Cancer Stem Cells and Nucleolin as Drivers of Carcinogenesis



Abstract
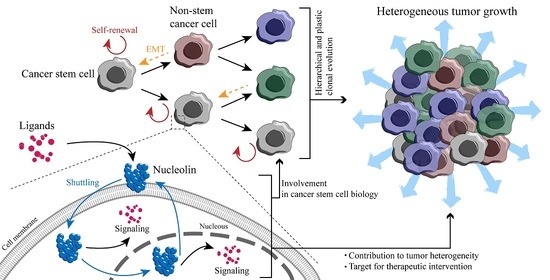
1. Introduction
2. The Established Hallmarks of Cancer
3. Cancer Stem Cells—Another Layer of Complexity
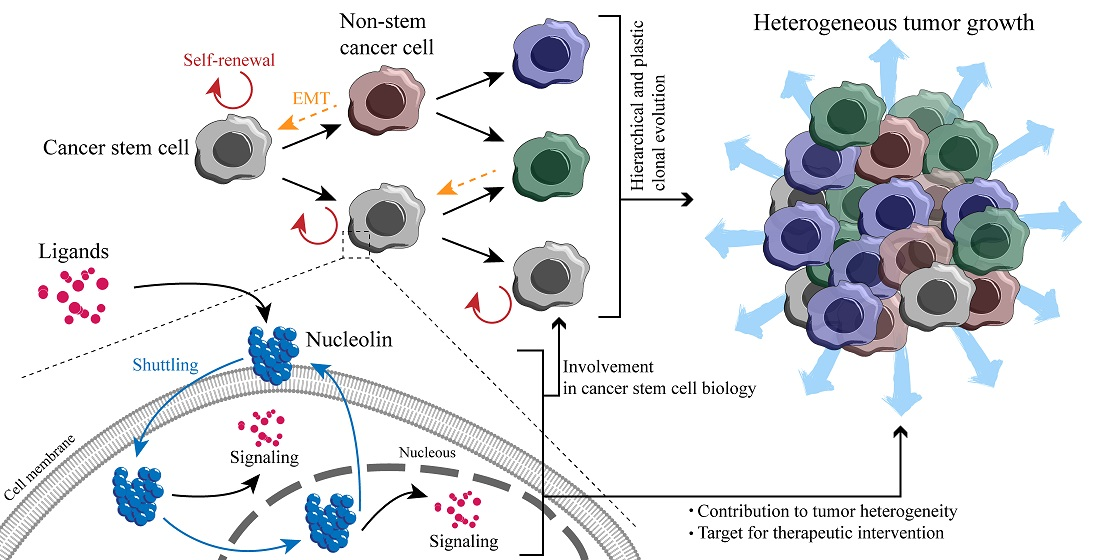
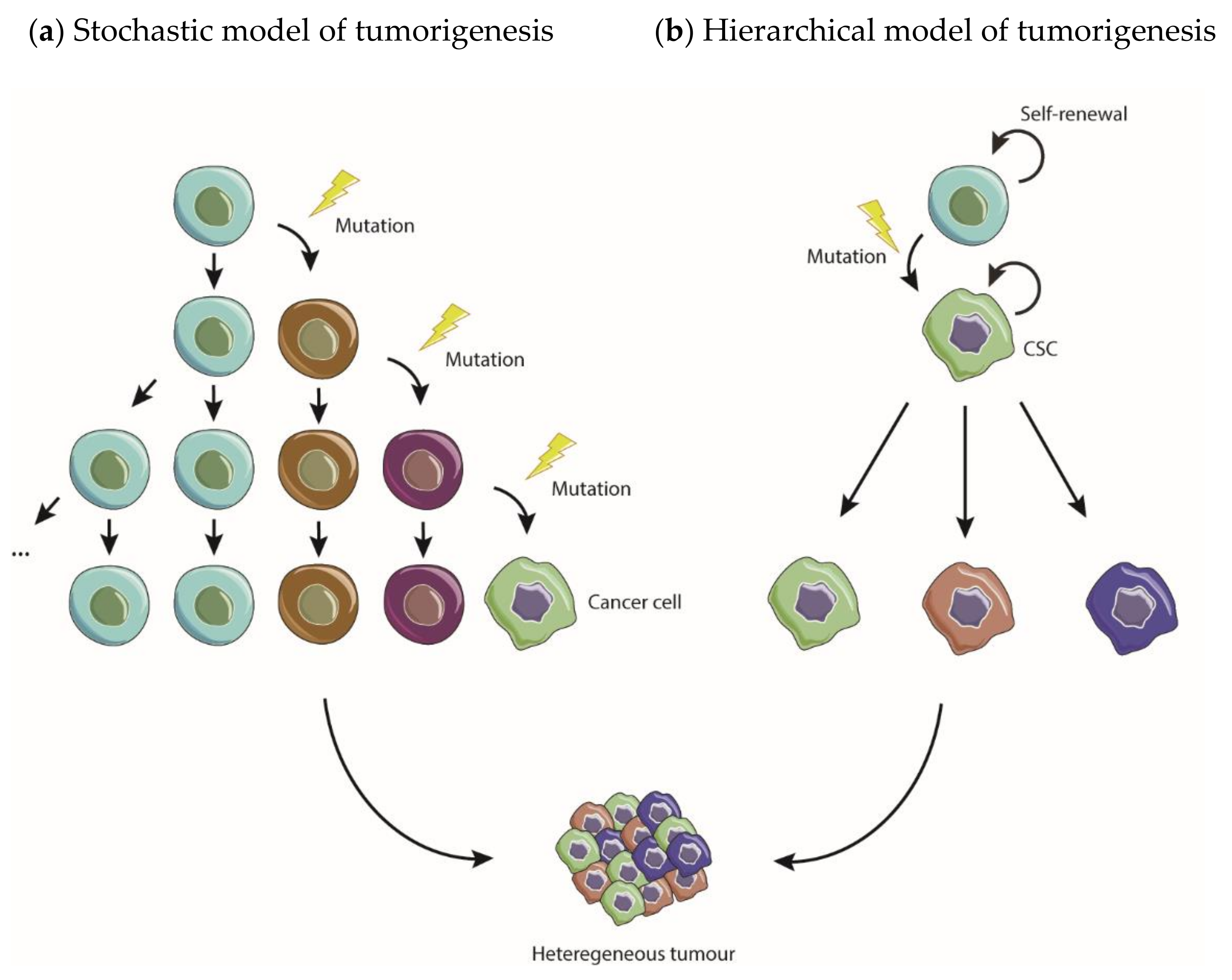
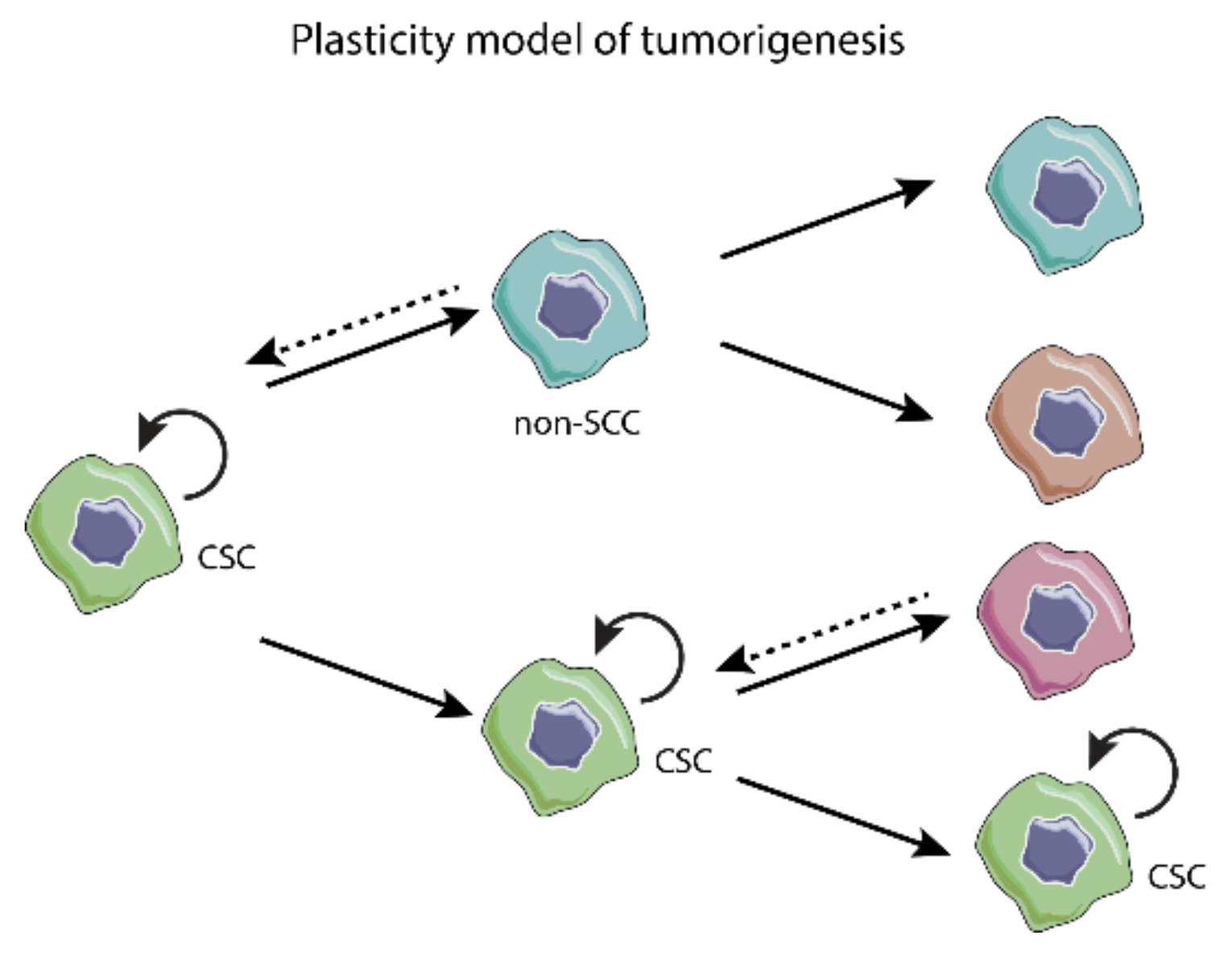
3.1. Models of Tumorigenesis—A New Paradigm Driven by CSC
3.2. Epithelial-to-Mesenchymal Transition
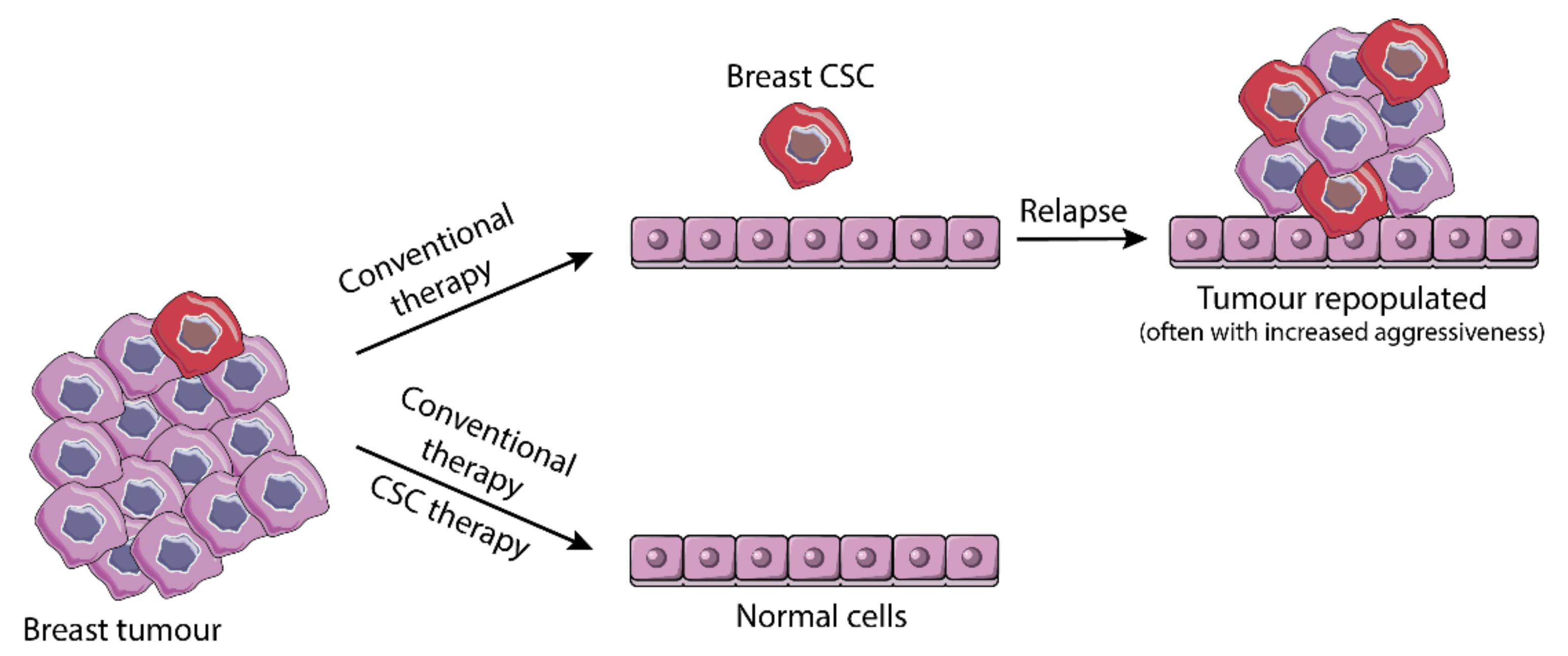
3.3. From Resistance to Standard Therapy to Stemness-Based Therapeutic Intervention
4. Multifunctional Protein Nucleolin—A Possible Driver of the Cancer Hallmarks?
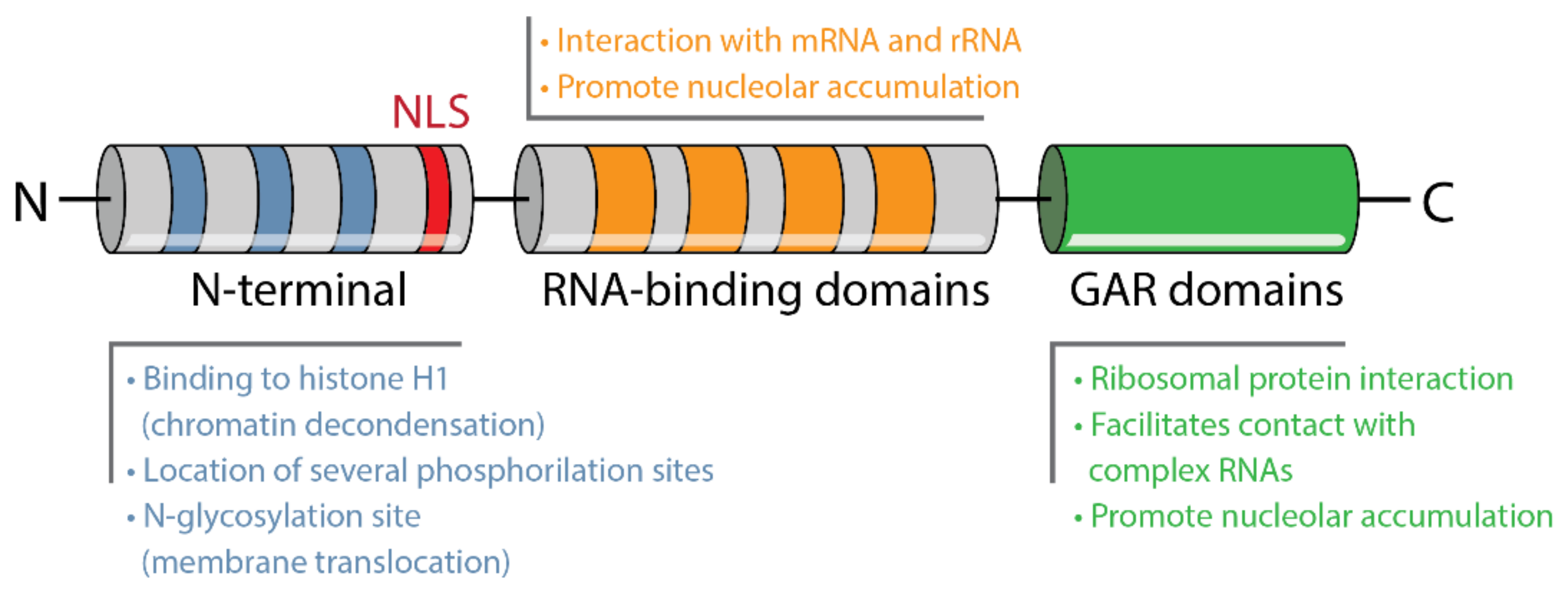
4.1. Structure and Localization
4.2. Role of Nucleolin on Ribosomes Biogenesis, Gene Transcription and Translation
4.3. Nucleolin as a Regulatory Protein of Proliferation, Cell Cycle and Cell Survival
4.4. Nucleolin in Tumor Initiation and Progression
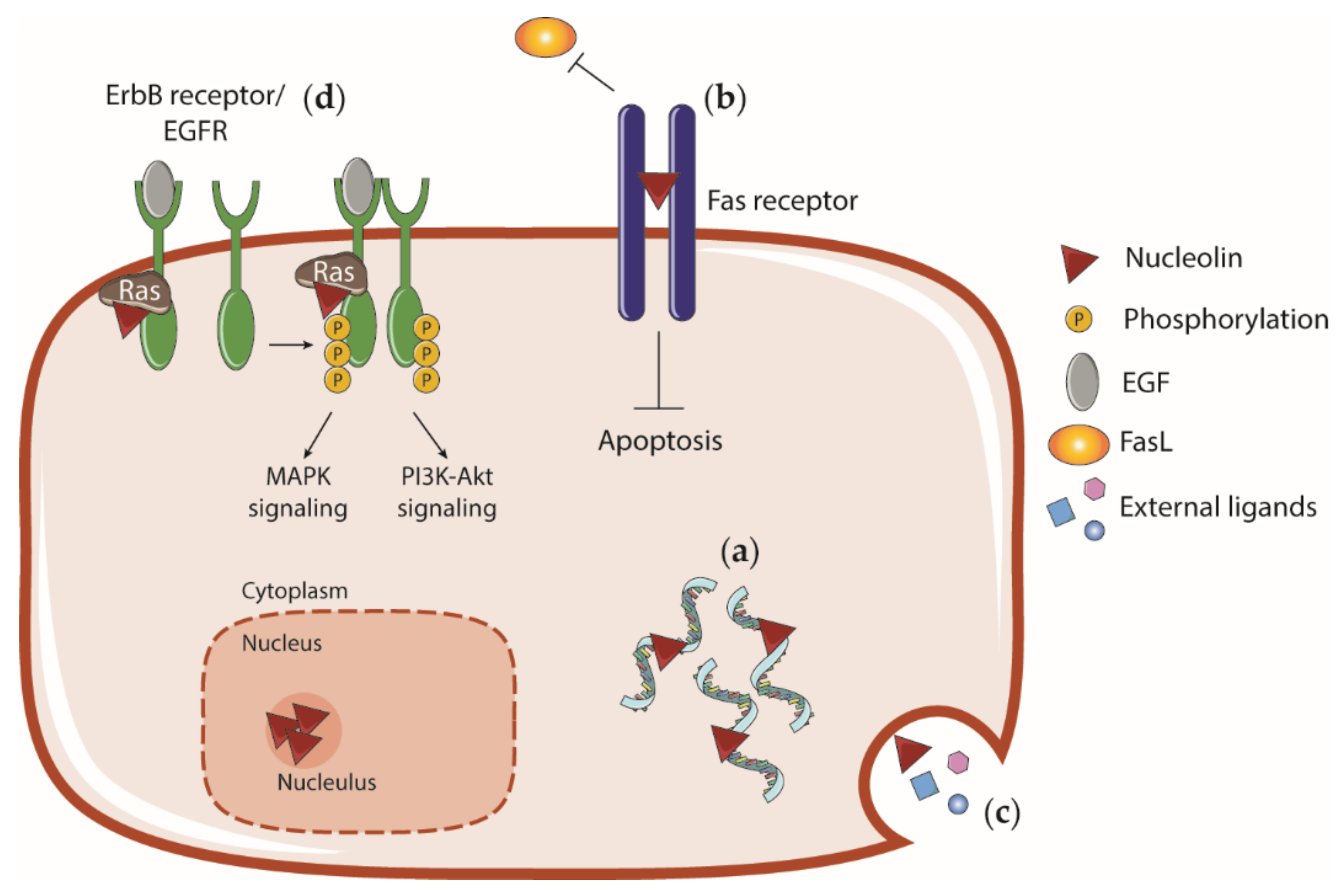
4.5. Cell Surface Nucleolin and Interaction with External Ligands
5. Role of Nucleolin on Stemness, Pluripotency and Differentiation: A Potential Target for Broad Anticancer Therapy
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Pineros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- De Sousa, E.M.F.; Vermeulen, L.; Fessler, E.; Medema, J.P. Cancer heterogeneity--a multifaceted view. EMBO Rep. 2013, 14, 686–695. [Google Scholar] [CrossRef]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef]
- Da Silva-Diz, V.; Lorenzo-Sanz, L.; Bernat-Peguera, A.; Lopez-Cerda, M.; Munoz, P. Cancer cell plasticity: Impact on tumor progression and therapy response. Semin. Cancer Biol. 2018, 53, 48–58. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Marjanovic, N.D.; Lee, T.; Bell, G.; Kleer, C.G.; Reinhardt, F.; D’Alessio, A.C.; Young, R.A.; Weinberg, R.A. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell 2013, 154, 61–74. [Google Scholar] [CrossRef]
- Fonseca, N.A.; Cruz, A.F.; Moura, V.; Simoes, S.; Moreira, J.N. The cancer stem cell phenotype as a determinant factor of the heterotypic nature of breast tumors. Crit. Rev. Oncol. Hematol. 2017, 113, 111–121. [Google Scholar] [CrossRef]
- Ginisty, H.; Sicard, H.; Roger, B.; Bouvet, P. Structure and functions of nucleolin. J. Cell Sci. 1999, 112, 761–772. [Google Scholar]
- Christian, S.; Pilch, J.; Akerman, M.E.; Porkka, K.; Laakkonen, P.; Ruoslahti, E. Nucleolin expressed at the cell surface is a marker of endothelial cells in angiogenic blood vessels. J. Cell Biol. 2003, 163, 871–878. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Xiong, L.; Yu, L.; Li, R.; Wang, Z.; Ren, B.; Dong, J.; Li, B.; Wang, D. Increased level of nucleolin confers to aggressive tumor progression and poor prognosis in patients with hepatocellular carcinoma after hepatectomy. Diagn. Pathol. 2014, 9, 175. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.Y.; Lu, S.; Xu, X.Y.; Hu, S.L.; Li, B.; Li, W.X.; Chang, J.Y. Prognostic significance of nuclear or cytoplasmic nucleolin expression in human non-small cell lung cancer and its relationship with DNA-PKcs. Tumour Biol. 2016, 37, 10349–10356. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, N.A.; Rodrigues, A.S.; Rodrigues-Santos, P.; Alves, V.; Gregorio, A.C.; Valerio-Fernandes, A.; Gomes-da-Silva, L.C.; Rosa, M.S.; Moura, V.; Ramalho-Santos, J.; et al. Nucleolin overexpression in breast cancer cell sub-populations with different stem-like phenotype enables targeted intracellular delivery of synergistic drug combination. Biomaterials 2015, 69, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Moura, V.; Lacerda, M.; Figueiredo, P.; Corvo, M.L.; Cruz, M.E.; Soares, R.; de Lima, M.C.; Simoes, S.; Moreira, J.N. Targeted and intracellular triggered delivery of therapeutics to cancer cells and the tumor microenvironment: Impact on the treatment of breast cancer. Breast Cancer Res. Treat. 2012, 133, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Kanwal, R.; Gupta, K.; Gupta, S. Cancer epigenetics: An introduction. Methods Mol. Biol. 2015, 1238, 3–25. [Google Scholar] [CrossRef]
- Peinado, H.; Aleckovic, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; Garcia-Santos, G.; Ghajar, C.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef]
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015, 17, 816–826. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Kolios, G.; Moodley, Y. Introduction to stem cells and regenerative medicine. Respiration 2013, 85, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Koren, E.; Fuchs, Y. The bad seed: Cancer stem cells in tumor development and resistance. Drug Resist. Update 2016, 28, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.Q.; Choi, Y.P.; Kang, S.; Youn, J.H.; Cho, N.H. CD24+ cells from hierarchically organized ovarian cancer are enriched in cancer stem cells. Oncogene 2010, 29, 2672–2680. [Google Scholar] [CrossRef] [PubMed]
- Mather, J.P.; Roberts, P.E.; Pan, Z.; Chen, F.; Hooley, J.; Young, P.; Xu, X.; Smith, D.H.; Easton, A.; Li, P.; et al. Isolation of cancer stem like cells from human adenosquamous carcinoma of the lung supports a monoclonal origin from a multipotential tissue stem cell. PLoS ONE 2013, 8, e79456. [Google Scholar] [CrossRef]
- Roesch, A.; Fukunaga-Kalabis, M.; Schmidt, E.C.; Zabierowski, S.E.; Brafford, P.A.; Vultur, A.; Basu, D.; Gimotty, P.; Vogt, T.; Herlyn, M. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 2010, 141, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Mirshahidi, S.; Simental, A.; Lee, S.C.; De Andrade Filho, P.A.; Peterson, N.R.; Cao, W.; Necochea-Campion, R.; Yang, H.; Duerksen-Hughes, P.; Yuan, X. Subpopulations of cancer stem cells found in papillary thyroid carcinoma. Exp. Cell Res. 2018, 362, 515–524. [Google Scholar] [CrossRef]
