1. Introduction
Nowadays, tablet remains the most commonly used dosage form on the pharmaceutical market. In recent years, at least half of the drugs approved by the U.S. Food and Drug Administration’s Center for Drug Evaluation and Research (CDER) have been in solid dosage form (54% in 2019, 53% in 2018, and 50% in 2017) [
1,
2,
3]. Among the general tablet manufacturing methods, direct compression (DC) stands the most prominent due to the cost-effectiveness, simplicity, and high stability it offers to the drug products [
4]. However, manufacturing without a granulation process and recent advancement in pharmaceutical tableting, such as continuous manufacturing and high-speed tableting machines, could limit this production method with the conventional excipients [
5,
6]. Simultaneously, it encourages the development of a new excipient to support tablet manufacturing under specific production conditions.
In the past few decades, the concept of a co-processed excipient has gained a lot of attention, as it is the simplest and fastest way to develop a new excipient without the requirement to study toxicity for regulatory approval [
7]. In general, a co-processed excipient outperforms the physical mixtures in terms of pharmaceutical properties because it synergizes the good properties of each excipient [
8,
9]. Moreover, a previous study has reported that the co-processed excipient improved content uniformity of dosage unit since the excipients were intimately combined, thus reducing the segregation problem [
8]. For these reasons, the co-processed excipient, which plays multiple roles in the formulation, could support tablet manufacturing with the DC technique.
In the previous study, we reported the ability of a novel rice starch-based, co-processed excipient (CS), prepared using a spray drying technique, as a multifunctional excipient for DC [
9]. Native rice starch (RS) is commonly used as a pharmaceutical excipient, but its application in DC is limited due to the poor flowability and the low disintegration property [
10]. Upon co-processing with cross-linked carboxymethyl starch (CCMS) and silicon dioxide, the functionality was improved. The CSs showed agglomerate spherical particles, thus improving the flowability of the material. The co-processing with CCMS, a superdisintegrant, provided rapid disintegration time of the co-processed tablet, while still preserving an adequate compressibility profile [
11]. Nevertheless, CS needed more investigation to be applied in DC formulation.
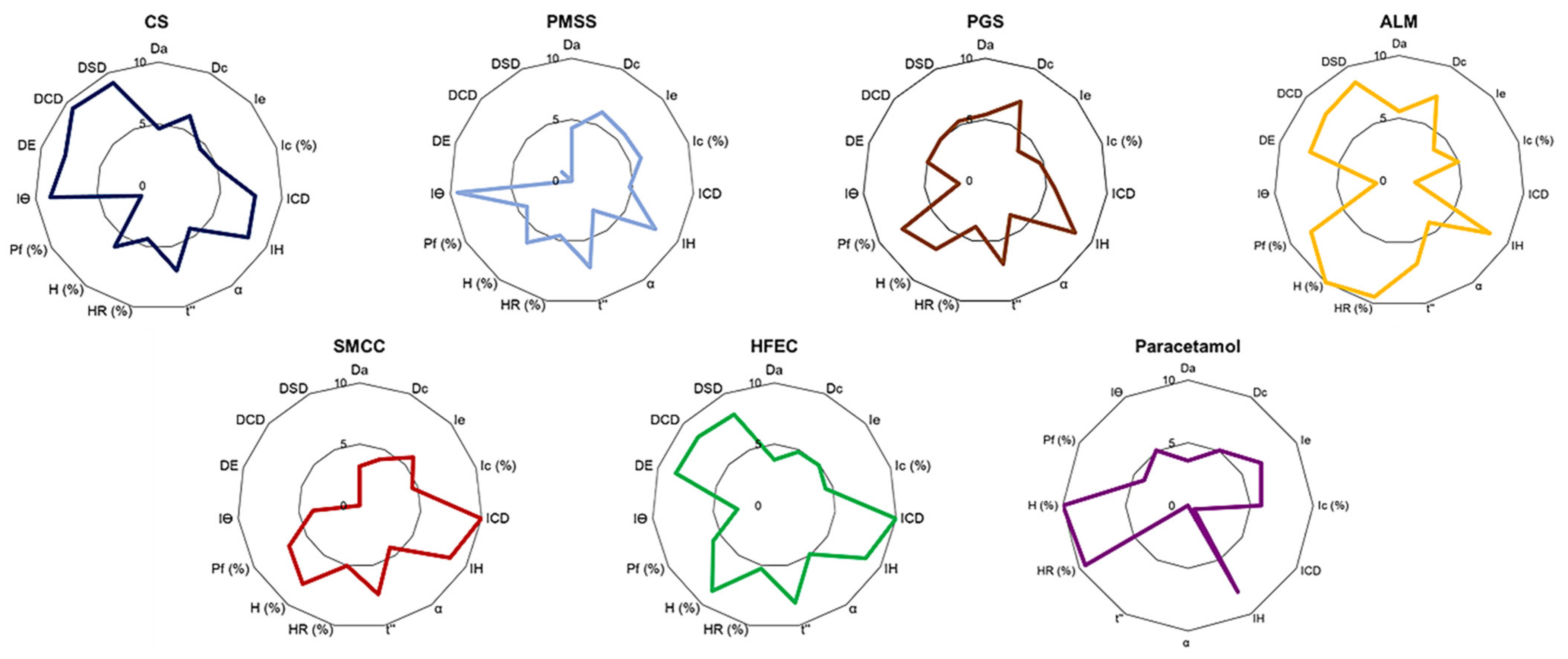
In the past, pharmaceutical formulation development depended mostly on experiments and research experience, which were time-consuming and costly processes. The SeDeM expert system developed by Carreras et al. [
