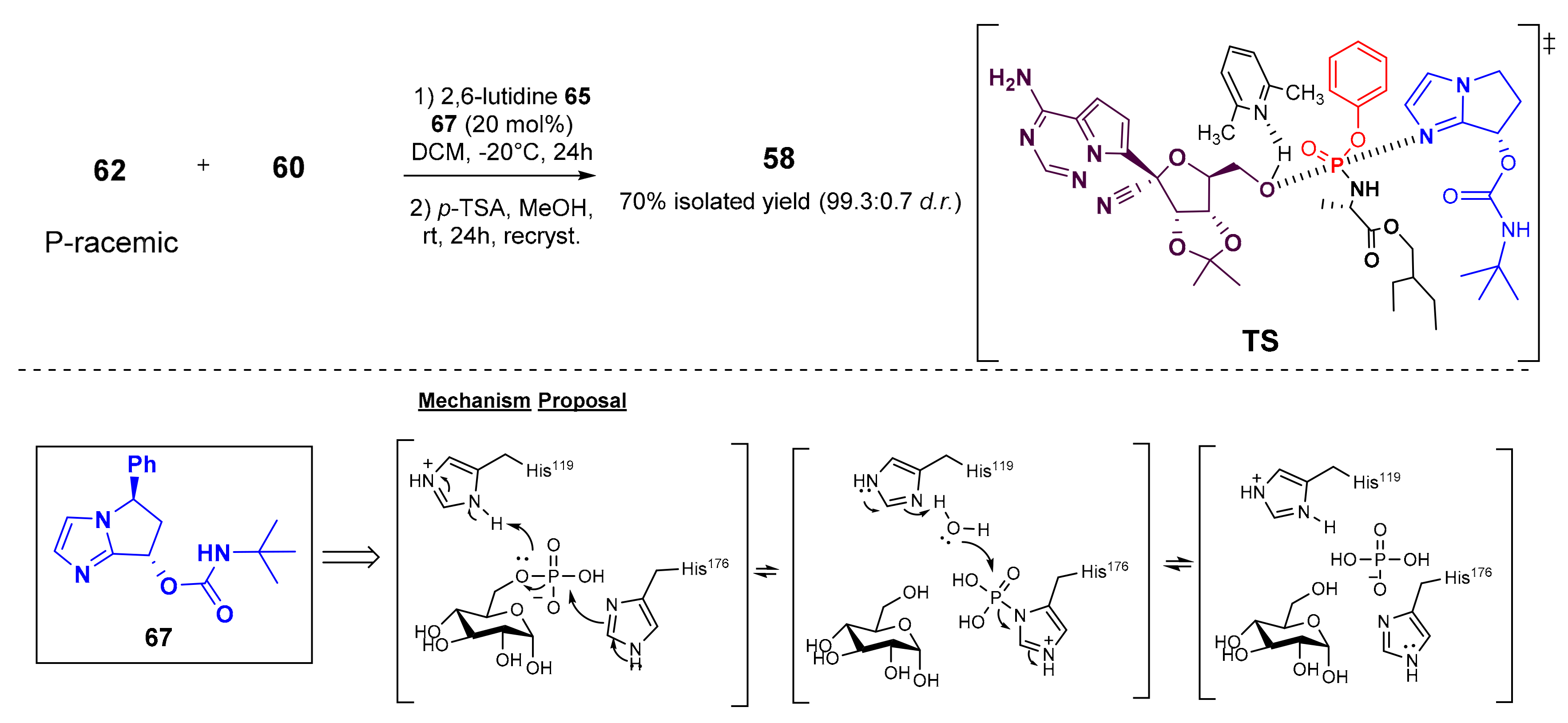
7. Synthesis of Remdesivir
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sagaya, J.R.; Khusro, A.; Agastian, P.; Alfarhan, A.; Al-Dhabi, N.A.; Arasu, M.V.; Rajagopal, R.; Barcelo, D.; Al-Tamini, A. Emerging paradigms of viral diseases and paramount role of natural resources as antiviral agents. Sci. Total Environ. 2021, 759, 143539. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Frutos-Beltran, E.; Kang, D.; Pannecouque, C.; Clercq, E.D.; Menendez-Arias, L.; Liu, X.; Zhan, P. Medicinal chemistry strategies for discovering antiviral effective against drig-resistant viruses. Chem. Soc. Rev. 2021, 50, 4514–4540. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO Coronavirus (COVID-19) Dashboard. 2021. Available online: https://covid19.who.int (accessed on 5 October 2021).
