NLRP1 Functions Downstream of the MAPK/ERK Signaling via ATF4 and Contributes to Acquired Targeted Therapy Resistance in Human Metastatic Melanoma


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
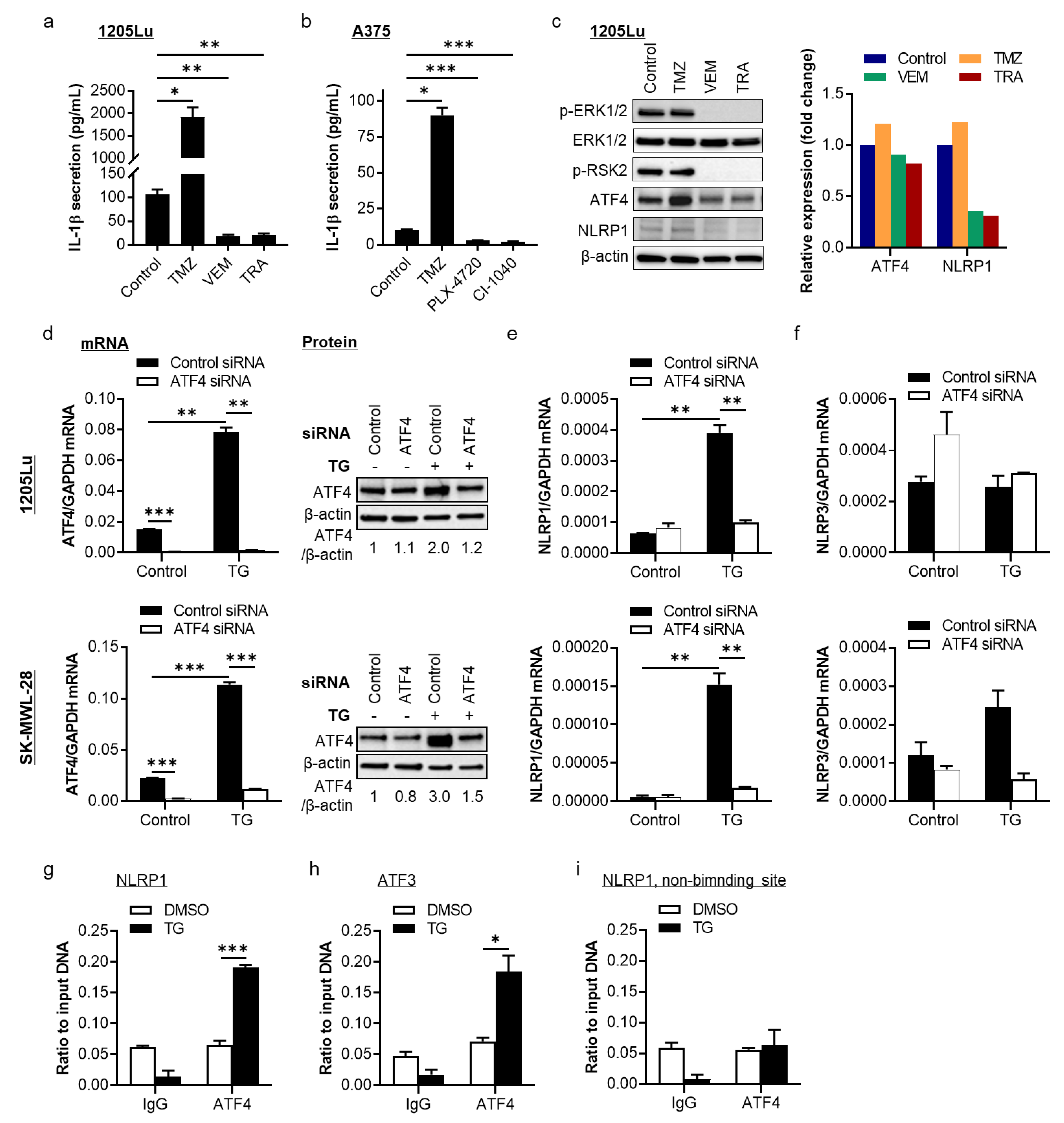
2.1. The MAPK/ERK Pathway Regulates NLRP1 Expression and IL-1β Secretion through Activating Transcription Factor 4 (ATF4) in BRAFV600E-Mutant Human Metastatic Melanoma Cells
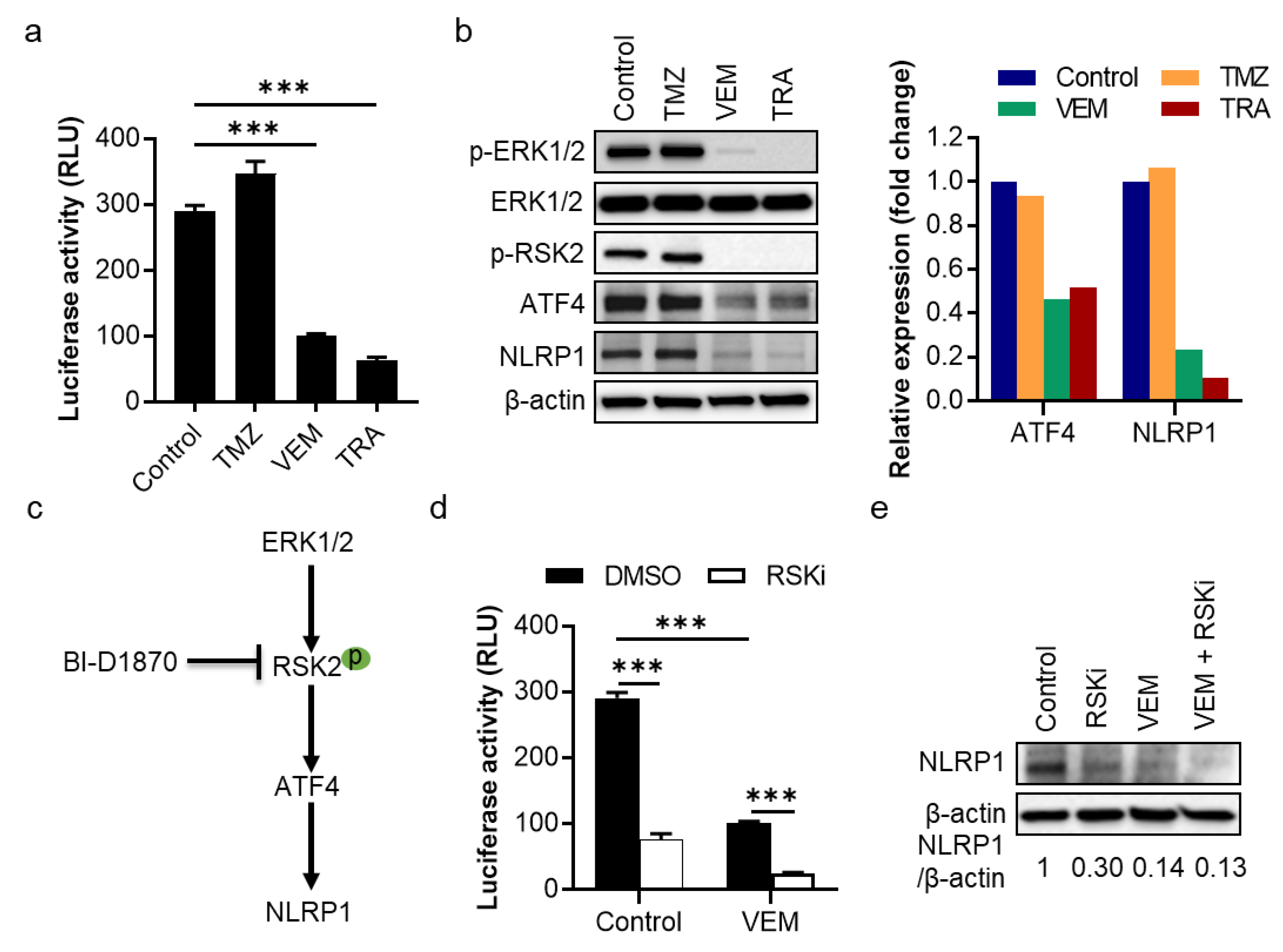
2.2. The MAPK/ERK Pathway Shows an RSK2-Dependent Regulation of NLRP1 Gene Promoter Activity and Protein Expression in Metastatic Melanoma Cells
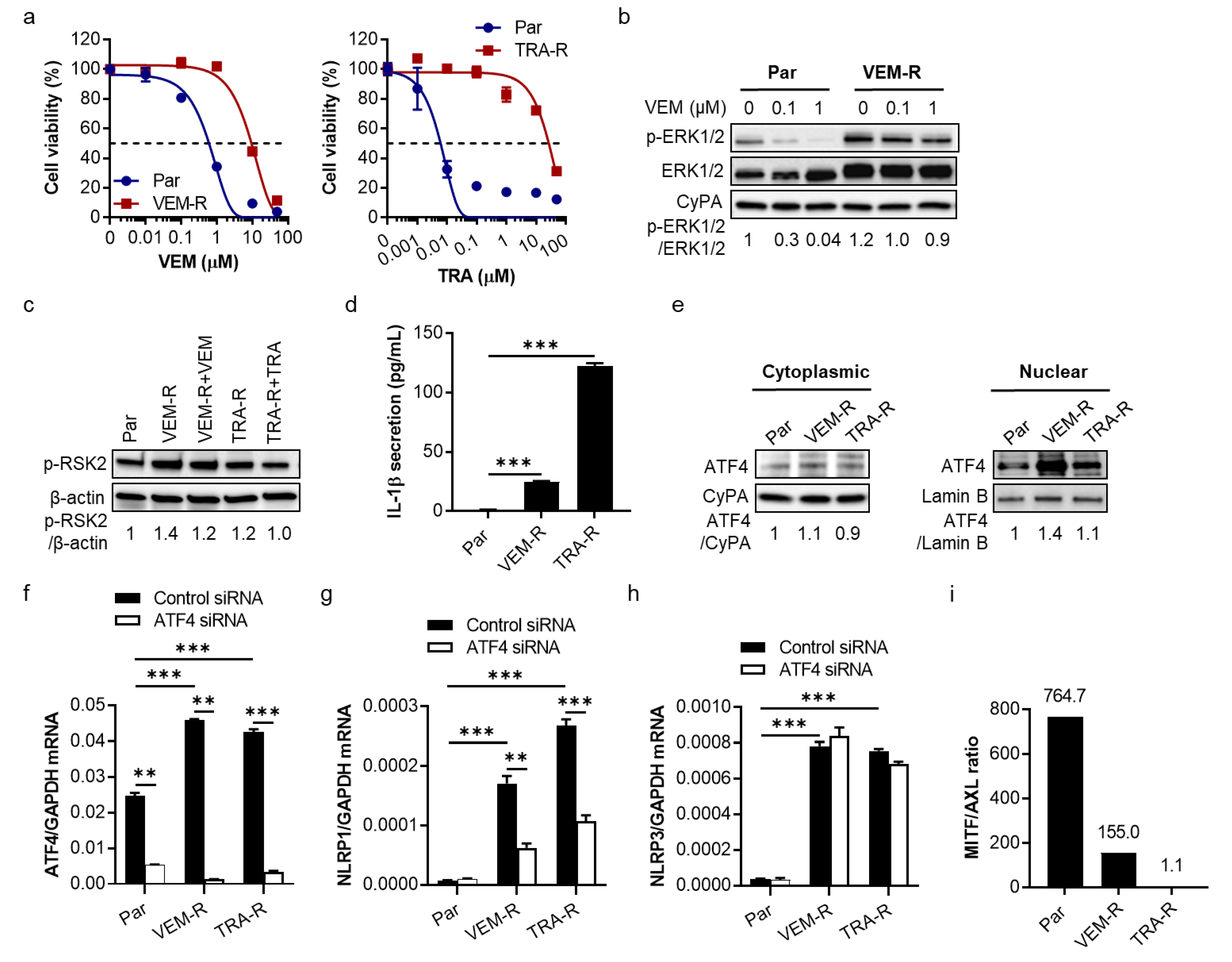
2.3. VEM- and TRA-Resistant Melanoma Cells Show Increased IL-1β Secretion, Upregulation of ATF4 and NLRP1 Gene Expression, and Downregulation of MITF/AXL Ratio
