
New Frontiers in Cancer Imaging and Therapy Based on Radiolabeled Fibroblast Activation Protein Inhibitors: A Rational Review and Current Progress

 ,
,
Abstract
1. Introduction
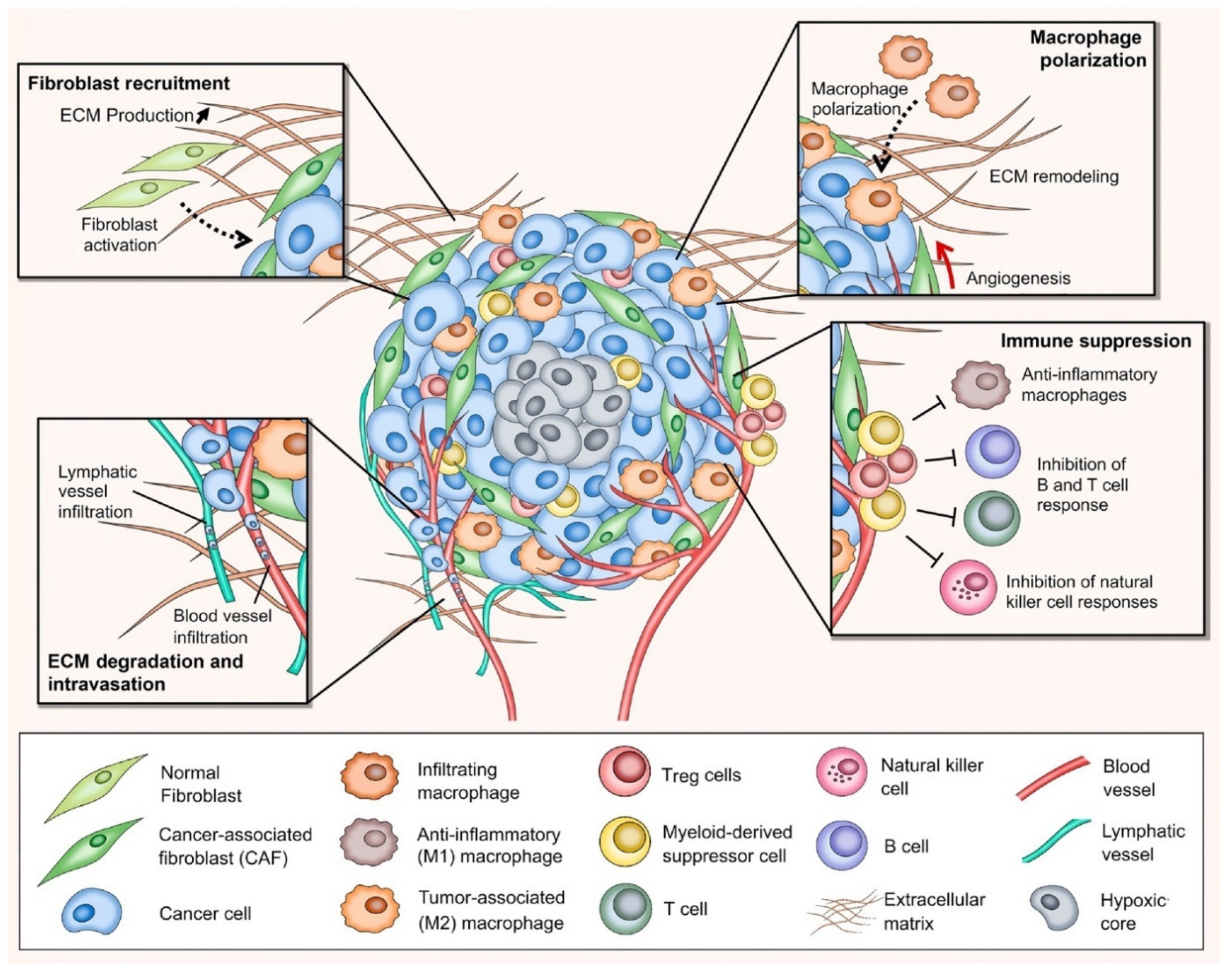
Tumor Microenvironment: A New Arena in Stromal Targeting
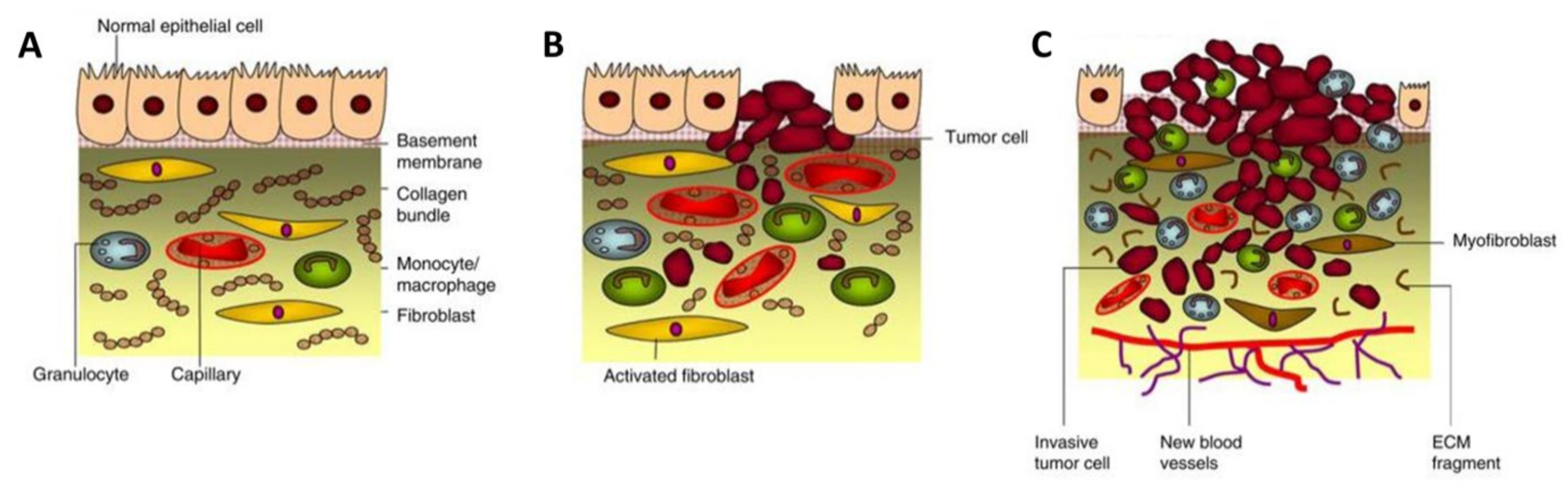
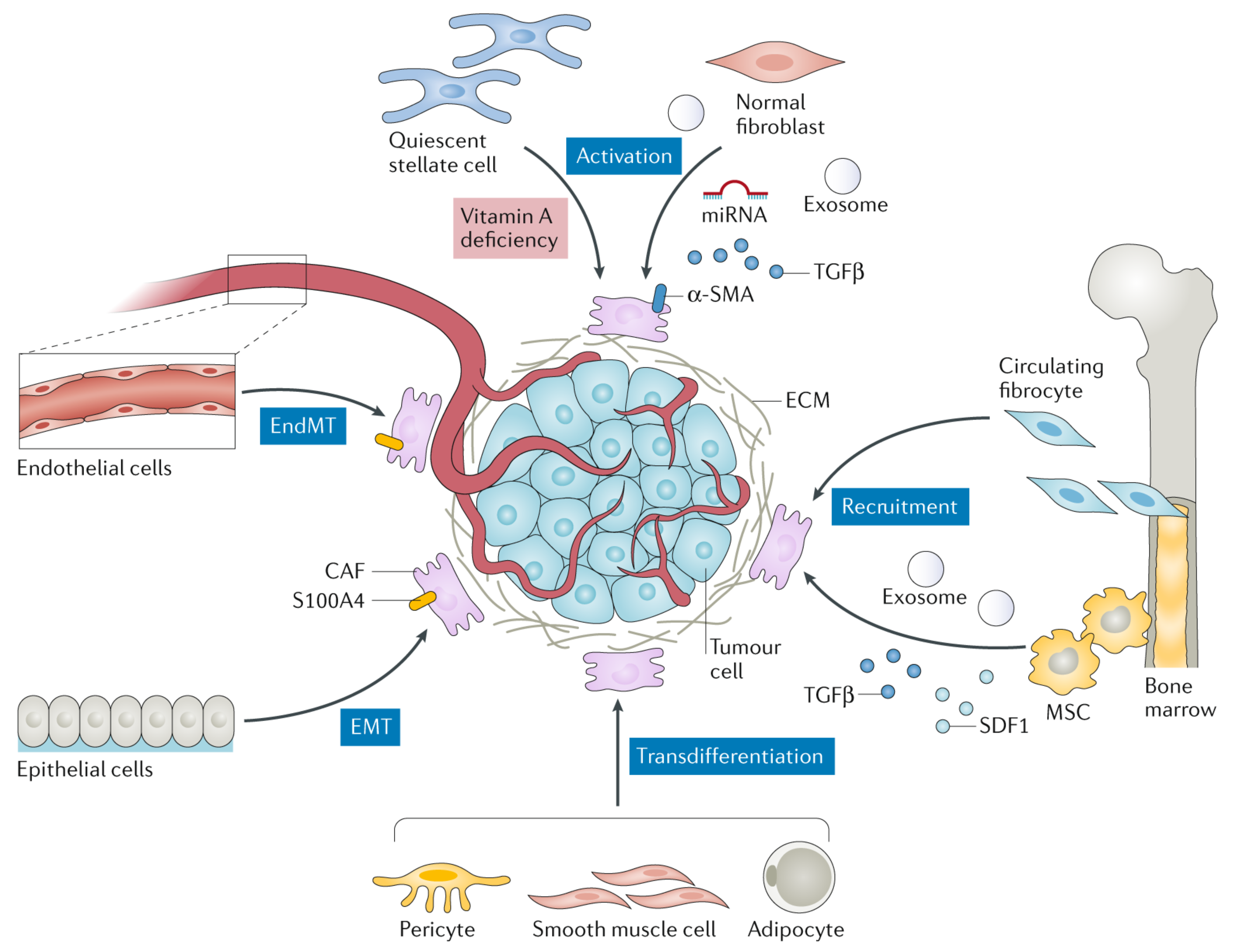
2. Cancer-Associated Fibroblasts: A Critical Mediator in Cancer Progression
3. Fibroblast Activation Protein: A Potential Diagnostic and Therapeutic Target across Stromal Barriers
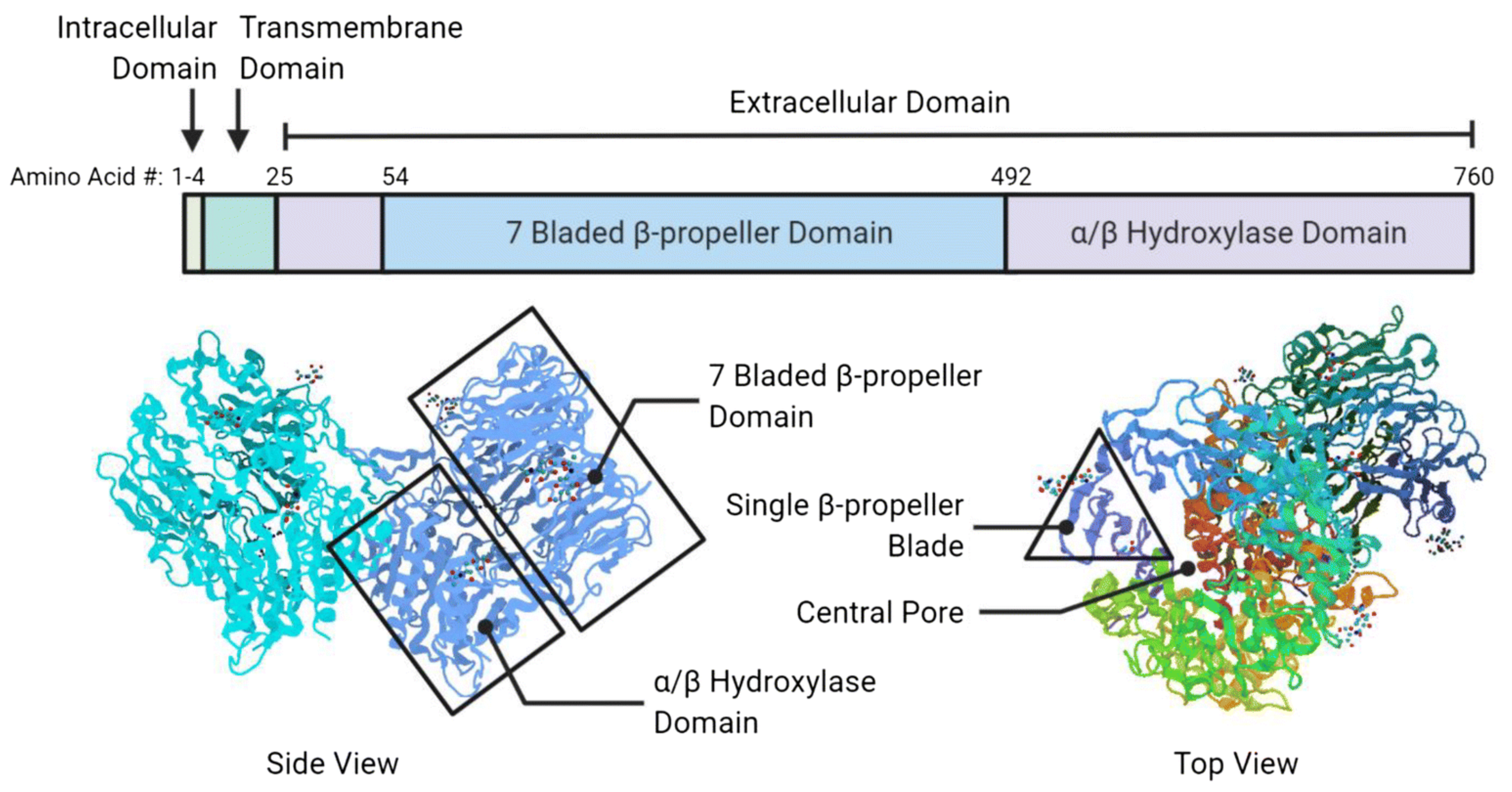
3.1. Biological Characteristics of FAP
3.2. Relationship between FAP and Immunosuppression in the TME
3.3. FAP as a Potential Target in Cancer
4. Development of Radiolabeled-Based FAP Tracers for Tumor Stroma Mediated Nuclear Imaging and Radionuclide-Based Therapy
4.1. Radiolabeled FAP-Targeted Antibodies
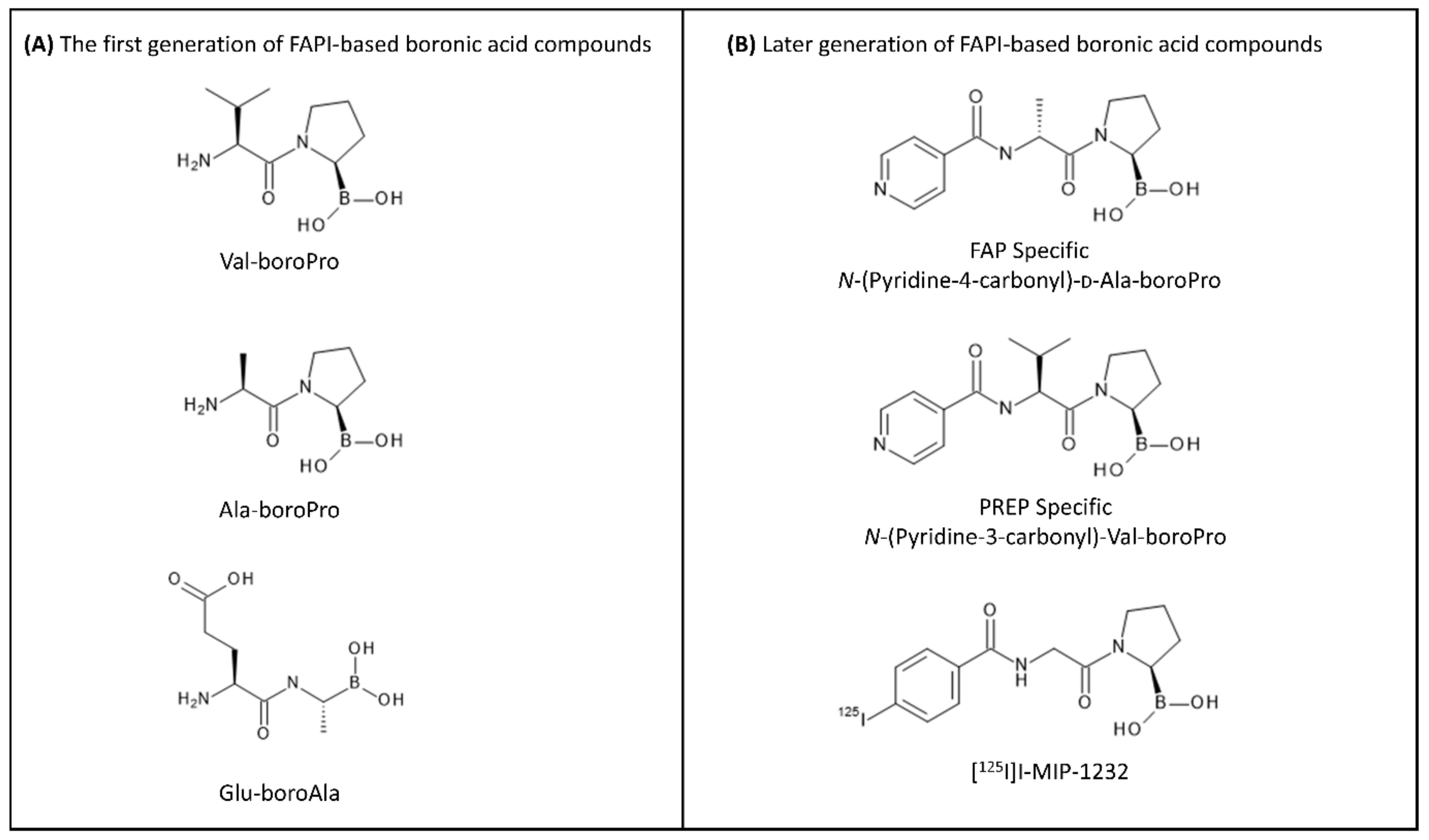
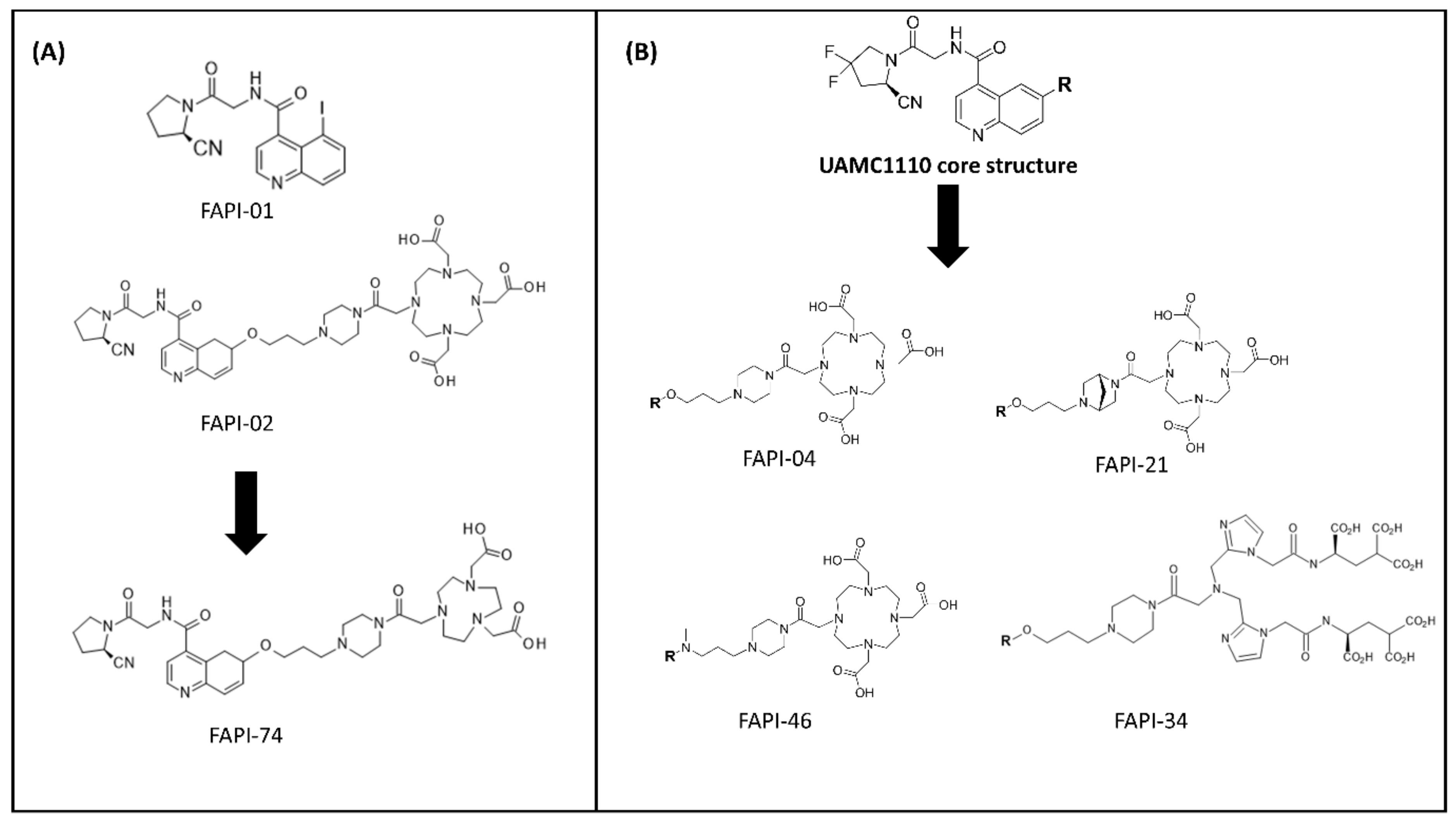
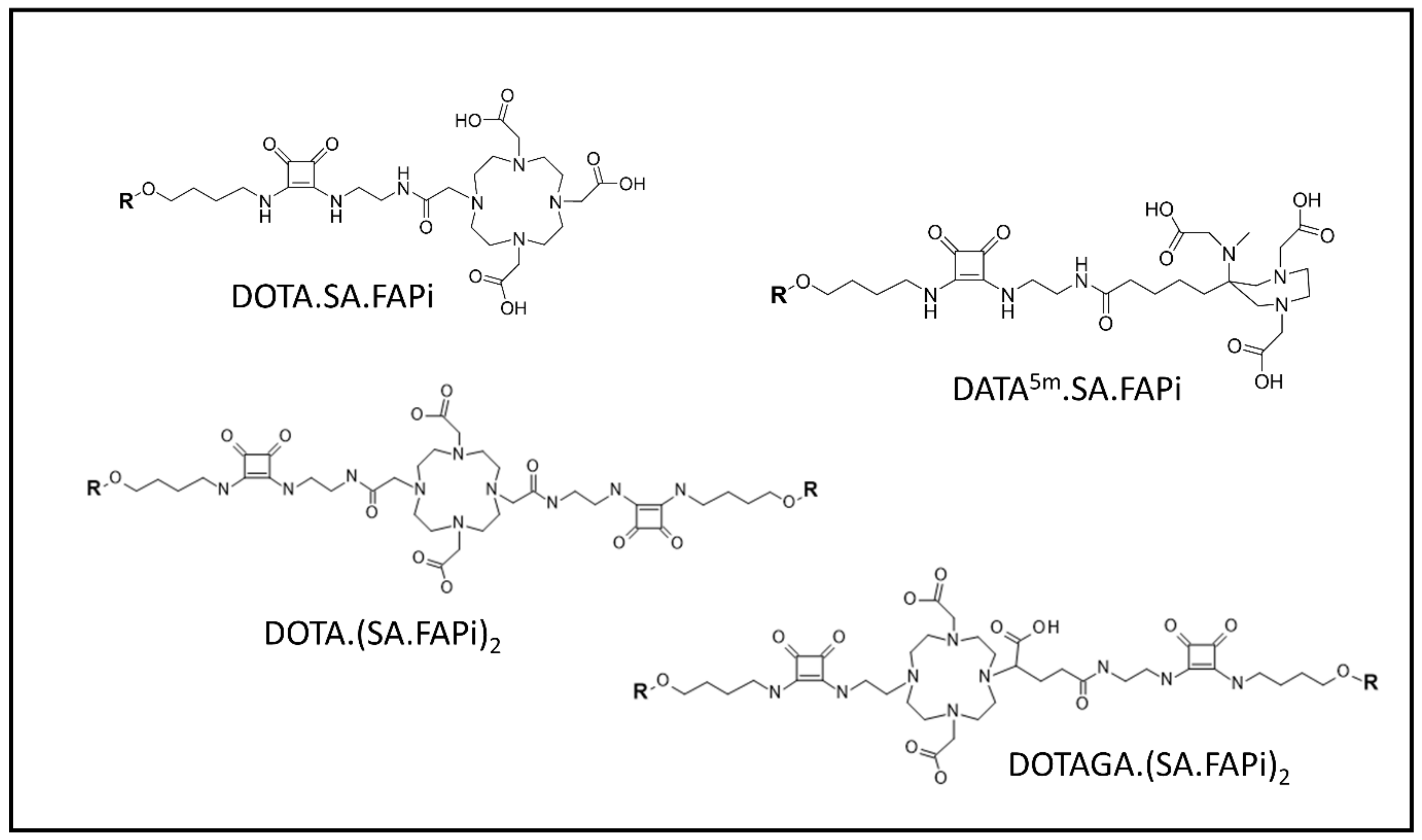
4.2. Radiolabeled FAP-Based Inhibitors
4.3. Radiolabeled FAP-Targeted Peptides
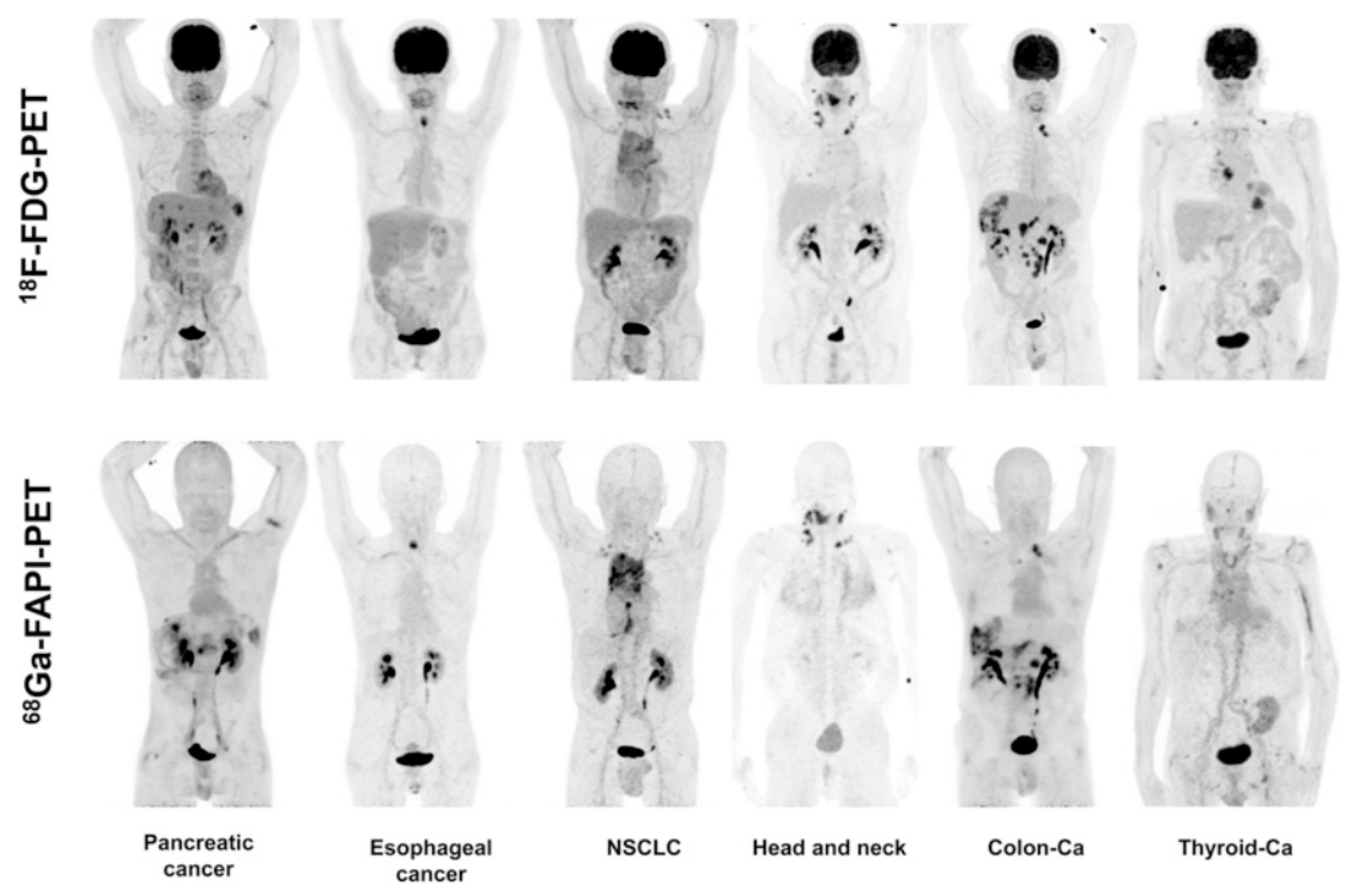
5. Clinical Studies of Radiolabeled-Based FAP Inhibitors
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Roma-Rodrigues, C.; Mendes, R.; Baptista, P.; Fernandes, A. Targeting Tumor Microenvironment for Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 840. [Google Scholar] [CrossRef]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar] [CrossRef]
- Lindner, T.; Loktev, A.; Giesel, F.; Kratochwil, C.; Altmann, A.; Haberkorn, U. Targeting of activated fibroblasts for imaging and therapy. EJNMMI Radiopharm. Chem. 2019, 4, 16. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Öhlund, D.; Elyada, E.; Tuveson, D. Fibroblast heterogeneity in the cancer wound. J. Exp. Med. 2014, 211, 1503–1523. [Google Scholar] [CrossRef] [PubMed]
- Smith, N.R.; Baker, D.; Farren, M.; Pommier, A.; Swann, R.; Wang, X.; Mistry, S.; McDaid, K.; Kendrew, J.; Womack, C.; et al. Tumor Stromal Architecture Can Define the Intrinsic Tumor Response to VEGF-Targeted Therapy. Clin. Cancer Res. 2013, 19, 6943–6956. [Google Scholar] [CrossRef] [PubMed]
- Neesse, A.; Michl, P.; Frese, K.K.; Feig, C.; Cook, N.; Jacobetz, M.A.; Lolkema, M.P.; Buchholz, M.; Olive, K.P.; Gress, T.M.; et al. Stromal biology and therapy in pancreatic cancer. Gut 2011, 60, 861–868. [Google Scholar] [CrossRef]
- Xing, F.; Saidou, J.; Watabe, K. Cancer associated fibroblasts (CAFs) in tumor microenvironment. Front. Biosci. Landmark Ed. 2010, 15, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Teijeiro, S.; García-Inclán, C.; Villaronga, M.; Casado, P.; Hermida-Prado, F.; Granda-Díaz, R.; Rodrigo, J.; Calvo, F.; del-Río-Ibisate, N.; Gandarillas, A.; et al. Factors Secreted by Cancer-Associated Fibroblasts that Sustain Cancer Stem Properties in Head and Neck Squamous Carcinoma Cells as Potential Therapeutic Targets. Cancers 2018, 10, 334. [Google Scholar] [CrossRef]
- Koustoulidou, S.; Hoorens, M.W.H.; Dalm, S.U.; Mahajan, S.; Debets, R.; Seimbille, Y.; de Jong, M. Cancer-Associated Fibroblasts as Players in Cancer Development and Progression and Their Role in Targeted Radionuclide Imaging and Therapy. Cancers 2021, 13, 1100. [Google Scholar] [CrossRef]
- Han, C.; Liu, T.; Yin, R. Biomarkers for cancer-associated fibroblasts. Biomark. Res. 2020, 8, 64. [Google Scholar] [CrossRef]
- Fitzgerald, A.A.; Weiner, L.M. The role of fibroblast activation protein in health and malignancy. Cancer Metastasis Rev. 2020, 39, 783–803. [Google Scholar] [CrossRef]
- Garcia, E.V. Physical attributes, limitations, and future potential for PET and SPECT. J. Nucl. Cardiol. 2012, 19, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Laverman, P.; van der Geest, T.; Terry, S.Y.A.; Gerrits, D.; Walgreen, B.; Helsen, M.M.; Nayak, T.K.; Freimoser-Grundschober, A.; Waldhauer, I.; Hosse, R.J.; et al. Immuno-PET and Immuno-SPECT of Rheumatoid Arthritis with Radiolabeled Anti-Fibroblast Activation Protein Antibody Correlates with Severity of Arthritis. J. Nucl. Med. 2015, 56, 778–783. [Google Scholar] [CrossRef] [PubMed]
- Van der Geest, T.; Laverman, P.; Gerrits, D.; Walgreen, B.; Helsen, M.M.; Klein, C.; Nayak, T.K.; Storm, G.; Metselaar, J.M.; Koenders, M.I.; et al. Liposomal Treatment of Experimental Arthritis Can Be Monitored Noninvasively with a Radiolabeled Anti–Fibroblast Activation Protein Antibody. J. Nucl. Med. 2017, 58, 151–155. [Google Scholar] [CrossRef]
- Toms, J.; Kogler, J.; Maschauer, S.; Daniel, C.; Schmidkonz, C.; Kuwert, T.; Prante, O. Targeting Fibroblast Activation Protein: Radiosynthesis and Preclinical Evaluation of an 18F-Labeled FAP Inhibitor. J. Nucl. Med. 2020, 61, 1806–1813. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Fang, R.; Xu, J.; Qiu, S.; Zhang, H.; Du, J.; Cai, S. Evaluation of the tumor targeting of a FAPα-based doxorubicin prodrug. J. Drug Target. 2011, 19, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Zboralski, D.; Osterkamp, F.; Simmons, A.D.; Bredenbeck, A.; Schumann, A.; Paschke, M.; Beindorff, N.; Mohan, A.-M.; Nguyen, M.; Xiao, J.; et al. 571P Preclinical evaluation of FAP-2286, a peptide-targeted radionuclide therapy (PTRT) to fibroblast activation protein alpha (FAP). Ann. Oncol. 2020, 31, S488. [Google Scholar] [CrossRef]