- Clercq, E.D. Antivirals and antiviral strategies. Nat. Rev. Microbiol. 2004, 2, 704–720. [Google Scholar] [CrossRef]
- Megank, R.M.; Baric, R.S. Developing therapeutic approaches for twenty-first-century emerging infectious viral diseases. Nat. Med. 2021, 27, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.L.; Yasuda, N.; Li, H.; McLaughlin, M.; Tschaen, D. Process Chemistry in Antiviral Research. Top. Curr. Chem. 2016, 374, 77. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.A.; He, H.; Pham-Huy, C. Chiral Drugs. An Overview. Int. J. Biomed. Sci. 2006, 2, 85–100. [Google Scholar]
- Abram, M.; Jakubiec, M.; Kaminski, K. Chirality as an Important Factor for the Development of New Antiepileptic Drugs. ChemMedChem 2019, 14, 1744–1761. [Google Scholar] [CrossRef]
- Calcaterra, A.; D’Acquarica, I. The market of chiral drugs: Chiral switches versus de novo enantiomerically pure compounds. J. Pharm. Biomed. Anal. 2018, 147, 323–340. [Google Scholar] [CrossRef]
- Eastgate, M.D.; Schmidt, M.A.; Fandrick, K.R. On the design of complex drug candidate syntheses in the pharmaceutical industry. Nat. Rev. Chem. 2017, 1, 0016. [Google Scholar] [CrossRef]
- Rogers, L.; Jensen, K.F. Continuous manufacturing—The Green Chemistry promise? Green Chem. 2019, 21, 3481–3498. [Google Scholar] [CrossRef] [Green Version]
- Reisinger, C.M.; Pan, S.C.; List, B. New Concepts for Catalysis. Organocatalysis 2008, 2, 35–64. [Google Scholar]
- Patel, R.N. Biocatalysis for synthesis of pharmaceuticals. Bioorg. Med. Chem. 2018, 26, 1252–1274. [Google Scholar] [CrossRef] [PubMed]
- Egorova, K.S.; Ananikov, V.P. Toxicity of Metal Compounds: Knowledge and Myths. Organometallics 2017, 36, 4071–4090. [Google Scholar] [CrossRef] [Green Version]
- Han, B.; He, X.H.; Liu, Y.Q.; He, G.; Peng, C.; Li, J.L. Asymmetric organocatalysis: An enabling technology for medicinal chemistry. Chem. Soc. Rev. 2021, 50, 1522–1586. [Google Scholar] [CrossRef] [PubMed]
- Moreira, N.M.; Martelli, L.S.R.; Corrêa, A.G. Asymmetric organocatalyzed synthesis of coumarin derivatives. Beilstein J. Org. Chem. 2021, 17, 1952–1980. [Google Scholar] [CrossRef] [PubMed]
- Holland, M.C.; Gilmour, R. Deconstructing covalent organocatalysis. Angew. Chem. Int. Ed. 2015, 54, 3862–3871. [Google Scholar] [CrossRef] [PubMed]
- Otvos, S.B.; Kappe, C.O. Continuous flow asymmetric synthesis of chiral active pharmaceutical ingredients and their advanced intermediates. Green Chem. 2021, 23, 6117–6138. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Bilcer, G.; Schiltz, G. Syntheses of FDA Approved HIV Protease Inhibitors. Synthesis 2001, 2001, 2203–2229. [Google Scholar] [CrossRef]
- Ganta, N.M.; Gedda, G.; Rathnakar, B.; Satyanarayana, M.; Yamajala, B.; Ahsan, M.J.; Jadav, S.S.; Balaraju, T. A review on HCV inhibitors: Significance of non-structural polyproteins. Eur. J. Med. Chem. 2019, 164, 576–601. [Google Scholar] [CrossRef] [PubMed]
- Voight, E.A.; Greszler, S.N.; Hartung, J.; Ji, J.G.; Klix, R.C.; Randolph, J.T.; Shelat, B.H.; Waters, J.E.; DeGoey, D.A. Desymmetrization of pibrentasvir for efficient prodrug synthesis. Chem. Sci. 2021, 12, 10076–10082. [Google Scholar] [CrossRef] [PubMed]
- Skwarecki, A.S.; Nowak, M.G.; Milewska, M.J. Amino Acid and Peptide-Based Antiviral Agents. ChemMedChem 2021, 16, 3106–3135. [Google Scholar] [CrossRef]