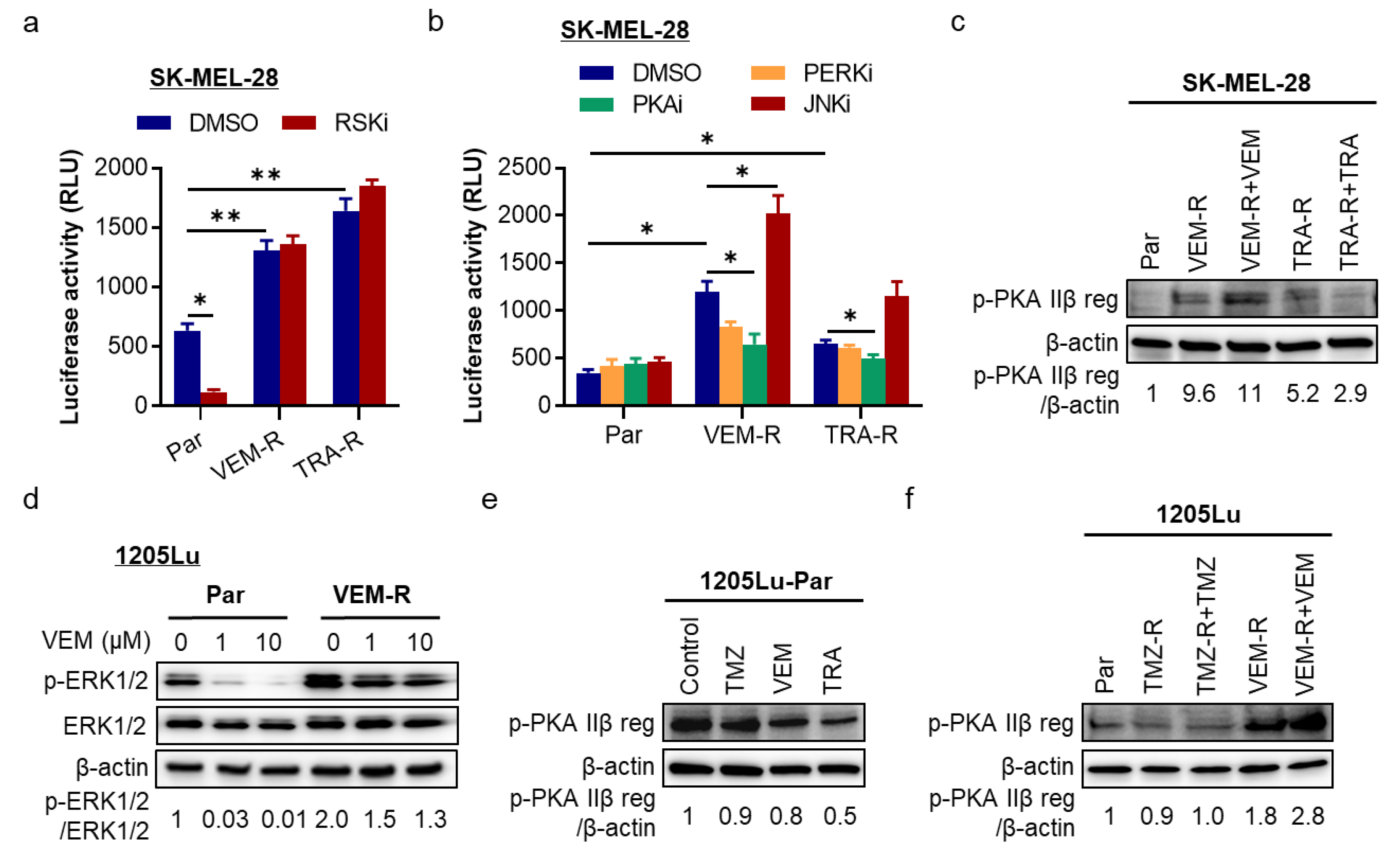
2.4. The cAMP/PKA Pathway Is a Regulator of ATF4/NLRP1 in Resistant Melanoma Cells
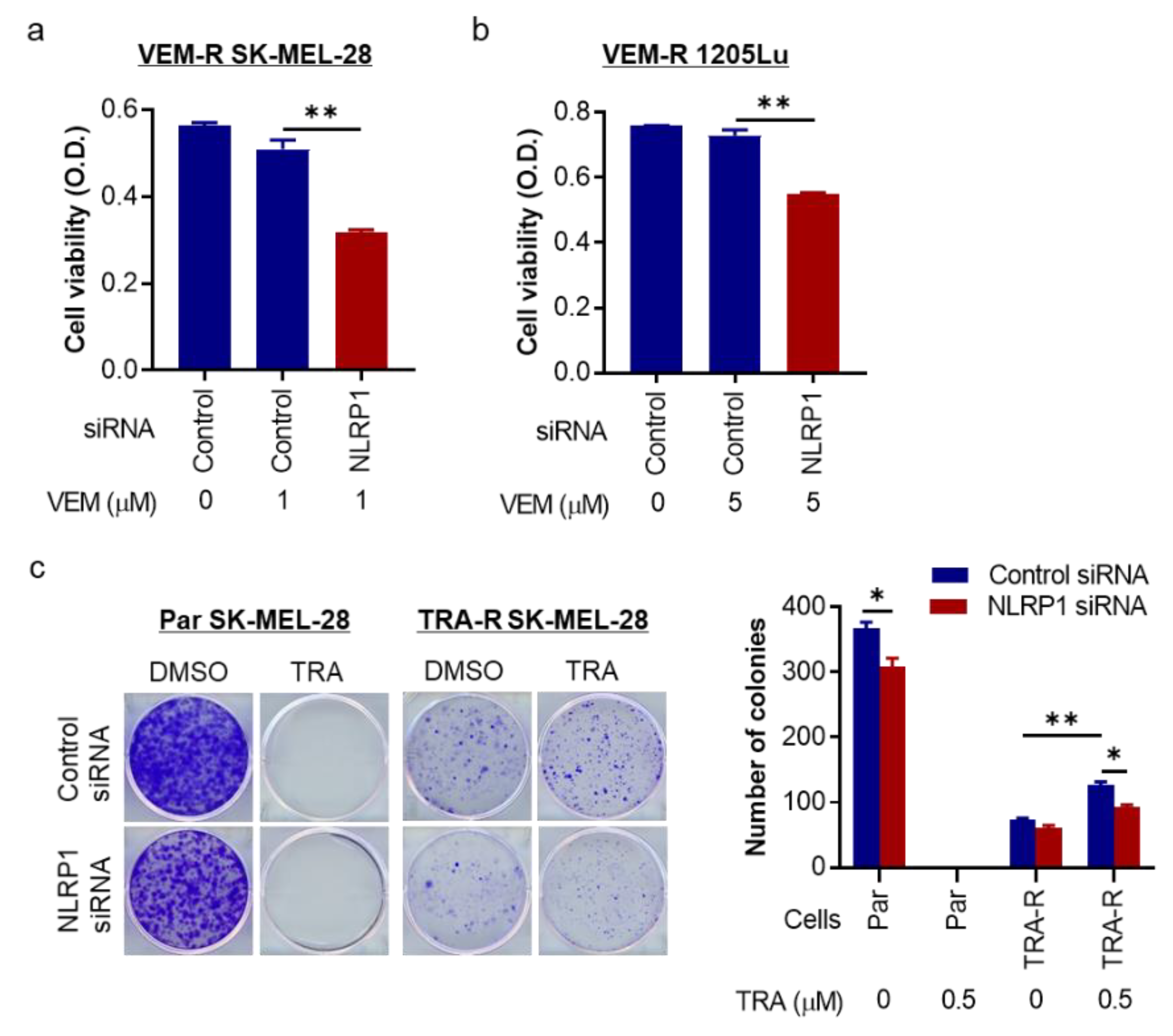
2.5. NLRP1 Is Required for Cell Growth and Colony Formation of Targeted Therapy-Resistant Melanoma Cells
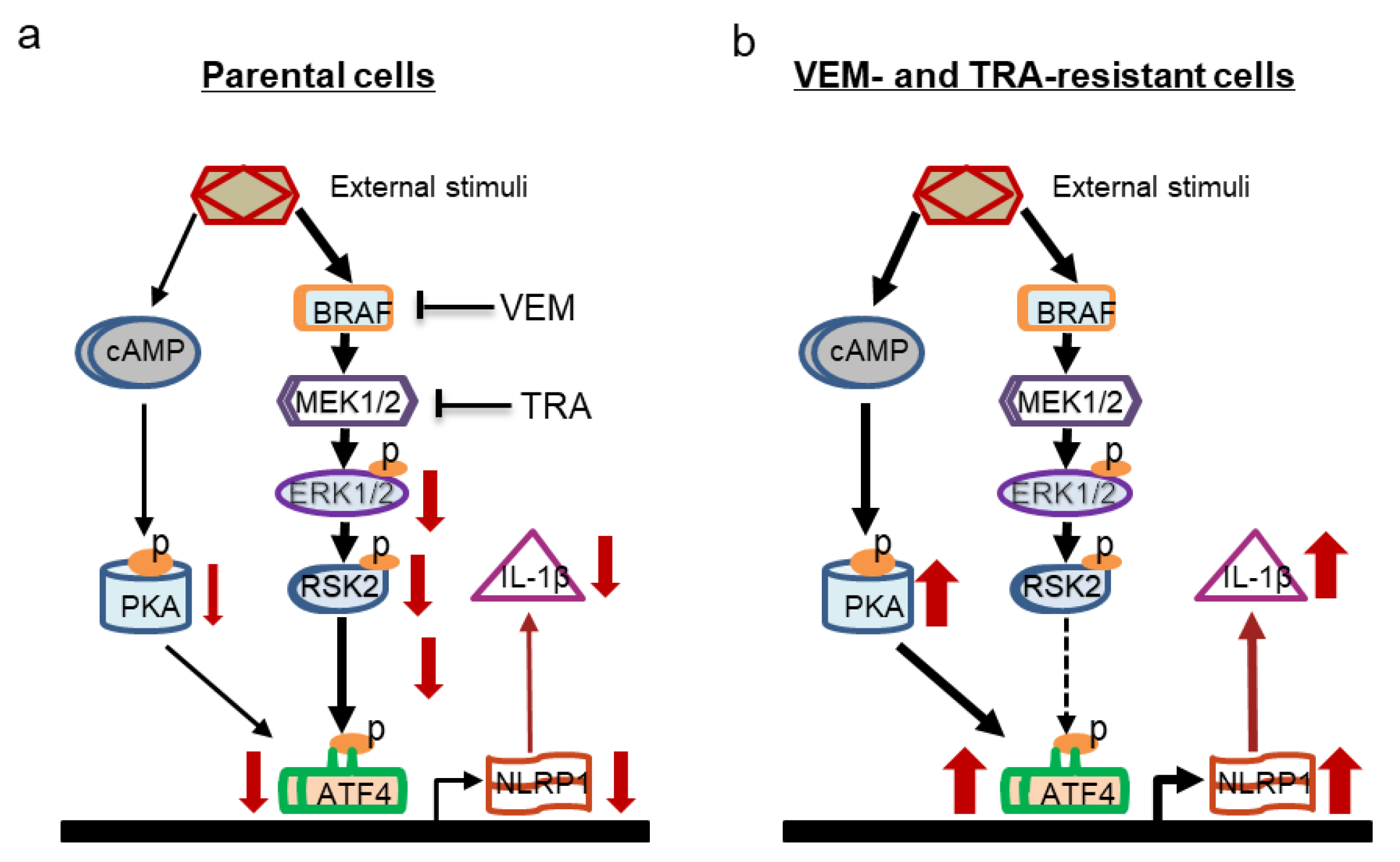
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture
4.3. Cell Growth Inhibition
4.4. Generation of Acquired Resistance
4.5. Enzyme-Linked Immunosorbent Assay (ELISA)
4.6. siRNA Transfection
4.7. Quantitative RT-PCR
4.8. Western Blot
4.9. Chromatin Immunoprecipitation (ChIP) Assay
4.10. Luciferase Reporter Assay
4.11. Clonogenic Assay