- Baum, R.P.; Schuchardt, C.; Singh, A.; Chantadisai, M.; Robiller, F.C.; Zhang, J.; Mueller, D.; Eismant, A.; Almaguel, F.; Zboralski, D.; et al. Feasibility, Biodistribution and Preliminary Dosimetry in Peptide-Targeted Radionuclide Therapy (PTRT) of Diverse Adenocarcinomas using 177Lu-FAP-2286: First-in-Human Results. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2021. [Google Scholar] [CrossRef]
- Jin, M.-Z.; Jin, W.-L. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct. Target. Ther. 2020, 5, 166. [Google Scholar] [CrossRef] [PubMed]
- McMillin, D.W.; Negri, J.M.; Mitsiades, C.S. The role of tumour-stromal interactions in modifying drug response: Challenges and opportunities. Nat. Rev. Drug Discov. 2013, 12, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, J.; Heinrich, M.A.; Teixeira, L.M.; Prakash, J. 3D In Vitro Model (R)evolution: Unveiling Tumor–Stroma Interactions. Trends Cancer 2021, 7, 249–264. [Google Scholar] [CrossRef] [PubMed]
- Mbeunkui, F.; Johann, D.J. Cancer and the tumor microenvironment: A review of an essential relationship. Cancer Chemother. Pharmacol. 2009, 63, 571–582. [Google Scholar] [CrossRef]
- Ramamonjisoa, N.; Ackerstaff, E. Characterization of the Tumor Microenvironment and Tumor–Stroma Interaction by Non-invasive Preclinical Imaging. Front. Oncol. 2017, 7, 3. [Google Scholar] [CrossRef]
- Liotta, L.A.; Kohn, E.C. The microenvironment of the tumour-host interface. Nature 2001, 411, 375–379. [Google Scholar] [CrossRef]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef]
- Liu, H.; Ni, S.; Wang, H.; Zhang, Q.; Weng, W. Charactering tumor microenvironment reveals stromal-related transcription factors promote tumor carcinogenesis in gastric cancer. Cancer Med. 2020, 9, 5247–5257. [Google Scholar] [CrossRef]
- Tania, M.; Khan, M.A.; Fu, J. Epithelial to mesenchymal transition inducing transcription factors and metastatic cancer. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2014, 35, 7335–7342. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.J.; Carroll, J.S. Transcription factors and chromatin proteins as therapeutic targets in cancer. Biochim. Biophys. Acta 2015, 1855, 183–192. [Google Scholar] [CrossRef]
- Vishnoi, K.; Viswakarma, N.; Rana, A.; Rana, B. Transcription Factors in Cancer Development and Therapy. Cancers 2020, 12, 2296. [Google Scholar] [CrossRef] [PubMed]
- Handsley, M.M.; Edwards, D.R. Metalloproteinases and their inhibitors in tumor angiogenesis. Int. J. Cancer 2005, 115, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Görögh, T.; Beier, U.H.; Bäumken, J.; Meyer, J.E.; Hoffmann, M.; Gottschlich, S.; Maune, S. Metalloproteinases and their inhibitors: Influence on tumor invasiveness and metastasis formation in head and neck squamous cell carcinomas. Head Neck 2006, 28, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Han, C.; Wang, S.; Fang, P.; Ma, Z.; Xu, L.; Yin, R. Cancer-associated fibroblasts: An emerging target of anti-cancer immunotherapy. J. Hematol. Oncol. 2019, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Wang, J.H.-C. Fibroblasts and myofibroblasts in wound healing: Force generation and measurement. J. Tissue Viability 2011, 20, 108–120. [Google Scholar] [CrossRef]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar]
- Byun, J.S.; Gardner, K. Wounds that will not heal: Pervasive cellular reprogramming in cancer. Am. J. Pathol. 2013, 182, 1055–1064. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Enomoto, A.; Woods, S.L.; Burt, A.D.; Takahashi, M.; Worthley, D.L. Cancer-associated fibroblasts in gastrointestinal cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 282–295. [Google Scholar] [CrossRef]
- Ziani, L.; Chouaib, S.; Thiery, J. Alteration of the Antitumor Immune Response by Cancer-Associated Fibroblasts. Front. Immunol. 2018, 9, 414. [Google Scholar] [CrossRef]
- Chen, X.; Song, E. Turning foes to friends: Targeting cancer-associated fibroblasts. Nat. Rev. Drug Discov. 2019, 18, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Pape, J.; Magdeldin, T.; Stamati, K.; Nyga, A.; Loizidou, M.; Emberton, M.; Cheema, U. Cancer-associated fibroblasts mediate cancer progression and remodel the tumouroid stroma. Br. J. Cancer 2020, 123, 1178–1190. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef]
- Mishra, R.; Haldar, S.; Suchanti, S.; Bhowmick, N.A. Epigenetic changes in fibroblasts drive cancer metabolism and differentiation. Endocr. Relat. Cancer 2019, 26, R673–R688. [Google Scholar] [CrossRef] [PubMed]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Omary, M.B.; Lugea, A.; Lowe, A.W.; Pandol, S.J. The pancreatic stellate cell: A star on the rise in pancreatic diseases. J. Clin. Invest. 2007, 117, 50–59. [Google Scholar] [CrossRef]
- Yin, C.; Evason, K.J.; Asahina, K.; Stainier, D.Y.R. Hepatic stellate cells in liver development, regeneration, and cancer. J. Clin. Invest. 2013, 123, 1902–1910. [Google Scholar] [CrossRef]
- Pereira, B.A.; Vennin, C.; Papanicolaou, M.; Chambers, C.R.; Herrmann, D.; Morton, J.P.; Cox, T.R.; Timpson, P. CAF Subpopulations: A New Reservoir of Stromal Targets in Pancreatic Cancer. Trends Cancer 2019, 5, 724–741. [Google Scholar] [CrossRef]