12] could improve this process based on the concept of pharmaceutical quality by design (QbD), which is a systematic approach to create a new pharmaceutical product based on a quality risk management and sound, logical science [
13]. This is a new galenic method that is used in tablet pre-formulation and formulation studies, particularly for the DC method. The powder characteristics of active pharmaceutical ingredients (APIs) and excipients are evaluated through the SeDeM diagram to determine whether the materials are suitable for DC manufacturing or need additional excipients for tableting. Recently, a SeDeM-ODT expert system has been developed to include disgregability factor, making it more advantageous than the old SeDeM method in the assessment of excipient properties [
14].
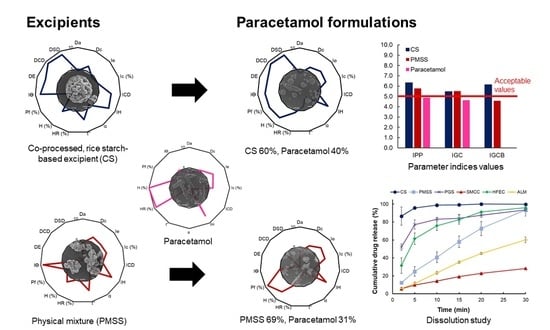
In this work, the SeDeM-ODT expert system is applied for the formulation study of CS, in comparison with the physical mixture and commercial excipients, as a multifunctional excipient for DC technique. Paracetamol is used as a model drug due to the unsatisfactory powder characteristics, which contribute to the difficulty in DC production. The powder characteristics of the excipients and the API are determined, and the paracetamol tablets are then produced based on the calculated percentage of a corrective excipient obtained from the SeDeM-ODT expert system.
3. Materials and Methods
3.1. Materials
Native rice starch (RS) (Lot no. 709161) was purchased from Thai Flour Industry Co., Ltd. (Bangkok, Thailand). Monochloroacetic acid (MCA, CAS No. 79-11-8, Product Code 8004121000) and sodium silicate (CAS No. 1344-09-8, Product Code 1056212500) were purchased from Merck (Hohenbrunn, Germany). Epichlorohydrin (ECH, CAS No. 106-89-8, Product Code E1055) was supplied by Sigma-Aldrich (Steinhiem, Germany). Agglomerated lactose monohydrate (ALM, Tablettose
® 80) (Product code: L104314615A552) was the product of Meggle pharma (Wasserburg, Germany). Silicified microcrystalline cellulose (SMCC 90, Prosolv
®) (Product code: P9D8L19) and high-functionality excipient composite (HFEC, Prosolv EasyTab
® SP) (Product code: 6809074049) were from JRS Pharma, Rosenberg, Germany). Paracetamol (Lot no. 01610131) was purchased from Vittayasom Sriracha Co., Ltd. (Chonburi, Thailand). Carboxymethyl rice starch cross-linked with ECH (CCMS) was produced using the method and conditions described in the previous study [
11]. The reaction time ratio of etherification and cross-linking was selected based on the disintegration property at 1:0.67.