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wan, P.T.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; Barford, D.; et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef]
- Kozar, I.; Margue, C.; Rothengatter, S.; Haan, C.; Kreis, S. Many ways to resistance: How melanoma cells evade targeted therapies. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 313–322. [Google Scholar] [CrossRef]
- Roesch, A. Tumor heterogeneity and plasticity as elusive drivers for resistance to MAPK pathway inhibition in melanoma. Oncogene 2015, 34, 2951–2957. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L. Overcoming therapeutic resistance by targeting cancer inflammation. Am. Soc. Clin. Oncol. Educ. Book 2016, 35, e168–e173. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, M.; Liu, W.; Luo, Y.; Tanaka, A.; Cai, X.; Norris, D.A.; Dinarello, C.A.; Fujita, M. Constitutively active inflammasome in human melanoma cells mediating autoinflammation via caspase-1 processing and secretion of interleukin-1beta. J. Biol. Chem. 2010, 285, 6477–6488. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Ekmekcioglu, S.; Liu, P.; Duncan, L.M.; Lizee, G.; Poindexter, N.; Grimm, E.A. Constitutive aberrant endogenous interleukin-1 facilitates inflammation and growth in human melanoma. Mol. Cancer Res. 2011, 9, 1537–1550. [Google Scholar] [CrossRef]
- Khalili, J.S.; Liu, S.; Rodriguez-Cruz, T.G.; Whittington, M.; Wardell, S.; Liu, C.; Zhang, M.; Cooper, Z.A.; Frederick, D.T.; Li, Y.; et al. Oncogenic BRAF(V600E) promotes stromal cell-mediated immunosuppression via induction of interleukin-1 in melanoma. Clin. Cancer Res. 2012, 18, 5329–5340. [Google Scholar] [CrossRef]