- Suva, M.L.; Riggi, N.; Stehle, J.C.; Baumer, K.; Tercier, S.; Joseph, J.M.; Suva, D.; Clement, V.; Provero, P.; Cironi, L.; et al. Identification of cancer stem cells in Ewing’s sarcoma. Cancer Res. 2009, 69, 1776–1781. [Google Scholar] [CrossRef]
- Barker, N.; Ridgway, R.A.; van Es, J.H.; van de Wetering, M.; Begthel, H.; van den Born, M.; Danenberg, E.; Clarke, A.R.; Sansom, O.J.; Clevers, H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009, 457, 608–611. [Google Scholar] [CrossRef]
- Schepers, A.G.; Snippert, H.J.; Stange, D.E.; van den Born, M.; van Es, J.H.; van de Wetering, M.; Clevers, H. Lineage tracing reveals Lgr5+ stem cell activity in mouse intestinal adenomas. Science 2012, 337, 730–735. [Google Scholar] [CrossRef]
- Rios, A.C.; Fu, N.Y.; Lindeman, G.J.; Visvader, J.E. In situ identification of bipotent stem cells in the mammary gland. Nature 2014, 506, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Rolny, C.; Mazzone, M.; Tugues, S.; Laoui, D.; Johansson, I.; Coulon, C.; Squadrito, M.L.; Segura, I.; Li, X.; Knevels, E.; et al. HRG inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of PlGF. Cancer Cell 2011, 19, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Lu, H.; Clauser, K.R.; Tam, W.L.; Frose, J.; Ye, X.; Eaton, E.N.; Reinhardt, F.; Donnenberg, V.S.; Bhargava, R.; Carr, S.A.; et al. A breast cancer stem cell niche supported by juxtacrine signalling from monocytes and macrophages. Nat. Cell Biol. 2014, 16, 1105–1117. [Google Scholar] [CrossRef] [PubMed]
- Miranda, A.; Hamilton, P.T.; Zhang, A.W.; Pattnaik, S.; Becht, E.; Mezheyeuski, A.; Bruun, J.; Micke, P.; de Reynies, A.; Nelson, B.H. Cancer stemness, intratumoral heterogeneity, and immune response across cancers. Proc. Natl. Acad. Sci. USA 2019, 116, 9020–9029. [Google Scholar] [CrossRef] [PubMed]
- Kreso, A.; Dick, J.E. Evolution of the cancer stem cell model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef]
- Eirew, P.; Steif, A.; Khattra, J.; Ha, G.; Yap, D.; Farahani, H.; Gelmon, K.; Chia, S.; Mar, C.; Wan, A.; et al. Dynamics of genomic clones in breast cancer patient xenografts at single-cell resolution. Nature 2015, 518, 422–426. [Google Scholar] [CrossRef]
- Abbaszadegan, M.R.; Bagheri, V.; Razavi, M.S.; Momtazi, A.A.; Sahebkar, A.; Gholamin, M. Isolation, identification, and characterization of cancer stem cells: A review. J. Cell. Physiol. 2017, 232, 2008–2018. [Google Scholar] [CrossRef]
- Liu, A.; Yu, X.; Liu, S. Pluripotency transcription factors and cancer stem cells: Small genes make a big difference. Chin. J. Cancer 2013, 32, 483–487. [Google Scholar] [CrossRef]
- Najafi, M.; Farhood, B.; Mortezaee, K. Cancer stem cells (CSCs) in cancer progression and therapy. J. Cell. Physiol. 2019, 234, 8381–8395. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, M.; D’Alterio, C.; Camerlingo, R.; Tirino, V.; Consales, C.; Riccio, A.; Ierano, C.; Cecere, S.C.; Losito, N.S.; Greggi, S.; et al. Identification of a distinct population of CD133(+)CXCR4(+) cancer stem cells in ovarian cancer. Sci. Rep. 2015, 5, 10357. [Google Scholar] [CrossRef] [PubMed]
- Prabavathy, D.; Swarnalatha, Y.; Ramadoss, N. Lung cancer stem cells-origin, characteristics and therapy. Stem Cell Investig. 2018, 5, 6. [Google Scholar] [CrossRef] [PubMed]
- Moltzahn, F.; Thalmann, G.N. Cancer stem cells in prostate cancer. Transl. Androl. Urol. 2013, 2, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Xia, L.; Wang, H.; Oyang, L.; Su, M.; Liu, Q.; Lin, J.; Tan, S.; Tian, Y.; Liao, Q.; et al. Cancer stem cells in progression of colorectal cancer. Oncotarget 2018, 9, 33403–33415. [Google Scholar] [CrossRef]
- Ishiwata, T.; Matsuda, Y.; Yoshimura, H.; Sasaki, N.; Ishiwata, S.; Ishikawa, N.; Takubo, K.; Arai, T.; Aida, J. Pancreatic cancer stem cells: Features and detection methods. Pathol. Oncol. Res. 2018, 24, 797–805. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Liu, Y.; Zhu, T.; Zhu, J.; Dimeco, F.; Vescovi, A.L.; Heth, J.A.; Muraszko, K.M.; Fan, X.; Lubman, D.M. CD90 is identified as a candidate marker for cancer stem cells in primary high-grade gliomas using tissue microarrays. Mol. Cell. Proteom. 2012, 11, M111.010744. [Google Scholar] [CrossRef]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Boiko, A.D.; Razorenova, O.V.; van de Rijn, M.; Swetter, S.M.; Johnson, D.L.; Ly, D.P.; Butler, P.D.; Yang, G.P.; Joshua, B.; Kaplan, M.J.; et al. Human melanoma-initiating cells express neural crest nerve growth factor receptor CD271. Nature 2010, 466, 133–137. [Google Scholar] [CrossRef]
- Quail, D.F.; Taylor, M.J.; Postovit, L.M. Microenvironmental regulation of cancer stem cell phenotypes. Curr. Stem Cell Res. Ther. 2012, 7, 197–216. [Google Scholar] [CrossRef]
- Malanchi, I.; Huelsken, J. Cancer stem cells: Never Went away from the niche. Curr. Opin. Oncol. 2009, 21, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hao, Y.; Mao, W.; Xue, X.; Xu, P.; Liu, L.; Yuan, J.; Zhang, D.; Li, N.; Chen, H.; et al. LincK contributes to breast tumorigenesis by promoting proliferation and epithelial-to-mesenchymal transition. J. Hematol. Oncol. 2019, 12, 19. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Gloushankova, N.A.; Zhitnyak, I.Y.; Rubtsova, S.N. Role of Epithelial-Mesenchymal Transition in Tumor Progression. Biochemistry 2018, 83, 1469–1476. [Google Scholar] [CrossRef] [PubMed]
- Bonde, A.K.; Tischler, V.; Kumar, S.; Soltermann, A.; Schwendener, R.A. Intratumoral macrophages contribute to epithelial-mesenchymal transition in solid tumors. BMC Cancer 2012, 12, 35. [Google Scholar] [CrossRef]
- Liu, C.Y.; Xu, J.Y.; Shi, X.Y.; Huang, W.; Ruan, T.Y.; Xie, P.; Ding, J.L. M2-polarized tumor-associated macrophages promoted epithelial-mesenchymal transition in pancreatic cancer cells, partially through TLR4/IL-10 signaling pathway. Lab. Investig. 2013, 93, 844–854. [Google Scholar] [CrossRef]
