Targeting Loss of Heterozygosity: A Novel Paradigm for Cancer Therapy


Abstract
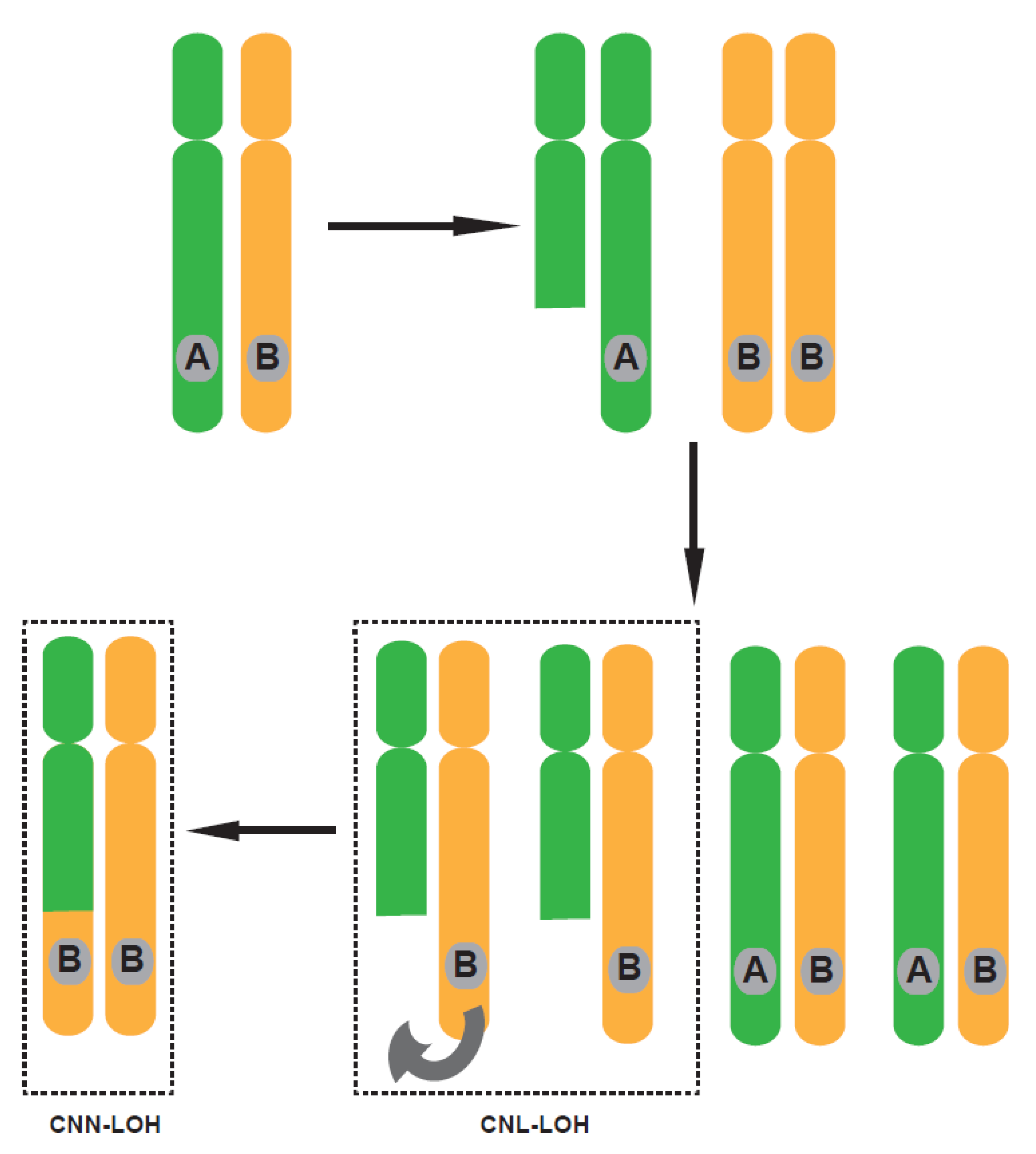
1. Introduction
2. Loss of Tumor Suppressor Genes by LOH
3. LOH Leads to Loss of Non-Driver Genes and Endows Cancer Cells with Unique Vulnerabilities
4. Loss of Heterozygosity Provides Novel Therapeutic Targets for Cancer Treatment
5. Loci Undergoing Loss of Heterozygosity as Clinical Biomarkers
5.1. Biomarkers on Chromosome 1q Associated with LOH
5.2. Biomarkers on Chromosome 3q Associated with LOH
5.3. Biomarkers on Chromosome 6q Associated with LOH
5.4. Biomarkers on Chromosome 16q Associated with LOH
5.5. Biomarkers on Chromosome 17q Associated with LOH
5.6. Biomarkers on Other Chromosomes Associated with LOH
6. Loss of Heterozygosity in Tumors Provides Up-and-Coming Possibilities to Understand the Underlying Mechanisms of Cancer Evolution
7. Future Perspective
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Cavenee, W.K.; Dryja, T.P.; Phillips, R.A.; Benedict, W.F.; Godbout, R.; Gallie, B.L.; Murphree, A.L.; Strong, L.C.; White, R.L. Expression of recessive alleles by chromosomal mechanisms in retinoblastoma. Nature 1983, 305, 779–784. [Google Scholar] [CrossRef] [PubMed]
- Fitzgibbon, J.; Smith, L.L.; Raghavan, M.; Smith, M.L.; Debernardi, S.; Skoulakis, S.; Lillington, D.; Lister, T.A.; Young, B.D. Association between acquired uniparental disomy and homozygous gene mutation in acute myeloid leukemias. Cancer Res. 2005, 65, 9152–9154. [Google Scholar] [CrossRef] [PubMed]
- Ryland, G.L.; Doyle, M.A.; Goode, D.; Boyle, S.E.; Choong, D.Y.; Rowley, S.M.; Li, J.; Australian Ovarian Cancer Study, G.; Bowtell, D.D.; Tothill, R.W.; et al. Loss of heterozygosity: What is it good for? BMC Med. Genom. 2015, 8, 45. [Google Scholar] [CrossRef] [PubMed]
- Tapial, S.; Garcia, J.L.; Corchete, L.; Holowatyj, A.N.; Perez, J.; Rueda, D.; Urioste, M.; Gonzalez-Sarmiento, R.; Perea, J. Copy neutral loss of heterozygosity (cnLOH) patterns in synchronous colorectal cancer. Eur. J. Hum. Genet. 2020. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.S.; Wang, Z.; Hu, Y.; Yang, H.H.; Gere, S.; Buetow, K.H.; Lee, M.P. Allelic variation in gene expression is common in the human genome. Genom. Res. 2003, 13, 1855–1862. [Google Scholar] [CrossRef]
- O’Keefe, C.; McDevitt, M.A.; Maciejewski, J.P. Copy neutral loss of heterozygosity: A novel chromosomal lesion in myeloid malignancies. Blood 2010, 115, 2731–2739. [Google Scholar] [CrossRef]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instabilities in human cancers. Nature 1998, 396, 643–649. [Google Scholar] [CrossRef]
- Murphree, A.L.; Benedict, W.F. Retinoblastoma: Clues to human oncogenesis. Science 1984, 223, 1028–1033. [Google Scholar] [CrossRef]
- Jiang, G.; Zhang, S.; Yazdanparast, A.; Li, M.; Pawar, A.V.; Liu, Y.; Inavolu, S.M.; Cheng, L. Comprehensive comparison of molecular portraits between cell lines and tumors in breast cancer. BMC Genom. 2016, 17 (Suppl. 7), 525. [Google Scholar] [CrossRef]
- Takai, E.; Nakamura, H.; Chiku, S.; Kubo, E.; Ohmoto, A.; Totoki, Y.; Shibata, T.; Higuchi, R.; Yamamoto, M.; Furuse, J.; et al. Whole-exome Sequencing Reveals New Potential Susceptibility Genes for Japanese Familial Pancreatic Cancer. Ann. Surg. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kaur, R.P.; Vasudeva, K.; Kumar, R.; Munshi, A. Role of p53 Gene in Breast Cancer: Focus on Mutation Spectrum and Therapeutic Strategies. Curr. Pharm. Des. 2018, 24, 3566–3575. [Google Scholar] [CrossRef]
- Lea, I.A.; Jackson, M.A.; Li, X.; Bailey, S.; Peddada, S.D.; Dunnick, J.K. Genetic pathways and mutation profiles of human cancers: Site- and exposure-specific patterns. Carcinogenesis 2007, 28, 1851–1858. [Google Scholar] [CrossRef] [PubMed]
- Chauffaille, M.; Zalcberg, I.; Barreto, W.G.; Bendit, I. Detection of somatic TP53 mutations and 17p deletions in patients with chronic lymphocytic leukemia: A review of the current methods. Hematol. Transfus. Cell Ther. 2020, 42, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Lebok, P.; Kopperschmidt, V.; Kluth, M.; Hube-Magg, C.; Ozden, C.; Taskin, B.; Hussein, K.; Mittenzwei, A.; Lebeau, A.; Witzel, I.; et al. Partial PTEN deletion is linked to poor prognosis in breast cancer. BMC Cancer 2015, 15, 963. [Google Scholar] [CrossRef]
- Neff, R.T.; Senter, L.; Salani, R. BRCA mutation in ovarian cancer: Testing, implications and treatment considerations. Ther. Adv. Med. Oncol 2017, 9, 519–531. [Google Scholar] [CrossRef]
- BRCA Gene Mutations: Cancer Risk and Genetic Testing. Available online: https://www.cancer.gov/about-cancer/causes-prevention/genetics/brca-fact-sheet (accessed on 19 November 2020).