- Terai, S.; Fushida, S.; Tsukada, T.; Kinoshita, J.; Oyama, K.; Okamoto, K.; Makino, I.; Tajima, H.; Ninomiya, I.; Fujimura, T.; et al. Bone marrow derived “fibrocytes” contribute to tumor proliferation and fibrosis in gastric cancer. Gastric Cancer 2015, 18, 306–313. [Google Scholar] [CrossRef]
- LeBleu, V.S.; Neilson, E.G. Origin and functional heterogeneity of fibroblasts. FASEB J. 2020, 34, 3519–3536. [Google Scholar] [CrossRef]
- Mishra, P.J.; Mishra, P.J.; Humeniuk, R.; Medina, D.J.; Alexe, G.; Mesirov, J.P.; Ganesan, S.; Glod, J.W.; Banerjee, D. Carcinoma-Associated Fibroblast–Like Differentiation of Human Mesenchymal Stem Cells. Cancer Res. 2008, 68, 4331–4339. [Google Scholar] [CrossRef] [PubMed]
- Kurashige, M.; Kohara, M.; Ohshima, K.; Tahara, S.; Hori, Y.; Nojima, S.; Wada, N.; Ikeda, J.; Miyamura, K.; Ito, M.; et al. Origin of cancer-associated fibroblasts and tumor-associated macrophages in humans after sex-mismatched bone marrow transplantation. Commun. Biol. 2018, 1, 131. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, W.; Sun, X.; Lin, Y.; Chen, W. Cancer-associated fibroblasts induce epithelial-mesenchymal transition through secreted cytokines in endometrial cancer cells. Oncol. Lett. 2018, 15, 5694–5702. [Google Scholar] [CrossRef]
- Yeon, J.H.; Jeong, H.E.; Seo, H.; Cho, S.; Kim, K.; Na, D.; Chung, S.; Park, J.; Choi, N.; Kang, J.Y. Cancer-derived exosomes trigger endothelial to mesenchymal transition followed by the induction of cancer-associated fibroblasts. Acta Biomater. 2018, 76, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Lu, Q.; Shen, B.; Huang, X.; Shen, L.; Zheng, X.; Huang, R.; Yan, J.; Guo, H. TGFβ1 secreted by cancer-associated fibroblasts induces epithelial-mesenchymal transition of bladder cancer cells through lncRNA-ZEB2NAT. Sci. Rep. 2015, 5, 11924. [Google Scholar] [CrossRef]
- Jopling, C.; Boue, S.; Izpisua Belmonte, J.C. Dedifferentiation, transdifferentiation and reprogramming: Three routes to regeneration. Nat. Rev. Mol. Cell Biol. 2011, 12, 79–89. [Google Scholar] [CrossRef]
- Nurmik, M.; Ullmann, P.; Rodriguez, F.; Haan, S.; Letellier, E. In search of definitions: Cancer-associated fibroblasts and their markers. Int. J. Cancer 2020, 146, 895–905. [Google Scholar] [CrossRef]
- Brennen, W.N.; Isaacs, J.T.; Denmeade, S.R. Rationale behind targeting fibroblast activation protein-expressing carcinoma-associated fibroblasts as a novel chemotherapeutic strategy. Mol. Cancer Ther. 2012, 11, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Busek, P.; Mateu, R.; Zubal, M.; Kotackova, L.; Sedo, A. Targeting fibroblast activation protein in cancer—Prospects and caveats. Front. Biosci. Landmark Ed. 2018, 23, 1933–1968. [Google Scholar]
- Fabre, M.; Ferrer, C.; Domínguez-Hormaetxe, S.; Bockorny, B.; Murias, L.; Seifert, O.; Eisler, S.A.; Kontermann, R.E.; Pfizenmaier, K.; Lee, S.Y.; et al. OMTX705, a Novel FAP-Targeting ADC Demonstrates Activity in Chemotherapy and Pembrolizumab-Resistant Solid Tumor Models. Clin. Cancer Res. 2020, 26, 3420–3430. [Google Scholar] [CrossRef]
- Wikberg, M.L.; Edin, S.; Lundberg, I.V.; Van Guelpen, B.; Dahlin, A.M.; Rutegård, J.; Stenling, R.; Oberg, A.; Palmqvist, R. High intratumoral expression of fibroblast activation protein (FAP) in colon cancer is associated with poorer patient prognosis. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2013, 34, 1013–1020. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.A.; Kraut, N.; Park, J.E.; Schubert, R.D.; Rettig, W.J.; Peter, R.U.; Garin-Chesa, P. Fibroblast activation protein: Differential expression and serine protease activity in reactive stromal fibroblasts of melanocytic skin tumors. J. Invest. Dermatol. 2003, 120, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Aertgeerts, K.; Levin, I.; Shi, L.; Snell, G.P.; Jennings, A.; Prasad, G.S.; Zhang, Y.; Kraus, M.L.; Salakian, S.; Sridhar, V.; et al. Structural and Kinetic Analysis of the Substrate Specificity of Human Fibroblast Activation Protein α. J. Biol. Chem. 2005, 280, 19441–19444. [Google Scholar] [CrossRef]
- Lee, K.N.; Jackson, K.W.; Christiansen, V.J.; Lee, C.S.; Chun, J.-G.; McKee, P.A. Antiplasmin-cleaving enzyme is a soluble form of fibroblast activation protein. Blood 2006, 107, 1397–1404. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.-M.; Xu, W.; Du, J.; Zhang, K.-S.; Zhang, Q.-G.; Wang, X.-W.; Liu, Z.-G.; Liu, S.-Q.; Xie, W.-Y.; Liu, H.-F.; et al. The application of the fibroblast activation protein α-targeted immunotherapy strategy. Oncotarget 2016, 7, 33472–33482. [Google Scholar] [CrossRef] [PubMed]
- Juillerat-Jeanneret, L.; Tafelmeyer, P.; Golshayan, D. Fibroblast activation protein-α in fibrogenic disorders and cancer: More than a prolyl-specific peptidase? Expert Opin. Ther. Targets 2017, 21, 977–991. [Google Scholar] [CrossRef]
- Park, J.E.; Lenter, M.C.; Zimmermann, R.N.; Garin-Chesa, P.; Old, L.J.; Rettig, W.J. Fibroblast Activation Protein, a Dual Specificity Serine Protease Expressed in Reactive Human Tumor Stromal Fibroblasts. J. Biol. Chem. 1999, 274, 36505–36512. [Google Scholar] [CrossRef]
- Fan, M.-H.; Zhu, Q.; Li, H.-H.; Ra, H.-J.; Majumdar, S.; Gulick, D.L.; Jerome, J.A.; Madsen, D.H.; Christofidou-Solomidou, M.; Speicher, D.W.; et al. Fibroblast Activation Protein (FAP) Accelerates Collagen Degradation and Clearance from Lungs in Mice. J. Biol. Chem. 2016, 291, 8070–8089. [Google Scholar] [CrossRef]