- Yi, Y.; Zeng, S.; Wang, Z.; Wu, M.; Ma, Y.; Ye, X.; Zhang, B.; Liu, H. Cancer-associated fibroblasts promote epithelial-mesenchymal transition and EGFR-TKI resistance of non-small cell lung cancers via HGF/IGF-1/ANXA2 signaling. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 793–803. [Google Scholar] [CrossRef]
- Wang, N.; He, Y.L.; Pang, L.J.; Zou, H.; Liu, C.X.; Zhao, J.; Hu, J.M.; Zhang, W.J.; Qi, Y.; Li, F. Down-regulated E-cadherin expression is associated with poor five-year overall survival in bone and soft tissue sarcoma: Results of a meta-analysis. PLoS ONE 2015, 10, e0121448. [Google Scholar] [CrossRef]
- Li, Z.; Yin, S.; Zhang, L.; Liu, W.; Chen, B. Prognostic value of reduced E-cadherin expression in breast cancer: A meta-analysis. Oncotarget 2017, 8, 16445–16455. [Google Scholar] [CrossRef]
- Hajra, K.M.; Chen, D.Y.; Fearon, E.R. The SLUG zinc-finger protein represses E-cadherin in breast cancer. Cancer Res. 2002, 62, 1613–1618. [Google Scholar]
- Batlle, E.; Sancho, E.; Franci, C.; Dominguez, D.; Monfar, M.; Baulida, J.; Garcia De Herreros, A. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2000, 2, 84–89. [Google Scholar] [CrossRef]
- Pan, Y.; Li, J.; Zhang, Y.; Wang, N.; Liang, H.; Liu, Y.; Zhang, C.Y.; Zen, K.; Gu, H. Slug-upregulated miR-221 promotes breast cancer progression through suppressing E-cadherin expression. Sci. Rep. 2016, 6, 25798. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Tillo, E.; Lazaro, A.; Torrent, R.; Cuatrecasas, M.; Vaquero, E.C.; Castells, A.; Engel, P.; Postigo, A. ZEB1 represses E-cadherin and induces an EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG1. Oncogene 2010, 29, 3490–3500. [Google Scholar] [CrossRef] [PubMed]
- Kroger, C.; Afeyan, A.; Mraz, J.; Eaton, E.N.; Reinhardt, F.; Khodor, Y.L.; Thiru, P.; Bierie, B.; Ye, X.; Burge, C.B.; et al. Acquisition of a hybrid E/M state is essential for tumorigenicity of basal breast cancer cells. Proc. Natl. Acad. Sci. USA 2019, 116, 7353–7362. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Banerjee, S.; Ahmad, A.; Li, Y.; Wang, Z.; Sethi, S.; Sarkar, F.H. Epithelial to mesenchymal transition is mechanistically linked with stem cell signatures in prostate cancer cells. PLoS ONE 2010, 5, e12445. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.; Samuel, S.; Evans, K.W.; Lu, J.; Xia, L.; Zhou, Y.; Sceusi, E.; Tozzi, F.; Ye, X.C.; Mani, S.A.; et al. Overexpression of snail induces epithelial-mesenchymal transition and a cancer stem cell-like phenotype in human colorectal cancer cells. Cancer Med. 2012, 1, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Long, H.; Xiang, T.; Qi, W.; Huang, J.; Chen, J.; He, L.; Liang, Z.; Guo, B.; Li, Y.; Xie, R.; et al. CD133+ ovarian cancer stem-like cells promote non-stem cancer cell metastasis via CCL5 induced epithelial-mesenchymal transition. Oncotarget 2015, 6, 5846–5859. [Google Scholar] [CrossRef]
- Tsai, J.H.; Donaher, J.L.; Murphy, D.A.; Chau, S.; Yang, J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 2012, 22, 725–736. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhu, X.; Cui, K.; Mancuso, J.; Federley, R.; Fischer, K.; Teng, G.; Mittal, V.; Gao, D.; Zhao, H.; et al. In Vivo Visualization and Characterization of Epithelial-Mesenchymal Transition in Breast Tumors. Cancer Res. 2016, 76, 2094–2104. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Hirschmann-Jax, C.; Foster, A.E.; Wulf, G.G.; Nuchtern, J.G.; Jax, T.W.; Gobel, U.; Goodell, M.A.; Brenner, M.K. A distinct "side population" of cells with high drug efflux capacity in human tumor cells. Proc. Natl. Acad. Sci. USA 2004, 101, 14228–14233. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Lee, T.K.; Zheng, B.J.; Chan, K.W.; Guan, X.Y. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene 2008, 27, 1749–1758. [Google Scholar] [CrossRef] [PubMed]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Ghisolfi, L.; Keates, A.C.; Zhang, J.; Xiang, S.; Lee, D.K.; Li, C.J. Induction of cancer cell stemness by chemotherapy. Cell Cycle 2012, 11, 2691–2698. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Tran, L.; Park, Y.; Chen, I.; Lan, J.; Xie, Y.; Semenza, G.L. Reciprocal Regulation of DUSP9 and DUSP16 Expression by HIF1 Controls ERK and p38 MAP Kinase Activity and Mediates Chemotherapy-Induced Breast Cancer Stem Cell Enrichment. Cancer Res. 2018, 78, 4191–4202. [Google Scholar] [CrossRef]
- Lee, H.H.; Bellat, V.; Law, B. Chemotherapy induces adaptive drug resistance and metastatic potentials via phenotypic CXCR4-expressing cell state transition in ovarian cancer. PLoS ONE 2017, 12, e0171044. [Google Scholar] [CrossRef]
- Wang, Y.; Zong, X.; Mitra, S.; Mitra, A.K.; Matei, D.; Nephew, K.P. IL-6 mediates platinum-induced enrichment of ovarian cancer stem cells. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Wu, X.; Tao, P.; Zhou, Q.; Li, J.; Yu, Z.; Wang, X.; Li, J.; Li, C.; Yan, M.; Zhu, Z.; et al. IL-6 secreted by cancer-associated fibroblasts promotes epithelial-mesenchymal transition and metastasis of gastric cancer via JAK2/STAT3 signaling pathway. Oncotarget 2017, 8, 20741–20750. [Google Scholar] [CrossRef]
- Hashida, S.; Yamamoto, H.; Shien, K.; Miyoshi, Y.; Ohtsuka, T.; Suzawa, K.; Watanabe, M.; Maki, Y.; Soh, J.; Asano, H.; et al. Acquisition of cancer stem cell-like properties in non-small cell lung cancer with acquired resistance to afatinib. Cancer Sci. 2015, 106, 1377–1384. [Google Scholar] [CrossRef] [PubMed]
- Poh, M.E.; Liam, C.K.; Rajadurai, P.; Chai, C.S. Epithelial-to-mesenchymal transition (EMT) causing acquired resistance to afatinib in a patient with epidermal growth factor receptor (EGFR)-mutant lung adenocarcinoma. J. Thorac. Dis. 2018, 10, E560–E563. [Google Scholar] [CrossRef] [PubMed]
- Ayob, A.Z.; Ramasamy, T.S. Cancer stem cells as key drivers of tumour progression. J. Biomed. Sci. 2018, 25, 20. [Google Scholar] [CrossRef] [PubMed]