- O’Brien, C. New tumor suppressor found in pancreatic cancer. Science 1996, 271, 294. [Google Scholar] [CrossRef]
- Hahn, S.A.; Schutte, M.; Hoque, A.T.; Moskaluk, C.A.; da Costa, L.T.; Rozenblum, E.; Weinstein, C.L.; Fischer, A.; Yeo, C.J.; Hruban, R.H.; et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 1996, 271, 350–353. [Google Scholar] [CrossRef]
- Lin, L.H.; Chang, K.W.; Cheng, H.W.; Liu, C.J. SMAD4 Somatic Mutations in Head and Neck Carcinoma Are Associated with Tumor Progression. Front. Oncol 2019, 9, 1379. [Google Scholar] [CrossRef]
- Hadžija, M.P.; Kapitanovic, S.; Radosevic, S.; Cacev, T.; Mirt, M.; Kovacevic, D.; Cacev, T.; Hadzija, M.; Spaventi, R.; Pavelic, K. Loss of heterozygosity of DPC4 tumor suppressor gene in human sporadic colon cancer. J. Mol. Med. (Berl.) 2001, 79, 128–132. [Google Scholar] [CrossRef]
- Boulay, J.L.; Stiefel, U.; Taylor, E.; Dolder, B.; Merlo, A.; Hirth, F. Loss of heterozygosity of TRIM3 in malignant gliomas. BMC Cancer 2009, 9, 71. [Google Scholar] [CrossRef]
- Boultwood, J. CUX1 in leukemia: Dosage matters. Blood 2013, 121, 869–871. [Google Scholar] [CrossRef] [PubMed]
- Mertens, F.; Heim, S.; Mandahl, N.; Johansson, B.; Mertens, O.; Persson, B.; Salemark, L.; Wennerberg, J.; Jonsson, N.; Mitelman, F. Cytogenetic analysis of 33 basal cell carcinomas. Cancer Res. 1991, 51, 954–957. [Google Scholar] [PubMed]
- Nichols, C.A.; Gibson, W.J.; Brown, M.S.; Kosmicki, J.A.; Busanovich, J.P.; Wei, H.; Urbanski, L.M.; Curimjee, N.; Berger, A.C.; Gao, G.F.; et al. Loss of heterozygosity of essential genes represents a widespread class of potential cancer vulnerabilities. Nat. Commun. 2020, 11, 2517. [Google Scholar] [CrossRef] [PubMed]
- Rendo, V.; Stoimenov, I.; Mateus, A.; Sjoberg, E.; Svensson, R.; Gustavsson, A.L.; Johansson, L.; Ng, A.; O’Brien, C.; Giannakis, M.; et al. Exploiting loss of heterozygosity for allele-selective colorectal cancer chemotherapy. Nat. Commun. 2020, 11, 1308. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.; Martens, J.; Van Hoeck, A.; Cuppen, E. Pan-cancer landscape of homologous recombination deficiency. Nat. Commun. 2020, 11, 5584. [Google Scholar] [CrossRef]
- Vosoughi, A.; Zhang, T.; Shohdy, K.S.; Vlachostergios, P.J.; Wilkes, D.C.; Bhinder, B.; Tagawa, S.T.; Nanus, D.M.; Molina, A.M.; Beltran, H.; et al. Common germline-somatic variant interactions in advanced urothelial cancer. Nat. Commun. 2020, 11, 6195. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.S.; De, S. Loss of heterozygosity preferentially occurs in early replicating regions in cancer genomes. Nucleic Acids Res. 2013, 41, 7615–7624. [Google Scholar] [CrossRef] [PubMed]
- Stafford, W.C.; Peng, X.; Olofsson, M.H.; Zhang, X.; Luci, D.K.; Lu, L.; Cheng, Q.; Tresaugues, L.; Dexheimer, T.S.; Coussens, N.P.; et al. Irreversible inhibition of cytosolic thioredoxin reductase 1 as a mechanistic basis for anticancer therapy. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Tantin, D. Oct transcription factors in development and stem cells: Insights and mechanisms. Development 2013, 140, 2857–2866. [Google Scholar] [CrossRef] [PubMed]
- Shakya, A.; Cooksey, R.; Cox, J.E.; Wang, V.; McClain, D.A.; Tantin, D. Oct1 loss of function induces a coordinate metabolic shift that opposes tumorigenicity. Nat. Cell Biol. 2009, 11, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Gemberling, M.; Nakamura, M.; Whitby, F.G.; Handa, H.; Fairbrother, W.G.; Tantin, D. A general mechanism for transcription regulation by Oct1 and Oct4 in response to genotoxic and oxidative stress. Genes Dev. 2009, 23, 208–222. [Google Scholar] [CrossRef] [PubMed]
- Moscow, J.A.; Schmidt, L.; Ingram, D.T.; Gnarra, J.; Johnson, B.; Cowan, K.H. Loss of heterozygosity of the human cytosolic glutathione peroxidase I gene in lung cancer. Carcinogenesis 1994, 15, 2769–2773. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Benya, R.V.; Carroll, R.E.; Diamond, A.M. Allelic loss of the gene for the GPX1 selenium-containing protein is a common event in cancer. J. Nutr. 2005, 135, 3021S–3024S. [Google Scholar] [CrossRef] [PubMed]
- Lenders, J.W.; Duh, Q.Y.; Eisenhofer, G.; Gimenez-Roqueplo, A.P.; Grebe, S.K.; Murad, M.H.; Naruse, M.; Pacak, K.; Young, W.F., Jr.; Endocrine, S. Pheochromocytoma and paraganglioma: An endocrine. society clinical practice guideline. J. Clin. Endocrinol. Metab. 2014, 99, 1915–1942. [Google Scholar] [CrossRef]
- Jochmanova, I.; Yang, C.; Zhuang, Z.; Pacak, K. Hypoxia-inducible factor signaling in pheochromocytoma: Turning the rudder in the right direction. J. Natl. Cancer Inst. 2013, 105, 1270–1283. [Google Scholar] [CrossRef]
