Memory Enhancers for Alzheimer’s Dementia: Focus on cGMP



Abstract
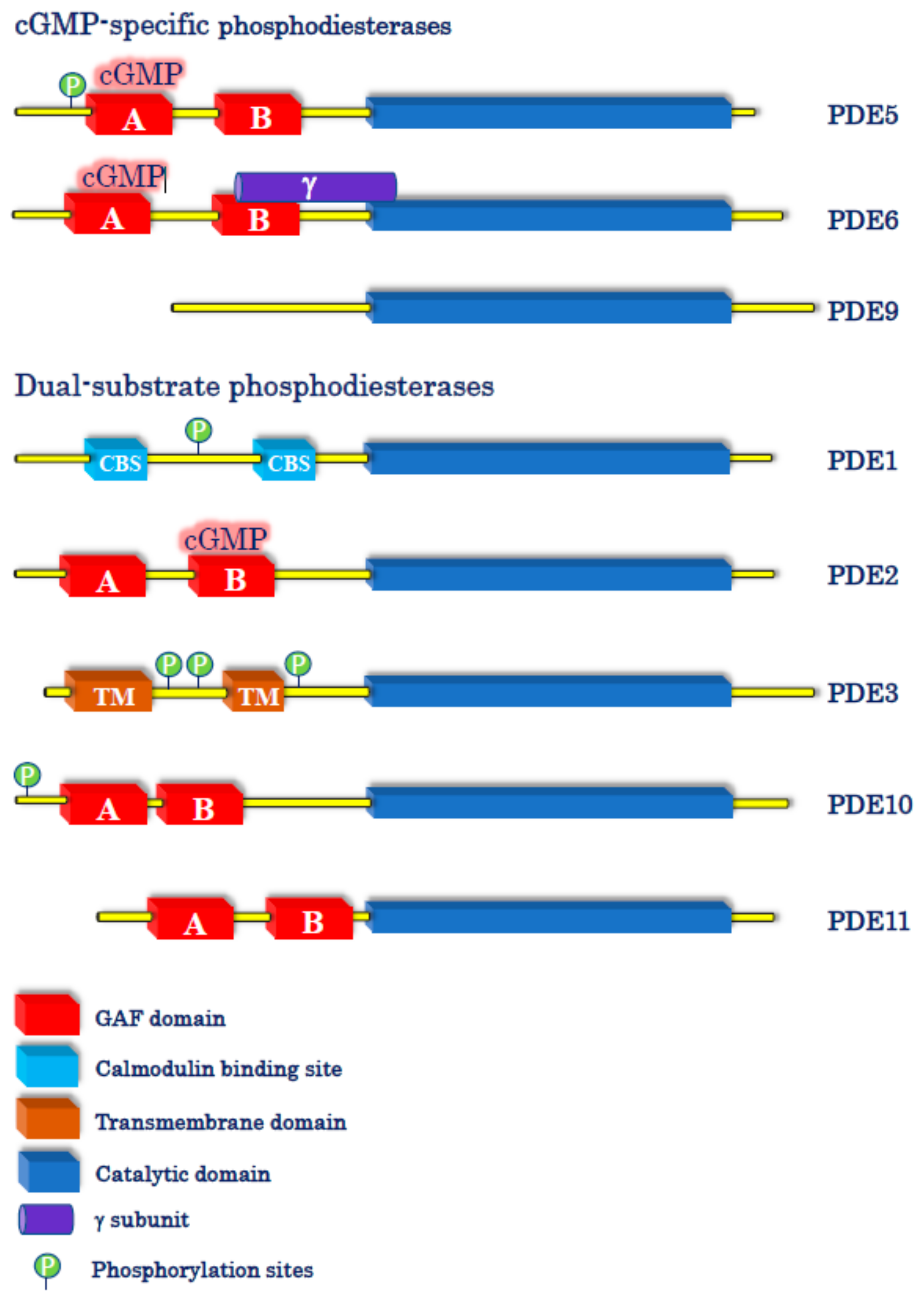
1. The cGMP Universe: Synthesis, Signaling, and Metabolism
2. The cGMP System in Memory Processes
3. The cGMP System in Alzheimer’s Disease
4. Clinical Studies on cGMP-Enhancers
4.1. sGC Stimulators
4.2. cGMP-Specific PDE Inhibitors
4.3. Dual-Substrate PDE Inhibitors
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ashman, D.F.; Lipton, R.; Melicow, M.M.; Price, T.D. Isolation of adenosine 3′,5′-monophosphate and guanosine 3′,5′-monophosphate from rat urine. Biochem. Biophys. Res. Commun. 1963, 11, 330–334. [Google Scholar] [CrossRef]
- Kimura, H.; Murad, F. Evidence for two different forms of guanylate cyclase in rat heart. J. Biol. Chem. 1974, 249, 6910–6916. [Google Scholar] [CrossRef]
- Chrisman, T.D.; Garbers, D.L.; Parks, M.A.; Hardman, J.G. Characterization of particulate and soluble guanylate cyclases from rat lung. J. Biol. Chem. 1975, 250, 374–381. [Google Scholar] [CrossRef]
- Kuhn, M. Molecular Physiology of Membrane Guanylyl Cyclase Receptors. Physiol. Rev. 2016, 96, 751–804. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.K.; Duda, T.; Makino, T.L. Integrative Signaling Networks of Membrane Guanylate Cyclases: Biochemistry and Physiology. Front. Mol. Neurosci. 2016, 9, 83. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, F. The cGMP system: Components and function. Biol. Chem. 2020, 401, 447–469. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, S.; Duda, T.; Pertzev, A.; Sharma, R.K. Membrane Guanylate Cyclase catalytic Subdomain: Structure and Linkage with Calcium Sensors and Bicarbonate. Front. Mol. Neurosci. 2017, 10, 173. [Google Scholar] [CrossRef]
- Montfort, W.R.; Wales, J.A.; Weichsel, A. Structure and activation of soluble guanylyl cyclase, the nitric oxide sensor. Antioxid. Redox Signal. 2017, 26, 107–121. [Google Scholar] [CrossRef]
- Horst, B.G.; Marletta, M.A. Physiological activation and deactivation of soluble guanylate cyclase. Nitric Oxide 2018, 77, 65–74. [Google Scholar] [CrossRef]
- Budworth, J.; Meillerais, S.; Charles, I.; Powell, K. Tissue distribution of the human soluble guanylate cyclases. Biochem. Biophys. Res. Commun. 1999, 263, 696–701. [Google Scholar] [CrossRef]
- Childers, K.C.; Garcin, E.D. Structure/function of the soluble guanylyl cyclase catalytic domain. Nitric Oxide 2018, 77, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, B.; Bertinetti, D.; Herberg, F.W. cAMP-Dependent Protein Kinase and cGMP-Dependent Protein Kinase as Cyclic Nucleotide Effectors. Handb. Exp. Pharmacol. 2017, 238, 105–122. [Google Scholar] [PubMed]
- Podda, M.V.; Grassi, C. New perspectives in cyclic nucleotide-mediated functions in the CNS: The emerging role of cyclic nucleotide-gated (CNG) channels. Pflug. Arch. 2014, 466, 1241–1257. [Google Scholar] [CrossRef] [PubMed]