CS was prepared as reported in the previous study [
9]. In brief, CCMS (10 g) was dispersed in distilled water, and stirred continuously until completely swelled. Then, RS (100 g) and sodium silicate solution (11 mL) were added and the mixture was homogenized. The co-processed particle was produced by spray drying of the mixture, using a B-290 mini spray dryer (Buchi, Flawil, Switzerland) with a 2.0 mm nozzle tip. The spray dry conditions were set at 190 °C, 95% of the aspirator, and 18% of the pump. A physical mixture (PMSS) was prepared by blending RS (88.5%), CCMS (8.9%), and spray dried sodium silicate (2.6%) in a plastic bag for 15 min. Pregelatinized rice starch (PGS) was produced by dispersing RS in distilled water (40%
w/
v). Starch slurry was heated at 60 °C for 10 min and dried in a hot air oven at 55 °C for 24 h.
3.2. Optimization of Excipients and APIs Powder Using SeDeM Expert System
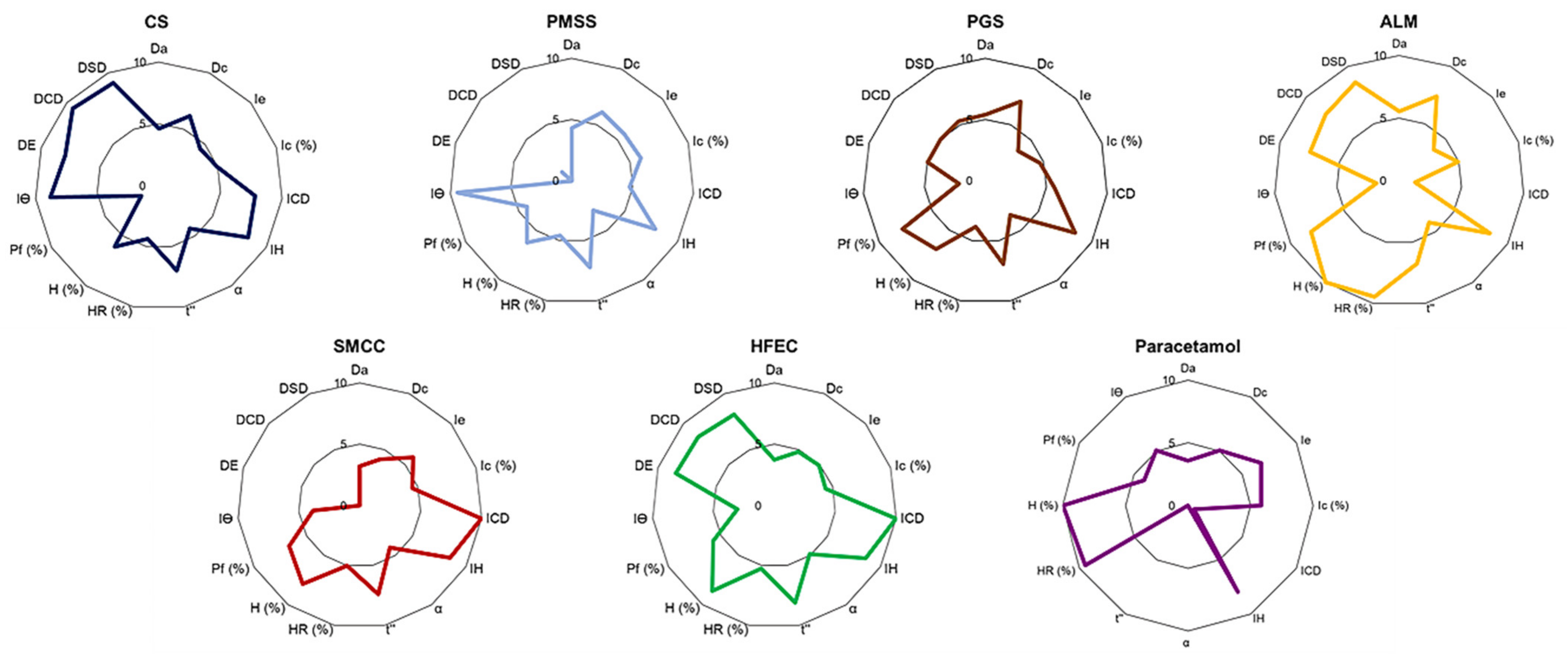
The SeDeM-ODT Expert System evaluated six factors, which were derived from 15 parameters pertinent to the physical characteristics and pharmaceutical functionality of the materials in order to investigate powder characteristics for DC manufacturing (
Table 9). These parameters were determined for excipients and API using the following methodologies, which were repeated at least in triplicate.
