
Assessing a Novel 3D Assay System for Drug Screening against OS Metastasis

 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
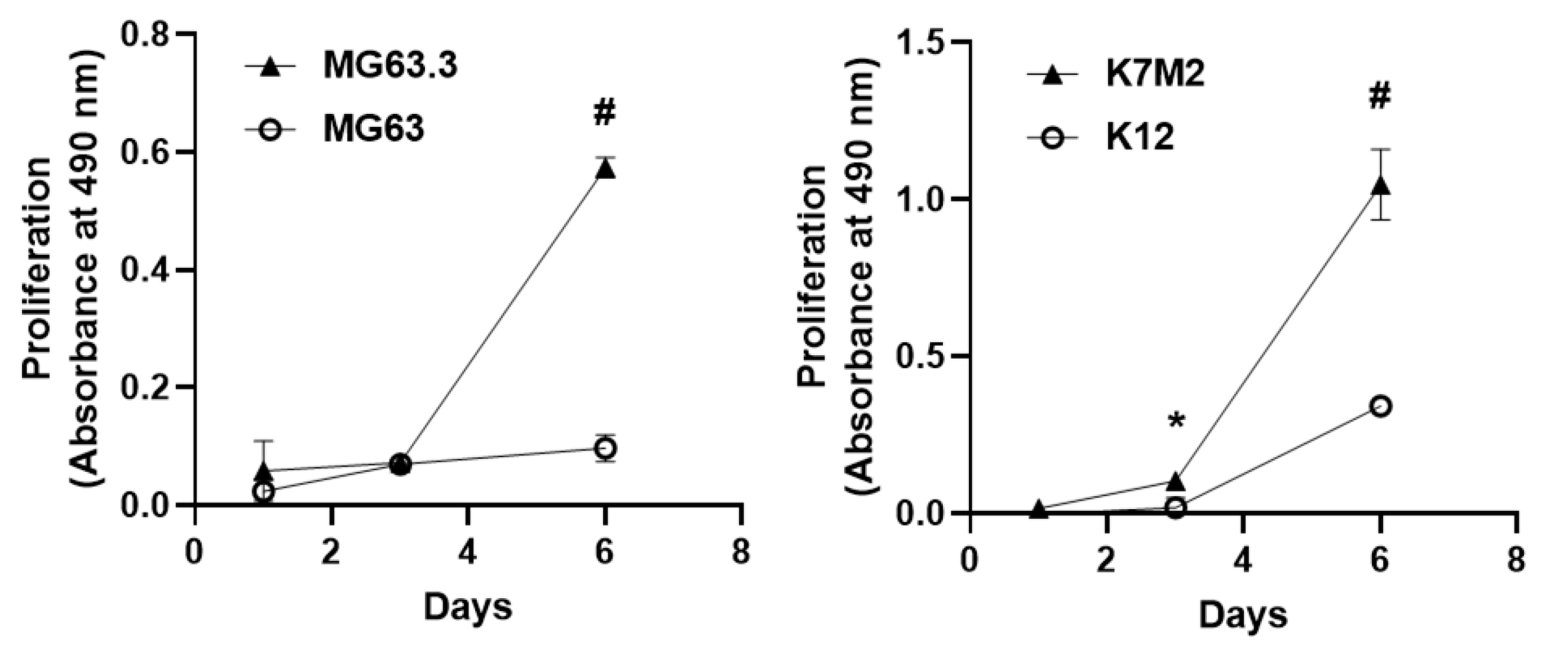
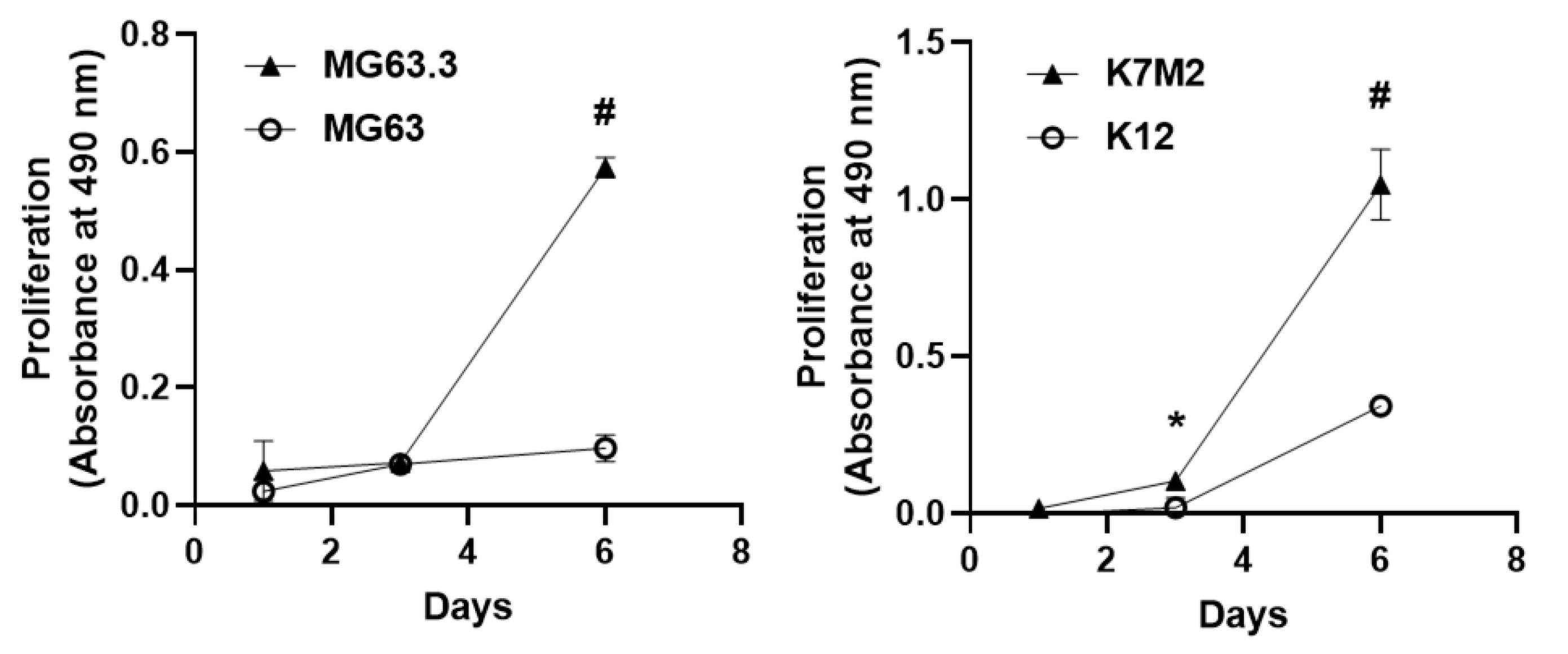
2.1. Validation of Matched High and Low Metastatic OS Cell Pairs in the 3D BME Assay
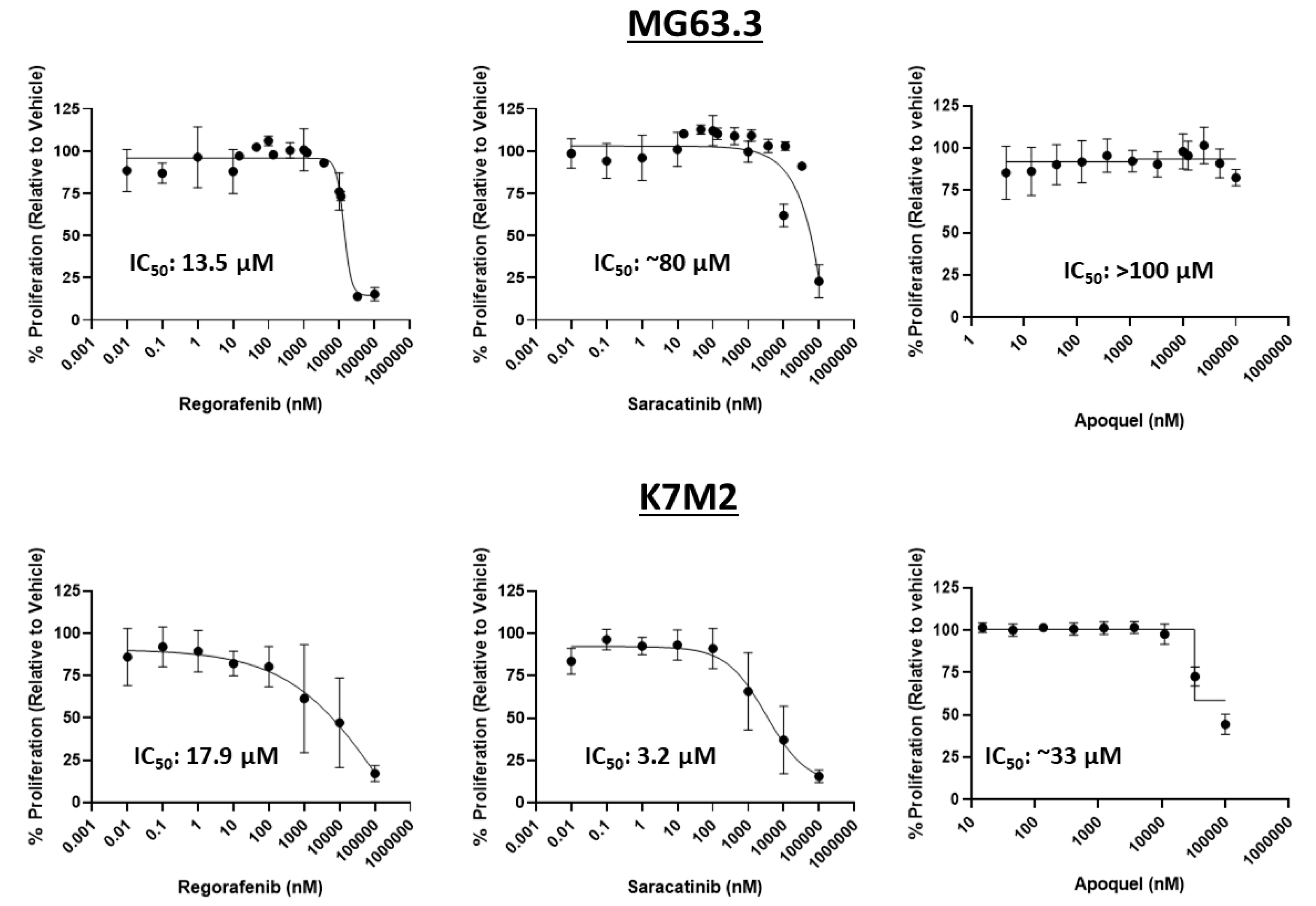
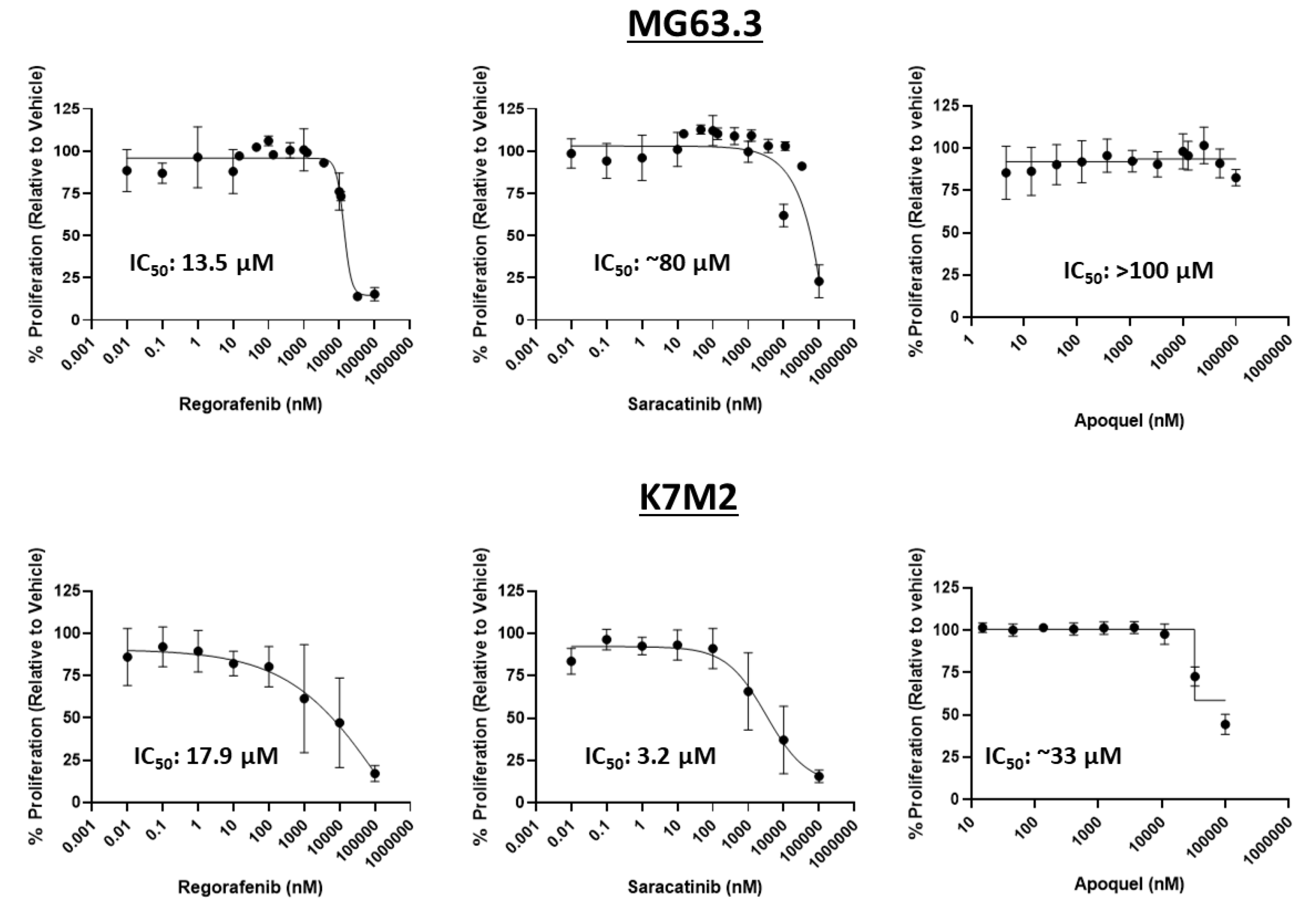
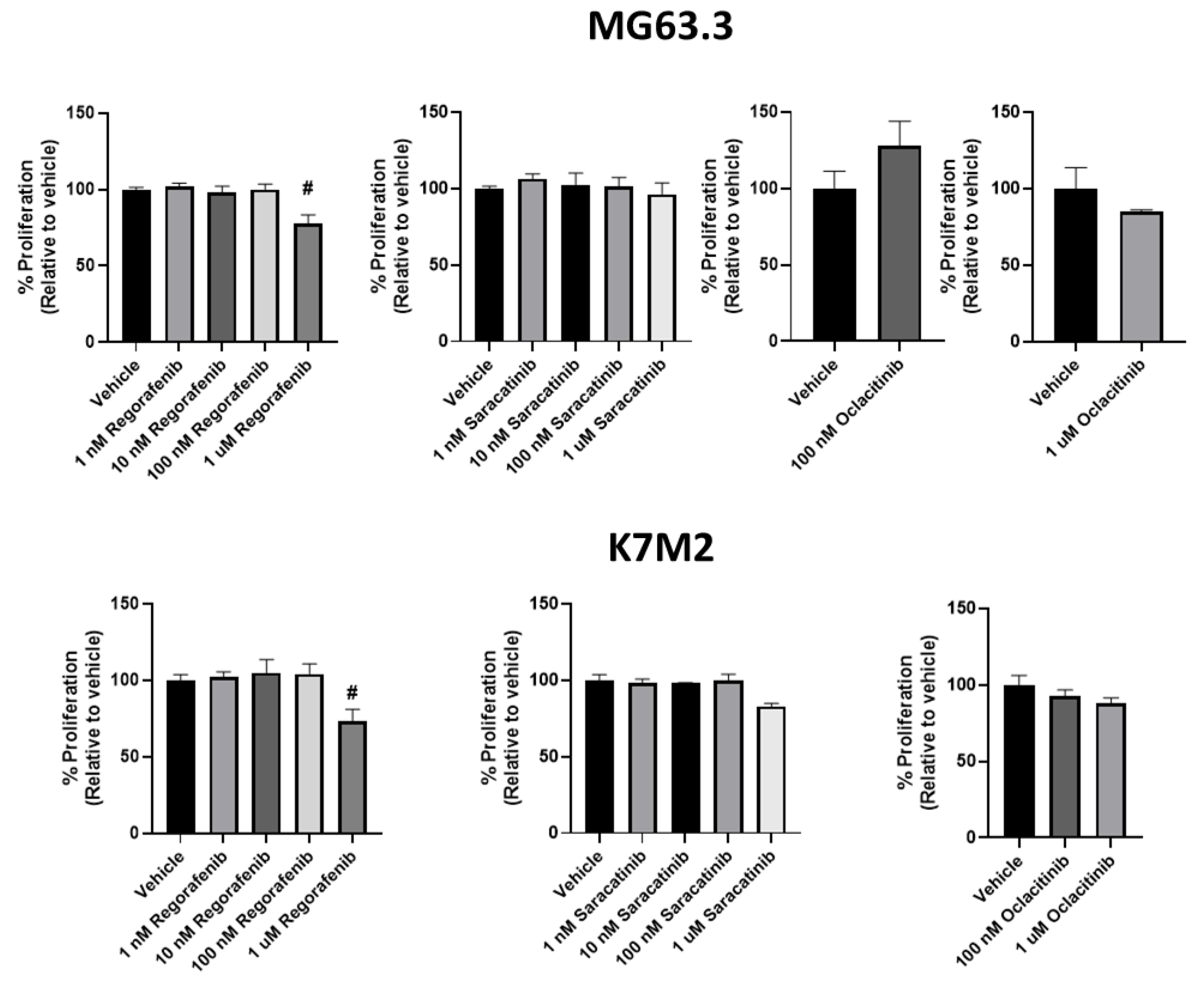
2.2. Assessment of Direct Drug Cytotoxicity by Cell Number
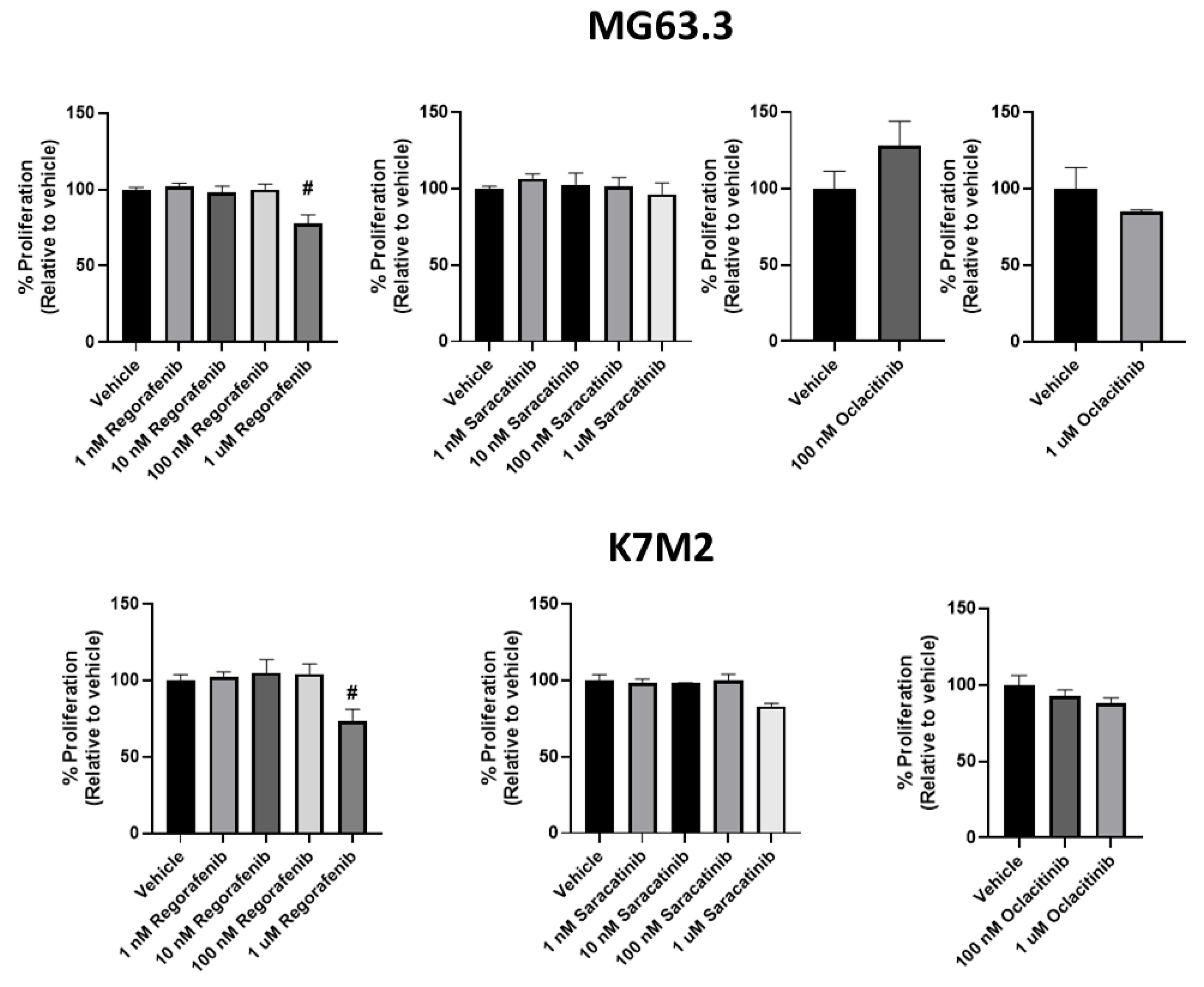
2.3. Effect of Drug Treatment on OS Cells in 3D BME
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Assessment of Cell Number and Cytotoxic Activity of Each Drug in 2D Culture
4.3. Assesment of ME and Drug Response of OS Cells in the 3D BME System
4.4. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Federman, N.; Bernthal, N.; Eilber, F.C.; Tap, W.D. The Multidisciplinary Management of Osteosarcoma. Curr. Treat. Options Oncol. 2009, 10, 82–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varan, A.; Yazici, N.; Aksoy, C.; Gedikoğlu, G.; Yalçin, B.; Akyüz, C.; Kutluk, T.; Büyükpamukçu, M. Treatment Results of Pediatric Osteosarcoma: Twenty-Year Experience. J. Pediatr. Orthop. 2007, 27, 241–246. [Google Scholar]