- Zhuang, Z.; Yang, C.; Lorenzo, F.; Merino, M.; Fojo, T.; Kebebew, E.; Popovic, V.; Stratakis, C.A.; Prchal, J.T.; Pacak, K. Somatic HIF2A gain-of-function mutations in paraganglioma with polycythemia. N. Engl. J. Med. 2012, 367, 922–930. [Google Scholar] [CrossRef]
- Sawyers, C. Targeted cancer therapy. Nature 2004, 432, 294–297. [Google Scholar] [CrossRef]
- Yang, B. Targeted therapy for cancer. Discov. Med. 2004, 4, 18–21. [Google Scholar]
- Martincorena, I.; Raine, K.M.; Gerstung, M.; Dawson, K.J.; Haase, K.; Van Loo, P.; Davies, H.; Stratton, M.R.; Campbell, P.J. Universal Patterns of Selection in Cancer and Somatic Tissues. Cell 2017, 171, 1029–1041. [Google Scholar] [CrossRef]
- Vousden, K.H.; Ryan, K.M. p53 and metabolism. Nat. Rev. Cancer 2009, 9, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Baugh, E.H.; Ke, H.; Levine, A.J.; Bonneau, R.A.; Chan, C.S. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. 2018, 25, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Litviakov, N.V.; Denisov, E.V.; Takhauov, R.M.; Karpov, A.B.; Skobel’skaja, E.V.; Vasil’eva, E.O.; Goncharik, O.O.; Ageeva, A.M.; Mamonova, N.V.; Mezheritskiy, S.A.; et al. Association between TP53 gene ARG72PRO polymorphism and chromosome aberrations in human cancers. Mol. Carcinog. 2010, 49, 521–524. [Google Scholar] [CrossRef] [PubMed]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef]
- Ciriello, G.; Miller, M.L.; Aksoy, B.A.; Senbabaoglu, Y.; Schultz, N.; Sander, C. Emerging landscape of oncogenic signatures across human cancers. Nat. Genet. 2013, 45, 1127–1133. [Google Scholar] [CrossRef]
- Carter, D.R.; Murray, J.; Cheung, B.B.; Gamble, L.; Koach, J.; Tsang, J.; Sutton, S.; Kalla, H.; Syed, S.; Gifford, A.J.; et al. Therapeutic targeting of the MYC signal by inhibition of histone chaperone FACT in neuroblastoma. Sci. Transl. Med. 2015, 7, 312ra176. [Google Scholar] [CrossRef]
- Rickman, D.S.; Schulte, J.H.; Eilers, M. The Expanding World of N-MYC-Driven Tumors. Cancer Discov. 2018, 8, 150–163. [Google Scholar] [CrossRef]
- Fan-Minogue, H.; Bodapati, S.; Solow-Cordero, D.; Fan, A.; Paulmurugan, R.; Massoud, T.F.; Felsher, D.W.; Gambhir, S.S. A c-Myc activation sensor-based high-throughput drug screening identifies an antineoplastic effect of nitazoxanide. Mol. Cancer Ther. 2013, 12, 1896–1905. [Google Scholar] [CrossRef]
- Bandopadhayay, P.; Bergthold, G.; Nguyen, B.; Schubert, S.; Gholamin, S.; Tang, Y.; Bolin, S.; Schumacher, S.E.; Zeid, R.; Masoud, S.; et al. BET bromodomain inhibition of MYC-amplified medulloblastoma. Clin. Cancer Res. 2014, 20, 912–925. [Google Scholar] [CrossRef]
- Demma, M.J.; Mapelli, C.; Sun, A.; Bodea, S.; Ruprecht, B.; Javaid, S.; Wiswell, D.; Muise, E.; Chen, S.; Zelina, J.; et al. Omomyc Reveals New Mechanisms To Inhibit the MYC Oncogene. Mol. Cell Biol. 2019, 39, e00248-19. [Google Scholar] [CrossRef]
- Villanueva, M.T. Long path to MYC inhibition approaches clinical trials. Nat. Rev. Cancer 2019, 19, 252. [Google Scholar] [CrossRef] [PubMed]
- Zinzalla, G. Targeting MYC: Is it getting any easier? Future Med. Chem. 2016, 8, 1899–1902. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Henricksen, L.A.; Wold, M.S.; Ingles, C.J. RPA involvement in the damage-recognition and incision steps of nucleotide excision repair. Nature 1995, 374, 566–569. [Google Scholar] [CrossRef]
- Brill, S.J.; Stillman, B. Replication factor-A from Saccharomyces cerevisiae is encoded by three essential genes coordinately expressed at S phase. Genes Dev. 1991, 5, 1589–1600. [Google Scholar] [CrossRef] [PubMed]
- Basilion, J.P.; Schievella, A.R.; Burns, E.; Rioux, P.; Olson, J.C.; Monia, B.P.; Lemonidis, K.M.; Stanton, V.P., Jr.; Housman, D.E. Selective killing of cancer cells based on loss of heterozygosity and normal variation in the human genome: A new paradigm for anticancer drug therapy. Mol. Pharmacol. 1999, 56, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Courtney, D.G.; Moore, J.E.; Atkinson, S.D.; Maurizi, E.; Allen, E.H.A.; Pedrioli, D.M.L.; McLean, W.H.I.; Nesbit, M.A.; Moore, C.B.T. CRISPR/Cas9 DNA cleavage at SNP-derived PAM enables both in vitro and in vivo KRT12 mutation-specific targeting. Gene Ther. 2015, 23, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Kilchert, C.; Wittmann, S.; Vasiljeva, L. The regulation and functions of the nuclear RNA exosome complex. Nat. Rev. Mol. Cell Biol. 2016, 17, 227–239. [Google Scholar] [CrossRef]
- Halling-Brown, M.D.; Bulusu, K.C.; Patel, M.; Tym, J.E.; Al-Lazikani, B. canSAR: An integrated cancer public translational research and drug discovery resource. Nucleic Acids Res. 2012, 40, D947–D956. [Google Scholar] [CrossRef]
- Bulusu, K.C.; Tym, J.E.; Coker, E.A.; Schierz, A.C.; Al-Lazikani, B. canSAR: Updated cancer research and drug discovery knowledgebase. Nucleic Acids Res. 2014, 42, D1040–D1047. [Google Scholar] [CrossRef]