- Michalakis, S.; Becirovic, E.; Biel, M. Retinal Cyclic Nucleotide-Gated Channels: From Pathophysiology to Therapy. Int. J. Mol. Sci. 2018, 19, 749. [Google Scholar] [CrossRef]
- Bender, A.T.; Beavo, J.A. Cyclic nucleotide phosphodiesterases: Molecular regulation to clinical use. Pharmacol. Rev. 2006, 58, 488–520. [Google Scholar] [CrossRef]
- Brescia, M.; Zaccolo, M. Modulation of Compartmentalised Cyclic Nucleotide Signalling via Local Inhibition of Phosphodiesterase Activity. Int. J. Mol. Sci. 2016, 17, 1672. [Google Scholar] [CrossRef]
- Bliss, T.V.; Lømo, T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J. Physiol. 1973, 232, 331–356. [Google Scholar] [CrossRef]
- Ricciarelli, R.; Fedele, E. cAMP, cGMP and Amyloid β: Three Ideal Partners for Memory Formation. Trends Neurosci. 2018, 41, 255–266. [Google Scholar] [CrossRef]
- Boulton, C.L.; Southam, E.; Garthwaite, J. Nitric oxide-dependent long-term potentiation is blocked by a specific inhibitor of soluble guanylyl cyclase. Neuroscience 1995, 69, 699–703. [Google Scholar] [CrossRef]
- Mutlu, O.; Akar, F.; Celikyurt, I.K.; Tanyeri, P.; Ulak, G.; Erden, F. 7-NI and ODQ Disturbs Memory in the Elevated Plus Maze, Morris Water Maze, and Radial Arm Maze Tests in Mice. Drug Target Insights 2015, 9, 1–8. [Google Scholar] [CrossRef]
- Chien, W.L.; Liang, K.C.; Teng, C.M.; Kuo, S.C.; Lee, F.Y.; Fu, W.M. Enhancement of long-term potentiation by a potent nitric oxide-guanylyl cyclase activator, 3-(5-hydroxymethyl-2-furyl)-1-benzyl-indazole. Mol. Pharmacol. 2003, 63, 1322–1328. [Google Scholar] [CrossRef] [PubMed]
- Komsuoglu Celikyurt, I.; Utkan, T.; Ozer, C.; Gacar, N.; Aricioglu, F. Effects of YC-1 on learning and memory functions of aged rats. Med. Sci. Monit. Basic Res. 2014, 20, 130–137. [Google Scholar] [PubMed]
- Zhuo, M.; Hu, Y.; Schultz, C.; Kandel, E.R.; Hawkins, R.D. Role of guanylyl cyclase and cGMP-dependent protein kinase in long-term potentiation. Nature 1994, 368, 635–639. [Google Scholar] [CrossRef] [PubMed]
- Arancio, O.; Antonova, I.; Gambaryan, S.; Lohmann, S.M.; Wood, J.S.; Lawrence, D.S.; Hawkins, R.D. Presynaptic role of cGMP-dependent protein kinase during long-lasting potentiation. J. Neurosci. 2000, 21, 143–149. [Google Scholar] [CrossRef]
- Serulle, Y.; Zhang, S.; Ninan, I.; Puzzo, D.; McCarthy, M.; Khatri, L.; Arancio, O.; Ziff, E.B. A GluR1-cGKII interaction regulates AMPA receptor trafficking. Neuron 2007, 56, 670–688. [Google Scholar] [CrossRef]
- Kleppisch, T.; Wolfsgruber, W.; Feil, S.; Allmann, R.; Wotjak, C.T.; Goebbels, S.; Nave, K.A.; Hofmann, F.; Feil, R. Hippocampal cGMP-dependent protein kinase I supports an age- and protein synthesis-dependent component of long-term potentiation but is not essential for spatial reference and contextual memory. J. Neurosci. 2003, 23, 6005–6012. [Google Scholar] [CrossRef]
