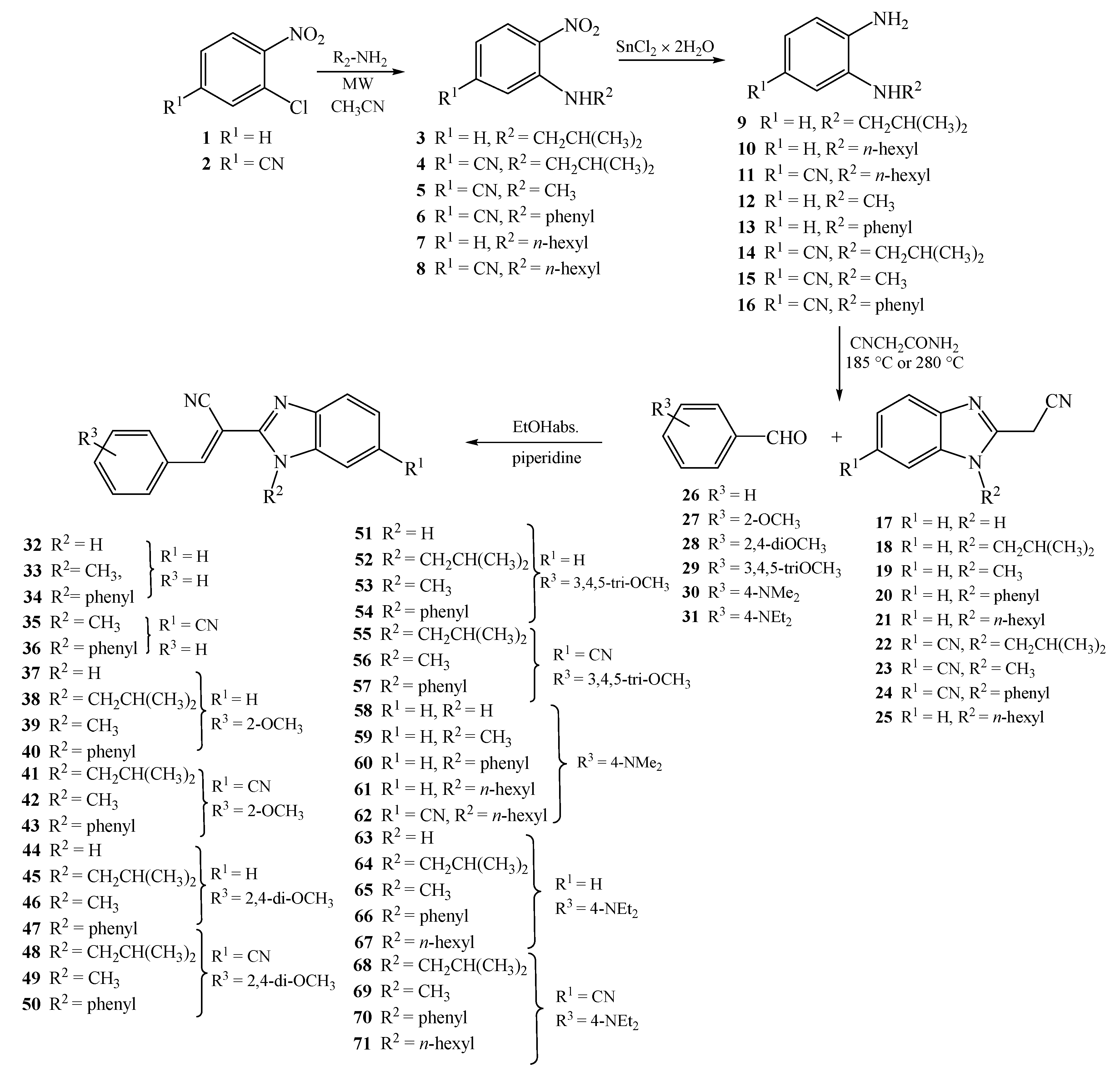
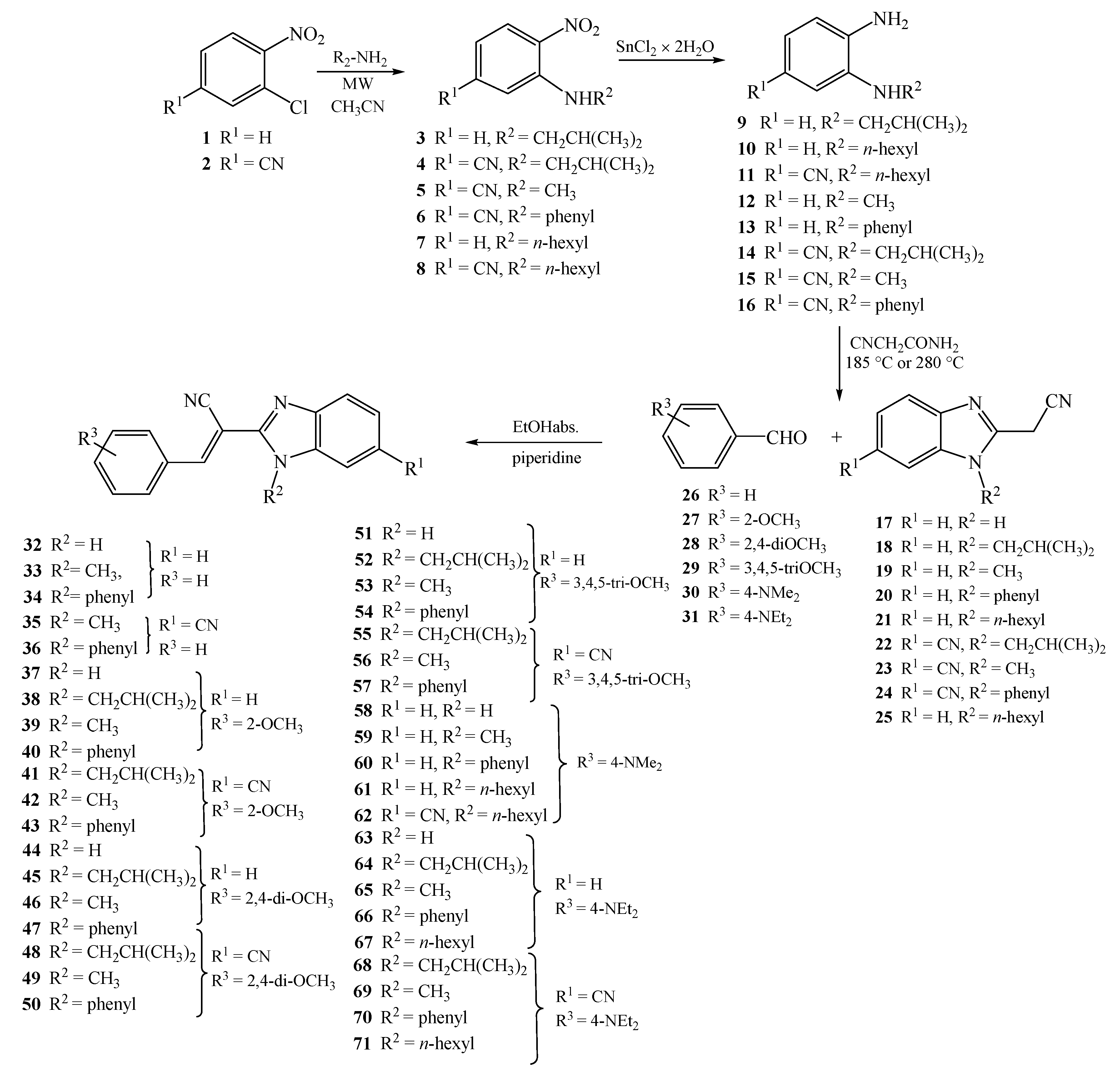
3.1.7. General Method for Preparation of Compounds 32–71
A solution of equimolar amounts of 2-(cyanomethyl)-benzimidazoles 17–25, corresponding aromatic aldehydes 25–30, and few drops of piperidine in absolute ethanol was refluxed for 2–4 h. The cooled reaction mixture was filtered, and if necessary the product was purified by column chromatography on SiO2 using dichloromethane/methanol at 200:1 as the eluent.
(E)-2-(1H-benzimidazol-2-yl)-3-phenylacrylonitrile 32
Compound 32 was prepared from 17 (0.10 g, 0.6 mmol) and 26 (0.07 g, 0.6 mmol) in absolute ethanol (2 mL) after refluxing for 2 h to yield 0.12 g (78%) of light yellow powder; m.p 224–228 °C; 1H NMR (DMSO-d6, 400 MHz): δ/ppm = 13.10 (s, 1H, NHbenz), 8.36 (s, 1H, Harom), 8.02–7.97 (m, 2H, Harom), 7.71 (d, 1H, J = 7.9 Hz, Harom), 7.64–7.55 (m, 4H, Harom), 7.30 (t, 1H, J = 7.6 Hz, Harom), 7.25 (t, 1H, J = 7.6 Hz, Harom); 13C NMR (DMSO-d6, 101 MHz): δ/ppm = 147.9, 145.8, 143.8, 135.3, 133.2, 132.2, 130.0 (2C), 129.9 (2C), 129.8, 124.2, 122.8, 119.7, 116.6, 112.0, 102.9; Anal. Calcd. for C16H11N3: C, 78.35; H, 4.52; N, 17.13. Found: C, 78.43; H, 4.61; N, 17.07%.
(E)-2-(N-methylbenzimidazol-2-yl)-3-phenylacrylonitrile 33
Compound 33 was prepared from 19 (0.10 g, 0.6 mmol) and 26 (0.06 g, 0.6 mmol) in absolute ethanol (2 mL) after refluxing for 2 h to yield 0.07 g (46%) of red oil. 1H NMR (DMSO-d6, 400 MHz): δ/ppm = 8.19 (s, 1H, Harom), 7.97–7.93 (m, 2H, Harom), 7.80–7.76 (m, 1H, Harom), 7.61–7.57 (m, 3H, Harom), 7.34–7.31 (m, 3H, Harom), 3.33 (s, 3H, CH3); 13C NMR (DMSO-d6, 101 MHz): δ/ppm = 166.8, 163.2, 151.0, 132.8, 132.4, 130.5, 130.2, 129.7, 129.5, 128.9, 128.3, 127.5, 119.3, 118.9, 116.9, 107.2, 30.3; Anal. Calcd. for C17H13N3: C, 78.74; H, 5.05; N, 16.20. Found: C, 78.69; H, 4.95; N, 16.24%.