- Komotar, R.J.; Otten, M.L.; Starke, R.M.; Anderson, R.C. Chromosome 1p and 11q deletions and outcome in neuroblastoma-a critical review. Clin. Med. Oncol. 2008, 2, 419–420. [Google Scholar] [CrossRef]
- Attiyeh, E.F.; London, W.B.; Mosse, Y.P.; Wang, Q.; Winter, C.; Khazi, D.; McGrady, P.W.; Seeger, R.C.; Look, A.T.; Shimada, H.; et al. Chromosome 1p and 11q deletions and outcome in neuroblastoma. N. Engl. J. Med. 2005, 353, 2243–2253. [Google Scholar] [CrossRef]
- Caron, H.; van Sluis, P.; de Kraker, J.; Bokkerink, J.; Egeler, M.; Laureys, G.; Slater, R.; Westerveld, A.; Voute, P.A.; Versteeg, R. Allelic loss of chromosome 1p as a predictor of unfavorable outcome in patients with neuroblastoma. N. Engl. J. Med. 1996, 334, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Spitz, R.; Hero, B.; Westermann, F.; Ernestus, K.; Schwab, M.; Berthold, F. Fluorescence in situ hybridization analyses of chromosome band 1p36 in neuroblastoma detect two classes of alterations. Genes Chromosomes Cancer 2002, 34, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Stein, C.K.; Glover, T.W.; Palmer, J.L.; Glisson, B.S. Direct correlation between FRA3B expression and cigarette smoking. Genes Chromosomes Cancer 2002, 34, 333–340. [Google Scholar] [CrossRef]
- Pylkkanen, L.; Wolff, H.; Stjernvall, T.; Tuominen, P.; Sioris, T.; Karjalainen, A.; Anttila, S.; Husgafvel-Pursiainen, K. Reduced Fhit protein expression and loss of heterozygosity at FHIT gene in tumours from smoking and asbestos-exposed lung cancer patients. Int. J. Oncol. 2002, 20, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.M.; Kong, K.L.; Chen, L.; Liu, M.; Wong, A.M.; Zhu, C.; Tsang, J.W.; Guan, X.Y. Characterization of CACNA2D3 as a putative tumor suppressor gene in the development and progression of nasopharyngeal carcinoma. Int. J. Cancer 2013, 133, 2284–2295. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.R.; Fu, L.; Sham, P.C.; Kwong, D.L.; Zhu, C.L.; Chu, K.K.; Li, Y.; Guan, X.Y. Single-nucleotide polymorphism-mass array reveals commonly deleted regions at 3p22 and 3p14.2 associate with poor clinical outcome in esophageal squamous cell carcinoma. Int. J. Cancer 2008, 123, 826–830. [Google Scholar] [CrossRef] [PubMed]
- Verbiest, A.; Lambrechts, D.; Van Brussel, T.; Couchy, G.; Wozniak, A.; Mejean, A.; Lerut, E.; Oudard, S.; Verkarre, V.; Job, S.; et al. Polymorphisms in the Von Hippel-Lindau Gene Are Associated with Overall Survival in Metastatic Clear-Cell Renal-Cell Carcinoma Patients Treated with VEGFR Tyrosine Kinase Inhibitors. Clin. Genitourin Cancer 2018, 16, 266–273. [Google Scholar] [CrossRef]
- Newey, A.; Griffiths, B.; Michaux, J.; Pak, H.S.; Stevenson, B.J.; Woolston, A.; Semiannikova, M.; Spain, G.; Barber, L.J.; Matthews, N.; et al. Immunopeptidomics of colorectal cancer organoids reveals a sparse HLA class I neoantigen landscape and no increase in neoantigens with interferon or MEK-inhibitor treatment. J. Immunother. Cancer 2019, 7, 309. [Google Scholar] [CrossRef]
- Rosenbaum, E.; Seier, K.; Bandlamudi, C.; Dickson, M.; Gounder, M.; Keohan, M.L.; Chi, P.; Kelly, C.; Movva, S.; Nacev, B.; et al. HLA Genotyping in Synovial Sarcoma: Identifying HLA-A*02 and Its Association with Clinical Outcome. Clin. Cancer Res. 2020, 26, 5448–5455. [Google Scholar] [CrossRef]
- Liang, Y.; Diehn, M.; Watson, N.; Bollen, A.W.; Aldape, K.D.; Nicholas, M.K.; Lamborn, K.R.; Berger, M.S.; Botstein, D.; Brown, P.O.; et al. Gene expression profiling reveals molecularly and clinically distinct subtypes of glioblastoma multiforme. Proc. Natl. Acad. Sci. USA 2005, 102, 5814–5819. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Koyanagi, K.; Narita, N.; Kawakami, Y.; Takata, M.; Uchiyama, A.; Nguyen, L.; Nguyen, T.; Ye, X.; Morton, D.L.; et al. Aberrant fatty acid-binding protein-7 gene expression in cutaneous malignant melanoma. J. Investig. Dermatol. 2010, 130, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Li, S.; Yang, Z.; Liu, J.; Han, Y. Loss of Heterozygosity of 9p Is Associated with Poorer Survival in Patients with Gliomas. Mol. Neurobiol. 2016, 53, 6407–6412. [Google Scholar] [CrossRef]
- Garcia, J.M.; Silva, J.M.; Dominguez, G.; Gonzalez, R.; Navarro, A.; Carretero, L.; Provencio, M.; Espana, P.; Bonilla, F. Allelic loss of the PTEN region (10q23) in breast carcinomas of poor pathophenotype. Breast Cancer Res. Treat. 1999, 57, 237–243. [Google Scholar] [CrossRef]
- Dolan, K.; Garde, J.; Gosney, J.; Sissons, M.; Wright, T.; Kingsnorth, A.N.; Walker, S.J.; Sutton, R.; Meltzer, S.J.; Field, J.K. Allelotype analysis of oesophageal adenocarcinoma: Loss of heterozygosity occurs at multiple sites. Br. J. Cancer 1998, 78, 950–957. [Google Scholar] [CrossRef]