3.2.1. Dimension
Bulk density is defined as the powder mass divided by the loosely packed powder volume. It was measured according to European Pharmacopeia (2.9.34). A known mass (g) of powder was poured into a 100 mL graduated cylinder (readable to 1 mL) [
48]. The appearance of bulk volume was read, and then bulk density was calculated using Equation (1):
where Da is the bulk density, M is the powder mass (g), and Va is the bulk volume (mL).
Tapped density is the powder mass divided by the powder volume after continuous tapping to a constant value. It was measured according to European Pharmacopeia (2.9.34) by mechanical tapping using a Jolting volumeter (Stav 2003, Erweka, Langen, Germany) of a 100 mL graduated cylinder (readable to 1 mL) containing the sample powder [
48]. The sample powder was continuously tapped and the volume was read after 10, 500, and 1250 taps. When the difference between V
500 and V
1250 was less than 2 mL, the tapped volume was read as V
1250. Then, tapped density was calculated using Equation (2):
where Dc is tapped density, M is powder mass (g), and Vc is tapped volume (mL).
3.2.2. Compressibility
The interparticle porosity represents the pore space between the particles. It was calculated from the bulk and tapped density values using Equation (3):
The Carr index is determined by the difference between the bulk and tapped densities. It measures the ability of powder to interact with other particles and resist powder flow. Thus, it indicates powder compressibility. The Carr index was calculated using Equation (4):
The cohesive index represents the compressibility of materials. The powder (500 mg) was compressed into a tablet with a 11.0-mm flat-face punch using a hydraulic press machine (C, Carver, Wabash, IN, USA) at 1.0 ton of compression force. In the case of uncompressible powder, 3.50% of standard lubricant was added, which included talc 2.36%, colloidal silicon dioxide 0.14%, and magnesium stearate 1.00%, and mixed for 2 min before compression. The tablet crushing strength (N) was determined using a tablet hardness tester (PTB-311, Pharmatest, Hainburg, Germany). This value indicated the maximum compressive stress that can be tolerated in the tablet without fracture.
3.2.3. Flowability
The Hausner ratio indirectly indicates powder flowability. It represents powder resistance against flow due to the particle interaction, which is based on powder density, size and shape, moisture content, and powder cohesiveness. It is calculated using bulk and tapped densities values using Equation (5):
The angle of repose is related to interparticular friction that limited particle movement. It is determined by a drained angle of repose method according to European Pharmacopeia (2.9.36) [
49]. The powder was poured through a funnel that was fixed with a stand and set 10 cm above the table surface [
24]. The height and the radius of the base of the powder bulk were measured, and then the angle of repose was calculated using Equation (6):
where Ө is the angle of repose (°), h is the height of the sample cone, and r is the radius of the base of the sample bulk.
This factor determines the rate of powder flow through an orifice, which varies depending on particle morphology or process. Powder flow was conducted according to European Pharmacopeia (2.9.16) using a typical apparatus, where a funnel was fixed to a stand and set at 10 cm above the table surface [
50]. The powder was poured steadily through a funnel. Powder flow was expressed in second related to 100 g of sample powder.
3.2.4. Stability
This factor determines the percentage of powder which can be volatilized under tested conditions. It was measured using a moisture analyzer (MX-50, A&D, Tokyo, Japan). Approximately one gram of powder was accurately weighed into the sample pan. Then, the sample was heated at 105 °C to constant weight and the percentage of weight loss was measured.