- Sauer, S.; Reed, D.R.; Ihnat, M.; Hurst, R.E.; Warshawsky, D.; Barkan, D. Innovative Approaches in the Battle Against Cancer Recurrence: Novel Strategies to Combat Dormant Disseminated Tumor Cells. Front. Oncol 2021, 11, 659963. [Google Scholar] [CrossRef] [PubMed]
- Stewart, S.; Barkan, D.; Warshawsky, D.; Khanna, C. Osteosarcoma Metastasis: An Unmet Clinical Need and Unique Drug Development Opportunity. Int. J. Orthop. 2019, 2, 004–0011. [Google Scholar]
- Barkan, D.; Green, J.E. An in Vitro System to Study Tumor Dormancy and the Switch to Metastatic Growth. J. Vis. Exp. 2011, 54, 2914. [Google Scholar] [CrossRef] [Green Version]
- Barkan, D.; Kleinman, H.; Simmons, J.L.; Asmussen, H.; Kamaraju, A.K.; Hoenorhoff, M.J.; Liu, Z.; Costes, S.V.; Cho, E.H.; Lockett, S.; et al. Inhibition of Metastatic Outgrowth from Single Dormant Tumor Cells by Targeting the Cytoskeleton. Cancer Res. 2008, 68, 6241–6250. [Google Scholar] [CrossRef] [Green Version]