- Orlando, E.J.; Han, X.; Tribouley, C.; Wood, P.A.; Leary, R.J.; Riester, M.; Levine, J.E.; Qayed, M.; Grupp, S.A.; Boyer, M.; et al. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat. Med. 2018, 24, 1504–1506. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Lane, D.P. p53 in health and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. [Google Scholar] [CrossRef]
- Aylon, Y.; Oren, M. New plays in the p53 theater. Curr. Opin. Genet. Dev. 2011, 21, 86–92. [Google Scholar] [CrossRef]
- Merajver, S.D.; Frank, T.S.; Xu, J.; Pham, T.M.; Calzone, K.A.; Bennett-Baker, P.; Chamberlain, J.; Boyd, J.; Garber, J.E.; Collins, F.S.; et al. Germline BRCA1 mutations and loss of the wild-type allele in tumors from families with early onset breast and ovarian cancer. Clin. Cancer Res. 1995, 1, 539–544. [Google Scholar]
- Sarli, L.; Bottarelli, L.; Bader, G.; Iusco, D.; Pizzi, S.; Costi, R.; D’Adda, T.; Bertolani, M.; Roncoroni, L.; Bordi, C. Association between recurrence of sporadic colorectal cancer, high level of microsatellite instability, and loss of heterozygosity at chromosome 18q. Dis. Colon Rectum 2004, 47, 1467–1482. [Google Scholar] [CrossRef]
- Popat, S.; Houlston, R.S. A systematic review and meta-analysis of the relationship between chromosome 18q genotype, DCC status and colorectal cancer prognosis. Eur. J. Cancer 2005, 41, 2060–2070. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Spitz, R.; Oberthur, A.; Westermann, F.; Berthold, F. Risk estimation of neuroblastoma patients using molecular markers. Klin. Padiatr. 2008, 220, 137–146. [Google Scholar] [CrossRef]
- Fischer, M.; Oberthuer, A.; Brors, B.; Kahlert, Y.; Skowron, M.; Voth, H.; Warnat, P.; Ernestus, K.; Hero, B.; Berthold, F. Differential expression of neuronal genes defines subtypes of disseminated neuroblastoma with favorable and unfavorable outcome. Clin. Cancer Res. 2006, 12, 5118–5128. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Diskin, S.; Rappaport, E.; Attiyeh, E.; Mosse, Y.; Shue, D.; Seiser, E.; Jagannathan, J.; Shusterman, S.; Bansal, M.; et al. Integrative genomics identifies distinct molecular classes of neuroblastoma and shows that multiple genes are targeted by regional alterations in DNA copy number. Cancer Res. 2006, 66, 6050–6062. [Google Scholar] [CrossRef]
- Hibi, K.; Takahashi, T.; Yamakawa, K.; Ueda, R.; Sekido, Y.; Ariyoshi, Y.; Suyama, M.; Takagi, H.; Nakamura, Y.; Takahashi, T. Three distinct regions involved in 3p deletion in human lung cancer. Oncogene 1992, 7, 445–449. [Google Scholar] [PubMed]
- Lee, Y.C.; Wu, C.T.; Shih, J.Y.; Jou, Y.S.; Chang, Y.L. Frequent allelic deletion at the FHIT locus associated with p53 overexpression in squamous cell carcinoma subtype of Taiwanese non-small-cell lung cancers. Br. J. Cancer 2004, 90, 2378–2383. [Google Scholar] [CrossRef]
- Sozzi, G.; Veronese, M.L.; Negrini, M.; Baffa, R.; Cotticelli, M.G.; Inoue, H.; Tornielli, S.; Pilotti, S.; De Gregorio, L.; Pastorino, U.; et al. The FHIT gene 3p14.2 is abnormal in lung cancer. Cell 1996, 85, 17–26. [Google Scholar] [CrossRef]
- Kurtz, A.; Zimmer, A.; Schnutgen, F.; Bruning, G.; Spener, F.; Muller, T. The expression pattern of a novel gene encoding brain-fatty acid binding protein correlates with neuronal and glial cell development. Development 1994, 120, 2637–2649. [Google Scholar]
- Anthony, T.E.; Mason, H.A.; Gridley, T.; Fishell, G.; Heintz, N. Brain lipid-binding protein is a direct target of Notch signaling in radial glial cells. Genes Dev. 2005, 19, 1028–1033. [Google Scholar] [CrossRef]
- Scheuermann, R.H.; Racila, E. CD19 antigen in leukemia and lymphoma diagnosis and immunotherapy. Leuk Lymphoma 1995, 18, 385–397. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wu, Z.; Liu, Y.; Han, W. New development in CAR-T cell therapy. J. Hematol. Oncol. 2017, 10, 53. [Google Scholar] [CrossRef] [PubMed]
- Riester, M.; Singh, A.P.; Brannon, A.R.; Yu, K.; Campbell, C.D.; Chiang, D.Y.; Morrissey, M.P. PureCN: Copy number calling and SNV classification using targeted short read sequencing. Source Code Biol. Med. 2016, 11, 13. [Google Scholar] [CrossRef] [PubMed]
- Benchimol, S.; Lamb, P.; Crawford, L.V.; Sheer, D.; Shows, T.B.; Bruns, G.A.; Peacock, J. Transformation associated p53 protein is encoded by a gene on human chromosome 17. Somat. Cell Mol. Genet. 1985, 11, 505–510. [Google Scholar] [CrossRef]
- El-Deiry, W.S.; Kern, S.E.; Pietenpol, J.A.; Kinzler, K.W.; Vogelstein, B. Definition of a consensus binding site for p53. Nat. Genet. 1992, 1, 45–49. [Google Scholar] [CrossRef]
- Bouaoun, L.; Sonkin, D.; Ardin, M.; Hollstein, M.; Byrnes, G.; Zavadil, J.; Olivier, M. TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum. Mutat. 2016, 37, 865–876. [Google Scholar] [CrossRef]