(E)-2-(N-phenylbenzimidazol-2-yl)-3-phenylacrylonitrile 34
Compound 34 was prepared from 20 (0.10 g, 0.4 mmol) and 26 (0.05 g, 0.4 mmol) in absolute ethanol (2 mL) after refluxing for 2 h to yield 0.04 g (%) of red oil. 1H NMR (DMSO-d6, 400 MHz): δ/ppm = 8.19 (s, 1H, Harom), 7.96–7.93 (m, 3H, Harom), 7.78 (bs, 1H, Harom), 7.61–7.56 (m, 4H, Harom), 7.50 (bs, 1H, Harom), 7.42–7.26 (m, 5H, Harom); 13C NMR (DMSO-d6, 101 MHz): δ/ppm = 166.8, 163.2, 151.0 (2C), 137.4, 132.8 (2C), 132.4, 130.5 (2C), 129.7 (2C), 129.5, 129.2, 129.1, 129.1, 128.9, 128.8, 128.3, 127.5, 118.9, 117.0, 107.2; Anal. Calcd. for C22H15N3: C, 82.22; H, 4.70; N, 13.08. Found: C, 82.15; H, 4.59; N, 13.24%.
(E)-2-(6-cyano-N-methylbenzimidazol-2-yl)-3-phenylacrylonitrile 35
Compound 35 was prepared from 23 (0.10 g, 0.5 mmol) and 26 (0.05 g, 0.6 mmol) in absolute ethanol (2 mL) after refluxing for 2 h to yield 0.11 g (77%) of light brown powder; m.p 197–202 °C; 1H NMR (DMSO-d6, 400 MHz): δ/ppm = 8.30 (d, 1H, J = 0.9 Hz, Harom), 8.27 (s, 1H, Harom), 8.09–8.04 (m, 2H, Harom), 7.91 (d, 1H, J = 8.1 Hz, Harom), 7.76 (dd, 1H, J = 8.5, 1.5 Hz, Harom), 7.65–7.59 (m, 3H, Harom), 4.07 (s, 3H, CH3); 13C NMR (DMSO-d6, 101 MHz): δ/ppm = 152.0, 150.6, 141.7, 139.9, 133.0, 132.7, 130.3 (2C), 129.7 (2C), 127.0, 124.8, 120.1, 116.9, 113.0, 105.4, 100.6, 32.6; Anal. Calcd. for C18H12N4: C, 76.04; H, 4.25; N, 19.71. Found: C, 76.11; H, 4.14; N, 19.68%.
(E)-2-(6-cyano-N-phenylbenzimidazol-2-yl)-3-phenylacrylonitrile 36
Compound 36 was prepared from 24 (0.10 g, 0.4 mmol) and 26 (0.04 g, 0.4 mmol) in absolute ethanol (2 mL) after refluxing for 2 h to yield 0.10 g (73%) of light yellow powder; m.p 195–200 °C; 1H NMR (DMSO-d6, 400 MHz): δ/ppm = 8.44 (s, 1H, Harom), 7.98 (s, 1H, Harom), 7.84–7.80 (m, 2H, Harom), 7.73 (dd, 1H, J = 8.4, 1.3 Hz, Harom), 7.71–7.66 (m, 5H, Harom), 7.59–7.53 (m, 3H, Harom), 7.39 (d, 1H, J = 8.5 Hz, Harom); 13C NMR (DMSO-d6, 101 MHz): δ/ppm = 152.3, 149.8, 141.9, 140.0, 134.8, 132.9, 132.6, 130.8 (2C), 130.5, 130.2 (2C), 129.7 (2C), 128.1 (2C), 128.0, 125.2, 119.9, 115.6, 112.8, 106.2, 100.3; Anal. Calcd. for C23H14N4: C, 79.75; H, 4.07; N, 16.17. Found: C, 79.71; H, 4.14; N, 16.22%.
(E)-2-(1H-benzimidazol-2-yl)-3-(2-methoxyphenyl)acrylonitrile 37
Compound 37 was prepared from 17 (0.10 g, 0.6 mmol) and 27 (0.09 g, 0.6 mmol) in absolute ethanol (3 mL) after refluxing for 3 h to yield 0.18 g (48%) of yellow powder; m.p 260–264 °C; 1H NMR (DMSO-d6, 600 MHz): δ/ppm = 13.11 (s, 1H, NHbenz), 8.54 (s, 1H, Harom), 8.10 (dd, 1H, J = 7.8, 1.2 Hz, Harom), 7.62 (bs, 1H, Harom), 7.57 (td, 1H, J = 7.9, 1.6 Hz, Harom), 7.28–7.24 (m, 2H, Harom), 7.22 (d, 1H, J = 8.4 Hz, Harom), 7.15 (t, 1H, J = 7.6 Hz, Harom), 3.94 (s, 3H, OCH3);
13C NMR (DMSO-d6, 75 MHz): δ/ppm = 158.5, 148.0, 140.8, 133.9, 128.7, 121.9, 121.2, 116.7, 112.3, 103.3, 56.4; Anal. Calcd. for C17H13N3O: C, 74.17; H, 4.76; N, 15.26; O, 5.81. Found: C, 74.11; H, 4.74; N, 15.31; O, 5.77%.
E(Z)-3-(2-methoxyphenyl)-2-(N-isobutylbenzimidazol-2-yl)acrylonitrile 38
Compound 38 was prepared from 18 (0.10 g, 0.5 mmol) and 27 (0.06 g, 0.5 mmol) in absolute ethanol (3 mL) after refluxing for 4.5 h to yield 0.15 g (67%) of yellow oil in the form of a mixture of E- and Z-isomers at the ratio of 38a/38b = 2:1; 38a: 1H NMR (DMSO-d6, 400 MHz): δ/ppm = 8.42 (s, 1H, Harom), 8.09 (dd, 1H, J = 7.8, 1.4 Hz, Harom), 7.74–7.71 (m, 2H, Harom), 7.62–7.58 (m, 1H, Harom), 7.32–7.30 (m, 1H, Harom), 7.28 (dd, 1H, J = 7.9, 1.3 Hz, Harom), 7.23 (d, 1H, J = 7.9 Hz, Harom), 7.18 (d, 1H, J = 7.7 Hz, Harom), 4.35 (d, 2H, J = 7.5 Hz, CH2), 3.92 (s, 3H, CH3), 2.24–2.15 (m, 1H, CH), 0.87 (d, 6H, J = 6.7 Hz, CH3); 13C NMR (DMSO-d6, 101 MHz): δ/ppm = 158.6, 147.2, 146.4, 142.2, 136.9, 134.2, 128.6, 123.8, 123.1, 121.8, 121.2, 119.8, 117.1, 112.4, 111.2, 102.5, 56.5, 51.3, 29.8, 20.0; 38b: 1H NMR (DMSO-d6, 400 MHz): δ/ppm = 8.15 (s, 1H, Harom), 7.76–7.73 (m, 1H, Harom), 7.63 (dd, 1H, J = 6.9, 1.4 Hz, Harom), 7.41 (td, 1H, J = 7.9, 1.7 Hz, Harom), 7.36–7.32 (m, 2H, Harom), 6.70 (t, 1H, J = 7.4 Hz, Harom), 6.57 (dd, 1H, J = 7.8, 1.6 Hz, Harom), 3.84 (s, 3H, CH3), 3.64 (d, 2H, J = 7.6 Hz, CH2), 2.06–2.00 (m, 1H, CH), 0.74 (d, 6H, J = 6.7 Hz, CH3); 13C NMR (DMSO-d6, 101 MHz): δ/ppm = 158.2, 146.8, 145.1, 142.8, 135.4, 134.0, 128.8, 124.0, 123.0, 121.8, 121.2, 121.0, 120.2, 118.6, 112.5, 112.1, 101.4, 56.5, 51.1, 29.0, 19.9; Anal. Calcd. for C21H21N3O: C, 76.11; H, 6.39; N, 12.68; O, 4.83. Found: C, 76.28; H, 6.35; N, 12.71; O, 4.74%.
(E)-3-(2-methoxyphenyl)-2-(N-methylbenzimidazol-2-yl)acrylonitrile 39
Compound 39 was prepared from 19 (0.10 g, 0.6 mmol) and 27 (0.08 g, 0.6 mmol) in absolute ethanol (3 mL) after refluxing for 3 h to yield 0.12 g (71%) of yellow powder; m.p 144–146 °C; 1H NMR (DMSO-d6, 400 MHz): δ/ppm = 8.33 (s, 1H, Harom), 8.12 (dd, 1H, J = 7.8, 1.4 Hz, Harom), 7.72 (d, 1H, J = 7.6 Hz, Harom), 7.65 (d, 1H, J = 7.9 Hz, Harom), 7.60 (td, 1H, J = 7.9, 1.4 Hz, Harom), 7.36 (td, 1H, J = 7.6, 1.2 Hz, Harom), 7.30 (td, 1H, J = 7.6, 1.1 Hz, Harom), 7.22 (d, 1H, J = 8.4 Hz, Harom), 7.17 (t, 1H, J = 7.6 Hz, Harom), 4.01 (s, 3H, OCH3), 3.92 (s, 3H, CH3); 13C NMR (DMSO-d6, 101 MHz): δ/ppm = 158.5, 147.8, 145.9, 142.3, 137.1, 134.1, 128.7, 123.8, 123.1, 121.9, 121.1, 119.7, 117.1, 112.3, 111.3, 101.4, 56.5, 32.1; Anal. Calcd. for C18H15N3O: C, 74.72; H, 5.23; N, 14.52; O, 5.53. Found: C, 74.86; H, 5.04; N, 14.31; O, 5.77%.
(E)-3-(2-methoxyphenyl)-2-(N-phenylbenzimidazol-2-yl)acrylonitrile 40
Compound 40 was prepared from 20 (0.10 g, 0.4 mmol) and 27 (0.06 g, 0.4 mmol) in absolute ethanol (3 mL) after refluxing for 2 h to yield 0.10 g (70%) of orange powder; m.p 169–173 °C; 1H NMR (DMSO-d6, 400 MHz): δ/ppm = 8.02 (dd, 1H, J = 7.8, 1.4 Hz, Harom), 7.98 (s, 1H, Harom), 7.84 (dd, 1H, J = 6.7, 1.5 Hz, Harom), 7.72–7.65 (m, 3H, Harom), 7.65–7.62 (m, 2H, Harom), 7.53 (td, 1H, J = 7.8, 1.5 Hz, Harom), 7.37 (td, 1H, J = 7.4, 1.5 Hz, Harom), 7.33 (td, 1H, J = 7.5, 1.5 Hz, Harom), 7.18 (dd, 1H, J = 6.9, 1.5 Hz, Harom), 7.11 (d, 1H, J = 7.9 Hz, Harom), 7.08 (d, 1H, J = 7.6 Hz, Harom), 3.75 (s, 3H, CH3); 13C NMR (DMSO-d6, 101 MHz): δ/ppm = 158.5, 147.2, 144.5, 142.4, 137.6, 135.8, 134.3, 130.8 (2C), 129.9, 128.1, 128.0 (2C), 126.0, 124.8, 123.8, 121.4 (2C), 121.1, 120.1, 116.3 (2C), 112.3, 111.0, 101.3, 56.2; Anal. Calcd. for C23H17N3O: C, 78.61; H, 4.88; N, 11.96; O, 4.55. Found: C, 78.51; H, 4.74; N, 11.81; O, 4.67%.