- Wincott, C.M.; Kim, S.; Titcombe, R.F.; Tukey, D.S.; Girma, H.K.; Pick, J.E.; Devito, L.M.; Hofmann, F.; Hoeffer, C.; Ziff, E.B. Spatial memory deficits and motor coordination facilitation in cGMP-dependent protein kinase type II-deficient mice. Neurobiol. Learn. Mem. 2013, 99, 32–37. [Google Scholar] [CrossRef]
- Parent, A.; Schrader, K.; Munger, S.D.; Reed, R.R.; Linden, D.J.; Ronnett, G.V. Synaptic transmission and hippocampal long-term potentiation in olfactory cyclic nucleotide-gated channel type 1 null mouse. J. Neurophysiol. 1998, 79, 3295–3301. [Google Scholar] [CrossRef]
- Michalakis, S.; Kleppisch, T.; Polta, S.A.; Wotjak, C.T.; Koch, S.; Rammes, G.; Matt, L.; Becirovic, E.; Biel, M. Altered synaptic plasticity and behavioral abnormalities in CNGA3-deficient mice. Genes Brain Behav. 2011, 10, 137–148. [Google Scholar] [CrossRef]
- Uthayathas, S.; Parameshwaran, K.; Karuppagounder, S.S.; Ahuja, M.; Dhanasekaran, M.; Suppiramaniam, V. Selective inhibition of phosphodiesterase 5 enhances glutamatergic synaptic plasticity and memory in mice. Synapse 2013, 67, 741–747. [Google Scholar] [CrossRef]
- Bollen, E.; Puzzo, D.; Rutten, K.; Privitera, L.; De Vry, J.; Vanmierlo, T.; Kenis, G.; Palmeri, A.; D’Hooge, R.; Balschun, D.; et al. Improved long-term memory via enhancing cGMP-PKG signaling requires cAMP-PKA signaling. Neuropsychopharmacology 2014, 39, 2497–2505. [Google Scholar] [CrossRef] [PubMed]
- Palmeri, A.; Ricciarelli, R.; Gulisano, W.; Rivera, D.; Rebosio, C.; Calcagno, E.; Tropea, M.R.; Conti, S.; Das, U.; Roy, S.; et al. Amyloid-β Peptide Is Needed for cGMP-Induced Long-Term Potentiation and Memory. J. Neurosci. 2017, 37, 6926–6937. [Google Scholar] [CrossRef] [PubMed]
- Argyrousi, E.K.; Heckman, P.R.A.; Prickaerts, J. Role of cyclic nucleotides and their downstream signaling cascades in memory function: Being at the right time at the right spot. Neurosci. Biobehav. Rev. 2020, 113, 12–38. [Google Scholar] [CrossRef] [PubMed]
- Dorner-Ciossek, C.; Kroker, K.S.; Rosenbrock, H. Role of PDE9 in Cognition. Adv. Neurobiol. 2017, 17, 231–254. [Google Scholar] [PubMed]
- Rosenbrock, H.; Giovannini, R.; Schänzle, G.; Koros, E.; Runge, F.; Fuchs, H.; Marti, A.; Reymann, K.G.; Schröder, U.H.; Fedele, E.; et al. The Novel Phosphodiesterase 9A Inhibitor BI 409306 Increases Cyclic Guanosine Monophosphate Levels in the Brain, Promotes Synaptic Plasticity, and Enhances Memory Function in Rodents. J. Pharmacol. Exp. Ther. 2019, 371, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Puzzo, D.; Gulisano, W.; Palmeri, A.; Arancio, O. Rodent models for Alzheimer’s disease drug discovery. Expert Opin. Drug Discov. 2015, 10, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Lee, S.H.; VandeVrede, L.; Qin, Z.; Ben Aissa, M.; Larson, J.; Teich, A.F.; Arancio, O.; D’Souza, Y.; Elharram, A.; et al. A multifunctional therapeutic approach to disease modification in multiple familial mouse models and a novel sporadic model of Alzheimer’s disease. Mol. Neurodegener. 2016, 11, 35. [Google Scholar] [CrossRef]