- Jabłońska-Trypuć, A.; Matejczyk, M.; Rosochacki, S. Matrix metalloproteinases (MMPs), the main extracellular matrix (ECM) enzymes in collagen degradation, as a target for anticancer drugs. J. Enzyme Inhib. Med. Chem. 2016, 31, 177–183. [Google Scholar] [CrossRef]
- Huang, C.-H.; Suen, C.-S.; Lin, C.-T.; Chien, C.-H.; Lee, H.-Y.; Chung, K.-M.; Tsai, T.-Y.; Jiaang, W.-T.; Hwang, M.-J.; Chen, X. Cleavage-site specificity of prolyl endopeptidase FAP investigated with a full-length protein substrate. J. Biochem. 2011, 149, 685–692. [Google Scholar] [CrossRef][Green Version]
- Zi, F.; He, J.; He, D.; Li, Y.; Yang, L.; Cai, Z. Fibroblast activation protein α in tumor microenvironment: Recent progression and implications (review). Mol. Med. Rep. 2015, 11, 3203–3211. [Google Scholar] [CrossRef]
- Jacob, M.; Chang, L.; Puré, E. Fibroblast activation protein in remodeling tissues. Curr. Mol. Med. 2012, 12, 1220–1243. [Google Scholar] [CrossRef]
- Hamson, E.J.; Keane, F.M.; Tholen, S.; Schilling, O.; Gorrell, M.D. Understanding fibroblast activation protein (FAP): Substrates, activities, expression and targeting for cancer therapy. Proteom. Clin. Appl. 2014, 8, 454–463. [Google Scholar] [CrossRef]
- Puré, E.; Blomberg, R. Pro-tumorigenic roles of fibroblast activation protein in cancer: Back to the basics. Oncogene 2018, 37, 4343–4357. [Google Scholar] [CrossRef] [PubMed]
- Ghersi, G.; Dong, H.; Goldstein, L.A.; Yeh, Y.; Hakkinen, L.; Larjava, H.S.; Chen, W.-T. Regulation of fibroblast migration on collagenous matrix by a cell surface peptidase complex. J. Biol. Chem. 2002, 277, 29231–29241. [Google Scholar] [CrossRef]
- Mueller, S.C.; Ghersi, G.; Akiyama, S.K.; Sang, Q.X.; Howard, L.; Pineiro-Sanchez, M.; Nakahara, H.; Yeh, Y.; Chen, W.T. A novel protease-docking function of integrin at invadopodia. J. Biol. Chem. 1999, 274, 24947–24952. [Google Scholar] [CrossRef]
- Sun, S.; Albright, C.F.; Fish, B.H.; George, H.J.; Selling, B.H.; Hollis, G.F.; Wynn, R. Expression, purification, and kinetic characterization of full-length human fibroblast activation protein. Protein Expr. Purif. 2002, 24, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Duan, Q.; Zhang, H.; Zheng, J.; Zhang, L. Turning Cold into Hot: Firing up the Tumor Microenvironment. Trends Cancer 2020, 6, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Qiu, X.; Wang, X.; He, J. FAP positive fibroblasts induce immune checkpoint blockade resistance in colorectal cancer via promoting immunosuppression. Biochem. Biophys. Res. Commun. 2017, 487, 8–14. [Google Scholar] [CrossRef]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.B.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef] [PubMed]
- Freedman, J.D.; Duffy, M.R.; Lei-Rossmann, J.; Muntzer, A.; Scott, E.M.; Hagel, J.; Campo, L.; Bryant, R.J.; Verrill, C.; Lambert, A.; et al. An Oncolytic Virus Expressing a T-cell Engager Simultaneously Targets Cancer and Immunosuppressive Stromal Cells. Cancer Res. 2018, 78, 6852–6865. [Google Scholar] [CrossRef]
- Coto-Llerena, M.; Ercan, C.; Kancherla, V.; Taha-Mehlitz, S.; Eppenberger-Castori, S.; Soysal, S.D.; Ng, C.K.Y.; Bolli, M.; von Flüe, M.; Nicolas, G.P.; et al. High Expression of FAP in Colorectal Cancer Is Associated With Angiogenesis and Immunoregulation Processes. Front. Oncol. 2020, 10, 979. [Google Scholar] [CrossRef]
- Huang, Y.; Simms, A.E.; Mazur, A.; Wang, S.; León, N.R.; Jones, B.; Aziz, N.; Kelly, T. Fibroblast activation protein-α promotes tumor growth and invasion of breast cancer cells through non-enzymatic functions. Clin. Exp. Metastasis 2011, 28, 567–579. [Google Scholar] [CrossRef]
- Li, M.; Cheng, X.; Rong, R.; Gao, Y.; Tang, X.; Chen, Y. High expression of fibroblast activation protein (FAP) predicts poor outcome in high-grade serous ovarian cancer. BMC Cancer 2020, 20, 1032. [Google Scholar] [CrossRef]
- Lee, H.-O.; Mullins, S.R.; Franco-Barraza, J.; Valianou, M.; Cukierman, E.; Cheng, J.D. FAP-overexpressing fibroblasts produce an extracellular matrix that enhances invasive velocity and directionality of pancreatic cancer cells. BMC Cancer 2011, 11, 245. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Zhan, F.; Barlogie, B.; Epstein, J.; Shaughnessy, J.; Yaccoby, S. Fibroblast activation protein (FAP) is upregulated in myelomatous bone and supports myeloma cell survival. Br. J. Haematol. 2006, 133, 83–92. [Google Scholar] [CrossRef]
- Wang, R.-F.; Zhang, L.-H.; Shan, L.-H.; Sun, W.-G.; Chai, C.-C.; Wu, H.-M.; Ibla, J.C.; Wang, L.-F.; Liu, J.-R. Effects of the fibroblast activation protein on the invasion and migration of gastric cancer. Exp. Mol. Pathol. 2013, 95, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Calais, J. FAP: The Next Billion Dollar Nuclear Theranostics Target? J. Nucl. Med. 2020, 61, 163–165. [Google Scholar] [CrossRef]
- Welt, S.; Divgi, C.R.; Scott, A.M.; Garin-Chesa, P.; Finn, R.D.; Graham, M.; Carswell, E.A.; Cohen, A.; Larson, S.M.; Old, L.J. Antibody targeting in metastatic colon cancer: A phase I study of monoclonal antibody F19 against a cell-surface protein of reactive tumor stromal fibroblasts. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1994, 12, 1193–1203. [Google Scholar] [CrossRef]