- Cruz, A.F.; Fonseca, N.A.; Moura, V.; Simoes, S.; Moreira, J.N. Targeting cancer stem cells and non-stem cancer cells: The potential of lipid-based nanoparticles. Curr. Pharm. Des. 2017. [Google Scholar] [CrossRef]
- Chaffer, C.L.; San Juan, B.P.; Lim, E.; Weinberg, R.A. EMT, cell plasticity and metastasis. Cancer Metastasis Rev. 2016, 35, 645–654. [Google Scholar] [CrossRef]
- Friedmann-Morvinski, D.; Verma, I.M. Dedifferentiation and reprogramming: Origins of cancer stem cells. EMBO Rep. 2014, 15, 244–253. [Google Scholar] [CrossRef]
- Ajani, J.A.; Song, S.; Hochster, H.S.; Steinberg, I.B. Cancer stem cells: The promise and the potential. Semin. Oncol. 2015, 42 (Suppl. 1), S3–S17. [Google Scholar] [CrossRef]
- Yang, X.G.; Zhu, L.C.; Wang, Y.J.; Li, Y.Y.; Wang, D. Current Advance of Therapeutic Agents in Clinical Trials Potentially Targeting Tumor Plasticity. Front. Oncol. 2019, 9, 887. [Google Scholar] [CrossRef]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target. Ther. 2020, 5, 8. [Google Scholar] [CrossRef]
- Chen, J.H.; Huang, W.C.; Bamodu, O.A.; Chang, P.M.; Chao, T.Y.; Huang, T.H. Monospecific antibody targeting of CDH11 inhibits epithelial-to-mesenchymal transition and represses cancer stem cell-like phenotype by up-regulating miR-335 in metastatic breast cancer, in vitro and in vivo. BMC Cancer 2019, 19, 634. [Google Scholar] [CrossRef]
- Fakiruddin, K.S.; Lim, M.N.; Nordin, N.; Rosli, R.; Zakaria, Z.; Abdullah, S. Targeting of CD133+ Cancer Stem Cells by Mesenchymal Stem Cell Expressing TRAIL Reveals a Prospective Role of Apoptotic Gene Regulation in Non-Small Cell Lung Cancer. Cancers 2019, 11, 1261. [Google Scholar] [CrossRef] [PubMed]
- Sreekumar, R.; Emaduddin, M.; Al-Saihati, H.; Moutasim, K.; Chan, J.; Spampinato, M.; Bhome, R.; Yuen, H.M.; Mescoli, C.; Vitale, A.; et al. Protein kinase C inhibitors override ZEB1-induced chemoresistance in HCC. Cell Death Dis. 2019, 10, 703. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Zhang, Y.; Zang, Y.; Chai, R.; Zhong, G.; Li, Z.; Duan, Z.; Ren, J.; Xu, Z. HP-1 inhibits the progression of ccRCC and enhances sunitinib therapeutic effects by suppressing EMT. Carbohydr. Polym. 2019, 223, 115109. [Google Scholar] [CrossRef] [PubMed]
- Porkka, K.; Laakkonen, P.; Hoffman, J.A.; Bernasconi, M.; Ruoslahti, E. A fragment of the HMGN2 protein homes to the nuclei of tumor cells and tumor endothelial cells in vivo. Proc. Natl. Acad. Sci. USA 2002, 99, 7444–7449. [Google Scholar] [CrossRef]
- Romano, S.; Moura, V.; Simoes, S.; Moreira, J.N.; Goncalves, J. Anticancer activity and antibody-dependent cell-mediated cytotoxicity of novel anti-nucleolin antibodies. Sci. Rep. 2018, 8, 7450. [Google Scholar] [CrossRef]
- Romano, S.; Fonseca, N.; Simoes, S.; Goncalves, J.; Moreira, J.N. Nucleolin-based targeting strategies for cancer therapy: From targeted drug delivery to cytotoxic ligands. Drug Discov. Today 2019, 24, 1985–2001. [Google Scholar] [CrossRef]
- Orrick, L.R.; Olson, M.O.; Busch, H. Comparison of nucleolar proteins of normal rat liver and Novikoff hepatoma ascites cells by two-dimensional polyacrylamide gel electrophoresis. Proc. Natl. Acad. Sci. USA 1973, 70, 1316–1320. [Google Scholar] [CrossRef]
- NCBI. NCL Nucleolin [Homo Sapiens (Human)]. Available online: https://www.ncbi.nlm.nih.gov/gene/4691 (accessed on 20 June 2019).
- Lapeyre, B.; Bourbon, H.; Amalric, F. Nucleolin, the major nucleolar protein of growing eukaryotic cells: An unusual protein structure revealed by the nucleotide sequence. Proc. Natl. Acad. Sci. USA 1987, 84, 1472–1476. [Google Scholar] [CrossRef]
- Jia, W.; Yao, Z.; Zhao, J.; Guan, Q.; Gao, L. New perspectives of physiological and pathological functions of nucleolin (NCL). Life Sci. 2017, 186, 1–10. [Google Scholar] [CrossRef]
- Srivastava, M.; Fleming, P.J.; Pollard, H.B.; Burns, A.L. Cloning and sequencing of the human nucleolin cDNA. FEBS Lett. 1989, 250, 99–105. [Google Scholar] [CrossRef]
- Erard, M.S.; Belenguer, P.; Caizergues-Ferrer, M.; Pantaloni, A.; Amalric, F. A major nucleolar protein, nucleolin, induces chromatin decondensation by binding to histone H1. Eur. J. Biochem. 1988, 175, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Tajrishi, M.M.; Tuteja, R.; Tuteja, N. Nucleolin: The most abundant multifunctional phosphoprotein of nucleolus. Commun. Integr. Biol. 2011, 4, 267–275. [Google Scholar] [CrossRef]
- Ugrinova, I.; Petrova, M.; Chalabi-Dchar, M.; Bouvet, P. Multifaceted Nucleolin Protein and Its Molecular Partners in Oncogenesis. Adv. Protein Chem. Struct. Biol. 2018, 111, 133–164. [Google Scholar] [CrossRef] [PubMed]
- Sirri, V.; Urcuqui-Inchima, S.; Roussel, P.; Hernandez-Verdun, D. Nucleolus: The fascinating nuclear body. Histochem. Cell Biol. 2008, 129, 13–31. [Google Scholar] [CrossRef] [PubMed]
- Borer, R.A.; Lehner, C.F.; Eppenberger, H.M.; Nigg, E.A. Major nucleolar proteins shuttle between nucleus and cytoplasm. Cell 1989, 56, 379–390. [Google Scholar] [CrossRef]
- Schmidt-Zachmann, M.S.; Nigg, E.A. Protein localization to the nucleolus: A search for targeting domains in nucleolin. J. Cell Sci. 1993, 105, 799–806. [Google Scholar]
- Schwab, M.S.; Dreyer, C. Protein phosphorylation sites regulate the function of the bipartite NLS of nucleolin. Eur. J. Cell Biol. 1997, 73, 287–297. [Google Scholar]
- Bouvet, P.; Diaz, J.J.; Kindbeiter, K.; Madjar, J.J.; Amalric, F. Nucleolin interacts with several ribosomal proteins through its RGG domain. J. Biol. Chem. 1998, 273, 19025–19029. [Google Scholar] [CrossRef]
- Carpentier, M.; Morelle, W.; Coddeville, B.; Pons, A.; Masson, M.; Mazurier, J.; Legrand, D. Nucleolin undergoes partial N- and O-glycosylations in the extranuclear cell compartment. Biochemistry 2005, 44, 5804–5815. [Google Scholar] [CrossRef]