- Mandala, D.; Thompson, W.A.; Watts, P. Synthesis routes to anti-HIV drugs. Tetrahedron 2016, 72, 3389–3420. [Google Scholar] [CrossRef]
- Gadakh, S.K.; Reddy, R.S.; Sudalai, A. Enantioselective synthesis of HIV protease inhibitor amprenavir via Co-catalyzed HKR of 2-(1-azido-2-phenylethyl)oxirane. Tetrahedron Asymmetry 2012, 23, 898–903. [Google Scholar] [CrossRef]
- Honda, Y.; Katayama, S.; Kojima, M.; Suzuki, T.; Kishibata, N.; Izawa, K. New approaches to the industrial synthesis of HIV protease inhibitors. Org. Biomol. Chem. 2004, 2, 2061–2070. [Google Scholar] [CrossRef]
- Evans, C.T.; Roberts, S.M.; Shoberu, K.A.; Sutherland, A.G. Potential use of carbocyclic nucleosides for the treatment of AIDS, chemoenzymatic synthesis of the enantiomers of carbovir. J. Chem. Soc. Perkin Trans. 1992, 1, 589–592. [Google Scholar] [CrossRef]
- Thieme, N.; Breit, B. Enantioselective and Regiodivergent Addition of Purines to Terminal Allenes: Synthesis of Abacavir. Angew. Chem. Int. Ed. 2017, 56, 1520–1524. [Google Scholar] [CrossRef]
- Xu, L.; Liu, H.; Murray, B.P.; Callebaut, C.; Lee, M.S.; Hong, A.; Strickley, R.G.; Tsai, L.K.; Stray, K.M.; Wang, Y.; et al. Cobicistat (GS-9350): A Potent and Selective Inhibitor of Human CYP3A as a Novel Pharmacoenhancer. ACS Med. Chem. Lett. 2010, 1, 209–213. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.H.; Liu, H.T.; Hong, A.; Vivian, R.; Murray, B.P.; Callebaut, C.; Choi, Y.C.; Lee, M.S.; Chau, J.; Tsai, L.K.; et al. Structure-activity relationships of diamine inhibitors of cytochrome P450 (CYP) 3A as novel pharmacoenhancers. Part II: P2/P3 region and discovery of cobicistat (GS-9350). Bioorg. Med. Chem. Lett. 2014, 24, 995–999. [Google Scholar] [CrossRef] [PubMed]
- Rana, A.; Mahajan, B.; Ghosh, S.; Srihari, P.; Singh, A.K. Integrated multi-step continuous flow synthesis of daclatasvir without intermediate purification and solvent exchange. React. Chem. Eng. 2020, 5, 2109–2114. [Google Scholar] [CrossRef]
- Sankareswaran, S.; Mannam, M.; Chakka, V.; Mandapati, S.R.; Kumar, P. Identification and Control of Critical Process Impurities: An Improved Process for the Preparation of Dolutegravir Sodium. Org. Process Res. Dev. 2016, 20, 1461–1468. [Google Scholar] [CrossRef]
- Radl, S.; Stach, J.; Pisa, O.; Cinibulk, J.; Havlicek, J.; Zajicova, M.; Pekareka, T. An Improved Synthesis of Elvitegravir. J. Heterocycl. Chem. 2016, 53, 1738–1749. [Google Scholar] [CrossRef]
- Mandala, D.; Watts, P. An Improved Synthesis of Lamivudine and Emtricitabine. ChemistrySelect 2017, 2, 1102–1105. [Google Scholar] [CrossRef]
- Askin, D.; Eng, K.K.; Rossen, K.; Purick, R.M.; Wells, K.M.; Volante, R.P.; Reider, P.J. Highly diastereoselective reaction of a chiral, non-racemic amide enolate with (S)-glycidyl tosylate. Synthesis of the orally active HIV-1 protease inhibitor L-735,524. Tetrahedron Lett. 1994, 35, 673–676. [Google Scholar] [CrossRef]
- Demir, A.S.; Hamamci, H.; Doganel, F.; Ozgul, E. Chemoenzymatic synthesis of (1S,2R)-1-amino-2-indanol, a key intermediate of HIV protease inhibitor, indinavir. J. Mol. Catal. B Enzym. 2000, 9, 157–161. [Google Scholar] [CrossRef]
- Cheng, Y.; Lu, Z.; Chapman, K.T.; Tata, J.R. Solid Phase Synthesis of Indinavir and Its Analogues. J. Comb. Chem. 2000, 2, 445–446. [Google Scholar] [CrossRef]