- Schmidt, A.; Müller, D.; Mersmann, M.; Wüest, T.; Gerlach, E.; Garin-Chesa, P.; Rettig, W.J.; Pfizenmaier, K.; Moosmayer, D. Generation of human high-affinity antibodies specific for the fibroblast activation protein by guided selection. Eur. J. Biochem. 2001, 268, 1730–1738. [Google Scholar] [CrossRef] [PubMed]
- Mersmann, M.; Schmidt, A.; Rippmann, J.F.; Wüest, T.; Brocks, B.; Rettig, W.J.; Garin-Chesa, P.; Pfizenmaier, K.; Moosmayer, D. Human antibody derivatives against the fibroblast activation protein for tumor stroma targeting of carcinomas. Int. J. Cancer 2001, 92, 240–248. [Google Scholar] [CrossRef]
- Wüest, T.; Moosmayer, D.; Pfizenmaier, K. Construction of a bispecific single chain antibody for recruitment of cytotoxic T cells to the tumour stroma associated antigen fibroblast activation protein. J. Biotechnol. 2001, 92, 159–168. [Google Scholar] [CrossRef]
- Ostermann, E.; Garin-Chesa, P.; Heider, K.H.; Kalat, M.; Lamche, H.; Puri, C.; Kerjaschki, D.; Rettig, W.J.; Adolf, G.R. Effective immunoconjugate therapy in cancer models targeting a serine protease of tumor fibroblasts. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 4584–4592. [Google Scholar] [CrossRef]
- Wäster, P.; Rosdahl, I.; Gilmore, B.F.; Seifert, O. Ultraviolet exposure of melanoma cells induces fibroblast activation protein-α in fibroblasts: Implications for melanoma invasion. Int. J. Oncol. 2011, 39, 193–202. [Google Scholar] [PubMed]
- Altmann, A.; Haberkorn, U.; Siveke, J. The Latest Developments in Imaging of Fibroblast Activation Protein. J. Nucl. Med. 2021, 62, 160–167. [Google Scholar] [CrossRef]
- Narra, K.; Mullins, S.R.; Lee, H.-O.; Strzemkowski-Brun, B.; Magalong, K.; Christiansen, V.J.; McKee, P.A.; Egleston, B.; Cohen, S.J.; Weiner, L.M.; et al. Phase II trial of single agent Val-boroPro (talabostat) inhibiting fibroblast activation protein in patients with metastatic colorectal cancer. Cancer Biol. Ther. 2007, 6, 1691–1699. [Google Scholar] [CrossRef]
- Eager, R.M.; Cunningham, C.C.; Senzer, N.N.; Stephenson, J.; Anthony, S.P.; O’Day, S.J.; Frenette, G.; Pavlick, A.C.; Jones, B.; Uprichard, M.; et al. Phase II assessment of talabostat and cisplatin in second-line stage IV melanoma. BMC Cancer 2009, 9, 263. [Google Scholar] [CrossRef]
- Eager, R.M.; Cunningham, C.C.; Senzer, N.; Richards, D.A.; Raju, R.N.; Jones, B.; Uprichard, M.; Nemunaitis, J. Phase II trial of talabostat and docetaxel in advanced non-small cell lung cancer. Clin. Oncol. 2009, 21, 464–472. [Google Scholar] [CrossRef]
- Jansen, K.; Heirbaut, L.; Cheng, J.D.; Joossens, J.; Ryabtsova, O.; Cos, P.; Maes, L.; Lambeir, A.-M.; De Meester, I.; Augustyns, K.; et al. Selective Inhibitors of Fibroblast Activation Protein (FAP) with a (4-Quinolinoyl)-glycyl-2-cyanopyrrolidine Scaffold. ACS Med. Chem. Lett. 2013, 4, 491–496. [Google Scholar] [CrossRef]
- Poplawski, S.E.; Lai, J.H.; Li, Y.; Jin, Z.; Liu, Y.; Wu, W.; Wu, Y.; Zhou, Y.; Sudmeier, J.L.; Sanford, D.G.; et al. Identification of selective and potent inhibitors of fibroblast activation protein and prolyl oligopeptidase. J. Med. Chem. 2013, 56, 3467–3477. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L.; Eckhardt, M.; Langkopf, E.; Tadayyon, M.; Himmelsbach, F.; Mark, M. (R)-8-(3-amino-piperidin-1-yl)-7-but-2-ynyl-3-methyl-1-(4-methyl-quinazolin-2-ylmethyl)-3,7-dihydro-purine-2,6-dione (BI 1356), a novel xanthine-based dipeptidyl peptidase 4 inhibitor, has a superior potency and longer duration of action compared with other dipeptidyl peptidase-4 inhibitors. J. Pharmacol. Exp. Ther. 2008, 325, 175–182. [Google Scholar]
- Tsai, T.-Y.; Yeh, T.-K.; Chen, X.; Hsu, T.; Jao, Y.-C.; Huang, C.-H.; Song, J.-S.; Huang, Y.-C.; Chien, C.-H.; Chiu, J.-H.; et al. Substituted 4-carboxymethylpyroglutamic acid diamides as potent and selective inhibitors of fibroblast activation protein. J. Med. Chem. 2010, 53, 6572–6583. [Google Scholar] [CrossRef] [PubMed]
- Tanswell, P.; Garin-Chesa, P.; Rettig, W.J.; Welt, S.; Divgi, C.R.; Casper, E.S.; Finn, R.D.; Larson, S.M.; Old, L.J.; Scott, A.M. Population pharmacokinetics of antifibroblast activation protein monoclonal antibody F19 in cancer patients. Br. J. Clin. Pharmacol. 2001, 51, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.M.; Wiseman, G.; Welt, S.; Adjei, A.; Lee, F.-T.; Hopkins, W.; Divgi, C.R.; Hanson, L.H.; Mitchell, P.; Gansen, D.N.; et al. A Phase I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein-positive cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2003, 9, 1639–1647. [Google Scholar]
- Fischer, E.; Chaitanya, K.; Wüest, T.; Wadle, A.; Scott, A.M.; van den Broek, M.; Schibli, R.; Bauer, S.; Renner, C. Radioimmunotherapy of fibroblast activation protein positive tumors by rapidly internalizing antibodies. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 6208–6218. [Google Scholar] [CrossRef] [PubMed]
- Connolly, B.A.; Sanford, D.G.; Chiluwal, A.K.; Healey, S.E.; Peters, D.E.; Dimare, M.T.; Wu, W.; Liu, Y.; Maw, H.; Zhou, Y.; et al. Dipeptide boronic acid inhibitors of dipeptidyl peptidase IV: Determinants of potency and in vivo efficacy and safety. J. Med. Chem. 2008, 51, 6005–6013. [Google Scholar] [CrossRef] [PubMed]
- Meletta, R.; Müller Herde, A.; Chiotellis, A.; Isa, M.; Rancic, Z.; Borel, N.; Ametamey, S.; Krämer, S.; Schibli, R. Evaluation of the Radiolabeled Boronic Acid-Based FAP Inhibitor MIP-1232 for Atherosclerotic Plaque Imaging. Molecules 2015, 20, 2081–2099. [Google Scholar] [CrossRef]