- Silwal-Pandit, L.; Vollan, H.K.; Chin, S.F.; Rueda, O.M.; McKinney, S.; Osako, T.; Quigley, D.A.; Kristensen, V.N.; Aparicio, S.; Borresen-Dale, A.L.; et al. TP53 mutation spectrum in breast cancer is subtype specific and has distinct prognostic relevance. Clin. Cancer Res. 2014, 20, 3569–3580. [Google Scholar] [CrossRef]
- Cho, Y.; Gorina, S.; Jeffrey, P.D.; Pavletich, N.P. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science 1994, 265, 346–355. [Google Scholar] [CrossRef]
- Kyndi, M.; Alsner, J.; Hansen, L.L.; Sorensen, F.B.; Overgaard, J. LOH rather than genotypes of TP53 codon 72 is associated with disease-free survival in primary breast cancer. Acta Oncol. 2006, 45, 602–609. [Google Scholar] [CrossRef]
- Thanendrarajan, S.; Tian, E.; Qu, P.; Mathur, P.; Schinke, C.; van Rhee, F.; Zangari, M.; Rasche, L.; Weinhold, N.; Alapat, D.; et al. The level of deletion 17p and bi-allelic inactivation of TP53 has a significant impact on clinical outcome in multiple myeloma. Haematologica 2017, 102, e364–e367. [Google Scholar] [CrossRef] [PubMed]
- Bepler, G.; Gautam, A.; McIntyre, L.M.; Beck, A.F.; Chervinsky, D.S.; Kim, Y.C.; Pitterle, D.M.; Hyland, A. Prognostic significance of molecular genetic aberrations on chromosome segment 11p15.5 in non-small-cell lung cancer. J. Clin. Oncol. 2002, 20, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Lopez, S.; Lim, E.L.; Horswell, S.; Haase, K.; Huebner, A.; Dietzen, M.; Mourikis, T.P.; Watkins, T.B.K.; Rowan, A.; Dewhurst, S.M.; et al. Interplay between whole-genome doubling and the accumulation of deleterious alterations in cancer evolution. Nat. Genet. 2020, 52, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Bielski, C.M.; Zehir, A.; Penson, A.V.; Donoghue, M.T.A.; Chatila, W.; Armenia, J.; Chang, M.T.; Schram, A.M.; Jonsson, P.; Bandlamudi, C.; et al. Genome doubling shapes the evolution and prognosis of advanced cancers. Nat. Genet. 2018, 50, 1189–1195. [Google Scholar] [CrossRef] [PubMed]
- Nei, M.; Gojobori, T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 1986, 3, 418–426. [Google Scholar] [CrossRef]
- Yang, Z.; Bielawski, J.P. Statistical methods for detecting molecular adaptation. Trends Ecol. Evol. 2000, 15, 496–503. [Google Scholar] [CrossRef]
- Tamborero, D.; Gonzalez-Perez, A.; Lopez-Bigas, N. OncodriveCLUST: Exploiting the positional clustering of somatic mu-tations to identify cancer genes. Bioinformatics 2013, 29, 2238–2244. [Google Scholar] [CrossRef]
- Zapata, L.; Pich, O.; Serrano, L.; Kondrashov, F.A.; Ossowski, S.; Schaefer, M.H. Negative selection in tumor genome evo-lution acts on essential cellular functions and the immunopeptidome. Genome Biol. 2018, 19, 67. [Google Scholar] [CrossRef]
- Nambara, S.; Masuda, T.; Tobo, T.; Kidogami, S.; Komatsu, H.; Sugimachi, K.; Saeki, H.; Oki, E.; Maehara, Y.; Mimori, K. Clinical significance of ZNF750 gene expression, a novel tumor suppressor gene, in esophageal squamous cell carcinoma. Oncol. Lett. 2017, 14, 1795–1801. [Google Scholar] [CrossRef]
- Hazawa, M.; Lin, D.C.; Handral, H.; Xu, L.; Chen, Y.; Jiang, Y.Y.; Mayakonda, A.; Ding, L.W.; Meng, X.; Sharma, A.; et al. ZNF750 is a lineage-specific tumour suppressor in squamous cell carcinoma. Oncogene 2017, 36, 2243–2254. [Google Scholar] [CrossRef]
- Sondka, Z.; Bamford, S.; Cole, C.G.; Ward, S.A.; Dunham, I.; Forbes, S.A. The COSMIC Cancer Gene Census: Describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 2018, 18, 696–705. [Google Scholar] [CrossRef] [PubMed]
- Zaccaria, S.; Raphael, B.J. Characterizing allele- and haplotype-specific copy numbers in single cells with CHISEL. Nat. Biotechnol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef]
- Aso, T.; Uozaki, H.; Morita, S.; Kumagai, A.; Watanabe, M. Loss of ARID1A, ARID1B, and ARID2 Expression during Progression of Gastric Cancer. Anticancer Res. 2015, 35, 6819–6827. [Google Scholar]
- Sim, J.C.; White, S.M.; Lockhart, P.J. ARID1B-mediated disorders: Mutations and possible mechanisms. Intractable Rare Dis. Res. 2015, 4, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Xu, B. Overcoming resistance to endocrine therapy in hormone receptor-positive human epidermal growth factor receptor 2-negative (HR+/HER2−) advanced breast cancer: A meta-analysis and systemic review of randomized clinical trials. Front. Med. 2020. [Google Scholar] [CrossRef]
- Hanker, A.B.; Sudhan, D.R.; Arteaga, C.L. Overcoming Endocrine Resistance in Breast Cancer. Cancer Cell 2020, 37, 496–513. [Google Scholar] [CrossRef]
- Nakao, M.; Fujiwara, S.; Iwase, H. Cancer Navigation Strategy for Endocrine Therapy-Resistant Breast Tumors. Trends Cancer 2018, 4, 404–407. [Google Scholar] [CrossRef]