- Hu, L.; Schaufelberger, F.; Zhang, Y.; Ramstrom, O. Efficient asymmetric synthesis of lamivudine via enzymatic dynamic kinetic resolution. Chem. Commun. 2013, 49, 10376–10378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snead, D.R.; Mcquade, D.T.; Ahmad, S.; Krack, R.; Stringham, R.W.; Burns, J.M.; Abdiaj, I.; Gopalsamuthiram, V.; Nelson, R.C.; Gupton, B.F. An Economical Route to Lamivudine Featuring a Novel Strategy for Stereospecific Assembly. Org. Process Res. Dev. 2020, 24, 1194–1198. [Google Scholar] [CrossRef] [Green Version]
- Aher, U.P.; Srivastava, D.; Jadhav, H.S.; Singh, G.P.; Jayashree, B.S.; Shenoy, G.G. Large-Scale Stereoselective Synthesis of 1,3-Oxathiolane Nucleoside, Lamivudine, via ZrCl4-Mediated N-Glycosylation. Org. Process Res. Dev. 2020, 24, 387–397. [Google Scholar] [CrossRef]
- Zhu, Y.J.; Li, H.Y.; Lin, K.L.; Wang, B.; Zhou, W.C. A novel and efficient asymmetric synthesis of anti-HIV drug maraviroc. Synth. Commun. 2019, 49, 1721–1728. [Google Scholar]
- Zhao, G.L.; Lin, S.; Korotvicka, A.; Deiana, L.; Kullberg, M.; Cordova, A. Asymmetric Synthesis of Maraviroc (UK-427,857). Adv. Synth. Catal. 2010, 352, 2291–2298. [Google Scholar] [CrossRef]
- Inaba, T.; Yamada, Y.; Abe, H.; Sagawa, S.; Cho, H. (1 S)-1-[(4 R)-2, 2-Dimethyl-1, 3-dioxolan-4-yl]-2-hydroxyethylammonium Benzoate, A Versatile Building Block for Chiral 2-Aminoalkanols: Concise Synthesis and Application to Nelfinavir, a Potent HIV-Protease Inhibitor. J. Org. Chem. 2000, 65, 1623–1628. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, S.; Krishnaiah, V.; Sridhar, B. Asymmetric Synthesis of the Potent HIV-Protease Inhibitor, Nelfinavir. J. Org. Chem. 2010, 75, 498–501. [Google Scholar] [CrossRef]
- Nagao, Y.; Hisanaga, T.; Utsumi, T.; Egami, H.; Kawato, Y.; Hamashima, Y. Enantioselective Synthesis of Nelfinavir via Asymmetric Bromocyclization of Bisallylic Amide. J. Org. Chem. 2018, 83, 7290–7295. [Google Scholar] [CrossRef]
- Kempf, D.J.; Sham, H.L.; Marsh, K.C.; Flentge, C.A.; Betebenner, D.; Green, B.E.; McDonald, E.; Vasavanonda, S.; Saldivar, A.; Wideburg, N.E.; et al. Discovery of Ritonavir, a Potent Inhibitor of HIV Protease with High Oral Bioavailability and Clinical Efficacy. J. Med. Chem. 1998, 41, 602–617. [Google Scholar] [CrossRef]
- Ramu, E.; Rao, B.V. A short approach to the synthesis of the ritonavir and lopinavir core and its C-3 epimer via cross metathesis. Tetrahedron Asymmetry 2009, 20, 2201–2204. [Google Scholar] [CrossRef]
- Göbring, W.; Gokbale, S.; Hilpert, H.; Roessler, F.; Schlageter, M.; Vogt, P. Synthesis of the HIV-Proteinase Inhibitor Saquinavir: A Challenge for Process Research. CHIMIA 1996, 50, 532–537. [Google Scholar]
- Rosenquist, A.; Samuelsson, B.; Johansson, P.O.; Cummings, M.D.; Lenz, O.; Raboisson, P.; Simmen, K.; Vendeville, S.; de Kock, H.; Nilsson, M.; et al. Discovery and Development of Simeprevir (TMC435), a HCV NS3/4A Protease Inhibitor. J. Med. Chem. 2014, 57, 1673–1693. [Google Scholar] [CrossRef]
- Horváth, A.; Depré, D.; Vermeulen, W.A.A.; Wuyts, S.L.; Harutyunyan, S.R.; Binot, G.; Cuypers, J.; Couck, W.; Van Den Heuvel, D. Ring-Closing Metathesis on Commercial Scale: Synthesis of HCV Protease Inhibitor Simeprevir. J. Org. Chem. 2019, 84, 4932–4939. [Google Scholar] [CrossRef]
- Moni, L.; Banfi, L.; Basso, A.; Carcone, L.; Rasparini, M.; Riva, R. Ugi and Passerini Reactions of Biocatalytically Derived Chiral Aldehydes: Application to the Synthesis of Bicyclic Pyrrolidines and of Antiviral Agent Telaprevir. J. Org. Chem. 2015, 80, 3411–3428. [Google Scholar] [CrossRef] [PubMed]