- Moon, E.S.; Elvas, F.; Vliegen, G.; De Lombaerde, S.; Vangestel, C.; De Bruycker, S.; Bracke, A.; Eppard, E.; Greifenstein, L.; Klasen, B.; et al. Targeting fibroblast activation protein (FAP): Next generation PET radiotracers using squaramide coupled bifunctional DOTA and DATA5m chelators. EJNMMI Radiopharm. Chem. 2020, 5, 19. [Google Scholar] [CrossRef]
- Jansen, K.; Heirbaut, L.; Verkerk, R.; Cheng, J.D.; Joossens, J.; Cos, P.; Maes, L.; Lambeir, A.-M.; De Meester, I.; Augustyns, K.; et al. Extended structure-activity relationship and pharmacokinetic investigation of (4-quinolinoyl)glycyl-2-cyanopyrrolidine inhibitors of fibroblast activation protein (FAP). J. Med. Chem. 2014, 57, 3053–3074. [Google Scholar] [CrossRef]
- Loktev, A.; Lindner, T.; Burger, E.-M.; Altmann, A.; Giesel, F.; Kratochwil, C.; Debus, J.; Marmé, F.; Jäger, D.; Mier, W.; et al. Development of Fibroblast Activation Protein-Targeted Radiotracers with Improved Tumor Retention. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2019, 60, 1421–1429. [Google Scholar] [CrossRef] [PubMed]
- Loktev, A.; Lindner, T.; Mier, W.; Debus, J.; Altmann, A.; Jäger, D.; Giesel, F.; Kratochwil, C.; Barthe, P.; Roumestand, C.; et al. A Tumor-Imaging Method Targeting Cancer-Associated Fibroblasts. J. Nucl. Med. 2018, 59, 1423–1429. [Google Scholar] [CrossRef] [PubMed]
- Jansen, K.; De Winter, H.; Heirbaut, L.; Cheng, J.D.; Joossens, J.; Lambeir, A.-M.; De Meester, I.; Augustyns, K.; Van der Veken, P. Selective inhibitors of fibroblast activation protein (FAP) with a xanthine scaffold. Med. Chem. Commun. 2014, 5, 1700–1707. [Google Scholar] [CrossRef]
- Lindner, T.; Loktev, A.; Altmann, A.; Giesel, F.; Kratochwil, C.; Debus, J.; Jäger, D.; Mier, W.; Haberkorn, U. Development of Quinoline-Based Theranostic Ligands for the Targeting of Fibroblast Activation Protein. J. Nucl. Med. 2018, 59, 1415–1422. [Google Scholar] [CrossRef] [PubMed]
- Lindner, T.; Altmann, A.; Krämer, S.; Kleist, C.; Loktev, A.; Kratochwil, C.; Giesel, F.; Mier, W.; Marme, F.; Debus, J.; et al. Design and Development of 99mTc-Labeled FAPI Tracers for SPECT Imaging and 188Re Therapy. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2020, 61, 1507–1513. [Google Scholar] [CrossRef] [PubMed]
- Roy, J.; Hettiarachchi, S.U.; Kaake, M.; Mukkamala, R.; Low, P.S. Design and validation of fibroblast activation protein alpha targeted imaging and therapeutic agents. Theranostics 2020, 10, 5778–5789. [Google Scholar] [CrossRef]
- Giesel, F.L.; Adeberg, S.; Syed, M.; Lindner, T.; Jiménez-Franco, L.D.; Mavriopoulou, E.; Staudinger, F.; Tonndorf-Martini, E.; Regnery, S.; Rieken, S.; et al. FAPI-74 PET/CT Using Either 18F-AlF or Cold-Kit 68Ga Labeling: Biodistribution, Radiation Dosimetry, and Tumor Delineation in Lung Cancer Patients. J. Nucl. Med. 2021, 62, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Dendl, K.; Finck, R.; Giesel, F.L.; Kratochwil, C.; Lindner, T.; Mier, W.; Cardinale, J.; Kesch, C.; Röhrich, M.; Rathke, H.; et al. FAP imaging in rare cancer entities-first clinical experience in a broad spectrum of malignancies. Eur. J. Nucl. Med. Mol. Imaging 2021. [Google Scholar] [CrossRef]
- Moon, E.S.; Ballal, S.; Yadav, M.P.; Bal, C.; Rymenant, Y.V.; Stephan, S.; Bracke, A.; der Veken, P.V.; Meester, I.D.; Roesch, F. Homodimeric Fibroblast Activation Protein (FAP) Targeting Radiotheranostics to Improve Tumor Uptake and Retention Time. Pharmaceuticals 2021. [Google Scholar] [CrossRef]
- Kelly, J.M.; Jeitner, T.M.; Ponnala, S.; Williams, C.; Nikolopoulou, A.; DiMagno, S.G.; Babich, J.W. A Trifunctional Theranostic Ligand Targeting Fibroblast Activation Protein-α (FAPα). Mol. Imaging Biol. 2021, 23, 686–696. [Google Scholar] [CrossRef] [PubMed]
- Millul, J.; Bassi, G.; Mock, J.; Elsayed, A.; Pellegrino, C.; Zana, A.; Dakhel Plaza, S.; Nadal, L.; Gloger, A.; Schmidt, E.; et al. An ultra-high-affinity small organic ligand of fibroblast activation protein for tumor-targeting applications. Proc. Natl. Acad. Sci. USA 2021, 118, e2101852118. [Google Scholar] [CrossRef]
- Eryilmaz, K.; Kilbas, B. Fully-automated synthesis of 177Lu labelled FAPI derivatives on the module modular lab-Eazy. EJNMMI Radiopharm. Chem. 2021, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- Watabe, T.; Liu, Y.; Kaneda-Nakashima, K.; Shirakami, Y.; Lindner, T.; Ooe, K.; Toyoshima, A.; Nagata, K.; Shimosegawa, E.; Haberkorn, U.; et al. Theranostics Targeting Fibroblast Activation Protein in the Tumor Stroma: 64Cu- and 225Ac-Labeled FAPI-04 in Pancreatic Cancer Xenograft Mouse Models. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2020, 61, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Slania, S.L.; Das, D.; Lisok, A.; Du, Y.; Jiang, Z.; Mease, R.C.; Rowe, S.P.; Nimmagadda, S.; Yang, X.; Pomper, M.G. Imaging of Fibroblast Activation Protein in Cancer Xenografts Using Novel (4-Quinolinoyl)-glycyl-2-cyanopyrrolidine-Based Small Molecules. J. Med. Chem. 2021, 64, 4059–4070. [Google Scholar] [CrossRef] [PubMed]
- Giesel, F.L.; Kratochwil, C.; Lindner, T.; Marschalek, M.M.; Loktev, A.; Lehnert, W.; Debus, J.; Jäger, D.; Flechsig, P.; Altmann, A.; et al. 68Ga-FAPI PET/CT: Biodistribution and Preliminary Dosimetry Estimate of 2 DOTA-Containing FAP-Targeting Agents in Patients with Various Cancers. J. Nucl. Med. 2019, 60, 386–392. [Google Scholar] [CrossRef]