- Acquarone, E.; Argyrousi, E.K.; van den Berg, M.; Gulisano, W.; Fa, M.; Staniszewski, A.; Calcagno, E.; Zuccarello, E.; D’Adamio, L.; Deng, S.X.; et al. Synaptic and memory dysfunction induced by tau oligomers is rescued by up-regulation of the nitric oxide cascade. Mol. Neurodegener. 2019, 14, 26. [Google Scholar] [CrossRef]
- Ribaudo, G.; Ongaro, A.; Zagotto, G.; Memo, M.; Gianoncelli, A. Therapeutic Potential of Phosphodiesterase Inhibitors against Neurodegeneration: The Perspective of the Medicinal Chemist. ACS Chem. Neurosci. 2020, 11, 1726–1739. [Google Scholar] [CrossRef]
- Sanders, O.; Rajagopal, L. Phosphodiesterase Inhibitors for Alzheimer’s Disease: A Systematic Review of Clinical Trials and Epidemiology with a Mechanistic Rationale. J. Alzheimers Dis. Rep. 2020, 4, 185–215. [Google Scholar] [CrossRef]
- Ugarte, A.; Gil-Bea, F.; García-Barroso, C.; Cedazo-Minguez, Á.; Ramírez, M.J.; Franco, R.; García-Osta, A.; Oyarzabal, J.; Cuadrado-Tejedor, M. Decreased levels of guanosine 3′,5′-monophosphate (cGMP) in cerebrospinal fluid (CSF) are associated with cognitive decline and amyloid pathology in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2015, 41, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Hesse, R.; Lausser, L.; Gummert, P.; Schmid, F.; Wahler, A.; Schnack, C.; Kroker, K.S.; Otto, M.; Tumani, H.; Kestler, H.A.; et al. Reduced cGMP levels in CSF of AD patients correlate with severity of dementia and current depression. Alzheimers Res. Ther. 2017, 9, 17. [Google Scholar] [CrossRef] [PubMed]
- Norris, P.J.; Faull, R.L.; Emson, P.C. Neuronal nitric oxide synthase (nNOS) mRNA expression and NADPH-diaphorase staining in the frontal cortex, visual cortex and hippocampus of control and Alzheimer’s disease brains. Brain Res. Mol. Brain Res. 1996, 41, 36–49. [Google Scholar] [CrossRef]
- Simic, G.; Lucassen, P.J.; Krsnik, Z.; Kruslin, B.; Kostovic, I.; Winblad, B.; Bogdanovi, N. nNOS expression in reactive astrocytes correlates with increased cell death related DNA damage in the hippocampus and entorhinal cortex in Alzheimer’s disease. Exp. Neurol. 2000, 165, 12–26. [Google Scholar] [CrossRef] [PubMed]
- Yew, D.T.; Wong, H.W.; Li, W.P.; Lai, H.W.; Yu, W.H. Nitric oxide synthase neurons in different areas of normal aged and Alzheimer’s brains. Neuroscience 1999, 89, 675–686. [Google Scholar] [CrossRef]
- Lüth, H.J.; Holzer, M.; Gärtner, U.; Staufenbiel, M.; Arendt, T. Expression of endothelial and inducible NOS-isoforms is increased in Alzheimer’s disease, in APP23 transgenic mice and after experimental brain lesion in rat: Evidence for an induction by amyloid pathology. Brain Res. 2001, 913, 57–67. [Google Scholar] [CrossRef]
- Liu, P.; Fleete, M.S.; Jing, Y.; Collie, N.D.; Curtis, M.A.; Waldvogel, H.J.; Faull, R.L.; Abraham, W.C.; Zhang, H. Altered arginine metabolism in Alzheimer’s disease brains. Neurobiol. Aging 2014, 35, 1992–2003. [Google Scholar] [CrossRef]