- Derstine, B.P.; Tomlin, J.W.; Peck, C.L.; Dietz, J.P.; Herrera, B.T.; Cardoso, F.S.P.; Paymode, D.J.; Yue, A.C.; Arduengo, A.J.; Opatz, T. An Efficient Synthesis of Tenofovir (PMPA): A Key Intermediate Leading to Tenofovir-Based HIV Medicines. Org. Process Res. Dev. 2020, 24, 1420–1427. [Google Scholar] [CrossRef]
- Yang, B.; Xie, H.M.; Rani, K.R.; Gant, Y.J. Efficient Synthesis and Resolution of Tenofovir Alafenamide. Lett. Org. Chem. 2018, 15, 10–14. [Google Scholar] [CrossRef]
- Vetukuri, P.R.V.N.K.V.; Vedantham, R.; Mathad, V.T.; Padi, P.R.; Ramasamy, V.A. A Concise Route to Valacyclovir Hydrochloride. Helv. Chim. Acta 2011, 94, 592–596. [Google Scholar] [CrossRef]
- Adkins, J.C.; Noble, S. Efavirenz. Drugs 1998, 56, 1055–1064. [Google Scholar] [CrossRef] [PubMed]
- Pierce, M.E.; Parsons, R.L.; Radesca, L.; Lo, Y.S.; Silverman, S.; Moore, J.R.; Islam, O.; Choudhury, A.; Fortunak, J.M.D.; Nguyen, D.; et al. Synthesis of Efavirenz via Asymmetric Alkynylation. J. Org. Chem. 1998, 63, 8536–8543. [Google Scholar] [CrossRef]
- Kawai, H.; Kitayama, T.; Tokunaga, E.; Shibata, N. A New Synthetic Approach to Efavirenz through Enantioselective Trifluoromethylation by Using the Ruppert–Prakash Reagent. Eur. J. Org. Chem. 2011, 2011, 5959–5961. [Google Scholar] [CrossRef]
- Okuso, S.; Kawai, H.; Yasuda, Y.; Sugita, Y.; Kitayama, T.; Tokunaga, E.; Shibata, N. Asymmetric Synthesis of Efavirenz via Organocatalyzed Enantioselective Trifluoromethylation. Asian J. Org. Chem. 2014, 3, 449–452. [Google Scholar] [CrossRef]
- Okuso, S.; Hirano, K.; Yasuda, Y.; Tanaka, J.; Tokunaga, E.; Fukaya, H.; Shibata, N. Alkynyl Cinchona Catalysts affect Enantioselective Trifluoromethylation for Efavirenz under Metal-Free Conditions. Org. Lett. 2016, 18, 5568–5571. [Google Scholar] [CrossRef]
- Silva, F.C. Síntese Total do (−)-Oseltamivir (Tamiflu®) por Reaçoes do Tipo Dominó. Rev. Virt. Quim. 2009, 1, 87–90. [Google Scholar]
- Rohloff, J.C.; Kent, K.M.; Postich, M.J.; Becker, M.W.; Chapman, H.H.; Kelly, D.E.; Lew, W.; Louie, M.S.; McGee, L.R.; Prisbe, E.J.; et al. Practical Total Synthesis of the Anti-Influenza Drug GS-4104. J. Org. Chem. 1998, 63, 4545–4550. [Google Scholar] [CrossRef]
- Abrecht, S.; Harrington, P.; Iding, H.; Karpf, M.; Trussardi, R.; Wirz, B.; Zutter, U. The Synthetic Development of the Anti-Influenza Neuraminidase Inhibitor Oseltamivir Phosphate (Tamiflu®): A Challenge for Synthesis & Process Research. CHIMIA 2004, 58, 621–629. [Google Scholar]
- Ishikawa, H.; Suzuki, T.; Hayashi, Y. High-Yielding Synthesis of the Anti-Influenza Neuramidase Inhibitor (−)-Oseltamivir by Three “One-Pot” Operations. Angew. Chem. Int. Ed. 2009, 48, 1304–1307. [Google Scholar] [CrossRef]
- Ishikawa, H.; Suzuki, T.; Orita, H.; Uchimaru, T.; Hayashi, Y. High-Yielding Synthesis of the Anti-Influenza Neuraminidase Inhibitor (−)-Oseltamivir by Two “One-Pot” Sequences. Chem. Eur. J. 2010, 16, 12616–12626. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Ogasawara, S. Time Economical Total Synthesis of (−)-Oseltamivir. Org. Lett. 2016, 18, 3426–3429. [Google Scholar] [CrossRef] [PubMed]
- Chapple, K.J.; Hendrick, A.E.; McCarthy, M.W. Zanamivir in the treatment and prevention of influenza. Ann. Pharmacother. 2000, 34, 798–801. [Google Scholar] [CrossRef]