- Baird, K.; Glod, J.; Steinberg, S.M.; Reinke, D.; Pressey, J.G.; Mascarenhas, L.; Federman, N.; Marina, N.; Chawla, S.; Lagmay, J.P.; et al. Results of a Randomized, Double-Blinded, Placebo-Controlled, Phase 2.5 Study of Saracatinib (AZD0530), in Patients with Recurrent Osteosarcoma Localized to the Lung. Sarcoma 2020, 2020, 1–6. [Google Scholar] [CrossRef]
- Tian, Z.; Niu, X.; Yao, W. Receptor Tyrosine Kinases in Osteosarcoma Treatment: Which Is the Key Target? Front. Oncol. 2020, 10, 1642. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, A.J.; Bowman, J.W.; Fici, G.J.; Zhang, M.; Mann, D.W.; Mitton-Fry, M. Oclacitinib (APOQUEL ®) Is a Novel Janus Kinase Inhibitor with Activity against Cytokines Involved in Allergy. J. Vet. Pharmacol. Therap. 2014, 37, 317–324. [Google Scholar] [CrossRef] [Green Version]
- Ren, L.; Mendoza, A.; Zhu, J.; Briggs, J.W.; Halsey, C.; Hong, E.S.; Burkett, S.S.; Morrow, J.J.; Lizardo, M.M.; Osborne, T.; et al. Characterization of the Metastatic Phenotype of a Panel of Established Osteosarcoma Cells. Oncotarget 2015, 6, 29469–29481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanna, C.; Khan, J.; Nguyen, P.; Prehn, J.; Caylor, J.; Yeung, C.; Trepel, J.; Meltzer, P.; Helman, L. Metastasis-Associated Differences in Gene Expression in a Murine Model of Osteosarcoma. Cancer Res. 2001, 61, 3750–3759. [Google Scholar] [PubMed]