- Bonkale, W.L.; Winblad, B.; Ravid, R.; Cowburn, R.F. Reduced nitric oxide responsive soluble guanylyl cyclase activity in the superior temporal cortex of patients with Alzheimer’s disease. Neurosci. Lett. 1995, 187, 5–8. [Google Scholar] [CrossRef]
- Baltrons, M.A.; Pifarré, P.; Ferrer, I.; Carot, J.M.; Guarcìa, A. Reduced expression of NO-sensitive guanylyl cyclase in reactive astrocytes of Alzheimer disease, Creutzfeldt–Jakob disease, and multiple sclerosis brains. Neurobiol. Dis. 2004, 17, 462–472. [Google Scholar] [CrossRef]
- Xu, M.; Zhang, D.F.; Luo, R.; Wu, Y.; Zhou, H.; Kong, L.L.; Bi, R.; Yao, Y.G. A systematic integrated analysis of brain expression profiles reveals YAP1 and other prioritized hub genes as important upstream regulators in Alzheimer’s disease. Alzheimers Dement. 2018, 14, 215–229. [Google Scholar] [CrossRef]
- Sanders, O. Sildenafil for the Treatment of Alzheimer’s Disease: A systematic Review. J. Alzheimers Dis. Rep. 2020, 4, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Irisarri, E.; Markerink-Van Ittersum, M.; Mengod, G.; de Vente, J. Expression of the cGMP-specific phosphodiesterase 2 and 9 in normal and Alzheimer’s disease human brains. Eur. J. Neurosci. 2007, 25, 3332–3338. [Google Scholar] [CrossRef] [PubMed]
- Prieto, G.A.; Trieu, B.H.; Dang, C.T.; Bilousova, T.; Gylys, K.H.; Berchtold, N.C.; Lynch, G.; Cotman, C.W. Pharmacological Rescue of Long-Term Potentiation in Alzheimer Diseased Synapses. J. Neurosci. 2017, 37, 1197–1212. [Google Scholar] [CrossRef] [PubMed]
- Hollas, M.A.; Ben Aissa, M.; Lee, S.H.; Gordon-Blake, J.M.; Thatcher, G.R.J. Pharmacological manipulation of cGMP and NO/cGMP in CNS drug discovery. Nitric Oxide 2019, 82, 59–74. [Google Scholar] [CrossRef]
- Borghans, L.G.J.M.; Sambeth, A.; Prickaerts, J.; Ramaekers, J.G.; Blokland, A. The effects of the soluble guanylate cyclase stimulator riociguat on memory performance in healthy volunteers with a biperiden-induced memory impairment. Psychopharmacology 2018, 235, 2407–2416. [Google Scholar] [CrossRef]
- Cyclerion Announces Positive Data from IW-6463 CNS Translational Pharmacology Study in Healthy Elderly Subjects. Available online: https://ir.cyclerion.com/news-releases/news-release-details/cyclerion-announces-positive-data-iw-6463-cns-translational (accessed on 1 December 2020).
- Schultheiss, D.; Müller, S.V.; Nager, W.; Stief, C.G.; Schlote, N.; Jonas, U.; Asvestis, C.; Johannes, S.; Münte, T.F. Central effects of sildenafil (Viagra) on auditory selective attention and verbal recognition memory in humans: A study with event-related brain potentials. World. J. Urol. 2001, 19, 46–50. [Google Scholar] [CrossRef]
- Grass, H.; Klotz, T.; Fathian-Sabet, B.; Berghaus, G.; Engelmann, U.; Käferstein, H. Sildenafil (Viagra): Is there an influence on psychological performance? Int. Urol. Nephrol. 2001, 32, 409–412. [Google Scholar] [CrossRef]