Hygroscopicity represents the ability of the powder to adsorb or absorb water from the environment. This parameter was determined at 76 ± 2% relative humidity (RH) at room temperature for 24 h. Powder (250 mg) was weighed into a 2.5 cm diameter, pre-weighed cup. The cups were placed in the tight containers which housed saturated salt solutions at the studied RH. After 24 h, the cups were re-weighed and the percentage of the weight gained was calculated.
3.2.5. Dosage
This parameter represents the percentage of particles that passed through a 50 um sieve. The test was conducted using a sieve vibrator (AS 200 control, Retsch, Haan, Germany) at 1.0 g for 10 min.
The Iθ value indicates the homogeneity of the particle size distribution of powder. This parameter was measured using a sieve vibrator with four different sieve sizes (45, 106, 212, and 355 µm). The powder sample (20 g) was determined with agitation at 1.0 g for 10 min. The Iθ value was calculated using Equation (7):
where Iθ is the relative homogeneity index, Fm, Fm + 1, and Fm − 1 are particle percentages remaining in the majority range, above the majority range, and below the majority range, respectively, dm, dm + 1, and dm − 1 are the mean diameter of particle in the majority range, above the majority range, and below the majority range, respectively, and n is the order number of the fraction studied under a series, relating to the major fraction.
3.2.6. Disgregability
The tablets were prepared by compression of the excipient powder (500 mg) with a 11.0-mm flat-face punch using a hydraulic press machine (C, Carver, Wabash, IN, USA) at 1.0 ton of compression force. The surface of punch and die were pre-lubricated with magnesium stearate solution (5% w/v) before compression.
This test determines the possibility of a tablet to be bucodispersible. The tablet (500 mg) was placed in a beaker containing an excess amount of distilled water at room temperature. At the point which the tablet completely disaggregated, the time was recorded (min).
This test imitates the mechanical movement inside the mount during the taking of a tablet, which promoted tablet disintegration. The test was conducted according to the standard USP method [
51]. The medium temperature was controlled at 37 ± 0.5 °C. The tablets (500 mg) were placed in a basket and covered with a disk, then tablet disintegration time was observed and recorded after the tablet completely disintegrated. Six tablets were determined simultaneously.
This test is specific for investigation of bucodispersible tablets. The test was conducted according to the standard USP method described for measuring DCD, except the disk was removed from the test. The tablet disintegration time was observed and recorded after the tablet disintegrated completely. Six tablets were determined simultaneously.
3.3. Conversion of Parameter Values to Radius Values of the SeDeM-ODT Diagram
The values of the parameters (v) obtained from topic 2.2 were calculated to convert into the radius values (r) of the SeDeM-ODT diagram (
Table 9). The SeDeM-ODT diagram, which consisted of 15 polygons of each parameter, was then plotted. The radius scale ranged from 0–10; a value of 5 was considered the minimum acceptable value of each parameter. The parameters were integrated into six incidence factors according to
Table 1. The incidence factor values were calculated as a mean of their respective integrated parameters.
3.4. The Determination of the Index Using SeDeM-ODT Diagram
The index parameter (IP), index profile parameter (IPP), index of good compressibility (IGC), and index of good compressibility and bucodispersibility (IGCB) were calculated to evaluate the suitability of powder for applying in DC manufacturing using the following equations:
where n° P > 5 is the number of parameters which radius values > 5, while n° Pt is the number of total studied parameters. The acceptability limit of IP index should correspond to >0.5.
The IGC was calculated based on 12 parameters, excluding the disgregability factors. The IGCB was obtained based on 15 parameters, including the three disgregability parameters. The acceptability limit of IPP, IGC, and IGCB should correspond to 5 or higher.
3.5. Calculation of the Number of Excipients Required to Adjust API Characteristics for DC Formulation
The flowability and compressibility of the API were corrected by combining it with a suitable excipient. The number of excipients required to adjust API characteristics was calculated using Equation (12), based on the SeDeM-ODT diagram. All factors can be adjusted using Equation (12). However, compressibility and flowability were given priority, as they represented the major important factors for DC production:
where CP is the percentage of a corrective excipient, RE is the average radius value of the corrective excipient, RP is the average radius value of the API to be corrected, and R represents the required average radius value of the mixture of excipient and API; thus, 5 is the minimum value allowed to correct API characteristics.