E(Z)-2-(6-cyano-N-isobutylbenzimidazol-2-yl)-3-(2-methoxyphenyl)acrylonitrile 41
Compound 41 was prepared from 22 (0.10 g, 0.4 mmol) and 27 (0.06 g, 0.4 mmol) in absolute ethanol (3 mL) after refluxing for 4.5 h to yield 0.15 g (30%) of yellow oil in the form of a mixture of E- and Z-isomers at a ratio of 41a/41b = 2:1; 41a: 1H NMR (DMSO-d6, 600 MHz): δ/ppm = 8.33 (d, 1H, J = 1.08 Hz, Harom), 8.21 (s, 1H, Harom), 7.88 (d, 1H, J = 8.45 Hz, Harom), 7.75–7.73 (m, 1H, Harom), 7.42 (td, 1H, J = 7.9, 1.6 Hz, Harom), 7.13 (d, 1H, J = 8.3 Hz, Harom), 6.73 (t, 1H, J = 7.5 Hz, Harom), 6.60 (dd, 1H, J = 7.8, 1.3 Hz, Harom), 3.77 (s, 3H, CH3), 3.69 (d, 2H, J = 7.6 Hz, CH2), 2.05–1.96 (m, 1H, CH), 0.73 (d, 6H, J = 6.6 Hz, CH3);
13C NMR (DMSO-d6, 151 MHz): δ/ppm = 158.2, 149.4, 147.2, 141.1, 139.3, 134.1, 128.2, 126.4, 124.5, 121.1, 120.7, 119.6, 116.3, 113.1, 112.0, 105.0, 100.0, 56.0, 51.1, 29.3, 19.4 (2C); 41b: 1H NMR (DMSO-d6, 600 MHz): δ/ppm = 8.51 (s, 1H, Harom), 8.30 (d, 1H, J = 1.0 Hz, Harom), 8.10 (dd, 1H, J1 = 7.7, 1.3 Hz, Harom), 7.96 (d, 1H, J = 8.6 Hz, Harom), 7.73–7.71 (m, 1H, Harom), 7.61 (td, 1H, J = 7.7, 1.6 Hz, Harom), 7.23 (d, 1H, J = 8.4 Hz, Harom), 7.17 (t, 1H, J = 7.5 Hz, Harom), 4.40 (d, 2H, J = 7.5 Hz, CH2), 3.92 (s, 3H, CH3), 2.22–2.16 (m, 1H, CH), 0.86 (d, 6H, J = 6.6 Hz, CH3); 13C NMR (DMSO-d6, 151 MHz): δ/ppm = 158.8, 148.8, 147.7, 141.7, 137.8, 133.8, 128.6, 126.7, 124.9, 121.1, 120.5, 119.4, 117.7, 113.2, 112.0, 105.0, 101.2, 56.0, 50.6, 28.6, 19.4 (2C); Anal. Calcd. for C22H20N4O: C, 74.14; H, 5.66; N, 15.72; O, 4.49. Found: C, 74.11; H, 5.74; N, 15.75; O, 4.54%.
(E)-2-(6-cyano-N-methylbenzimidazol-2-yl)-3-(2-methoxyphenyl)acrylonitrile 42
Compound 42 was prepared from 23 (0.10 g, 0.5 mmol) and 27 (0.10 g, 0.5 mmol) in absolute ethanol (3 mL) after refluxing for 3 h to yield 0.13 g (80%) of brown powder; m.p 178–181 °C; 1H NMR (DMSO-d6, 400 MHz): δ/ppm = 8.41 (s, 1H, Harom), 8.30 (d, 1H, J = 1.0 Hz, Harom), 8.13 (dd, 1H, J = 7.4, 1.4 Hz, Harom), 7.88 (d, 1H, J = 8.4 Hz, Harom), 7.75 (dd, 1H, J = 8.5, 1.4 Hz, Harom), 7.62 (td, 1H, J = 7.9, 1.4 Hz, Harom), 7.23 (d, 1H, J = 8.1 Hz, Harom), 7.18 (t, 1H, J = 7.6 Hz, Harom), 4.05 (s, 3H, OCH3), 3.92 (s, 3H, CH3); 13C NMR (DMSO-d6, 101 MHz): δ/ppm = 158.7, 150.6, 147.1, 141.7, 139.9, 134.5, 128.7, 126.9, 124.8, 121.7, 121.2, 120.2, 116.8, 113.0, 112.4, 105.3, 100.7, 56.5, 32.6; Anal. Calcd. for C19H14N4O: C, 72.60; H, 4.49; N, 17.82; O, 5.09. Found: C, 72.53; H, 4.41; N, 17.75; O, 4.94%.
(E)-2-(6-cyano-N-phenylbenzimidazol-2-yl)-3-(2-methoxyphenyl)acrylonitrile 43
Compound 43 was prepared from 24 (0.10 g, 0.4 mmol) and 27 (0.05 g, 0.4 mmol) in absolute ethanol (3 mL) after refluxing for 2 h to yield 0.15 g (77%) of yellow powder; m.p 222–225 °C; 1H NMR (DMSO-d6, 400 MHz): δ/ppm = 8.44 (d, 1H, J = 0.9 Hz, Harom), 8.06–8.01 (m, 2H, Harom), 7.73–7.67 (m, 6H, Harom), 7.55 (td, 1H, J = 7.9, 1.4 Hz, Harom), 7.32 (dd, 1H, J = 8.4, 0.5 Hz, Harom), 7.12 (d, 1H, J = 8.6 Hz, Harom), 7.08 (d, 1H, J = 7.5 Hz, Harom), 3.75 (s, 3H, CH3); 13C NMR (DMSO-d6, 101 MHz): δ/ppm = 158.6, 149.9, 145.8, 141.9, 140.3, 135.1, 134.7, 130.9 (2C), 130.5, 128.2, 128.1 (2C), 127.9, 126.1, 125.1, 121.1, 119.9, 116.0, 112.6, 112.4, 106.0 (2C), 100.5, 56.2; Anal. Calcd. for C24H16N4O: C, 76.58; H, 4.28; N, 14.88; O, 4.25. Found: C, 76.61; H, 4.24; N, 15.05; O, 4.34%.
(E)-2-(1H-benzimidazol-2-yl)-3-(2,4-dimethoxyphenyl)acrylonitrile 44
Compound 44 was prepared from 17 (0.10 g, 0.6 mmol) and 28 (0.10 g, 0.6 mmol) in absolute ethanol (3 mL) after refluxing for 3 h to yield 0.19 g (80%) of yellow powder; m.p 205–209 °C; 1H NMR (DMSO-d6, 600 MHz): δ/ppm = 13.00 (s, 1H, NHbenz), 8.47 (s, 1H, Harom), 8.17 (d, 1H, J = 8.7 Hz, Harom), 7.62 (bs, 2H, Harom), 7.23 (bs, 2H, Harom), 6.76 (dd, 1H, J = 8.8, 2.3 Hz, Harom), 6.74 (d, 1H, J = 2.3 Hz, Harom), 3.95 (s, 3H, OCH3), 3.88 (s, 3H, OCH3); 13C NMR (DMSO-d6, 151 MHz): δ/ppm = 164.0, 160.0, 148.0, 139.5, 129.2, 116.9, 114.3, 106.4, 98.9, 98.4, 56.1, 55.7; Anal. Calcd. for C18H15N3O2: C, 70.81; H, 4.95; N, 13.76; O, 10.48. Found: C, 70.78; H, 4.74; N, 13.75; O, 10.54%.
E(Z)-3-(2,4-dimethoxyphenyl)-2-(N-isobutylbenzimidazol-2-yl)acrylonitrile 45
Compound 45 was prepared from 18 (0.10 g, 0.5 mmol) and 28 (0.08 g, 0.5 mmol) in absolute ethanol (3 mL) after refluxing for 4 h to yield 0.12 g (70%) of yellow oil in the form of a mixture of E- and Z-isomers at a ratio of 45a/45b = 5:1; 45a: 1H NMR (DMSO-d6, 600 MHz): δ/ppm = 8.33 (s, 1H, Harom), 8.16 (d, 1H, J = 8.8 Hz, Harom), 7.67–7.63 (m, 2H, Harom), 7.28–7.26 (m, 1H, Harom), 7.25–7.21 (m, 1H, Harom), 6.75 (dd, 1H, J = 8.8, 2.3 Hz, Harom), 6.71 (d, 1H, J = 2.3 Hz, Harom), 4.29 (d, 2H, J = 7.4 Hz, CH2), 3.88 (s, 3H, OCH3), 3.86 (s, 3H, OCH3), 2.19–2.11 (m, 1H, CH), 0.82 (d, 6H, J = 6.6 Hz, CH3); 13C NMR (DMSO-d6, 151 MHz): δ/ppm = 164.3, 160.2, 147.4, 144.7, 141.7, 136.4, 129.2, 123.0, 122.5, 119.1, 117.4, 114.1, 111.3, 106.5, 98.4, 96.6, 56.1, 55.8, 50.8, 29.2, 19.5 (2C); 45b: 1H NMR (DMSO-d6, 600 MHz): δ/ppm = 7.98 (s, 1H, Harom), 7.70 (d, 1H, J = 7.8 Hz, Harom), 7.61 (d, 1H, J = 8.0 Hz, Harom), 7.31–7.28 (m, 2H, Harom), 6.61 (d, 1H, J = 2.3 Hz, Harom), 6.43 (d, 1H, J = 8.8 Hz, Harom), 6.29 (dd, 1H, J = 8.8, 2.3 Hz, Harom), 6.71 (d, 1H, J = 2.3 Hz, Harom), 3.81 (s, 3H, OCH3), 3.71 (s, 3H, OCH3), 3.68 (d, 2H, J = 7.4 Hz, CH2), 2.05–1.98 (m, 1H, CH), 0.72 (d, 6H, J = 6.6 Hz, CH3); 13C NMR (DMSO-d6, 151 MHz): δ/ppm = 164.0, 159.8, 145.1, 144.9, 142.4, 134.9, 129.5, 129.2, 123.0, 119.7, 118.7, 114.0, 111.5, 106.5, 98.4, 56.2, 55.6, 50.7, 28.6, 19.5 (2C); Anal. Calcd. for C22H23N3O2: C, 73.11; H, 6.41; N, 11.63; O, 8.85. Found: C, 73.19; H, 6.51; N, 11.58; O, 8.75%.