- Goff, D.C.; Cather, C.; Freudenreich, O.; Henderson, D.C.; Evins, A.E.; Culhane, M.A.; Walsh, J.P. A placebo-controlled study of sildenafil effects on cognition in schizophrenia. Psychopharmacology 2009, 202, 411–417. [Google Scholar] [CrossRef]
- Sheng, M.; Lu, H.; Liu, P.; Li, Y.; Ravi, H.; Peng, S.L.; Diaz-Arrastia, R.; Devous, M.D.; Womack, K.B. Sildenafil Improves Vascular and Metabolic Function in Patients with Alzheimer’s Disease. J. Alzheimers Dis. 2017, 60, 1351–1364. [Google Scholar] [CrossRef]
- Samudra, N.; Motes, M.; Lu, H.; Sheng, M.; Diaz-Arrastia, R.; Devous, M.; Hart, J.; Womack, K.B. A Pilot Study of Changes in Medial Temporal Lobe Fractional Amplitude of Low Frequency Fluctuations after Sildenafil Administration in Patients with Alzheimer’s Disease. J. Alzheimers Dis. 2019, 70, 163–170. [Google Scholar] [CrossRef]
- Reneerkens, O.A.; Sambeth, A.; Van Duinen, M.A.; Blokland, A.; Steinbusch, H.W.; Prickaerts, J. The PDE5 inhibitor vardenafil does not affect auditory sensory gating in rats and humans. Psychopharmacology 2013, 225, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Reneerkens, O.A.; Sambeth, A.; Ramaekers, J.G.; Steinbusch, H.W.; Blokland, A.; Prickaerts, J. The effects of the phosphodiesterase type 5 inhibitor vardenafil on cognitive performance in healthy adults: A behavioral-electroencephalography study. J. Psychopharmacol. 2013, 27, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Shim, Y.S.; Pae, C.U.; Kim, S.W.; Kim, H.W.; Kim, J.C.; Koh, J.S. Effects of repeated dosing with Udenafil (Zydena) on cognition, somatization and erection in patients with erectile dysfunction: A pilot study. Int. J. Impot. Res. 2011, 23, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Shim, Y.S.; Pae, C.U.; Cho, K.J.; Kim, S.W.; Kim, J.C.; Koh, J.S. Effects of daily low-dose treatment with phosphodiesterase type 5 inhibitor on cognition, depression, somatization and erectile function in patients with erectile dysfunction: A double-blind, placebo-controlled study. Int. J. Impot. Res. 2014, 26, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Schwam, E.M.; Nicholas, T.; Chew, R.; Billing, C.B.; Davidson, W.; Ambrose, D.; Altstiel, L.D. A multicenter, double-blind, placebo-controlled trial of the PDE9A inhibitor, PF-04447943, in Alzheimer’s disease. Curr. Alzheimer Res. 2014, 11, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Frölich, L.; Wunderlich, G.; Thamer, C.; Roehrle, M.; Garcia, M., Jr.; Dubois, B. Evaluation of the efficacy, safety and tolerability of orally administered BI 409306, a novel phosphodiesterase type 9 inhibitor, in two randomised controlled phase II studies in patients with prodromal and mild Alzheimer’s disease. Alzheimers Res. Ther. 2019, 11, 18. [Google Scholar] [CrossRef]
- Zhang, Y.S.; Li, J.D.; Yan, C. An update on vinpocetine: New discoveries and clinical implications. Eur. J. Pharmacol. 2018, 819, 30–34. [Google Scholar] [CrossRef]