3.6. Formulation Study
3.6.1. Tablet Preparation
Tablets were prepared using a direct compression method. Paracetamol powder was homogenously blended with excipients using a geometric dilution technique for 15 min. After that, magnesium stearate (1%) was added and mixed for 2 min. The powder mixture (100 g) was compressed into tablets using a single punch tableting machine (CMT 12, Charatchai, Bangkok, Thailand), housed with a 10.3 mm round, flat-face punch, at a tableting speed of 2640 tab/h. The target weight of a tablet was set at 300 mg.
3.6.2. Evaluation of Tablet Properties
Weight variation was determined, according to the USP method [
52], by weighing and recording the weight of 30 individual tablets. Tablet hardness was measured using a tablet hardness tester and conducted on 10 tablets. The friability test was conducted according to the USP method. Approximately 6.5 g of the tablets was sampled and dedusted before the test [
53]. The tablets were accurately weighed and placed in a friability tester. The test was conducted on the rotation at 25 rpm for 4 min. The tested tablets were dedusted and re-weighed. Tablet friability was calculated from the weight difference. The acceptable friability of tablets should be less than 1.0%. The disintegration test was determined in at least six tablets using a basket apparatus according to the USP method [
51]. Each tablet was placed in a basket tube and covered with a disc. The test was carried out in a distilled water medium, maintained at a temperature of 37 ± 0.5 °C throughout the test. The disintegration time was recorded after the entire tablet completely disintegrated. The drug content percentage (DCP) of paracetamol tablet was determined according to the BP method, which should be in the range of 95.0–105.0% of the stated amount [
54].
3.6.3. Tablet Criteria
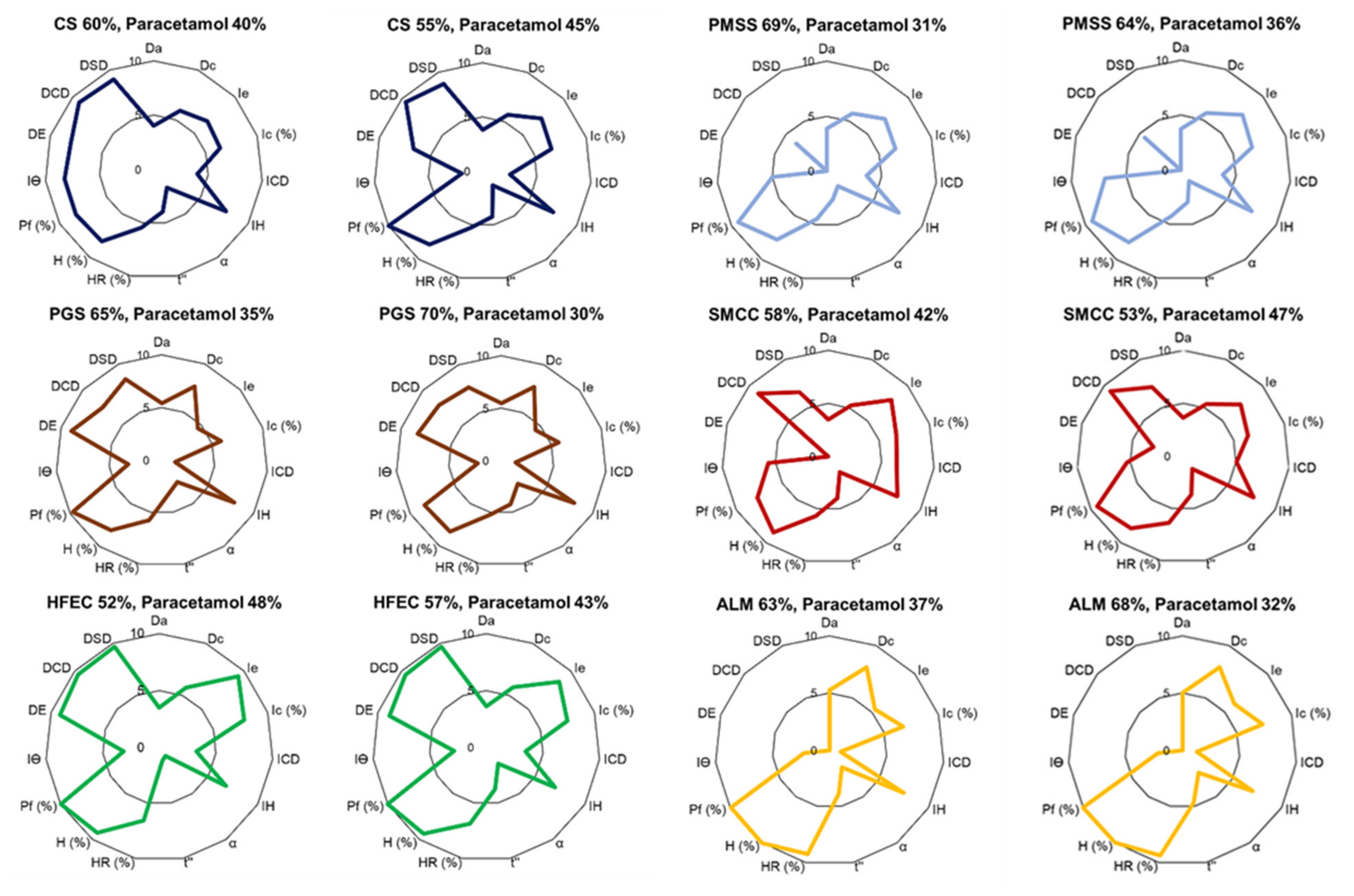