(E)-3-(2,4-dimethoxyphenyl)-2-(N-methylbenzimidazol-2-yl)acrylonitrile 46
Compound 46 was prepared from 19 (0.10 g, 0.6 mmol) and 28 (0.09 g, 0.6 mmol) in absolute ethanol (3 mL) after refluxing for 3 h to yield 0.19 g (89%) of yellow powder; m.p 168–171 °C; 1H NMR (DMSO-d6, 600 MHz): δ/ppm = 8.26 (s, 1H, Harom), 8.20 (d, 1H, J = 8.7 Hz, Harom), 7.68 (d, 1H, J = 8.0 Hz, Harom), 7.61 (d, 1H, J = 8.0 Hz, Harom), 7.32 (td, 1H, J = 7.7, 1.0 Hz, Harom), 7.27 (td, 1H, J = 7.8, 1.0 Hz, Harom), 6.78 (dd, 1H, J = 8.8, 2.4 Hz, Harom), 6.73 (d, 1H, J = 2.3 Hz, Harom), 3.97 (s, 3H, OCH3), 3.91 (s, 3H, OCH3), 3.89 (s, 3H, CH3) 13C NMR (DMSO-d6, 151 MHz): δ/ppm = 164.2, 160.1, 148.0, 144.4, 141.9, 136.6, 129.2, 123.0, 122.5, 119.0, 117.3, 114.2, 110.6, 106.5, 98.4, 96.7, 56.1, 55.7, 31.5; Anal. Calcd. for C19H17N3O2: C, 71.46; H, 5.37; N, 13.16; O, 10.02. Found: C, 71.39; H, 5.43; N, 13.07; O, 10.13%.
(E)-3-(2,4-dimethoxyphenyl)-2-(N-phenylbenzimidazol-2-yl)acrylonitrile 47
Compound 47 was prepared from 20 (0.10 g, 0.4 mmol) and 28 (0.07 g, 0.4 mmol) in absolute ethanol (3 mL) after refluxing for 4 h to yield 0.16 g (75%) of yellow powder; m.p 164–166 °C; 1H NMR (DMSO-d6, 600 MHz): δ/ppm = 8.10 (d, 1H, J = 8.8 Hz, Harom), 7.95 (s, 1H, Harom), 7.80 (d, 1H, J = 7.9 Hz, Harom), 7.69–7.62 (m, 3H, Harom), 7.59 (dd, 2H, J = 6.9, 1.5 Hz, Harom), 7.34 (td, 1H, J = 8.1, 1.1 Hz, Harom), 7.30 (td, 1H, J = 7.9, 1.0 Hz, Harom), 7.14 (d, 1H, J = 7.9 Hz, Harom), 6.70 (dd, 1H, J = 8.9, 2.3 Hz, Harom), 6.63 (d, 1H, J = 2.3 Hz, Harom), 3.85 (s, 3H, OCH3), 3.75 (s, 3H, OCH3); 13C NMR (DMSO-d6, 75 MHz): δ/ppm = 164.7, 160.5, 147.9, 143.5, 142.4, 137.6, 136.0, 130.7, 129.9, 129.2, 128.0, 124.5, 123.7, 119.9, 117.0, 114.3, 110.9, 107.0, 98.7, 97.2, 56.4, 56.2, 56.1; Anal. Calcd. for C24H19N3O2: C, 75.57; H, 5.02; N, 11.02; O, 8.39. Found: C, 75.51; H, 4.92; N, 11.08; O, 8.22%.
(E)-2-(6-cyano-N-isobutylbenzimidazol-2-yl)-3-(2,4-dimethoxyphenyl)acrylonitrile 48
Compound 48 was prepared from 22 (0.10 g, 0.4 mmol) and 28 (0.07 g, 0.4 mmol) in absolute ethanol (3 mL) after refluxing for 4 h to yield 0.16 g (84%) of orange powder; m.p 120–125 °C; 1H NMR (DMSO-d6, 600 MHz): δ/ppm = 8.43 (s, 1H, Harom), 8.23 (d, 1H, J = 0.9 Hz, Harom), 8.18 (d, 1H, J = 8.8 Hz, Harom), 7.89 (d, 1H, J = 8.5 Hz, Harom), 7.67 (dd, 1H, J = 8.5, 1.4 Hz, Harom), 6.75 (dd, 1H, J = 8.8, 2.3 Hz, Harom), 6.71 (d, 1H, J = 2.3 Hz, Harom), 4.35 (d, 2H, J = 7.6 Hz, CH2), 3.89 (s, 3H, OCH3), 3.86 (s, 3H, OCH3), 2.18–2.10 (m, 1H, CH), 0.82 (d, 6H, J = 6.6 Hz, CH3); 13C NMR (DMSO-d6, 151 MHz): δ/ppm = 164.2, 159.8, 148.2, 146.2, 141.8, 137.8, 129.9, 126.6, 124.8, 119.5, 118.4, 113.8, 113.2, 106.7, 104.9, 98.4, 97.2, 56.2, 55.7, 50.9, 28.7, 19.4 (2C); Anal. Calcd. for C23H22N4O2: C, 71.48; H, 5.74; N, 14.50; O, 8.28. Found: C, 71.54; H, 5.61; N, 14.58; O, 8.15%.
(E)-2-(6-cyano-N-methylbenzimidazol-2-yl)-3-(2,4-dimethoxyphenyl)acrylonitrile 49
Compound 49 was prepared from 23 (0.10 g, 0.5 mmol) and 28 (0.08 g, 0.5 mmol) in absolute ethanol (3 mL) after refluxing for 2 h to yield 0.17 g (77%) of yellow powder; m.p 228–231 °C; 1H NMR (DMSO-d6, 400 MHz): δ/ppm = 8.37 (s, 1H, Harom), 8.26 (d, 1H, J = 0.9 Hz, Harom), 8.23 (d, 1H, J = 8.8 Hz, Harom), 7.85 (d, 1H, J = 8.4 Hz, Harom), 7.72 (dd, 1H, J = 8.5, 1.4 Hz, Harom), 6.80 (dd, 1H, J = 8.8, 2.3 Hz, Harom), 6.75 (d, 1H, J = 2.4 Hz, Harom), 4.03 (s, 3H, OCH3), 3.93 (s, 3H, OCH3), 3.91 (s, 3H, CH3); 13C NMR (DMSO-d6, 151 MHz): δ/ppm = 165.1, 160.9, 151.3, 146.0, 141.8, 140.0, 129.9, 126.6, 124.5, 120.2, 117.6, 114.5, 112.8, 107.1, 105.1, 98.9, 96.2, 56.7, 56.3, 32.5; Anal. Calcd. for C20H16N4O2: C, 69.76; H, 4.68; N, 16.27; O, 9.29. Found: C, 69.71; H, 4.51; N, 16.38; O, 9.15%.
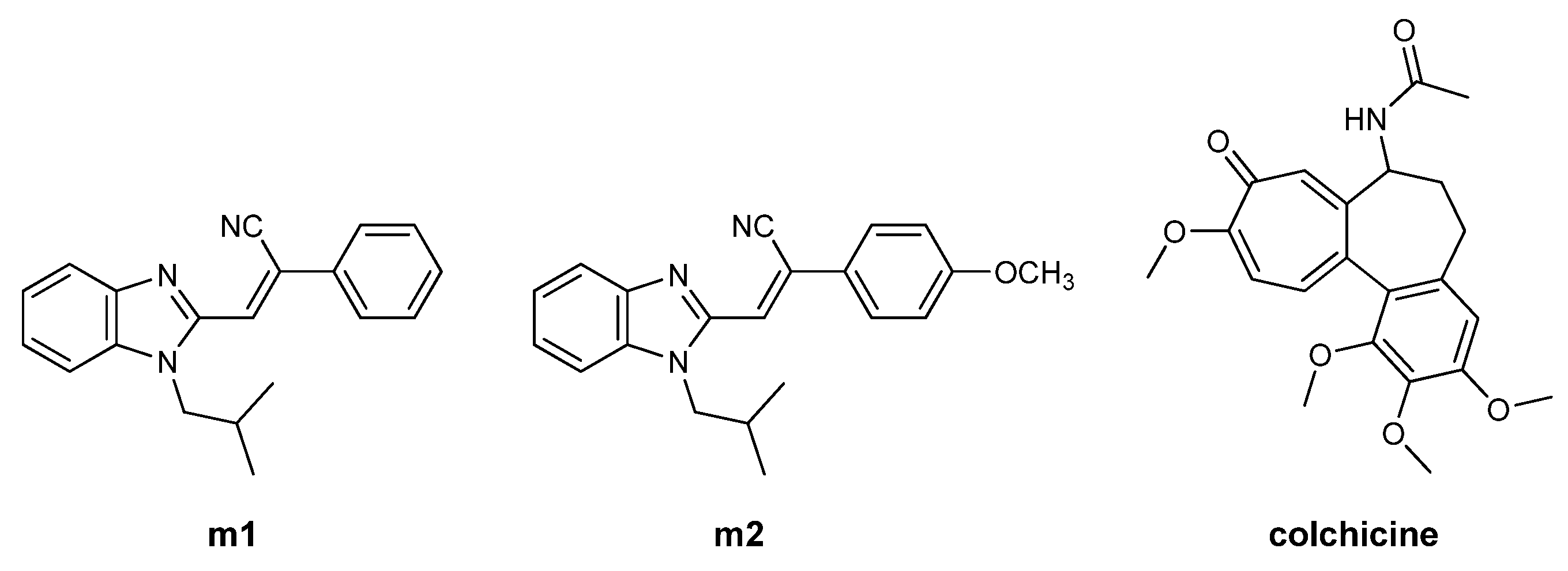
(E)-2-(6-cyano-N-phenylbenzimidazol-2-yl)-3-(2,4-dimethoxyphenyl)acrylonitrile 50
Compound 50 was prepared from 24 (0.10 g, 0.4 mmol) and 28 (0.06 g, 0.4 mmol) in absolute ethanol (3 mL) after refluxing for 2 h to yield 0.16 g (98%) of yellow powder; m.p 213–217 °C; 1H NMR (DMSO-d6, 400 MHz): δ/ppm = 8.39 (d, 1H, J = 0.8 Hz, Harom), 8.13 (d, 1H, J = 8.9 Hz, Harom), 8.02 (s, 1H, Harom), 7.70–7.65 (m, 6H, Harom), 7.28 (d, 1H, J = 8.5 Hz, Harom), 6.71 (dd, 1H, J = 8.9, 2.3 Hz, Harom), 6.64 (d, 1H, J = 2.4 Hz, Harom), 3.86 (s, 3H, OCH3), 3.76 (s, 3H, OCH3); 13C NMR (DMSO-d6, 151 MHz): δ/ppm = 164.4, 153.4, 150.9, 133.1 (2C), 119.2, 118.7, 112.1 (2C), 97.7, 40.0 (2C); Anal. Calcd. for C25H18N4O2: C, 73.88; H, 4.46; N, 13.78; O, 7.87. Found: C, 73.73; H, 4.51; N, 13.69; O, 7.75%.