- Fenger, J.M.; London, C.A.; Kisseberth, W.C. Canine Osteosarcoma: A Naturally Occurring Disease to Inform Pediatric Oncology. ILAR J. 2014, 55, 69–85. [Google Scholar] [CrossRef] [Green Version]
- Mendoza, A.; Hong, S.-H.; Osborne, T.; Khan, M.A.; Campbell, K.; Briggs, J.; Eleswarapu, A.; Buquo, L.; Ren, L.; Hewitt, S.M.; et al. Modeling Metastasis Biology and Therapy in Real Time in the Mouse Lung. J. Clin. Investig. 2010, 120, 2979–2988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, H.; Battaglin, F.; Wang, J.; Lo, J.H.; Soni, S.; Zhang, W.; Lenz, H.-J. Molecular Insight of Regorafenib Treatment for Colorectal Cancer. Cancer Treat. Rev. 2019, 81, 101912. [Google Scholar] [CrossRef] [PubMed]
- Crona, D.J.; Keisler, M.D.; Walko, C.M. Regorafenib: A Novel Multitargeted Tyrosine Kinase Inhibitor for Colorectal Cancerand Gastrointestinal Stromal Tumors. Ann. Pharmacother. 2013, 47, 1685–1696. [Google Scholar] [CrossRef]
- Eisen, T.; Joensuu, H.; Nathan, P.D.; Harper, P.G.; Wojtukiewicz, M.Z.; Nicholson, S.; Bahl, A.; Tomczak, P.; Pyrhonen, S.; Fife, K.; et al. Regorafenib for Patients with Previously Untreated Metastatic or Unresectable Renal-Cell Carcinoma: A Single-Group Phase 2 Trial. Lancet Oncol. 2012, 13, 1055–1062. [Google Scholar] [CrossRef]