- Subhan, Z.; Hindmarch, I. Psychopharmacological effects of vinpocetine in normal healthy volunteers. Eur. J. Clin. Pharmacol. 1985, 28, 567–571. [Google Scholar] [CrossRef]
- Thal, L.J.; Salmon, D.P.; Lasker, B.; Bower, D.; Klauber, M.R. The safety and lack of efficacy of vinpocetine in Alzheimer’s disease. J. Am. Geriatr. Soc. 1989, 37, 515–520. [Google Scholar] [CrossRef]
- Szatmari, S.Z.; Whitehouse, P.J. Vinpocetine for cognitive impairment and dementia. Cochrane Database Syst. Rev. 2003, 1, CD003119. [Google Scholar] [CrossRef]
- Valikovics, A. Investigation of the effect of vinpocetine on cerebral blood flow and cognitive functions. Ideggyogy. Szle. 2007, 60, 301–310. [Google Scholar]
- Valikovics, A.; Csányi, A.; Németh, L. Study of the effects of vinpocetin on cognitive functions. Ideggyogy. Szle. 2012, 65, 115–120. [Google Scholar]
- Ogunrin, A.O. Effect of Vinpocetine (Cognitol™) on Cognitive Performances of a Nigerian Population. Ann. Med. Health Sci. Res. 2014, 4, 654–661. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Tsuji, M. Pharmacological Potential of Cilostazol for Alzheimer’s Disease. Front. Pharmacol. 2019, 10, 559. [Google Scholar] [CrossRef]
- Arai, H.; Takahashi, T. A combination therapy of donepezil and cilostazol for patients with moderate Alzheimer disease: Pilot follow-up study. Am. J. Geriatr. Psychiatry 2009, 14, 353–354. [Google Scholar] [CrossRef]
- Sakurai, H.; Hanyu, H.; Sato, T.; Kume, K.; Hirao, K.; Kanetaka, H.; Iwamoto, T. Effects of cilostazol on cognition and regional cerebral blood flow in patients with Alzheimer’s disease and cerebrovascular disease: A pilot study. Geriatr. Gerontol. Int. 2013, 13, 90–97. [Google Scholar] [CrossRef]
- Taguchi, A.; Takata, Y.; Ihara, M.; Kasahara, Y.; Tsuji, M.; Nishino, M.; Stern, D.; Okada, M. Cilostazol improves cognitive function in patients with mild cognitive impairment: A retrospective analysis. Psychogeriatrics 2013, 13, 164–169. [Google Scholar] [CrossRef]
- Hishikawa, N.; Fukui, Y.; Sato, K.; Ohta, Y.; Yamashita, T.; Abe, K. Comprehensive effects of galantamine and cilostazol combination therapy on patients with Alzheimer’s disease with asymptomatic lacunar infarction. Geriatr. Gerontol. Int. 2017, 17, 1384–1391. [Google Scholar] [CrossRef]
- Tai, S.Y.; Chen, C.H.; Chien, C.Y.; Yang, Y.H. Cilostazol as an add-on therapy for patients with Alzheimer’s disease in Taiwan: A case control study. BMC Neurol. 2017, 17, 40. [Google Scholar] [CrossRef]
- Saito, S.; Kojima, S.; Oishi, N.; Kakuta, R.; Maki, T.; Yasuno, F.; Nagatsuka, K.; Yamamoto, H.; Fukuyama, H.; Fukushima, M.; et al. A multicenter, randomized, placebo-controlled trial for cilostazol in patients with mild cognitive impairment: The COMCID study protocol. Alzheimers Dement. 2016, 2, 250–257. [Google Scholar] [CrossRef]
- NCT02491268. Available online: https://clinicaltrials.com (accessed on 2 December 2020).