(E)-2-(1H-benzimidazol-2-yl)-3-(3,4,5-trimethoxyphenyl)acrylonitrile 51
Compound 51 was prepared from 17 (0.10 g, 0.6 mmol) and 29 (0.12 g, 0.6 mmol) in absolute ethanol (3 mL) after refluxing for 2.5 h to yield 0.05 g (22%) of yellow powder; m.p 188–193 °C; 1H NMR (DMSO-d6, 400 MHz): δ/ppm = 8.17 (s, 1H, Harom), 7.85 (bs, 1H, Harom), 7.74 (bs, 2H, Harom), 7.36 (s, 3H, Harom), 3.83 (s, 6H, OCH3), 3.77 (s, 3H, OCH3); 13C NMR (DMSO-d6, 101 MHz): δ/ppm = 163.2, 153.4, 151.3, 141.5, 127.6, 117.3, 108.4, 105.5, 60.8, 56.5; Anal. Calcd. for C19H17N3O3: C, 68.05; H, 5.11; N, 12.53; O, 14.31. Found: C, 68.11; H, 4.96; N, 12.38; O, 14.36%.
(E)-2-(N-isobutylbenzimidazol-2-yl)-3-(3,4,5-trimethoxyphenyl)acrylonitrile 52
Compound 52 was prepared from 18 (0.10 g, 0.5 mmol) and 29 (0.09 g, 0.5 mmol) in absolute ethanol (3 mL) after refluxing for 4 h to yield 0.18 g (64%) of orange oil; 1H NMR (DMSO-d6, 400 MHz): δ/ppm = 8.25 (s, 1H, Harom), 7.72 (t, 2H, J = 8.3 Hz, Harom), 7.50 (s, 1H, Harom), 7.35–7.28 (m, 2H, Harom), 4.36 (d, 2H, J = 7.4 Hz, CH2), 3.87 (s, 6H, OCH3), 3.79 (s, 3H, OCH3), 2.21–2.13 (m, 1H, CH), 0.84 (d, 6H, J = 6.7 Hz, CH3); 13C NMR (DMSO-d6, 101 MHz): δ/ppm = 153.4, 153.0, 151.5, 147.3, 142.2, 136.9, 128.4, 128.1, 123.8, 123.1, 119.7, 117.6, 112.0, 108.2, 107.8, 99.1, 60.8, 56.5 (2C), 55.7, 51.3, 20.0 (2C); Anal. Calcd. for C23H25N3O3: C, 70.57; H, 6.44; N, 10.73; O, 12.26. Found: C, 70.62; H, 6.41; N, 10.81; O, 12.36%.
(E)-3-(3,4,5-trimethoxyphenyl)-2-(N-methylbenzimidazol-2-yl)acrylonitrile 53
Compound 53 was prepared from 19 (0.10 g, 0.6 mmol) and 29 (0.11 g, 0.6 mmol) in absolute ethanol (3 mL) after refluxing for 4 h to yield 0.20 g (49%) of yellow powder; m.p 134–137 °C; 1H NMR (DMSO-d6, 400 MHz): δ/ppm = 8.13 (s, 1H, Harom), 7.71 (d, 1H, J = 7.7 Hz, Harom), 7.67 (d, 1H, J = 7.9 Hz, Harom), 7.49 (s, 2H, Harom), 7.36 (td, 1H, J = 7.6, 1.1 Hz, Harom), 7.30 (td, 1H, J = 7.5, 1.1 Hz, Harom), 4.02 (s, 3H, OCH3), 3.87 (s, 3H, OCH3), 3.79 (s, 3H, OCH3); 13C NMR (DMSO-d6, 101 MHz): δ/ppm = 153.4, 150.6, 147.9, 142.4, 141.2, 137.1, 128.5, 123.8, 123.1, 119.7, 117.6, 111.3, 108.1, 99.7, 60.8, 56.5 (2C), 32.2; Anal. Calcd. for C20H19N3O3: C, 68.75; H, 5.48; N, 12.03; O, 13.74. Found: C, 68.72; H, 5.36; N, 12.08; O, 13.66%.
(E)-2-(N-phenylbenzimidazol-2-yl)-3-(3,4,5-trimethoxyphenyl)acrylonitrile 54
Compound 54 was prepared from 20 (0.10 g, 0.4 mmol) and 29 (0.08 g, 0.4 mmol) in absolute ethanol (3 mL) after refluxing for 4 h to yield 0.17 g (71%) of orange oil; 1H NMR (DMSO-d6, 300 MHz): δ/ppm = 7.99 (s, 1H, Harom), 7.81 (d, 1H, J = 6.8 Hz, Harom), 7.67–7.62 (m, 3H, Harom), 7.51–7.46 (m, 1H, Harom), 7.38–7.33 (m, 2H, Harom), 7.27 (s, 2H, Harom), 7.22 (d, 1H, J = 7.2 Hz, Harom), 7.13–7.10 (m, 1H, Harom), 3.79 (s, 3H, OCH3), 3.75 (s, 3H, OCH3); 13C NMR (DMSO-d6, 101 MHz): δ/ppm = 153.3, 153.0, 152.5, 151.0, 135.5, 134.6, 130.7, 130.6, 130.2, 130.2, 129.2, 128.0, 126.4, 126.3, 125.2, 124.0, 123.9, 120.4, 107.6, 107.5, 60.8, 60.7, 56.5, 55.8, 55.7; Anal. Calcd. for C25H21N3O3: C, 72.98; H, 5.14; N, 10.21; O, 11.67. Found: C, 72.86; H, 5.06; N, 10.18; O, 11.59%.
(E)-2-(6-cyano-N-isobutylbenzimidazol-2-yl)-3-(3,4,5-trimethoxyphenyl)acrylonitrile 55
Compound 55 was prepared from 22 (0.10 g, 0.4 mmol) and 29 (0.08 g, 0.4 mmol) in absolute ethanol (3 mL) after refluxing for 4 h to yield 0.17 g (56%) of yellow powder; m.p 163–167 °C; 1H NMR (DMSO-d6, 600 MHz): δ/ppm = 8.33 (s, 1H, Harom), 8.28 (d, 1H, J = 1.2 Hz, Harom), 7.98 (d, 1H, J = 8.4 Hz, Harom), 7.74 (dd, 1H, J = 8.5, 1.4 Hz, Harom), 7.51 (s, 2H, Harom), 4.43 (d, 2H, J = 7.6 Hz, CH2), 3.86 (s, 6H, OCH3), 3.80 (s, 3H, OCH3), 2.20–2.15 (m, 1H, CH), 0.84 (d, 6H, J = 6.6 Hz, CH3); 13C NMR (DMSO-d6, 151 MHz): δ/ppm = 152.9, 152.3, 149.5, 141.2, 141.1, 139.3, 127.6, 126.4, 124.3, 119.5, 116.7, 113.1, 108.0, 105.0, 97.7, 60.3, 56.0 (2C), 51.0, 29.3, 19.4; Anal. Calcd. for C24H24N4O3: C, 69.21; H, 5.81; N, 13.45; O, 11.52. Found: C, 69.19; H, 5.86; N, 13.38; O, 11.56%.
(E)-2-(6-cyano-N-methylbenzimidazol-2-yl)-3-(3,4,5-trimethoxyphenyl)acrylonitrile 56
Compound 56 was prepared from 23 (0.10 g, 0.5 mmol) and 29 (0.09 g, 0.5 mmol) in absolute ethanol (3 mL) after refluxing for 4 h to yield 0.16 g (88%) of yellow powder; m.p 259–262 °C; 1H NMR (DMSO-d6, 400 MHz): δ/ppm = 8.28 (d, 1H, J = 1.0 Hz, Harom), 8.19 (s, 1H, Harom), 7.89 (d, 1H, J = 8.5 Hz, Harom), 7.75 (dd, 1H, J = 8.5, 1.5 Hz, Harom), 7.50 (s, 2H, Harom), 4.06 (s, 3H, OCH3), 3.87 (s, 3H, OCH3), 3.80 (s, 3H, OCH3); 13C NMR (DMSO-d6, 101 MHz): δ/ppm = 153.4, 151.8, 150.7, 141.7, 141.5, 139.9, 128.2, 126.9, 124.7, 120.2, 117.3, 113.0, 108.3, 105.3, 98.9, 60.8, 56.5 (2C), 32.6; Anal. Calcd. for C21H18N4O3: C, 67.37; H, 4.85; N, 14.96; O, 12.82. Found: C, 67.29; H, 4.89; N, 14.88; O, 12.86%.
(E)-2-(6-cyano-N-phenylbenzimidazol-2-yl)-3-(3,4,5-trimethoxyphenyl)acrylonitrile 57
Compound 57 was prepared from 24 (0.10 g, 0.4 mmol) and 29 (0.08 g, 0.4 mmol) in absolute ethanol (3 mL) after refluxing for 4 h to yield 0.17 g (83%) of yellow powder; m.p 147–150 °C; 1H NMR (DMSO-d6, 600 MHz): δ/ppm = 8.39 (d, 1H, J = 0.9 Hz, Harom), 8.06 (s, 1H, Harom), 7.71 (dd, 1H, J = 8.5, 1.4 Hz, Harom), 7.69–7.66 (m, 5H, Harom), 7.36 (d, 1H, J = 8.4 Hz, Harom), 7.29 (s, 2H, Harom), 3.79 (s, 3H, OCH3), 3.76 (s, 3H, OCH3); 13C NMR (DMSO-d6, 75 MHz): δ/ppm = 153.4, 152.3, 145.0, 141.9, 140.1, 134.8, 130.7, 130.5, 128.1, 127.9, 127.9, 124.9, 119.9, 115.9, 112.7, 108.2, 106.1, 98.4, 60.8, 56.5 (2C); Anal. Calcd. for C26H20N4O3: C, 71.55; H, 4.62; N, 12.84; O, 11.00. Found: C, 71.48; H, 4.65; N, 12.78; O, 11.07%.