- Berry, V.; Basson, L.; Bogart, E.; Mir, O.; Blay, J.-Y.; Italiano, A.; Bertucci, F.; Chevreau, C.; Clisant-Delaine, S.; Liegl-Antzager, B.; et al. REGOSARC: Regorafenib versus Placebo in Doxorubicin-Refractory Soft-Tissue Sarcoma-A Quality-Adjusted Time without Symptoms of Progression or Toxicity Analysis: Regorafenib vs Placebo: Q-TWiST Analysis. Cancer 2017, 123, 2294–2302. [Google Scholar] [CrossRef] [Green Version]
- Duffaud, F.; Mir, O.; Boudou-Rouquette, P.; Piperno-Neumann, S.; Penel, N.; Bompas, E.; Delcambre, C.; Kalbacher, E.; Italiano, A.; Collard, O.; et al. Efficacy and Safety of Regorafenib in Adult Patients with Metastatic Osteosarcoma: A Non-Comparative, Randomised, Double-Blind, Placebo-Controlled, Phase 2 Study. Lancet Oncol 2019, 20, 120–133. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Dumas, J.; Adnane, L.; Lynch, M.; Carter, C.A.; Schütz, G.; Thierauch, K.-H.; Zopf, D. Regorafenib (BAY 73-4506): A New Oral Multikinase Inhibitor of Angiogenic, Stromal and Oncogenic Receptor Tyrosine Kinases with Potent Preclinical Antitumor Activity. Int. J. Cancer 2011, 129, 245–255. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, L.; Wang, Y.; He, A.; Hu, H.; Zhang, J.; Han, M.; Huang, Y. Curcumin Inhibits the Proliferation and Invasion of MG-63 Cells through Inactivation of the p-JAK2/p-STAT3 Pathway. OncoTargets Ther. 2019, 12, 2011–2021. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Chu, Y.; Yue, B.; Ma, X.; Zhang, G.; Xiang, H.; Liu, Y.; Wang, T.; Wu, X.; Chen, B. Adipose-Derived Mesenchymal Stem Cells Promote Osteosarcoma Proliferation and Metastasis by Activating the STAT3 Pathway. Oncotarget 2017, 8, 23803–23816. [Google Scholar] [CrossRef]
- Banovic, F.; Tarigo, J.; Gordon, H.; Barber, J.P.; Gogal, R.M. Immunomodulatory In Vitro Effects of Oclacitinib on Canine T-Cell Proliferation and Cytokine Production. Vet. Dermatol. 2019, 30, 17-e6. [Google Scholar] [CrossRef] [Green Version]
- Jasiecka-Mikołajczyk, A.; Jaroszewski, J.J.; Maślanka, T. Oclacitinib Depletes Canine CD4+ and CD8+ T Cells in Vitro. Res. Vet. Sci. 2018, 121, 124–129. [Google Scholar] [CrossRef]