Tablet formulation was determined as a successful formulation based on three parameters, including weight variation, friability (%), and drug content (%). These parameters reflected the ability of the powder blend to fill the dies during tablet compression and provided a suitable tablet strength during the production and throughout its shelf life. DCP represented the uniformity of the powder blend. To be classified as a successful formulation, a tablet formulation must meet all the requirements. After that, the excipient percentage in the formulation was increased by 5% for the failed formulations or decreased by 5% for the successful formulations based on the reliability factor of SeDeM diagram (f = 0.952). The paracetamol tablets were formulated, and the tablet properties were evaluated using the procedure described above.
3.6.4. Dissolution Study
The successful formulations were selected and subjected to dissolution tests using a paddle apparatus according to the standard USP method [
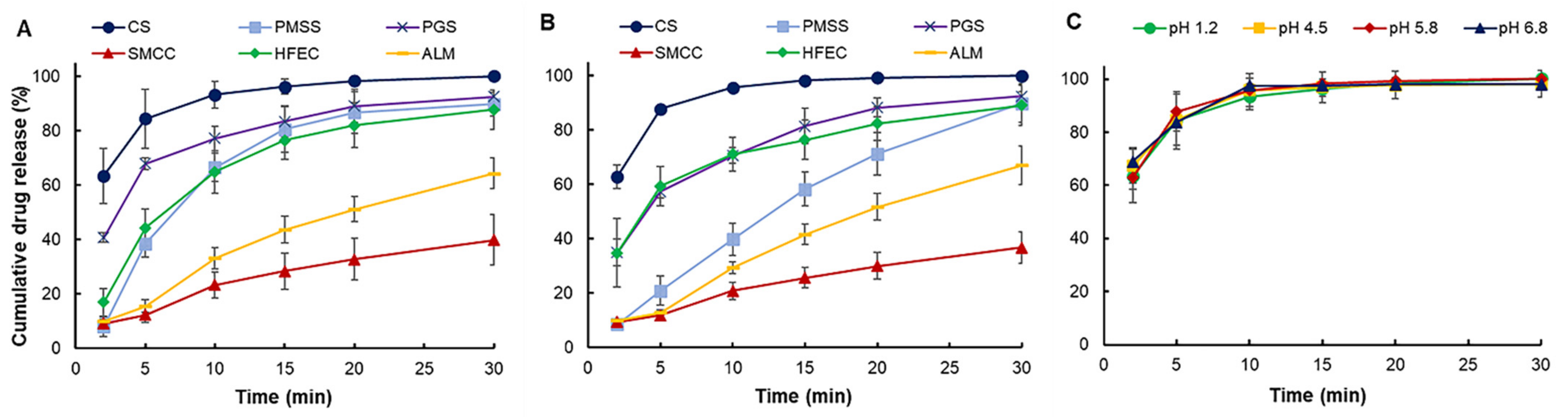
55]. For PGS and ALM formulations, the one that contained a higher number of excipients was selected to enter the test. The test was carried out for 30 min, using different dissolution mediums (hydrochloric acid pH 1.2 and phosphate buffer pH 5.8), at a controlled temperature of 37 ± 0.5 °C throughout the test, where the paddle speed was set at 50 rpm. CS formulation was also conducted with additional dissolution mediums (acetate buffer pH 4.5 and phosphate buffer pH 6.8) to evaluate CS dissolution behavior along the gastrointestinal tract. Five ml of sample was taken from the vessel at 2, 5, 10, 15, 20, and 30 min, with equal volume replacement of fresh medium for each sampling. The samples were analyzed using a UV-Vis 2450 spectrophotometer (Shimadzu, Japan) at 243 nm. The cumulative drug release percentage was then calculated. The test was repeated at least in triplicate.