(E)-2-(1H-benzimidazol-2-yl)-3-(4-N,N-dimethylaminophenyl)acrylonitrile 58
Compound 58 was prepared from 17 (0.10 g, 0.6 mmol) and 30 (0.09 g, 0.6 mmol) in absolute ethanol (2 mL) after refluxing for 3 h to yield 0.14 g (76%) of orange powder; m.p 272–277 °C; 1H NMR (DMSO-d6, 300 MHz): δ/ppm = 12.78 (s, 1H, NHbenz), 8.13 (s, 1H, Harom), 7.91 (d, 1H, J = 9.0 Hz, Harom), 7.63 (d, 1H, J = 7.4 Hz, Harom), 7.50 (d, 1H, J = 7.1 Hz, Harom), 7.25–7.16 (m, 2H, Harom), 6.87 (d, 2H, J = 9.1 Hz, Harom), 3.07 (s, 6H, CH3); 13C NMR (DMSO-d6, 75 MHz): δ/ppm = 164.4, 152.9, 150.9, 149.4, 145.9, 133.1 (2C), 132.3, 120.3, 119.2, 118.2, 112.3 (2C), 112.1 (2C), 94.2; Anal. Calcd. for C18H16N4: C, 74.98; H, 5.59; N, 19.43. Found: C, 74.91; H, 5.76; N, 19.38%.
(E)-3-(4-N,N-dimethylaminophenyl)-2-(N-methylbenzimidazol-2-yl)acrylonitrile 59
Compound 59 was prepared from 19 (0.10 g, 0.6 mmol) and 30 (0.09 g, 0.6 mmol) in absolute ethanol (2 mL) after refluxing for 3.5 h to yield 0.10 g (58%) of red powder; m.p 202–206 °C; 1H NMR (DMSO-d6, 400 MHz): δ/ppm = 7.97 (s, 1H, Harom), 7.86 (d, 3H, J = 9.1 Hz, Harom), 7.60–7.45 (m, 2H, Harom), 6.83 (d, 3H, J = 9.1 Hz, Harom), 3.06 (s, 9H, CH3); 13C NMR (DMSO-d6, 101 MHz): δ/ppm = 164.4, 153.4, 150.9, 133.1 (2C), 119.2 (2C), 118.7 (2C), 112.1 (2C), 97.7, 40.0 (3C); Anal. Calcd. for C19H18N4: C, 75.47; H, 6.00; N, 18.53. Found: C, 75.56; H, 5.94; N, 18.47%.
(E)-3-(4-N,N-dimethylaminophenyl)-2-(N-phenylbenzimidazol-2-yl)acrylonitrile 60
Compound 60 was prepared from 20 (0.10 g, 0.4 mmol) and 30 (0.06 g, 0.4 mmol) in absolute ethanol (2 mL) after refluxing for 3.5 h to yield 0.11 g (69%) of yellow powder; m.p 206–209 °C; 1H NMR (DMSO-d6, 300 MHz): δ/ppm = 7.77 (dd, 1H, J = 7.0, 1.1 Hz, Harom), 7.73 (d, 3H, J = 8.8 Hz, Harom), 7.68–7.56 (m, 5H, Harom), 7.34 (td, 1H, J = 7.4, 1.4 Hz, Harom), 7.27 (td, 1H, J = 7.9, 1.4 Hz, Harom), 7.17 (dd, 1H, J = 7.1, 1.4 Hz, Harom), 6.79 (d, 2H, J = 9.1 Hz, Harom), 3.04 (s, 3H, CH3); 13C NMR (DMSO-d6, 75 MHz): δ/ppm = 153.1, 150.6, 148.7, 142.6, 137.4, 136.0, 133.1, 132.4, 130.6, 129.8, 128.0, 124.0, 123.5, 120.0, 119.5, 117.6, 112.1, 110.8, 91.9, 40.0 (2C); Anal. Calcd. for C24H20N4: C, 79.10; H, 5.53; N, 15.37. Found: C, 79.17; H, 5.47; N, 15.43%.
E(Z)-3-(4-N,N-dimethylaminophenyl)-2-(N-hexylbenzimidazol-2-yl)acrylonitrile 61
Compound 61 was prepared from 21 (0.07 g, 0.3 mmol) and 30 (0.04 g, 0.3 mmol) in absolute ethanol (2 mL) after refluxing for 3 h to yield 0.10 g (94%) of brown oil in the form of a mixture of E- and Z-isomers at a ratio of 61a/61b = 2:1; 61a: 1H NMR (DMSO-d6, 400 MHz): δ/ppm = 7.99 (s, 1H, Harom), 7.97 (d, 1H, J = 9.0 Hz, Harom), 7.67 (d, 1H, J = 7.6 Hz, Harom), 7.63 (d, 1H, J = 7.9 Hz, Harom), 7.33–7.27 (m, 1H, Harom), 7.27–7.23 (m, 1H, Harom), 6.88 (t, 2H, J = 8.7 Hz, Harom), 6.56 (d, 1H, J = 9.1 Hz, Harom), 4.43 (t, 2H, J = 7.5 Hz, CH2), 3.08 (s, 6H, CH3), 1.84–1.76 (m, 2H, CH2), 1.30–1.20 (m, 6H, CH2), 0.80 (t, 6H, J = 7.0 Hz, CH3); 13C NMR (DMSO-d6, 101 MHz): δ/ppm = 152.8, 151.7, 146.0, 143.1, 135.2, 132.6 (2C), 123.8, 122.8, 120.2, 120.1, 119.9, 112.1 (2C), 111.9, 93.3, 44.2, 31.1, 29.6, 26.1, 22.4, 14.2 (2C); 61b: 1H NMR (DMSO-d6, 400 MHz): δ/ppm = 7.97 (d, 2H, J = 9.0 Hz, Harom), 7.87 (s, 1H, Harom), 7.74 (d, 1H, J = 7.8 Hz, Harom), 7.63 (d, 1H, J = 7.9 Hz, Harom), 7.38–7.32 (m, 1H, Harom), 7.33–7.27 (m, 1H, Harom), 6.88 (t, 2H, J = 8.7 Hz, Harom), 4.03 (t, 2H, J = 7.1 Hz, CH2), 2.92 (s, 6H, CH3), 1.65–1.59 (m, 2H, CH2), 1.14–1.08 (m, 6H, CH2), 0.76 (t, 6H, J = 6.7 Hz, CH3); 13C NMR (DMSO-d6, 101 MHz): δ/ppm = 153.1, 151.3, 148.7, 142.6, 136.4, 132.4 (2C), 123.8, 123.2, 120.3, 119.9, 119.4, 111.9 (2C), 111.8, 91.2, 44.4, 31.1, 29.7, 26.2, 22.3, 14.2 (2C); Anal. Calcd. for C24H28N4: C, 77.38; H, 7.58; N, 15.04. Found: C, 77.41; H, 7.66; N, 15.08%.
(E)-2-(5-cyano-N-hexylbenzimidazol-2-yl)-3-(4-N,N-dimethylaminophenyl)acrylonitrile 62
Compound 62 was prepared from 25 (0.05 g, 0.2 mmol) and 30 (0.03 g, 0.2 mmol) in absolute ethanol (1.5 mL) after refluxing for 3 h to yield 0.3 g (76%) of orange oil; 1H NMR (DMSO-d6, 600 MHz): δ/ppm = 8.20 (d, 1H, J = 1.0 Hz, Harom), 8.08 (s, 1H, Harom), 7.98 (d, 2H, J = 9.1 Hz, Harom), 7.87 (d, 1H, J = 9.1 Hz, Harom), 7.69 (dd, 1H, J = 8.4, 1.4 Hz, Harom), 6.87 (d, 2H, J = 9.1 Hz, Harom), 4.49 (t, 2H, J = 7.5 Hz, CH2), 3.08 (s, 6H, CH3), 1.81–1.78 (m, 2H, CH2), 1.26–1.23 (m, 6H, CH2), 0.80 (t, 3H, J = 6.7 Hz, CH3); 13C NMR (DMSO-d6, 151 MHz): δ/ppm = 153.4, 152.4, 151.5, 142.0, 139.3, 133.0 (2C), 126.4, 124.2, 120.2, 120.1, 118.6, 112.8, 112.2 (2C), 105.1, 89.9, 44.8, 31.0, 29.7, 26.0, 22.4, 14.2 (2C); Anal. Calcd. for C25H27N5: C, 75.54; H, 6.85; N, 17.62. Found: C, 75.61; H, 6.83; N, 17.58%.
(E)-2-(1H-benzimidazol-2-yl)-3-(4-N,N-diethylaminophenyl)acrylonitrile 63
Compound 63 was prepared from 17 (0.10 g, 0.6 mmol) and 31 (0.11 g, 0.6 mmol) in absolute ethanol (2 mL) after refluxing for 3 h to yield 0.14 g (68%) of orange powder; m.p 151–156 °C; 1H NMR (DMSO-d6, 300 MHz): δ/ppm = 12.75 (s, 1H, NHbenz), 8.10 (s, 1H, Harom), 7.97–7.81 (m, 3H, Harom), 7.53 (bs, 2H, Harom), 7.23–7.17 (m, 1H, Harom), 6.87–6.77 (m, 2H, Harom), 3.47 (q, 4H, J = 6.4 Hz, CH2), 1.15 (t, 6H, J = 6.7 Hz, CH3); 13C NMR (DMSO-d6, 151 MHz): δ/ppm = 150.6, 149.5, 145.9, 144.0, 135.3, 133.5, 132.8, 123.2, 122.3, 119.6, 119.0, 118.4 (2C), 111.8, 111.6, 93.4, 44.4 (2C), 12.9 (2C); Anal. Calcd. for C20H20N4: C, 75.92; H, 6.37; N, 17.71. Found: C, 75.97; H, 6.46; N, 17.76%.