- Ryan, D.M.; Ticehurst, J.; Dempsey, M.H.; Penn, C.R. Inhibition of influenza virus replication in mice by GG167 (4-guanidino-2,4-dideoxy-2,3-dehydro-N-acetylneuraminic acid) is consistent with extracellular activity of viral neuraminidase (sialidase). Antimicrob. Agents Chemother. 1994, 38, 2270–2275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandler, M.; Bamford, M.J.; Conroy, R.; Lamont, B.; Patel, B.; Patel, V.K.; Steeples, I.P.; Storer, R.; Weir, N.G.; Wright, M.; et al. Synthesis of the potent influenza neuraminidase inhibitor 4-guanidino Neu5Ac2en. X-ray molecular structure of 5-acetamido-4-amino-2,6-anhydro-3,4,5-trideoxy-d-erythro-l-gluco-nononic acid. J. Chem. Soc. 1995, 1173–1180. [Google Scholar] [CrossRef]
- Tian, J.; Zhong, J.; Li, Y.; Ma, D. Organocatalytic and Scalable Synthesis of the Anti-Influenza Drugs Zanamivir, Laninamivir, and CS-8958. Angew. Chem. Int. Ed. 2014, 126, 14105–14108. [Google Scholar] [CrossRef]
- André, C.; Bolte, J.; Demuynck, C. Syntheses of l-threose and d-erythrose analogues modified at position 2. Tetrahedron Asymmetry 1998, 9, 1359–1367. [Google Scholar] [CrossRef]
- Humprey, G.R.; Dalby, S.M.; Andreani, T.; Xiang, B.; Luzung, M.R.; Song, Z.J.; Shevlin, M.; Christensen, M.; Belyk, K.M.; Tschaen, D.M. Synthesis of Letermovir by an Asymmetric Aza-Michael Reaction. Org. Process Res. Dev. 2016, 20, 1097–1103. [Google Scholar] [CrossRef] [Green Version]
- Krishna, B.A.; Wills, M.R.; Sinclair, J.H. Advances in the treatment of cytomegalovirus. Br. Med. Bull. 2019, 131, 5–7. [Google Scholar] [CrossRef]
- Chung, C.K.; Liu, Z.; Lexa, K.; Andreani, T.; Xu, Y.; Ji, Y.; DiRocco, D.A.; Humphrey, G.R.; Ruck, R.T. Asymmetric Hydrogen Bonding Catalysis for the Synthesis of Dihydroquinazoline-containing Antiviral, Letermovir. J. Am. Chem. Soc. 2017, 31, 10637–10640. [Google Scholar] [CrossRef]
- Bandini, M.; Bottoni, A.; Eichholzer, A.; Miscione, G.P.; Stenta, M. Asymmetric Phase-Transfer-Catalyzed Intramolecular N-Alkylation of Indoles and Pyrroles: A Combined Experimental and Theoretical Investigation. Chem. Eur. J. 2010, 16, 12462–12473. [Google Scholar] [CrossRef]
- Coricello, A.; Mesiti, F.; Lupia, A.; Maruca, A.; Alcaro, S. Inside Perspective of the Synthetic and Computational Toolbox of JAK Inhibitors: Recent Updates. Molecules 2020, 25, 3321. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Meloni, D.; Pan, Y.; Xia, M.; Rodgers, J.; Shepard, S.; Li, M.; Galya, L.; Metcalf, B.; Yue, T.Y.; et al. Enantioselective Synthesis of Janus Kinase Inhibitor INCB018424 via an Organocatalytic Aza-Michael Reaction. Org. Lett. 2009, 11, 1999–2002. [Google Scholar] [CrossRef] [PubMed]
- Diner, P.; Nielsen, M.; Marigo, M.; Jorgensen, K.A. Enantioselective Organocatalytic Conjugate Addition of N Heterocycles to a,b-Unsaturated Aldehydes. Angew. Chem. Int. Ed. 2007, 46, 1983–1987. [Google Scholar] [CrossRef] [PubMed]
- Haydl, A.M.; Xu, K.; Breit, B. Regio- and Enantioselective Synthesis of N-Substituted Pyrazoles by Rhodium-Catalyzed Asymmetric Addition to Allenes. Angew. Chem. Int. Ed. 2015, 127, 7255–7259. [Google Scholar] [CrossRef]
- Warren, T.K.; Jordan, R.; Bavari, S. Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature 2013, 531, 381–385. [Google Scholar] [CrossRef] [PubMed]
- US Food and Drug Administration. Remdesivir EUA Letter of Authorization. 2020. Available online: https://www.fda.gov/media/137564/ (accessed on 1 August 2021).