- Kreppel, B.; Gärtner, F.C.; Marinova, M.; Attenberger, U.; Meisenheimer, M.; Toma, M.; Kristiansen, G.; Feldmann, G.; Moon, E.S.; Roesch, F.; et al. [68Ga]Ga-DATA5m.SA.FAPi PET/CT: Specific Tracer-uptake in Focal Nodular Hyperplasia and potential Role in Liver Tumor Imaging. Nukl. Nucl. Med. 2020, 59, 387–389. [Google Scholar] [CrossRef]
- Ballal, S.; Yadav, M.P.; Moon, E.S.; Kramer, V.S.; Roesch, F.; Kumari, S.; Tripathi, M.; ArunRaj, S.T.; Sarswat, S.; Bal, C. Biodistribution, pharmacokinetics, dosimetry of [68Ga]Ga-DOTA.SA.FAPi, and the head-to-head comparison with [18F]F-FDG PET/CT in patients with various cancers. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 1915–1931. [Google Scholar] [CrossRef]
- Ballal, S.; Yadav, M.P.; Kramer, V.; Moon, E.S.; Roesch, F.; Tripathi, M.; Mallick, S.; ArunRaj, S.T.; Bal, C. A theranostic approach of [68Ga]Ga-DOTA.SA.FAPi PET/CT-guided [177Lu]Lu-DOTA.SA.FAPi radionuclide therapy in an end-stage breast cancer patient: New frontier in targeted radionuclide therapy. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 942–944. [Google Scholar] [CrossRef]
- Kreppel, B.; Gonzalez-Carmona, M.A.; Feldmann, G.; Küppers, J.; Moon, E.S.; Marinova, M.; Bundschuh, R.A.; Kristiansen, G.; Essler, M.; Roesch, F.; et al. Fibroblast activation protein inhibitor (FAPi) positive tumour fraction on PET/CT correlates with Ki-67 in liver metastases of neuroendocrine tumours. Nuklearmedizin 2021, 60, 344–354. [Google Scholar] [CrossRef]
- Röhrich, M.; Loktev, A.; Wefers, A.K.; Altmann, A.; Paech, D.; Adeberg, S.; Windisch, P.; Hielscher, T.; Flechsig, P.; Floca, R.; et al. IDH-wildtype glioblastomas and grade III/IV IDH-mutant gliomas show elevated tracer uptake in fibroblast activation protein-specific PET/CT. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 2569–2580. [Google Scholar] [CrossRef]
- Kratochwil, C.; Giesel, F.L.; Rathke, H.; Fink, R.; Dendl, K.; Debus, J.; Mier, W.; Jäger, D.; Lindner, T.; Haberkorn, U. [153Sm]Samarium-labeled FAPI-46 radioligand therapy in a patient with lung metastases of a sarcoma. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 3011–3013. [Google Scholar] [CrossRef]
- Rathke, H.; Fuxius, S.; Giesel, F.L.; Lindner, T.; Debus, J.; Haberkorn, U.; Kratochwil, C. Two Tumors, One Target: Preliminary Experience With 90Y-FAPI Therapy in a Patient With Metastasized Breast and Colorectal Cancer. Clin. Nucl. Med. 2021, 46, 842–844. [Google Scholar] [CrossRef]
- Ferdinandus, J.; Fragoso Costa, P.; Kessler, L.; Weber, M.; Hirmas, N.; Kostbade, K.; Bauer, S.; Schuler, M.; Ahrens, M.; Schildhaus, H.-U.; et al. Initial clinical experience with 90Y-FAPI-46 radioligand therapy for advanced stage solid tumors: A case series of nine patients. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2021, jnumed.121.262468. [Google Scholar] [CrossRef]
- Fearon, D.T. The carcinoma-associated fibroblast expressing fibroblast activation protein and escape from immune surveillance. Cancer Immunol. Res. 2014, 2, 187–193. [Google Scholar] [CrossRef]
- Kratochwil, C.; Flechsig, P.; Lindner, T.; Abderrahim, L.; Altmann, A.; Mier, W.; Adeberg, S.; Rathke, H.; Röhrich, M.; Winter, H.; et al. 68Ga-FAPI PET/CT: Tracer Uptake in 28 Different Kinds of Cancer. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2019, 60, 801–805. [Google Scholar] [CrossRef]
- Chen, H.; Pang, Y.; Wu, J.; Zhao, L.; Hao, B.; Wu, J.; Wei, J.; Wu, S.; Zhao, L.; Luo, Z.; et al. Comparison of [68Ga]Ga-DOTA-FAPI-04 and [18F] FDG PET/CT for the diagnosis of primary and metastatic lesions in patients with various types of cancer. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 1820–1832. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Xing, H.; Yang, X.; Li, F.; Yao, S.; Zhang, H.; Zhao, H.; Hacker, M.; Huo, L.; Li, X. Fibroblast imaging of hepatic carcinoma with 68Ga-FAPI-04 PET/CT: A pilot study in patients with suspected hepatic nodules. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Dahlbom, M.; Lindner, T.; Vauclin, S.; Mona, C.; Slavik, R.; Czernin, J.; Haberkorn, U.; Calais, J. Radiation Dosimetry and Biodistribution of 68Ga-FAPI-46 PET Imaging in Cancer Patients. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2020, 61, 1171–1177. [Google Scholar] [CrossRef]
- Koerber, S.A.; Staudinger, F.; Kratochwil, C.; Adeberg, S.; Haefner, M.F.; Ungerechts, G.; Rathke, H.; Winter, E.; Lindner, T.; Syed, M.; et al. The Role of 68Ga-FAPI PET/CT for Patients with Malignancies of the Lower Gastrointestinal Tract: First Clinical Experience. J. Nucl. Med. 2020, 61, 1331–1336. [Google Scholar] [CrossRef] [PubMed]
- Koerber, S.A.; Finck, R.; Dendl, K.; Uhl, M.; Lindner, T.; Kratochwil, C.; Röhrich, M.; Rathke, H.; Ungerechts, G.; Adeberg, S.; et al. Novel FAP ligands enable improved imaging contrast in sarcoma patients due to FAPI-PET/CT. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 3918–3924. [Google Scholar] [CrossRef] [PubMed]
- Dendl, K.; Koerber, S.A.; Finck, R.; Mokoala, K.M.G.; Staudinger, F.; Schillings, L.; Heger, U.; Röhrich, M.; Kratochwil, C.; Sathekge, M.; et al. 68Ga-FAPI-PET/CT in patients with various gynecological malignancies. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 4089–4100. [Google Scholar] [CrossRef]