- van der Leun, A.M.; Thommen, D.S.; Schumacher, T.N. CD8+ T Cell States in Human Cancer: Insights from Single-Cell Analysis. Nat. Rev. Cancer 2020, 20, 218–232. [Google Scholar] [CrossRef] [PubMed]
- Rettew, A.N.; Young, E.D.; Lev, D.C.; Kleinerman, E.S.; Abdul-Karim, F.W.; Getty, P.J.; Greenfield, E.M. Multiple Receptor Tyrosine Kinases Promote the in Vitro Phenotype of Metastatic Human Osteosarcoma Cell Lines. Oncogenesis 2012, 1, e34. [Google Scholar] [CrossRef] [PubMed]
- Präbst, K.; Engelhardt, H.; Ringgeler, S.; Hübner, H. Basic Colorimetric Proliferation Assays: MTT, WST, and Resazurin. In Cell Viability Assays; Gilbert, D.F., Friedrich, O., Eds.; Springer: New York, NY, USA, 2017; Volume 1601, pp. 1–17. ISBN 9781493969593. [Google Scholar]
- Briggs, J.W.; Ren, L.; Chakrabarti, K.R.; Tsai, Y.C.; Weissman, A.M.; Hansen, R.J.; Gustafson, D.L.; Khan, Y.A.; Dinman, J.D.; Khanna, C. Activation of the Unfolded Protein Response in Sarcoma Cells Treated with Rapamycin or Temsirolimus. PLoS ONE 2017, 12, e0185089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, X.; Mendoza, A.; Khanna, C.; Helman, L.J. Rapamycin Inhibits Ezrin-Mediated Metastatic Behavior in a Murine Model of Osteosarcoma. Cancer Res. 2005, 65, 