- Kokic, G.; Hillen, H.S.; Tegunov, D.; Dienemann, C.; Seitz, F.; Schmitzova, J.; Farnung, L.; Siewert, A.; Höbartner, C.; Cramer, P. Mechanism of SARS-CoV-2 polymerase stalling by remdesivir. Nat. Commun. 2021, 12, 279. [Google Scholar] [CrossRef]
- Oka, N.; Wada, T. Stereocontrolled synthesis of oligonucleotide analogs containing chiral internucleotidic phosphorus atoms. Chem. Soc. Rev. 2011, 40, 5829–5843. [Google Scholar] [CrossRef] [PubMed]
- Yi-Ren, Z.; Kui, L.; Jin-Sheng, Y.; Jian, Z. Recent Advances in Catalytic Asymmetric Synthesis of P-Chiral Phosphine Oxides. Acta Chim. Sin. 2020, 78, 193–216. [Google Scholar]
- Siegel, D.; Hui, H.C.; Doerffler, E.; Clarke, M.O.; Chun, K.; Zhang, L.; Neville, S.; Carra, E.; Lew, W.; Ross, B.; et al. Discovery and Synthesis of a Phosphoramidate Prodrug of a Pyrrolo[2,1-f][triazin-4-amino] Adenine C-Nucleoside (GS-5734) for the Treatment of Ebola and Emerging Viruses. J. Med. Chem. 2017, 60, 1648–1661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Zhang, L.; Huo, X.; Zhang, Z.; Yuan, Q.; Li, P.; Chen, J.; Zou, Y.; Wu, Z.; Zhang, W. Catalytic Asymmetric Synthesis of the anti-COVID-19 Drug Remdesivir. Angew. Chem. Int. Ed. 2020, 59, 20814–20819. [Google Scholar] [CrossRef] [PubMed]
- Gannedi, V.; Kumar, B.K.; Reddy, S.N.; Ku, C.C.; Wong, C.H.; Hung, S.C. Practical Remdesivir Synthesis through One-Pot Organocatalyzed Asymmetric (S)-P-Phosphoramidation. J. Org. Chem. 2021, 86, 4977–4985. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | API | Chemical Structure | Treatment or Indications and Mode of Action | Enantioselective Synthesis: Chiral Pool or Key Step |
|---|---|---|---|---|
| 1 | Amprenavir | 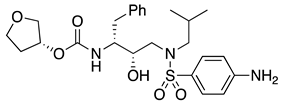 | AIDS, HIV protease inhibitor | (a) Co(salen) catalyzed kinetic resolution of 2-(1-azido-2-phenylethyl)oxirane [24] (b) from L-acid malic and L-phenylalanine [25] |
| 2 | Abacavir | 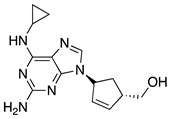 | AIDS and hepatitis B virus (HBV), reverse transcriptase inhibitor | (a) chiral resolution using γ-lactamase [26] (b) Oppolzer-sultam as auxiliary then asymmetric rhodium-catalyzed coupling using Josiphos 003-1 [27] |
| 3 | Cobicistat | 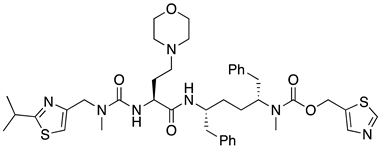 | AIDS, human CYP3A proteins inhibitor | chiral amino acids as starting materials [28,29] |
| 4 | Daclatasvir | 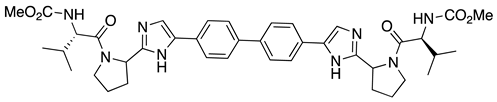 | chronic hepatitis C virus (HCV) genotype 3, inhibitor of the NS5A | continuous-flow synthesis with chiral amino acids as starting materials [30] |
| 5 | Dolutegravir | 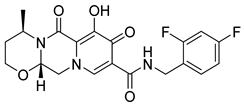 | AIDS, integrase strand transfer inhibitor | (R)-3-amino-1-butanol as starting material [31] |
| 6 | Elvitegravir | 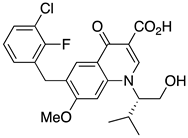 | AIDS, HIV integrase inhibitor | (S)-valinol as starting material [32] |
| 7 | Emtricitabine | 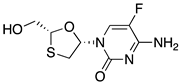 | AIDS and HBV, reverse transcriptase inhibitor | L-menthol as chiral auxiliary [33] |