- Losfeld, M.E.; Khoury, D.E.; Mariot, P.; Carpentier, M.; Krust, B.; Briand, J.P.; Mazurier, J.; Hovanessian, A.G.; Legrand, D. The cell surface expressed nucleolin is a glycoprotein that triggers calcium entry into mammalian cells. Exp. Cell Res. 2009, 315, 357–369. [Google Scholar] [CrossRef]
- Bouche, G.; Caizergues-Ferrer, M.; Bugler, B.; Amalric, F. Interrelations between the maturation of a 100 kDa nucleolar protein and pre rRNA synthesis in CHO cells. Nucleic Acids Res. 1984, 12, 3025–3035. [Google Scholar] [CrossRef] [PubMed]
- Bourbon, H.; Bugler, B.; Caizergues-Ferrer, M.; Amalric, F. Role of phosphorylation on the maturation pathways of a 100 kDa nucleolar protein. FEBS Lett. 1983, 155, 218–222. [Google Scholar] [CrossRef]
- Egyhazi, E.; Pigon, A.; Chang, J.H.; Ghaffari, S.H.; Dreesen, T.D.; Wellman, S.E.; Case, S.T.; Olson, M.O. Effects of anti-C23 (nucleolin) antibody on transcription of ribosomal DNA in Chironomus salivary gland cells. Exp. Cell Res. 1988, 178, 264–272. [Google Scholar] [CrossRef]
- Roger, B.; Moisand, A.; Amalric, F.; Bouvet, P. Repression of RNA polymerase I transcription by nucleolin is independent of the RNA sequence that is transcribed. J. Biol. Chem. 2002, 277, 10209–10219. [Google Scholar] [CrossRef]
- Rickards, B.; Flint, S.J.; Cole, M.D.; LeRoy, G. Nucleolin is required for RNA polymerase I transcription in vivo. Mol. Cell. Biol. 2007, 27, 937–948. [Google Scholar] [CrossRef] [PubMed]
- Ghisolfi-Nieto, L.; Joseph, G.; Puvion-Dutilleul, F.; Amalric, F.; Bouvet, P. Nucleolin is a sequence-specific RNA-binding protein: Characterization of targets on pre-ribosomal RNA. J. Mol. Biol. 1996, 260, 34–53. [Google Scholar] [CrossRef] [PubMed]
- Cong, R.; Das, S.; Bouvet, P. The multiple properties and functions of nucleolin. In The Nucleolus, 1st ed.; Olson, M.O.J., Ed.; Springer: New York, NY, USA, 2011; Volume 15, pp. 185–212. [Google Scholar]
- Ginisty, H.; Amalric, F.; Bouvet, P. Nucleolin functions in the first step of ribosomal RNA processing. EMBO J. 1998, 17, 1476–1486. [Google Scholar] [CrossRef]
- Ginisty, H.; Serin, G.; Ghisolfi-Nieto, L.; Roger, B.; Libante, V.; Amalric, F.; Bouvet, P. Interaction of nucleolin with an evolutionarily conserved pre-ribosomal RNA sequence is required for the assembly of the primary processing complex. J. Biol. Chem. 2000, 275, 18845–18850. [Google Scholar] [CrossRef]
- Takagi, M.; Absalon, M.J.; McLure, K.G.; Kastan, M.B. Regulation of p53 translation and induction after DNA damage by ribosomal protein L26 and nucleolin. Cell 2005, 123, 49–63. [Google Scholar] [CrossRef]
- Xie, Q.; Guo, X.; Gu, J.; Zhang, L.; Jin, H.; Huang, H.; Li, J.; Huang, C. p85alpha promotes nucleolin transcription and subsequently enhances EGFR mRNA stability and EGF-induced malignant cellular transformation. Oncotarget 2016, 7, 16636–16649. [Google Scholar] [CrossRef]
- Sengupta, T.K.; Bandyopadhyay, S.; Fernandes, D.J.; Spicer, E.K. Identification of nucleolin as an AU-rich element binding protein involved in bcl-2 mRNA stabilization. J. Biol. Chem. 2004, 279, 10855–10863. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.H.; Childress, M.O.; Geahlen, R.L. Syk interacts with and phosphorylates nucleolin to stabilize Bcl-x(L) mRNA and promote cell survival. Mol. Cell. Biol. 2014, 34, 3788–3799. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, S.H.; Malter, J.S. Nucleolin and heterogeneous nuclear ribonucleoprotein C proteins specifically interact with the 3’-untranslated region of amyloid protein precursor mRNA. J. Biol. Chem. 1995, 270, 17292–17298. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.B.; Rishal, I.; Doron-Mandel, E.; Kalinski, A.L.; Medzihradszky, K.F.; Terenzio, M.; Alber, S.; Koley, S.; Lin, A.; Rozenbaum, M.; et al. Nucleolin-Mediated RNA Localization Regulates Neuron Growth and Cycling Cell Size. Cell Rep. 2016, 16, 1664–1676. [Google Scholar] [CrossRef]
- Miniard, A.C.; Middleton, L.M.; Budiman, M.E.; Gerber, C.A.; Driscoll, D.M. Nucleolin binds to a subset of selenoprotein mRNAs and regulates their expression. Nucleic Acids Res. 2010, 38, 4807–4820. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Gherzi, R.; Andersen, J.S.; Gaietta, G.; Jurchott, K.; Royer, H.D.; Mann, M.; Karin, M. Nucleolin and YB-1 are required for JNK-mediated interleukin-2 mRNA stabilization during T-cell activation. Genes Dev. 2000, 14, 1236–1248. [Google Scholar]
- Liao, X.; Huang, C.; Zhang, D.; Wang, J.; Li, J.; Jin, H.; Huang, C. Mitochondrial catalase induces cells transformation through nucleolin-dependent Cox-2 mRNA stabilization. Free Radic. Biol. Med. 2017, 113, 478–486. [Google Scholar] [CrossRef]
- Abdelmohsen, K.; Tominaga, K.; Lee, E.K.; Srikantan, S.; Kang, M.J.; Kim, M.M.; Selimyan, R.; Martindale, J.L.; Yang, X.; Carrier, F.; et al. Enhanced translation by Nucleolin via G-rich elements in coding and non-coding regions of target mRNAs. Nucleic Acids Res. 2011, 39, 8513–8530. [Google Scholar] [CrossRef]
- Derenzini, M.; Sirri, V.; Trere, D.; Ochs, R.L. The quantity of nucleolar proteins nucleolin and protein B23 is related to cell doubling time in human cancer cells. Lab. Investig. 1995, 73, 497–502. [Google Scholar]
- Sirri, V.; Roussel, P.; Trere, D.; Derenzini, M.; Hernandez-Verdun, D. Amount variability of total and individual Ag-NOR proteins in cells stimulated to proliferate. J. Histochem. Cytochem. 1995, 43, 887–893. [Google Scholar] [CrossRef]
- Ma, N.; Matsunaga, S.; Takata, H.; Ono-Maniwa, R.; Uchiyama, S.; Fukui, K. Nucleolin functions in nucleolus formation and chromosome congression. J. Cell Sci. 2007, 120, 2091–2105. [Google Scholar] [CrossRef]
- Gaume, X.; Tassin, A.M.; Ugrinova, I.; Mongelard, F.; Monier, K.; Bouvet, P. Centrosomal nucleolin is required for microtubule network organization. Cell Cycle 2015, 14, 902–919. [Google Scholar] [CrossRef]