- Lawrence, B.; Blenkiron, C.; Parker, K.; Tsai, P.; Fitzgerald, S.; Shields, P.; Robb, T.; Yeong, M.L.; Kramer, N.; James, S.; et al. Recurrent loss of heterozygosity correlates with clinical outcome in pancreatic neuroendocrine cancer. NPJ Genom. Med. 2018, 3, 18. [Google Scholar] [CrossRef]
- Amin, R.; Tripathi, K.; Sanderson, R.D. Nuclear Heparanase Regulates Chromatin Remodeling, Gene Expression and PTEN Tumor Suppressor Function. Cells 2020, 9, 2038. [Google Scholar] [CrossRef]
- Yang, J.; Yin, Y. PTEN in Chromatin Remodeling. Cold Spring Harb Perspect Med. 2020, 10, a036160. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Li, N.; Dong, B.; Guo, W.; Wei, H.; Chen, Q.; Yuan, H.; Han, Y.; Chang, H.; Kan, S.; et al. Chromatin remodeling ATPase BRG1 and PTEN are synthetic lethal in prostate cancer. J. Clin. Investig. 2019, 129, 759–773. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.A.; Fraser, M.; Rebello, R.J.; Boutros, P.C.; Murphy, D.G.; Bristow, R.G.; Risbridger, G.P. The influence of BRCA2 mutation on localized prostate cancer. Nat. Rev. Urol 2019, 16, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Kwong, A.; Shin, V.Y.; Ho, J.C.; Kang, E.; Nakamura, S.; Teo, S.H.; Lee, A.S.; Sng, J.H.; Ginsburg, O.M.; Kurian, A.W.; et al. Comprehensive spectrum of BRCA1 and BRCA2 deleterious mutations in breast cancer in Asian countries. J. Med. Genet. 2016, 53, 15–23. [Google Scholar] [CrossRef]
- Berry, J.L.; Polski, A.; Cavenee, W.K.; Dryja, T.P.; Murphree, A.L.; Gallie, B.L. The RB1 Story: Characterization and Cloning of the First Tumor Suppressor Gene. Genes 2019, 10, 879. [Google Scholar] [CrossRef]
- Dyson, N.J. RB1: A prototype tumor suppressor and an enigma. Genes Dev. 2016, 30, 1492–1502. [Google Scholar] [CrossRef]
- Palin, K.; Pitkanen, E.; Turunen, M.; Sahu, B.; Pihlajamaa, P.; Kivioja, T.; Kaasinen, E.; Valimaki, N.; Hanninen, U.A.; Cajuso, T.; et al. Contribution of allelic imbalance to colorectal cancer. Nat. Commun. 2018, 9, 3664. [Google Scholar] [CrossRef]
- Bhagirath, D.; Yang, T.L.; Tabatabai, Z.L.; Shahryari, V.; Majid, S.; Dahiya, R.; Tanaka, Y.; Saini, S. Role of a novel race-related tumor suppressor microRNA located in frequently deleted chromosomal locus 8p21 in prostate cancer progression. Carcinogenesis 2019, 40, 633–642. [Google Scholar] [CrossRef]
- Kang, X.L.; Zou, H.; Pang, L.J.; Hu, W.H.; Zhao, J.; Qi, Y.; Liu, C.X.; Hu, J.M.; Tang, J.X.; Li, H.A.; et al. Chromosomal imbalances revealed in primary renal cell carcinomas by comparative genomic hybridization. Int. J. Clin. Exp. Pathol. 2015, 8, 3636–3647. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Sima, J.; Gilbert, D.M. Complex correlations: Replication timing and mutational landscapes during cancer and genome evolution. Curr. Opin. Genet. Dev. 2014, 25, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Guren, M.G. The global challenge of colorectal cancer. Lancet Gastroenterol. Hepatol. 2019, 4, 894–895. [Google Scholar] [CrossRef]
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef]
- Ongen, H.; Andersen, C.L.; Bramsen, J.B.; Oster, B.; Rasmussen, M.H.; Ferreira, P.G.; Sandoval, J.; Vidal, E.; Whiffin, N.; Planchon, A.; et al. Putative cis-regulatory drivers in colorectal cancer. Nature 2014, 512, 87–90. [Google Scholar] [CrossRef]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Campoli, M.; Ferrone, S. HLA antigen changes in malignant cells: Epigenetic mechanisms and biologic significance. Oncogene 2008, 27, 5869–5885. [Google Scholar] [CrossRef]
- Seliger, B.; Ferrone, S. HLA Class I Antigen Processing Machinery Defects in Cancer Cells-Frequency, Functional Significance, and Clinical Relevance with Special Emphasis on Their Role in T Cell-Based Immunotherapy of Malignant Disease. Methods Mol. Biol. 2020, 2055, 325–350. [Google Scholar] [CrossRef]
- Durgeau, A.; Virk, Y.; Gros, G.; Voilin, E.; Corgnac, S.; Djenidi, F.; Salmon, J.; Adam, J.; de Montpreville, V.; Validire, P.; et al. Human preprocalcitonin self-antigen generates TAP-dependent and -independent epitopes triggering optimised T-cell responses toward immune-escaped tumours. Nat. Commun. 2018, 9, 5097. [Google Scholar] [CrossRef]
- Anichini, A.; Perotti, V.E.; Sgambelluri, F.; Mortarini, R. Immune Escape Mechanisms in Non Small Cell Lung Cancer. Cancers 2020, 12, 3605. [Google Scholar] [CrossRef]
- Yu, S.; Zhao, Z.; Chen, L.; Gu, T.; Yu, H.; Tang, H.; Wang, Q.; Wu, Y. HLA loss of heterozygosity-mediated discordant responses to immune checkpoint blockade in squamous cell lung cancer with renal metastasis. Immunotherapy 2020. [Google Scholar] [CrossRef]