| 8 | Indinavir | 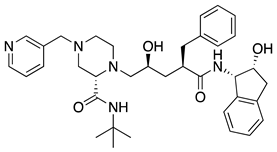 | AIDS, HIV-1 protease inhibitor | (a) (R)-glycidol as starting material [34] (b) from indanone via enantioselective ester hydrolysis with Rhizopus oryzae [35] (c) (1S,2R)-cis-aminoindanol as starting material [36] |
| 9 | Lamivudine | 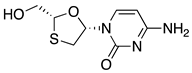 | AIDS and HBV, reverse transcriptase inhibitor | (a) enzymatic dynamic kinetic resolution [37] (b) (S)-lactic acid as starting material [38] (c) L-menthol as chiral auxiliary [39] |
| 10 | Maraviroc | 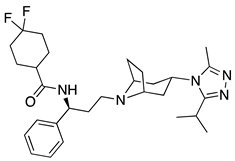 | AIDS, antagonist of the CCR5 receptor | (a) (S)-tert-butanesulfinamide as chiral auxiliary [40] (b) aza-Michael/hemiacetal reaction using the Hayashi–Jørgensen organocatalyst [41] |
| 11 | Nelfinavir |  | AIDS, HIV protease inhibitor | (a) Ti catalyzed asymmetric aminolysis of epoxide [42] (b) starting from chiral sulfoxide [43] (c) bromocyclization of bisallylic catalyzed by (S)-BINAP [44] |
| 12 | Ritonavir | 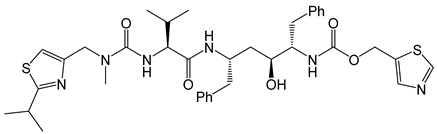 | AIDS, HIV protease inhibitor | chiral amino acids as starting materials [45,46] |
| 13 | Saquinavir |  | AIDS, HIV protease inhibitor | chiral amino acids as starting materials [47] |
| 14 | Simeprevir |  | HCV, NS3/4A protease inhibitor | (a) chiral resolution using pig liver esterase [48] (b) chiral resolution using cinchonidine [49] |
| 15 | Telaprevir | 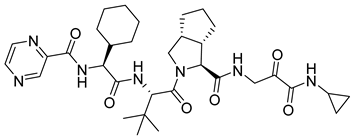 | HCV, NS3/4A protease inhibitor | lipase mediated desymmetrization of 1,2-cyclopentanedimethanol and chiral amino acids [50] |
| 16 | Tenofoviralafenamide | 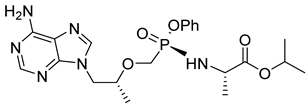 | AIDS and HBV, nucleotide reverse transcriptase inhibitor | (R)-9-[2-(phosphonomethoxy) propyl] adenine (PMPA) [51] from L-threonine and L-alanine as starting materials [52] |
| 17 | Valacyclovir | 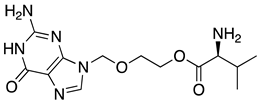 | Herpes, prodrug converted to acyclovir, a DNA polymerase inhibitor | from L-valine [53] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
da Silva, E.M.; Vidal, H.D.A.; Corrêa, A.G. Advances on Greener Asymmetric Synthesis of Antiviral Drugs via Organocatalysis. Pharmaceuticals 2021, 14, 1125. https://doi.org/10.3390/ph14111125
da Silva EM, Vidal HDA, Corrêa AG. Advances on Greener Asymmetric Synthesis of Antiviral Drugs via Organocatalysis. Pharmaceuticals. 2021; 14(11):1125. https://doi.org/10.3390/ph14111125
Chicago/Turabian Styleda Silva, Everton M., Hérika D. A. Vidal, and Arlene G. Corrêa. 2021. "Advances on Greener Asymmetric Synthesis of Antiviral Drugs via Organocatalysis" Pharmaceuticals 14, no. 11: 1125. https://doi.org/10.3390/ph14111125
APA Styleda Silva, E. M., Vidal, H. D. A., & Corrêa, A. G. (2021). Advances on Greener Asymmetric Synthesis of Antiviral Drugs via Organocatalysis. Pharmaceuticals, 14(11), 1125. https://doi.org/10.3390/ph14111125