- Ugrinova, I.; Monier, K.; Ivaldi, C.; Thiry, M.; Storck, S.; Mongelard, F.; Bouvet, P. Inactivation of nucleolin leads to nucleolar disruption, cell cycle arrest and defects in centrosome duplication. BMC Mol. Biol. 2007, 8, 66. [Google Scholar] [CrossRef]
- Wang, W.; Luo, J.; Xiang, F.; Liu, X.; Jiang, M.; Liao, L.; Hu, J. Nucleolin down-regulation is involved in ADP-induced cell cycle arrest in S phase and cell apoptosis in vascular endothelial cells. PLoS ONE 2014, 9, e110101. [Google Scholar] [CrossRef]
- Sun, H.; Huang, L.; Liang, P.; Tang, Y.; Chen, C.; Chen, H.; Lin, X.; Luo, Z.; Li, Y.; Jiang, B.; et al. Nucleolin regulates the proliferation of vascular smooth muscle cells in atherosclerotic via Aurora B. J. Cell. Mol. Med. 2020. [Google Scholar] [CrossRef]
- Belenguer, P.; Caizergues-Ferrer, M.; Labbe, J.C.; Doree, M.; Amalric, F. Mitosis-specific phosphorylation of nucleolin by p34cdc2 protein kinase. Mol. Cell. Biol. 1990, 10, 3607–3618. [Google Scholar] [CrossRef]
- Yang, A.; Shi, G.; Zhou, C.; Lu, R.; Li, H.; Sun, L.; Jin, Y. Nucleolin maintains embryonic stem cell self-renewal by suppression of p53 protein-dependent pathway. J. Biol. Chem. 2011, 286, 43370–43382. [Google Scholar] [CrossRef]
- Hoja-Lukowicz, D.; Przybylo, M.; Pochec, E.; Drabik, A.; Silberring, J.; Kremser, M.; Schadendorf, D.; Laidler, P.; Litynska, A. The new face of nucleolin in human melanoma. Cancer Immunol. Immunother. 2009, 58, 1471–1480. [Google Scholar] [CrossRef]
- Marcel, V.; Catez, F.; Berger, C.M.; Perrial, E.; Plesa, A.; Thomas, X.; Mattei, E.; Hayette, S.; Saintigny, P.; Bouvet, P.; et al. Expression Profiling of Ribosome Biogenesis Factors Reveals Nucleolin as a Novel Potential Marker to Predict Outcome in AML Patients. PLoS ONE 2017, 12, e0170160. [Google Scholar] [CrossRef]
- Qiu, W.; Zhou, F.; Zhang, Q.; Sun, X.; Shi, X.; Liang, Y.; Wang, X.; Yue, L. Overexpression of nucleolin and different expression sites both related to the prognosis of gastric cancer. APMIS 2013, 121, 919–925. [Google Scholar] [CrossRef]
- Galzio, R.; Rosati, F.; Benedetti, E.; Cristiano, L.; Aldi, S.; Mei, S.; D’Angelo, B.; Gentile, R.; Laurenti, G.; Cifone, M.G.; et al. Glycosilated nucleolin as marker for human gliomas. J. Cell. Biochem. 2012, 113, 571–579. [Google Scholar] [CrossRef]
- Hammoudi, A.; Song, F.; Reed, K.R.; Jenkins, R.E.; Meniel, V.S.; Watson, A.J.; Pritchard, D.M.; Clarke, A.R.; Jenkins, J.R. Proteomic profiling of a mouse model of acute intestinal Apc deletion leads to identification of potential novel biomarkers of human colorectal cancer (CRC). Biochem. Biophys. Res. Commun. 2013, 440, 364–370. [Google Scholar] [CrossRef]
- Chen, C.; Chen, L.; Yao, Y.; Qin, Z.; Chen, H. Nucleolin overexpression is associated with an unfavorable outcome for ependymoma: A multifactorial analysis of 176 patients. J. Neurooncol. 2016, 127, 43–52. [Google Scholar] [CrossRef]
- Wolfson, E.; Goldenberg, M.; Solomon, S.; Frishberg, A.; Pinkas-Kramarski, R. Nucleolin-binding by ErbB2 enhances tumorigenicity of ErbB2-positive breast cancer. Oncotarget 2016, 7, 65320–65334. [Google Scholar] [CrossRef]
- Nguyen Van Long, F.; Lardy-Cleaud, A.; Bray, S.; Chabaud, S.; Dubois, T.; Diot, A.; Thompson, A.M.; Bourdon, J.C.; Perol, D.; Bouvet, P.; et al. Druggable Nucleolin Identifies Breast Tumours Associated with Poor Prognosis That Exhibit Different Biological Processes. Cancers 2018, 10, 390. [Google Scholar] [CrossRef]
- Wise, J.F.; Berkova, Z.; Mathur, R.; Zhu, H.; Braun, F.K.; Tao, R.H.; Sabichi, A.L.; Ao, X.; Maeng, H.; Samaniego, F. Nucleolin inhibits Fas ligand binding and suppresses Fas-mediated apoptosis in vivo via a surface nucleolin-Fas complex. Blood 2013, 121, 4729–4739. [Google Scholar] [CrossRef]
- Fogal, V.; Sugahara, K.N.; Ruoslahti, E.; Christian, S. Cell surface nucleolin antagonist causes endothelial cell apoptosis and normalization of tumor vasculature. Angiogenesis 2009, 12, 91–100. [Google Scholar] [CrossRef]
- Reyes-Reyes, E.M.; Akiyama, S.K. Cell-surface nucleolin is a signal transducing P-selectin binding protein for human colon carcinoma cells. Exp. Cell Res. 2008, 314, 2212–2223. [Google Scholar] [CrossRef]
- Di Segni, A.; Farin, K.; Pinkas-Kramarski, R. Identification of nucleolin as new ErbB receptors- interacting protein. PLoS ONE 2008, 3, e2310. [Google Scholar] [CrossRef]
- Farin, K.; Schokoroy, S.; Haklai, R.; Cohen-Or, I.; Elad-Sfadia, G.; Reyes-Reyes, M.E.; Bates, P.J.; Cox, A.D.; Kloog, Y.; Pinkas-Kramarski, R. Oncogenic synergism between ErbB1, nucleolin, and mutant Ras. Cancer Res. 2011, 71, 2140–2151. [Google Scholar] [CrossRef]
- Shin, S.H.; Lee, G.Y.; Lee, M.; Kang, J.; Shin, H.W.; Chun, Y.S.; Park, J.W. Aberrant expression of CITED2 promotes prostate cancer metastasis by activating the nucleolin-AKT pathway. Nat. Commun. 2018, 9, 4113. [Google Scholar] [CrossRef]
- Yang, Y.; Yang, C.; Zhang, J. C23 protein meditates bone morphogenetic protein-2-mediated EMT via up-regulation of Erk1/2 and Akt in gastric cancer. Med. Oncol. 2015, 32, 76. [Google Scholar] [CrossRef]
- Hung, C.Y.; Yang, W.B.; Wang, S.A.; Hsu, T.I.; Chang, W.C.; Hung, J.J. Nucleolin enhances internal ribosomal entry site (IRES)-mediated translation of Sp1 in tumorigenesis. Biochim. Biophys. Acta 2014, 1843, 2843–2854. [Google Scholar] [CrossRef]
- Pichiorri, F.; Palmieri, D.; De Luca, L.; Consiglio, J.; You, J.; Rocci, A.; Talabere, T.; Piovan, C.; Lagana, A.; Cascione, L.; et al. In vivo NCL targeting affects breast cancer aggressiveness through miRNA regulation. J. Exp. Med. 2013, 210, 951–968. [Google Scholar] [CrossRef]
- Sheetz, T.; Mills, J.; Tessari, A.; Pawlikowski, M.; Braddom, A.E.; Posid, T.; Zynger, D.L.; James, C.; Embrione, V.; Parbhoo, K.; et al. NCL Inhibition Exerts Antineoplastic Effects against Prostate Cancer Cells by Modulating Oncogenic MicroRNAs. Cancers 2020, 12, 1861. [Google Scholar] [CrossRef]