2406–2411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barkan, D.; Green, J.E.; Chambers, A.F. Extracellular Matrix: A Gatekeeper in the Transition from Dormancy to Metastatic Growth. Eur. J. Cancer 2010, 46, 1181–1188. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koons, N.; Amato, N.; Sauer, S.; Warshawsky, D.; Barkan, D.; Khanna, C. Assessing a Novel 3D Assay System for Drug Screening against OS Metastasis. Pharmaceuticals 2021, 14, 971. https://doi.org/10.3390/ph14100971
Koons N, Amato N, Sauer S, Warshawsky D, Barkan D, Khanna C. Assessing a Novel 3D Assay System for Drug Screening against OS Metastasis. Pharmaceuticals. 2021; 14(10):971. https://doi.org/10.3390/ph14100971
Chicago/Turabian StyleKoons, Natalie, Nicole Amato, Scott Sauer, David Warshawsky, Dalit Barkan, and Chand Khanna. 2021. "Assessing a Novel 3D Assay System for Drug Screening against OS Metastasis" Pharmaceuticals 14, no. 10: 971. https://doi.org/10.3390/ph14100971
APA StyleKoons, N., Amato, N., Sauer, S., Warshawsky, D., Barkan, D., & Khanna, C. (2021). Assessing a Novel 3D Assay System for Drug Screening against OS Metastasis. Pharmaceuticals, 14(10), 971. https://doi.org/10.3390/ph14100971