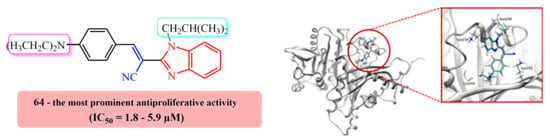
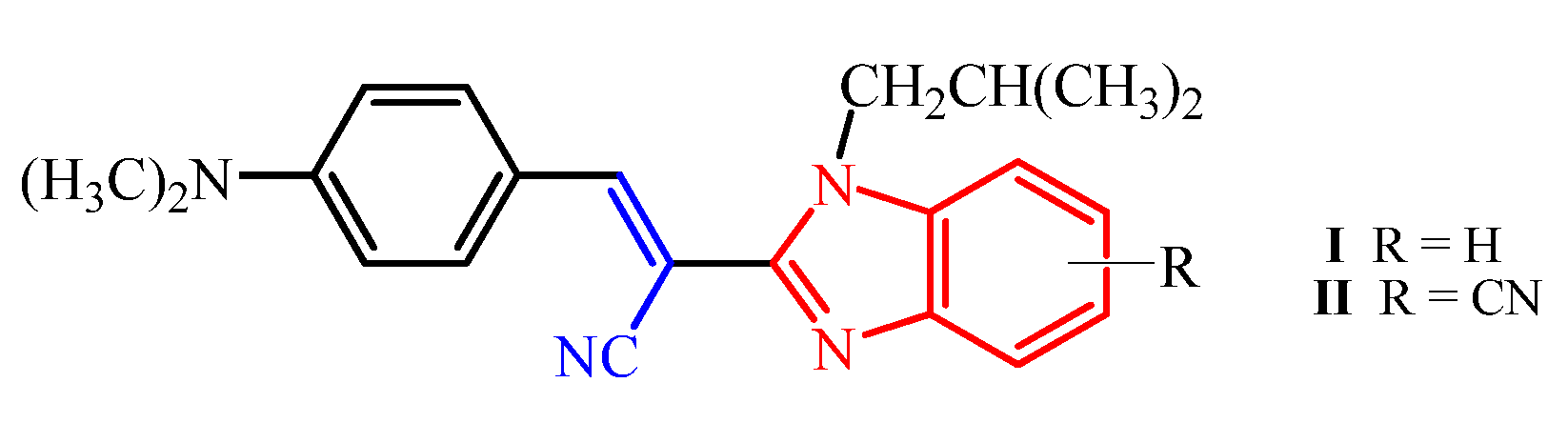
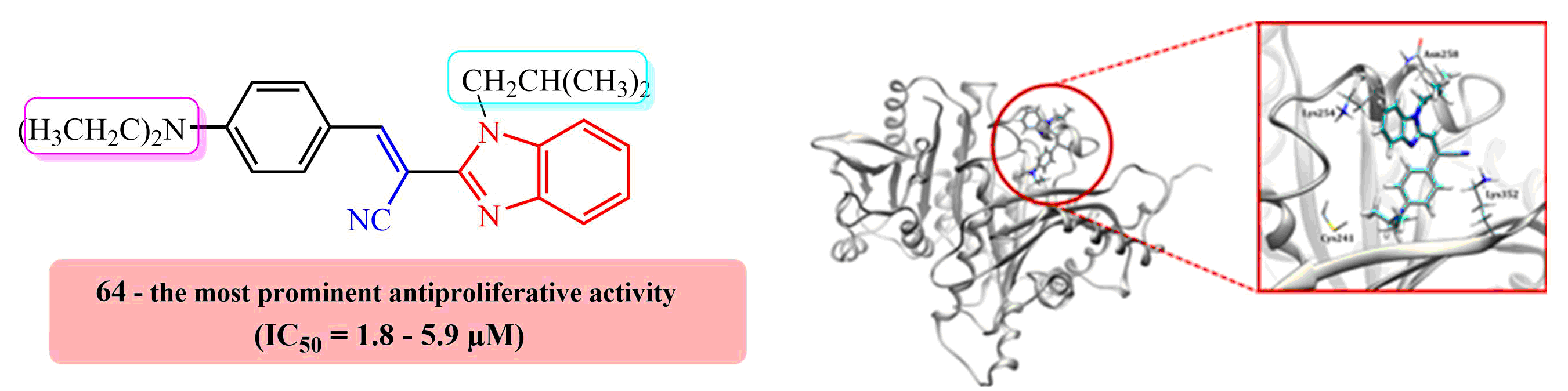
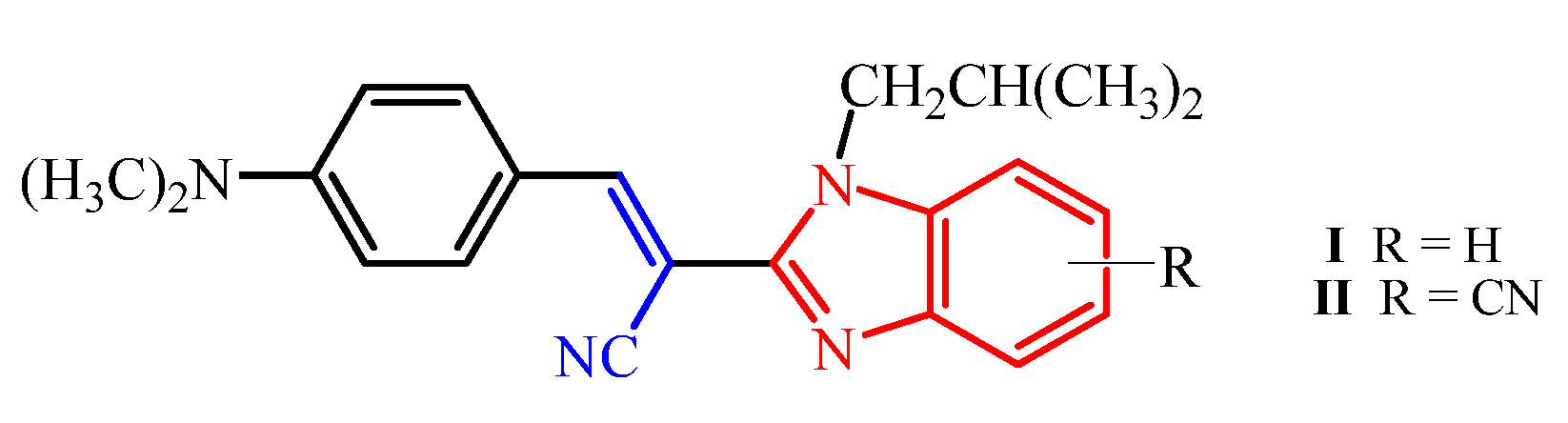
E(Z)-3-(4-N,N-diethylaminophenyl)-2-(N-isobutylbenzimidazol-2-yl)acrylonitrile 64
Compound 64 was prepared from 18 (0.10 g, 0.5 mmol) and 31 (0.08 g, 0.5 mmol) in absolute ethanol (2 mL) after refluxing for 3 h to yield 0.10 g (61%) of light red oil in the form of a mixture of E- and Z-isomers at a ratio of 64a/64b = 2:1; 64a: 1H NMR (DMSO-d6, 600 MHz): δ/ppm = 8.02 (s, 1H, Harom), 7.95 (d, 2H, J = 9.1 Hz, Harom), 7.67–7.63 (m, 2H, Harom), 7.31–7.29 (m, 2H, Harom), 7.24 (td, 1H, J = 7.6, 1.1 Hz, Harom), 6.85–6.81 (m, 1H, Harom), 6.52 (d, 1H, J = 9.2 Hz, Harom), 4.31 (d, 2H, J = 7.5 Hz, CH2), 3.47 (q, 4H, J = 7.0 Hz, CH2), 2.22–2.09 (m, 1H, CH), 1.15 (t, 6H, J = 6.9 Hz, CH3), 0.83 (d, 6H, J = 6.7 Hz, CH3); 13C NMR (DMSO-d6, 151 MHz): δ/ppm = 151.3, 150.9, 148.8, 142.4, 136.9, 133.0 (2C), 123.1, 122.7, 119.7, 119.3, 119.0, 111.7 (2C), 111.5, 90.6, 51.2, 44.4 (2C), 29.6, 20.1 (2C), 12.9 (2C); 64b: 1H NMR (DMSO-d6, 600 MHz): δ/ppm = 7.79 (s, 1H, Harom), 7.73 (d, 1H, J = 7.9 Hz, Harom), 7.69 (d, 1H, J = 8.0 Hz, Harom), 7.35 (td, 1H, J = 7.5, 1.0 Hz, Harom), 7.31–7.29 (m, 1H, Harom), 6.85–6.81 (m, 4H, Harom), 3.84 (d, 2H, J = 7.6 Hz, CH2), 3.31 (q, 4H, J = 6.8 Hz, CH2), 2.12–2.08 (m, 1H, CH), 1.02 (t, 6H, J = 6.9 Hz, CH3), 0.79 (d, 6H, J = 6.6 Hz, CH3); 13C NMR (DMSO-d6, 151 MHz): δ/ppm = 151.3, 150.5, 146.3, 143.0, 135.5, 132.8 (2C), 123.7, 122.8, 120.2, 120.1, 119.4, 112.1, 111.6 (2C), 92.6, 51.3, 44.2 (2C), 29.1, 20.2 (2C), 12.8 (2C); Anal. Calcd. for C24H28N4: C, 77.38; H, 7.58; N, 15.04. Found: C, 77.42; H, 7.63; N, 15.10%.
(E)-3-(4-N,N-diethylaminophenyl)-2-(N-methylbenzimidazol-2-yl)acrylonitrile 65
Compound 65 was prepared from 19 (0.10 g, 0.6 mmol) and 31 (0.10 g, 0.6 mmol) in absolute ethanol (2 mL) after refluxing for 2.5 h to yield 0.12 g (65%) of orange oil; 1H NMR (DMSO-d6, 400 MHz): δ/ppm = 7.95 (s, 1H, Harom), 7.84 (d, 3H, J = 9.1 Hz, Harom), 7.60–7.45 (m, 2H, Harom), 6.80 (d, 3H, J = 9.1 Hz, Harom), 3.45 (q, 4H, J = 7.0 Hz, Harom), 1.13 (t, 6H, J = 7.0 Hz, Harom); 13C NMR (DMSO-d6, 101 MHz): δ/ppm = 164.5, 151.2, 150.8 (2C), 147.5, 133.5 (2C), 133.2, 118.9, 118.6, 111.7 (2C), 111.2, 96.9, 44.4 (2C), 12.9 (3C); Anal. Calcd. for C21H22N4: C, 76.33; H, 6.71; N, 16.96. Found: C, 76.27; H, 6.76; N, 16.88%.
(E)-3-(4-N,N-diethylaminophenyl)-2-(N-phenylbenzimidazol-2-yl)acrylonitrile 66
Compound 66 was prepared from 20 (0.10 g, 0.4 mmol) and 31 (0.08 g, 0.4 mmol) in absolute ethanol (2.5 mL) after refluxing for 2 h to yield 0.06 g (50%) of red powder; m.p 142–147 °C; 1H NMR (DMSO-d6, 600 MHz): δ/ppm = 7.75 (d, 1H, J = 7.9 Hz, Harom), 7.70 (d, 2H, J = 9.0 Hz, Harom), 7.67–7.63 (m, 3H, Harom), 7.61–7.59 (m, 1H, Harom), 7.59–7.56 (m, 2H, Harom), 7.32 (td, 1H, J = 7.6, 0.9 Hz, Harom), 7.27 (td, 1H, J = 8.1, 1.0 Hz, Harom), 7.15 (d, 1H, J = 8.0 Hz, Harom), 6.75 (d, 2H, J = 9.0 Hz, Harom), 3.43 (q, 4H, J = 6.9 Hz, CH2), 1.11 (t, 6H, J = 7.0 Hz, CH3); 13C NMR (DMSO-d6, 151 MHz): δ/ppm = 150.8, 150.4, 148.9, 142.6, 137.4, 136.1, 132.9 (2C), 130.6 (2C), 129.7, 128.0 (2C), 124.0, 123.5, 119.5, 119.5, 117.8, 111.7 (2C), 110.7, 91.2, 44.4 (2C), 12.9 (2C); Anal. Calcd. for C26H24N4: C, 79.56; H, 6.16; N, 14.27. Found: C, 79.51; H, 6.26; N, 14.19%.