- Goldson, T.M.; Turner, K.L.; Huang, Y.; Carlson, G.E.; Caggiano, E.G.; Oberhauser, A.F.; Fennewald, S.M.; Burdick, M.M.; Resto, V.A. Nucleolin mediates the binding of cancer cells to L-selectin under conditions of lymphodynamic shear stress. Am. J. Physiol. Cell Physiol. 2020, 318, C83–C93. [Google Scholar] [CrossRef]
- Joo, E.J.; Wasik, B.R.; Parrish, C.; Paz, H.; Mupsilonhlenhoff, M.; Abdel-Azim, H.; Groffen, J.; Heisterkamp, N. Pre-B acute lymphoblastic leukemia expresses cell surface nucleolin as a 9-O-acetylated sialoglycoprotein. Sci. Rep. 2018, 8, 17174. [Google Scholar] [CrossRef]
- Huang, Y.; Shi, H.; Zhou, H.; Song, X.; Yuan, S.; Luo, Y. The angiogenic function of nucleolin is mediated by vascular endothelial growth factor and nonmuscle myosin. Blood 2006, 107, 3564–3571. [Google Scholar] [CrossRef]
- Ding, Y.; Song, N.; Liu, C.; He, T.; Zhuo, W.; He, X.; Chen, Y.; Song, X.; Fu, Y.; Luo, Y. Heat shock cognate 70 regulates the translocation and angiogenic function of nucleolin. Arterioscler. Thromb. Vasc. Biol. 2012, 32, e126–e134. [Google Scholar] [CrossRef]
- Shi, H.; Huang, Y.; Zhou, H.; Song, X.; Yuan, S.; Fu, Y.; Luo, Y. Nucleolin is a receptor that mediates antiangiogenic and antitumor activity of endostatin. Blood 2007, 110, 2899–2906. [Google Scholar] [CrossRef]
- Hovanessian, A.G.; Puvion-Dutilleul, F.; Nisole, S.; Svab, J.; Perret, E.; Deng, J.S.; Krust, B. The cell-surface-expressed nucleolin is associated with the actin cytoskeleton. Exp. Cell Res. 2000, 261, 312–328. [Google Scholar] [CrossRef]
- Dumler, I.; Stepanova, V.; Jerke, U.; Mayboroda, O.A.; Vogel, F.; Bouvet, P.; Tkachuk, V.; Haller, H.; Gulba, D.C. Urokinase-induced mitogenesis is mediated by casein kinase 2 and nucleolin. Curr. Biol. 1999, 9, 1468–1476. [Google Scholar] [CrossRef]
- Legrand, D.; Vigie, K.; Said, E.A.; Elass, E.; Masson, M.; Slomianny, M.C.; Carpentier, M.; Briand, J.P.; Mazurier, J.; Hovanessian, A.G. Surface nucleolin participates in both the binding and endocytosis of lactoferrin in target cells. Eur. J. Biochem. 2004, 271, 303–317. [Google Scholar] [CrossRef]
- Wang, Y.; Mao, M.; Xu, J.C. Cell-surface nucleolin is involved in lipopolysaccharide internalization and signalling in alveolar macrophages. Cell Biol. Int. 2011, 35, 677–685. [Google Scholar] [CrossRef]
- Hirano, K.; Miki, Y.; Hirai, Y.; Sato, R.; Itoh, T.; Hayashi, A.; Yamanaka, M.; Eda, S.; Beppu, M. A multifunctional shuttling protein nucleolin is a macrophage receptor for apoptotic cells. J. Biol. Chem. 2005, 280, 39284–39293. [Google Scholar] [CrossRef]
- Chan, C.M.; Chu, H.; Zhang, A.J.; Leung, L.H.; Sze, K.H.; Kao, R.Y.; Chik, K.K.; To, K.K.; Chan, J.F.; Chen, H.; et al. Hemagglutinin of influenza A virus binds specifically to cell surface nucleolin and plays a role in virus internalization. Virology 2016, 494, 78–88. [Google Scholar] [CrossRef]
- Watanabe, T.; Hirano, K.; Takahashi, A.; Yamaguchi, K.; Beppu, M.; Fujiki, H.; Suganuma, M. Nucleolin on the cell surface as a new molecular target for gastric cancer treatment. Biol. Pharm. Bull. 2010, 33, 796–803. [Google Scholar] [CrossRef]
- Su, P.Y.; Wang, Y.F.; Huang, S.W.; Lo, Y.C.; Wang, Y.H.; Wu, S.R.; Shieh, D.B.; Chen, S.H.; Wang, J.R.; Lai, M.D.; et al. Cell surface nucleolin facilitates enterovirus 71 binding and infection. J. Virol. 2015, 89, 4527–4538. [Google Scholar] [CrossRef]
- Tayyari, F.; Marchant, D.; Moraes, T.J.; Duan, W.; Mastrangelo, P.; Hegele, R.G. Identification of nucleolin as a cellular receptor for human respiratory syncytial virus. Nat. Med. 2011, 17, 1132–1135. [Google Scholar] [CrossRef]
- Johansson, H.; Svensson, F.; Runnberg, R.; Simonsson, T.; Simonsson, S. Phosphorylated nucleolin interacts with translationally controlled tumor protein during mitosis and with Oct4 during interphase in ES cells. PLoS ONE 2010, 5, e13678. [Google Scholar] [CrossRef]
- Percharde, M.; Lin, C.J.; Yin, Y.; Guan, J.; Peixoto, G.A.; Bulut-Karslioglu, A.; Biechele, S.; Huang, B.; Shen, X.; Ramalho-Santos, M. A LINE1-Nucleolin Partnership Regulates Early Development and ESC Identity. Cell 2018, 174, 391–405.e19. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Reister, S.; Mahotka, C.; Meisel, R.; Borkhardt, A.; Grinstein, E. Control of AC133/CD133 and impact on human hematopoietic progenitor cells through nucleolin. Leukemia 2015, 29, 2208–2220. [Google Scholar] [CrossRef] [PubMed]
- Reister, S.; Mahotka, C.; van den Hofel, N.; Grinstein, E. Nucleolin promotes Wnt signaling in human hematopoietic stem/progenitor cells. Leukemia 2019, 33, 1052–1054. [Google Scholar] [CrossRef] [PubMed]
- Turck, N.; Lefebvre, O.; Gross, I.; Gendry, P.; Kedinger, M.; Simon-Assmann, P.; Launay, J.F. Effect of laminin-1 on intestinal cell differentiation involves inhibition of nuclear nucleolin. J. Cell. Physiol. 2006, 206, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Ognibene, M.; Pezzolo, A. Roniciclib down-regulates stemness and inhibits cell growth by inducing nucleolar stress in neuroblastoma. Sci. Rep. 2020, 10, 12902. [Google Scholar] [CrossRef] [PubMed]
- Gregorio, A.C.; Lacerda, M.; Figueiredo, P.; Simoes, S.; Dias, S.; Moreira, J.N. Meeting the needs of breast cancer: A nucleolin’s perspective. Crit. Rev. Oncol. Hematol. 2018, 125, 89–101. [Google Scholar] [CrossRef]
- Bates, P.J.; Laber, D.A.; Miller, D.M.; Thomas, S.D.; Trent, J.O. Discovery and development of the G-rich oligonucleotide AS1411 as a novel treatment for cancer. Exp. Mol. Pathol. 2009, 86, 151–164. [Google Scholar] [CrossRef]
- Girvan, A.C.; Teng, Y.; Casson, L.K.; Thomas, S.D.; Juliger, S.; Ball, M.W.; Klein, J.B.; Pierce, W.M., Jr.; Barve, S.S.; Bates, P.J. AGRO100 inhibits activation of nuclear factor-kappaB (NF-kappaB) by forming a complex with NF-kappaB essential modulator (NEMO) and nucleolin. Mol. Cancer Ther. 2006, 5, 1790–1799. [Google Scholar] [CrossRef]