- Tran, E.; Robbins, P.F.; Lu, Y.C.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Pasetto, A.; Zheng, Z.; Ray, S.; Groh, E.M.; et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med. 2016, 375, 2255–2262. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Rosenthal, R.; Hiley, C.T.; Rowan, A.J.; Watkins, T.B.K.; Wilson, G.A.; Birkbak, N.J.; Veeriah, S.; Van Loo, P.; Herrero, J.; et al. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell 2017, 171, 1259–1271.e11. [Google Scholar] [CrossRef]
- Negrao, M.V.; Quek, K.; Zhang, J.; Sepesi, B. TRACERx: Tracking tumor evolution to impact the course of lung cancer. J. Thorac. Cardiovasc. Surg. 2018, 155, 1199–1202. [Google Scholar] [CrossRef]
- Jamal-Hanjani, M.; Hackshaw, A.; Ngai, Y.; Shaw, J.; Dive, C.; Quezada, S.; Middleton, G.; de Bruin, E.; Le Quesne, J.; Shafi, S.; et al. Tracking genomic cancer evolution for precision medicine: The lung TRACERx study. PLoS Biol. 2014, 12, e1001906. [Google Scholar] [CrossRef] [PubMed]
- Brastianos, P.K.; Carter, S.L.; Santagata, S.; Cahill, D.P.; Taylor-Weiner, A.; Jones, R.T.; Van Allen, E.M.; Lawrence, M.S.; Horowitz, P.M.; Cibulskis, K.; et al. Genomic Characterization of Brain Metastases Reveals Branched Evolution and Potential Therapeutic Targets. Cancer Discov. 2015, 5, 1164–1177. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Fearon, E.R.; Kern, S.E.; Hamilton, S.R.; Preisinger, A.C.; Nakamura, Y.; White, R. Allelotype of colorectal carcinomas. Science 1989, 244, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Watkins, T.B.K.; Lim, E.L.; Petkovic, M.; Elizalde, S.; Birkbak, N.; Wilson, G.A.; Moore, D.A.; Grönroos, E.; Rowan, A.; Dewhurst, S.M.; et al. Pervasive chromosomal instability and karyotype order in tumour evolution. Nat. Cell Biol. 2020, 587, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Linjama, T.; Impola, U.; Niittyvuopio, R.; Kuittinen, O.; Kaare, A.; Rimpilainen, J.; Volin, L.; Perasaari, J.; Jaatinen, T.; Lauronen, J.; et al. Conflicting HLA assignment by three different typing methods due to the ap-parent loss of heterozygosity in the MHC region. HLA 2016, 87, 350–355. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Chromosome Associated with LOH | Gene Name | LOH Position | Predictive Biomarker | Reference |
|---|---|---|---|---|
| 1p | N/A | 1p36 | Significantly associated with prognostic markers of aggressive neuroblastoma when patients are diagnosed | [61,62,63,64] |
| 3p | FRA3B | 3p13–3p21 | Predicted early event in the genesis of smoking-related cancers. | [65,66] |
| CACNA2D3 | 3p21 | Associated with poor clinical outcome in esophageal cancer. | [67,68] | |
| VHL | 3p25 | Predictive biomarker for clinical outcome in clear-cell renal-cell carcinoma (ccRCC) patients. | [69] | |
| 6q | HLA | 6p21 | Leads to a poor response to immune checkpoint inhibitors. | [70] |
| Predictive biomarker for patients with synovial sarcoma and is prognostic of poor clinical outcome. | [71] | |||
| FABP7 | 6q22 | Correlated with survival in patients with glioblastoma. | [72] | |
| A potential diagnostic biomarker of early-stage melanoma systemic spreading in blood. | [73] | |||
| 9q | N/A | 9p13 | Significantly associated with poorer prognosis of glioma patients. | [74] |
| 10q | PTEN | 10q23 | Functionally related to the development of breast cancer, associated with poor prognosis | [75] |
| 11q | NELL1 | 11p15 | An association between LOH at 11p15.5 and poor survival in 180 lung cancer patients. | [76] |
| 16q | CD19 | 16p11 | Irreversible loss of heterozygosity in CD19 could be used as a biomarker for an outcome prediction after the CAR T cells therapy CTL019. | [77] |
| 17q | TP53 | 17p13 | One of the most frequent genetic alterations leading to human cancers. | [78,79] |
| BRCA1 | 17q21 | A germline LOH on BRCA1 confront an 85% lifetime risk of breast cancer and a greatly elevated risk of ovarian cancer | [80] | |
| 18q | DPC4 (SMAD4) | 18q21 | Assist in predicting the clinical outcome after therapies in colorectal cancer (CRC) patients. | [81,82] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Sjöblom, T. Targeting Loss of Heterozygosity: A Novel Paradigm for Cancer Therapy. Pharmaceuticals 2021, 14, 57. https://doi.org/10.3390/ph14010057
Zhang X, Sjöblom T. Targeting Loss of Heterozygosity: A Novel Paradigm for Cancer Therapy. Pharmaceuticals. 2021; 14(1):57. https://doi.org/10.3390/ph14010057
Chicago/Turabian StyleZhang, Xiaonan, and Tobias Sjöblom. 2021. "Targeting Loss of Heterozygosity: A Novel Paradigm for Cancer Therapy" Pharmaceuticals 14, no. 1: 57. https://doi.org/10.3390/ph14010057
APA StyleZhang, X., & Sjöblom, T. (2021). Targeting Loss of Heterozygosity: A Novel Paradigm for Cancer Therapy. Pharmaceuticals, 14(1), 57. https://doi.org/10.3390/ph14010057