- Röhrich, M.; Naumann, P.; Giesel, F.L.; Choyke, P.L.; Staudinger, F.; Wefers, A.; Liew, D.P.; Kratochwil, C.; Rathke, H.; Liermann, J.; et al. Impact of 68Ga-FAPI PET/CT Imaging on the Therapeutic Management of Primary and Recurrent Pancreatic Ductal Adenocarcinomas. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2021, 62, 779–786. [Google Scholar]
- Giesel, F.L.; Heussel, C.P.; Lindner, T.; Röhrich, M.; Rathke, H.; Kauczor, H.-U.; Debus, J.; Haberkorn, U.; Kratochwil, C. FAPI-PET/CT improves staging in a lung cancer patient with cerebral metastasis. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 1754–1755. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biomarker | Function | Surface Expression | Related Cancer Model | Shared Expression with Other Cells |
|---|---|---|---|---|
| Cytoskeleton Marker | ||||
| α-SMA | Structure Contractility Motility | No | Pancreatic Liver Breast | Normal fibroblasts Smooth muscle cells Pericytes |
| S100A4 or FSP-1 | Collagen induction Fibrosis Motility | No | Breast | Normal fibroblasts Macrophages Epithelial cells |
| Vimentin | Structure Motility | No | Breast Prostate | Neurons Epithelial cells Endothelial cells |
| Membrane-bound protein and receptor | ||||
| FAP | Fibrogenesis ECM remodeling | Yes | >90% of all cancers | Stromal fibroblasts Immune cells |
| PDGFRα/β | Tyrosine kinase activity receptor | Yes | Cervical Colorectal | Normal fibroblasts Skeletal muscle Pericytes Vascular Smooth muscle |
| ECM Component | ||||
| Tenascin C | Cell adhesion | No | Breast Malignant glioma | Cancer cells |
| Radionuclide | Inhibitor | Quality of Radiation | Evaluation Phase | Reference | |
|---|---|---|---|---|---|
| Imaging | Radiotherapy | ||||
| 18F | FAPI-74 | PET | - | Clinical: patients with lung cancer | [116] |
| Glc-FAPI-04 | Preclinical: fibrosarcoma and glioblastoma xenografts | [17] | |||
| 68Ga | FAPI-02 | PET | - | Clinical: various cancers | [124] |
| FAPI-04 | Clinical: various cancers | [110,111,113,124] | |||
| FAPI-20 | Preclinical: fibrosarcoma xenograft | ||||
| FAPI-21 | Clinical: various cancers | ||||
| FAPI-22 | Preclinical: fibrosarcoma xenograft | ||||
| FAPI-31 | |||||
| FAPI-35 | |||||
| FAPI-36 | |||||
| FAPI-37 | |||||
| FAPI-46 | Clinical: various cancers | ||||
| FAPI-74 | Clinical: patients with lung cancer | ||||
| DOTA.SA.FAPi | Preclinical: colorectal adenocarcinoma xenograftClinical: various cancer patients | [108,125,126,127,128] | |||
| DATA5m.SA.FAPi | Preclinical: in vitro modelsClinical: restaging of tumor manifestation, liver tumor and metastases imaging | ||||
| DOTA.(SA.FAPi)2 | Clinical: patient with thyroid and pancreatic neuroendocrine tumors | [118] | |||
| DOTAGA.(SA.FAPi)2 | |||||
| RPS-309 | Preclinical: liposarcoma xenograft | [119] | |||
| 111In | QCP02 | SPECT | - | Preclinical: glioblastoma xenograft | [123] |
| 99mTc | FAPI-34 | SPECT | - | Clinical: patients with ovarian metastasis and pancreatic cancer | [114] |
| FL-L3 | Preclinical: breast cancer xenograft | [115] | |||
| 225Ac | FAPI-04 | - | Yes | Preclinical: pancreatic cancer xenograft | [122] |
| 64Cu | FAPI-04 | PET | Yes | ||
| 177Lu | FAPI-02 | SPECT | Yes | Preclinical: glioblastoma xenograft | [111,129] |
| FAPI-04 | |||||
| FAPI-46 | Fully automated radiosynthesis unit | [121] | |||
| RPS-309 | Preclinical: liposarcoma xenograft | [119] | |||
| OncoFAP | Preclinical: renal carcinoma and fibrosarcoma xenografts | [120] | |||
| FAP-2286 | Preclinical: HEK-FAP tumor bearing animalsClinical: Patients with diverse adenocarcinomas | [19,20] | |||
| 153Sm | FAPI-46 | Scintigraphy | Yes | Clinical: Patient with lung metastatic, fibrous spindle cell soft tissue sarcoma | [130] |
| 90Y | FAPI-04 | - | Yes | Clinical: metastatic breast cancer patient | [113] |
| FAPI-46 | Clinical: patient with metastasized breast and colorectal cancers | [131] | |||
| Clinical: patients with metastatic soft tissue or bone sarcoma, and pancreatic cancer | [132] | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Imlimthan, S.; Moon, E.S.; Rathke, H.; Afshar-Oromieh, A.; Rösch, F.; Rominger, A.; Gourni, E. New Frontiers in Cancer Imaging and Therapy Based on Radiolabeled Fibroblast Activation Protein Inhibitors: A Rational Review and Current Progress. Pharmaceuticals 2021, 14, 1023. https://doi.org/10.3390/ph14101023
Imlimthan S, Moon ES, Rathke H, Afshar-Oromieh A, Rösch F, Rominger A, Gourni E. New Frontiers in Cancer Imaging and Therapy Based on Radiolabeled Fibroblast Activation Protein Inhibitors: A Rational Review and Current Progress. Pharmaceuticals. 2021; 14(10):1023. https://doi.org/10.3390/ph14101023
Chicago/Turabian StyleImlimthan, Surachet, Euy Sung Moon, Hendrik Rathke, Ali Afshar-Oromieh, Frank Rösch, Axel Rominger, and Eleni Gourni. 2021. "New Frontiers in Cancer Imaging and Therapy Based on Radiolabeled Fibroblast Activation Protein Inhibitors: A Rational Review and Current Progress" Pharmaceuticals 14, no. 10: 1023. https://doi.org/10.3390/ph14101023
APA StyleImlimthan, S., Moon, E. S., Rathke, H., Afshar-Oromieh, A., Rösch, F., Rominger, A., & Gourni, E. (2021). New Frontiers in Cancer Imaging and Therapy Based on Radiolabeled Fibroblast Activation Protein Inhibitors: A Rational Review and Current Progress. Pharmaceuticals, 14(10), 1023. https://doi.org/10.3390/ph14101023