3.7. Validation of CS Functionality
The characteristics of ibuprofen powder (model drug) were investigated using the SeDeM expert system, and the poor characteristics (flowability and/or compressibility) were corrected with CS. The number of excipients required to compensate for ibuprofen characteristics (CP, %) was calculated. The ibuprofen formulation was prepared according to the CP value by DC technique; then, the SeDeM-ODT expert system was applied to determine formulation characteristics. Ibuprofen tablets (300 mg/tablet) were produced using a single punch tableting machine with a 10.3 mm round, flat-face punch. The tablet properties were evaluated as described earlier. After that, CS concentration was varied by a 5% increase from the calculated CP value until the tablet properties met the requirements. The dissolution property of the successful formulation was determined according to the standard USP method.
3.8. Stability Study
The successful CS formulation was subjected to stability study under accelerated condition(s) according to the International Conference on Harmonization (ICH) guidelines. The tablets were stored at 40 ± 2 °C and 75 ± 5% RH for 45 days. Stability samples were taken at 0, 30, and 45 days to determine tablet characteristics, which included weight variation, breaking force, friability, disintegration time, drug content, and dissolution study. A difference factor (f
1) and similarity factor (f
2) were applied to determine the sameness of the dissolution profile [
47]:
where n is the number of sampling time point, Rt is the average of cumulative drug release of day 0 tablets at time t, and Tt represents the average of cumulative drug release of day 30 or day 45 tablets at that similar time point.
3.9. Statistics
All tests were conducted at least in triplicate and the data are presented as average values. Statistical analysis was performed using a one-way analysis of variance (ANOVA) in SPSS (version 19.0). Significance tests were analyzed using Tukey’s honestly significant difference (HSD) multiple range test at a 95% confidence level (p < 0.05).
4. Conclusions
The formulation study of a co-processed, rice starch-based excipient (CS) was evaluated using the SeDeM expert system for application in direct compression (DC) formulation. CS exhibited acceptable powder characteristics based on the values of six incidence factors. The comprehensive indices (IGC and IGCB) of CS surpassed the acceptable values, suggesting that CS was suitable for DC manufacturing without the addition of a disintegrant to the tablet formulation. The CP value of the excipient required to compensate for the property of the model drug paracetamol was selected based on flowability. The formulation study revealed that CS can be diluted up to 60% with an API of poor flow and compressibility in the paracetamol formulation. At this percentage, CS tablets showed narrow tablet weight variation, low friability, suitable drug content, and fast disintegration time. The dissolution study showed that CS can release more than 80% of the drug content in less than 5 min at all tested pH conditions, which was superior to other excipients. CS was comparable to HFEC, and can be used to compensate the inadequate flow and compressibility of APIs. Other excipients, such as SMCC, required the addition of a disintegrant, while PGS and ALM may not be suitable for poorly compressible APIs. PMSS, a simple, physical mixture of various excipients, could not significantly improve the API properties and, thus, it was not considered as a multifunctional excipient. The CS formulated with 50% of ibuprofen showed satisfactory tablet properties, which implied the wide range of CS functionality to formulate with various APIs. The stability study of the CS tablets showed no significant change after 45 days storage. Overall, CS exhibited properties and characteristics as a multifunctional excipient for direct compression of pharmaceutical tablets.



{kind=link}
{kind=link}
{kind=link}
{kind=link}