E(Z)-3-(4-N,N-diethylaminophenyl)-2-(N-hexylbenzimidazol-2-yl)acrylonitrile 67
Compound 67 was prepared from 21 (0.07 g, 0.3 mmol) and 31 (0.05 g, 0.3 mmol) in absolute ethanol (2 mL) after refluxing for 3 h to yield 0.10 g (87%) of light red oil in the form of a mixture of E- and Z-isomers at a ratio of 67a/67b = 2:1; 67a: 1H NMR (DMSO-d6, 400 MHz): δ/ppm = 7.96 (s, 2H, Harom), 7.69–7.65 (m, 1H, Harom), 7.65–7.61 (m, 1H, Harom), 7.27–7.22 (m, 1H, Harom), 6.87 (d, 1H, J = 9.1 Hz, Harom), 6.83 (d, 2H, J = 9.1 Hz, Harom), 6.52 (d, 1H, J = 9.2 Hz, Harom), 4.43 (t, 2H, J = 7.4 Hz, CH2), 3.48 (q, 4H, J = 7.0 Hz, CH2), 1.84–1.75 (m, 2H, CH2), 1.30–1.20 (m, 6H, CH2), 1.15 (t, 6H, J = 7.0 Hz, CH3), 0.80 (t, 3H, J = 7.0 Hz, CH3); 13C NMR (DMSO-d6, 151 MHz): δ/ppm = 151.2, 150.9, 148.8, 142.6, 136.4, 133.0 (2C), 123.2, 122.8, 119.7, 119.3, 118.9, 111.7 (2C), 111.2, 90.4, 44.4 (2C), 31.1, 29.7, 26.1, 22.4, 14.2, 12.9 (2C); 67b: 1H NMR (DMSO-d6, 400 MHz): δ/ppm = 7.94 (s, 2H, Harom), 7.83 (s, 1H, Harom), 7.74 (d, 1H, J = 7.4 Hz, Harom), 7.65–7.61 (m, 1H, Harom), 7.38–7.33 (m, 1H, Harom), 7.32–7.30 (m, 1H, Harom), 7.30–7.27 (m, 2H, Harom), 4.06 (t, 2H, J = 7.1 Hz, CH2), 1.67–1.57 (m, 2H, CH2), 1.12–1.08 (m, 6H, CH2), 1.02 (t, 6H, J = 7.0 Hz, CH3), 0.74 (t, 3H, J = 6.7 Hz, CH3); 13C NMR (DMSO-d6, 151 MHz): δ/ppm = 151.5, 150.5, 146.1, 143.2, 135.2, 132.8 (2C), 123.7, 122.8, 120.2, 120.0, 119.5, 111.4 (2C), 111.0, 92.4, 44.2 (2C), 31.2, 29.5, 26.2, 22.3, 14.2, 12.8 (2C); Anal. Calcd. for C26H32N4: C, 77.96; H, 8.05; N, 13.99. Found: C, 77.91; H, 8.16; N, 14.03%.
E(Z)-2-(6-cyano-N-butylbenzimidazol-2-yl)-3-(4-N,N-diethylaminophenyl)acrylonitrile 68
Compound 68 was prepared from 22 (0.10 g, 0.4 mmol) and 31 (0.07 g, 0.4 mmol) in absolute ethanol (2 mL) after refluxing for 3 h to yield 0.10 g (61%) of red oil in the form of a mixture of E- and Z-isomers at a ratio of 68a/68b = 2:1; 68a: 1H NMR (DMSO-d6, 600 MHz): δ/ppm = 8.20 (d, 1H, J = 1.2 Hz, Harom), 8.13 (s, 1H, Harom), 7.97 (d, 2H, J = 9.1 Hz, Harom), 7.68 (dd, 1H, J = 8.4, 1.4 Hz, Harom), 6.85–6.81 (m, 2H, Harom), 6.54 (d, 1H, J = 9.2 Hz, Harom), 4.39 (d, 2H, J = 7.6 Hz, CH2), 3.48 (q, 4H, J = 7.0 Hz, CH2), 2.18–2.13 (m, 1H, CH), 1.15 (t, 6H, J = 7.0 Hz, CH3), 0.83 (d, 6H, J = 6.6 Hz, CH3);
13C NMR (DMSO-d6, 151 MHz): δ/ppm = 152.3, 151.6, 151.2, 141.8, 139.9, 133.5 (2C), 126.3, 124.1, 120.2, 119.6, 118.7, 113.1, 111.8 (2C), 105.0, 89.4, 51.4, 44.5 (2C), 29.7, 19.9 (2C), 12.9 (2C); 68b: 1H NMR (DMSO-d6, 600 MHz): δ/ppm = 8.33 (d, 1H, J = 1.0 Hz, Harom), 7.95 (d, 1H, J = 8.6 Hz, Harom), 7.90 (d, 2H, J = 8.4 Hz, Harom), 7.86 (s, 1H, Harom), 7.76 (dd, 1H, J = 8.5, 1.5 Hz, Harom), 6.85–6.81 (m, 2H, Harom), 3.91 (d, 2H, J = 7.6 Hz, CH2), 3.32 (q, 4H, J = 6.8 Hz, CH2), 2.10–2.05 (m, 1H, CH), 1.02 (t, 6H, J = 7.0 Hz, CH3), 0.79 (d, 6H, J = 6.6 Hz, CH3); 13C NMR (DMSO-d6, 151 MHz): δ/ppm = 152.1, 150.7, 149.3, 142.4, 138.4, 132.9 (2C), 127.0, 125.3, 120.1, 119.8, 119.1, 113.8, 111.6 (2C), 105.3, 91.2, 51.5, 44.2 (2C), 29.2, 20.0 (2C), 12.8 (2C); Anal. Calcd. for C25H27N5: C, 75.54; H, 6.85; N, 17.62. Found: C, 75.50; H, 6.89; N, 17.57%.
(E)-2-(6-cyano-N-methylbenzimidazol-2-yl)-3-(4-N,N-diethylaminophenyl)acrylonitrile 69
Compound 69 was prepared from 23 (0.10 g, 0.5 mmol) and 31 (0.09 g, 0.5 mmol) in absolute ethanol (2 mL) after refluxing for 2.5 h to yield 0.15 g (84%) of red powder; m.p 164–169 °C; 1H NMR (DMSO-d6, 300 MHz): δ/ppm = 8.18 (s, 1H, Harom), 7.99 (d, 2H, J = 4.7 Hz, Harom), 7.95 (s, 1H, Harom), 7.82 (d, 1H, J = 8.4 Hz, Harom), 7.68 (dd, 1H, J = 8.4, 1.4 Hz, Harom), 6.84 (d, 2H, J = 9.1 Hz, Harom), 4.00 (s, 3H, CH3), 3.48 (q, 4H, J = 7.0 Hz, Harom), 1.15 (t, 6H, J = 7.0 Hz, Harom); 13C NMR (DMSO-d6, 151 MHz): δ/ppm = 152.3, 151.7, 151.1, 142.0, 140.0, 133.4 (2C), 126.3, 124.0, 120.3, 119.6, 118.7, 112.5, 111.7 (2C), 104.9, 89.8, 44.5 (2C), 32.5, 13.0 (2C); Anal. Calcd. for C22H21N5: C, 74.34; H, 5.96; N, 19.70. Found: C, 74.28; H, 5.86; N, 19.78%.
(E)-2-(6-cyano-N-phenylbenzimidazol-2-yl)-3-(4-N,N-diethylaminophenyl)acrylonitrile 70
Compound 70 was prepared from 24 (0.10 g, 0.4 mmol) and 31 (0.07 g, 0.4 mmol) in absolute ethanol (2 mL) after refluxing for 2 h to yield 0.14 g (86%) of red powder; m.p 210–214 °C; 1H NMR (DMSO-d6, 300 MHz): δ/ppm = 8.31 (d, 1H, J = 0.8 Hz, Harom), 7.74 (s, 2H, Harom), 7.70 (s, 1H, Harom), 7.69–7.57 (m, 6H, Harom), 7.29 (d, 1H, J = 8.5 Hz, Harom), 6.77 (d, 2H, J = 9.1 Hz, Harom), 3.45 (q, 4H, J = 6.9 Hz, CH2), 1.13 (t, 6H, J = 7.0 Hz, CH3); 13C NMR (DMSO-d6, 75 MHz): δ/ppm = 151.7, 151.5, 151.2, 142.2, 140.3, 135.3, 133.2 (2C), 130.7 (2C), 130.3, 128.1 (2C), 127.2, 124.2, 120.1, 119.3, 117.4, 112.2, 111.8, 105.7, 90.1, 44.5 (2C), 12.7 (2C); Anal. Calcd. for C27H23N5: C, 77.67; H, 5.55; N, 16.77. Found: C, 77.61; H, 5.66; N, 16.73%.
(E)-2-(6-cyano-N-hexylbenzimidazol-2-yl)-3-(4-N,N-diethylaminophenyl)acrylonitrile 71
Compound 71 was prepared from 25 (0.05 g, 0.2 mmol) and 31 (0.03 g, 0.2 mmol) in absolute ethanol (1.5 mL) after refluxing for 3 h to yield 0.03 g (40%) of orange powder; m.p 118–122 °C; 1H NMR (DMSO-d6, 400 MHz): δ/ppm = 8.19 (d, 1H, J = 1.0 Hz, Harom), 8.05 (s, 1H, Harom), 7.97 (d, 2H, J = 9.1 Hz, Harom), 7.87 (d, 1H, J = 8.5 Hz, Harom), 7.69 (dd, 1H, J = 8.4, 1.4 Hz, Harom), 6.84 (d, 2H, J = 9.2 Hz, Harom), 4.49 (t, 2H, J = 7.4 Hz, CH2), 3.49 (q, 4H, J = 6.9 Hz, CH2), 1.84–1.75 (m, 2H, CH2), 1.30–1.20 (m, 6H, CH2), 1.15 (t, 6H, J = 6.7 Hz, CH3), 0.80 (t, 3H, J = 6.7 Hz, CH3); 13C NMR (DMSO-d6, 151 MHz): δ/ppm = 152.2, 151.6, 151.2, 142.0, 139.4, 133.5 (2C), 126.4, 124.1, 120.3, 119.5, 118.7, 112.8, 111.8 (2C), 105.0, 89.2, 44.8, 44.5 (2C), 31.0, 29.7, 26.0, 22.4, 14.2, 12.9 (2C); Anal. Calcd. for C27H31N5: C, 76.20; H, 7.34; N, 16.46. Found: C, 76.23; H, 7.28; N, 16.38%.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}