- Prickaerts, J.; Van Goethem, N.P.; Gulisano, W.; Argyrousi, E.K.; Palmeri, A.; Puzzo, D. Physiological and pathological processes of synaptic plasticity and memory in drug discovery: Do not forget the dose-response curve. Eur. J. Pharmacol. 2017, 817, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Kelly, M.P. Cyclic nucleotide signaling changes associated with normal aging and age-related diseases of the brain. Cell. Signal. 2018, 42, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Ricciarelli, R.; Fedele, E. Phosphodiesterase 4D: An enzyme to remember. Br. J. Pharmacol. 2015, 172, 4785–4789. [Google Scholar] [CrossRef] [PubMed]
- Ricciarelli, R.; Brullo, C.; Prickaerts, J.; Arancio, O.; Villa, C.; Rebosio, C.; Calcagno, E.; Balbi, M.; van Hagen, B.T.; Argyrousi, E.K.; et al. Memory-enhancing effects of GEBR-32a, a new PDE4D inhibitor holding promise for the treatment of Alzheimer’s disease. Sci. Rep. 2017, 7, 46320. [Google Scholar] [CrossRef] [PubMed]
- Bollen, E.; Akkerman, S.; Puzzo, D.; Gulisano, W.; Palmeri, A.; D’Hooge, R.; Balschun, D.; Steinbusch, H.W.; Blokland, A.; Prickaerts, J. Object memory enhancement by combining sub-efficacious doses of specific phosphodiesterase inhibitors. Neuropharmacology 2015, 95, 361–366. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Drug | Mechanism | Main Outcomes | Refs |
|---|---|---|---|
| Riociguat | sGC activator | No effect | [55] |
| IW-6462 | sGC activator | Positive effects | [56] |
| Sildenafil | PDE5 inhibitor | Not conclusive effects | [57,58,59,60,61] |
| Vardenafil | PDE5 inhibitor | No effect | [62,63] |
| Udenafil | PDE5 inhibitor | Positive effects | [64,65] |
| PF-04447943 | PDE9 inhibitor | No effect | [66] |
| BI 409306 | PDE9 inhibitor | No effect | [67] |
| Vinpocetine | PDE1 inhibitor | Possible positive effects | [68,69,70,71,72,73,74] |
| Cilostazol | PDE3 inhibitor | Positive effects | [75,76,77,78,79,80] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fedele, E.; Ricciarelli, R. Memory Enhancers for Alzheimer’s Dementia: Focus on cGMP. Pharmaceuticals 2021, 14, 61. https://doi.org/10.3390/ph14010061
Fedele E, Ricciarelli R. Memory Enhancers for Alzheimer’s Dementia: Focus on cGMP. Pharmaceuticals. 2021; 14(1):61. https://doi.org/10.3390/ph14010061
Chicago/Turabian StyleFedele, Ernesto, and Roberta Ricciarelli. 2021. "Memory Enhancers for Alzheimer’s Dementia: Focus on cGMP" Pharmaceuticals 14, no. 1: 61. https://doi.org/10.3390/ph14010061
APA StyleFedele, E., & Ricciarelli, R. (2021). Memory Enhancers for Alzheimer’s Dementia: Focus on cGMP. Pharmaceuticals, 14(1), 61. https://doi.org/10.3390/ph14010061