- Soundararajan, S.; Chen, W.; Spicer, E.K.; Courtenay-Luck, N.; Fernandes, D.J. The nucleolin targeting aptamer AS1411 destabilizes Bcl-2 messenger RNA in human breast cancer cells. Cancer Res. 2008, 68, 2358–2365. [Google Scholar] [CrossRef]
- Birmpas, C.; Briand, J.P.; Courty, J.; Katsoris, P. The pseudopeptide HB-19 binds to cell surface nucleolin and inhibits angiogenesis. Vasc. Cell 2012, 4, 21. [Google Scholar] [CrossRef]
- Gomes-da-Silva, L.C.; Ramalho, J.S.; Pedroso de Lima, M.C.; Simoes, S.; Moreira, J.N. Impact of anti-PLK1 siRNA-containing F3-targeted liposomes on the viability of both cancer and endothelial cells. Eur. J. Pharm. Biopharm. 2013, 85, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Gomes-da-Silva, L.C.; Fernandez, Y.; Abasolo, I.; Schwartz, S., Jr.; Ramalho, J.S.; Pedroso de Lima, M.C.; Simoes, S.; Moreira, J.N. Efficient intracellular delivery of siRNA with a safe multitargeted lipid-based nanoplatform. Nanomedicine 2013, 8, 1397–1413. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, N.A.; Gomes-da-Silva, L.C.; Moura, V.; Simoes, S.; Moreira, J.N. Simultaneous active intracellular delivery of doxorubicin and C6-ceramide shifts the additive/antagonistic drug interaction of non-encapsulated combination. J. Control. Release 2014, 196, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Pesarrodona, M.; Sanchez-Garcia, L.; Seras-Franzoso, J.; Sanchez-Chardi, A.; Balta-Foix, R.; Camara-Sanchez, P.; Gener, P.; Jara, J.J.; Pulido, D.; Serna, N.; et al. Engineering a Nanostructured Nucleolin-Binding Peptide for Intracellular Drug Delivery in Triple-Negative Breast Cancer Stem Cells. ACS Appl. Mater. Interfaces 2020, 12, 5381–5388. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Bae, C.; Kim, M.J.; Song, I.H.; Ryu, J.H.; Choi, J.H.; Lee, C.J.; Nam, J.S.; Kim, J.I. A novel nucleolin-binding peptide for Cancer Theranostics. Theranostics 2020, 10, 9153–9171. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumor | Markers | References |
|---|---|---|
| Acute Myeloid Leukemia | CD34+/CD38− | [6,21] |
| Breast | CD44high/CD24low | [20] |
| Ovary | CD24+, CD133+, CXCR4+ | [25,42] |
| Ewing’s Sarcoma | CD133+ | [29] |
| Lung | CD133+, CD90+ | [43] |
| Prostate | CD44+/α2β1integrinhigh/CD133+ | [44] |
| Colorectal | CD44+, CD133+, CD166+, Lgr5+, EpCAM+ | [45] |
| Pancreas | CD44+, CD24+, EpCAM+ | [46] |
| Brain | CD90+, CD133+ | [47,48] |
| Melanoma | CD271+ | [49] |
| Interaction with | Attributed Functions/Impact | References |
|---|---|---|
| 5′UTR of p53 mRNA | Suppression of p53 translation and induction after DNA damage | [121] |
| EGFR mRNA | Stabilization of EGFR mRNA and increased expression of the receptor involved in cell malignization | [122] |
| AU-rich element of BCL-2 mRNA | Stabilization of BCL-2 mRNA and decreased apoptosis | [123] |
| BCL-XL mRNA (when phosphorylated) | Stabilization of BCL-XL mRNA and decreased apoptosis | [124] |
| 3′UTR APP mRNA | Stabilization of APP mRNA and consequent accumulation of APP protein in Alzheimer’s disease | [125] |
| Kinesins and importin β1 mRNA | Transportation of importin β1 mRNA to specific sites in cells to control cell growth | [126] |
| Selenoproteins mRNA | Selective enhancing of a subset of selonoproteins at the level of translation | [127] |
| IL-2 mRNA | Stabilization of IL-2 mRNA during T-cells activation | [128] |
| COX-2 mRNA | Stabilization of COX-2 mRNA leading to COX-2 upregulation and consequent malignant transformation | [129] |
| Ligands | Attributed Functions/Impact | References |
|---|---|---|
| F3 peptide (synthetically derived from HMGN2) | Targeting of tumor endothelial cells and tumor cells; possible deliverer of therapeutic molecules. | [11,14,15,94] |
| Urokinase | Formation of a complex that includes nucleolin, urokinase receptor and CK2 that mediates the mitogenic activity of urokinase. | [164] |
| Lactoferrin | Internalization of lactoferrin and induction of recycling/degradation pathway or nucleolus translocation. | [165] |
| P-selectin | Interaction with P-selectin on the cell surface of human colon carcinoma cells and formation of a signaling complex that includes phosphorylated surface nucleolin, PI3K and p38 MAPK. This complex regulates cell adhesion and spreading which are implicated in carcinogenesis. | [150] |
| LPS | Internalization of LPS on activated alveolar macrophages and consequent mediation of the inflammatory response to bacterial infection. | [166] |
| Apoptotic cells | Interaction of macrophage surface nucleolin with apoptotic cells signalized to phagocytosis. | [167] |
| Influenza A viruses | Internalization of several subtypes of influenza A viruses thus mediating infection. | [168] |
| Tipα | Internalization, on gastric cancer cells, of Tipα (carcinogenic factor of Helicobacter pylori). | [169] |
| Enterovirus 71 | Mediation of enterovirus 71 cell infection. | [170] |
| Respiratory syncytial virus | Interaction with respiratory syncytial virus at the apical membrane and mediation of infection. | [171] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carvalho, L.S.; Gonçalves, N.; Fonseca, N.A.; Moreira, J.N. Cancer Stem Cells and Nucleolin as Drivers of Carcinogenesis. Pharmaceuticals 2021, 14, 60. https://doi.org/10.3390/ph14010060
Carvalho LS, Gonçalves N, Fonseca NA, Moreira JN. Cancer Stem Cells and Nucleolin as Drivers of Carcinogenesis. Pharmaceuticals. 2021; 14(1):60. https://doi.org/10.3390/ph14010060
Chicago/Turabian StyleCarvalho, Laura Sofia, Nélio Gonçalves, Nuno André Fonseca, and João Nuno Moreira. 2021. "Cancer Stem Cells and Nucleolin as Drivers of Carcinogenesis" Pharmaceuticals 14, no. 1: 60. https://doi.org/10.3390/ph14010060
APA StyleCarvalho, L. S., Gonçalves, N., Fonseca, N. A., & Moreira, J. N. (2021). Cancer Stem Cells and Nucleolin as Drivers of Carcinogenesis. Pharmaceuticals, 14(1), 60. https://doi.org/10.3390/ph14010060