- Whipple, C.A.; Brinckerhoff, C.E. BRAF(V600E) melanoma cells secrete factors that activate stromal fibroblasts and enhance tumourigenicity. Br. J. Cancer 2014, 111, 1625–1633. [Google Scholar] [CrossRef]
- Young, H.L.; Rowling, E.J.; Bugatti, M.; Giurisato, E.; Luheshi, N.; Arozarena, I.; Acosta, J.C.; Kamarashev, J.; Frederick, D.T.; Cooper, Z.A.; et al. An adaptive signaling network in melanoma inflammatory niches confers tolerance to MAPK signaling inhibition. J. Exp. Med. 2017, 214, 1691–1710. [Google Scholar] [CrossRef]
- Zhai, Z.; Liu, W.; Kaur, M.; Luo, Y.; Domenico, J.; Samson, J.M.; Shellman, Y.G.; Norris, D.A.; Dinarello, C.A.; Spritz, R.A.; et al. NLRP1 promotes tumor growth by enhancing inflammasome activation and suppressing apoptosis in metastatic melanoma. Oncogene 2017, 36, 3820–3830. [Google Scholar] [CrossRef]
- Zhong, F.L.; Mamai, O.; Sborgi, L.; Boussofara, L.; Hopkins, R.; Robinson, K.; Szeverenyi, I.; Takeichi, T.; Balaji, R.; Lau, A.; et al. Germline NLRP1 mutations cause skin inflammatory and cancer susceptibility syndromes via inflammasome activation. Cell 2016, 167, 187–202 e17. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Z.; Samson, J.M.; Yamauchi, T.; Vaddi, P.K.; Matsumoto, Y.; Dinarello, C.A.; Ravindran Menon, D.; Fujita, M. Inflammasome sensor NLRP1 confers acquired drug resistance to temozolomide in human melanoma. Cancers 2020, 12, 2518. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, I.; Koido, M.; Tani, Y.; Tsukahara, S.; Kunimasa, K.; Tomida, A. Disrupting ATF4 expression mechanisms provides an effective strategy for BRAF-targeted melanoma therapy. iScience 2020, 23, 101028. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, P.; Richmond, A. A novel NF-kappa B-inducing kinase-MAPK signaling pathway up-regulates NF-kappa B activity in melanoma cells. J. Biol. Chem. 2002, 277, 7920–7928. [Google Scholar] [CrossRef]
- Liu, J.; Suresh Kumar, K.G.; Yu, D.; Molton, S.A.; McMahon, M.; Herlyn, M.; Thomas-Tikhonenko, A.; Fuchs, S.Y. Oncogenic BRAF regulates beta-Trcp expression and NF-kappaB activity in human melanoma cells. Oncogene 2007, 26, 1954–1958. [Google Scholar] [CrossRef]
- Yang, X.; Matsuda, K.; Bialek, P.; Jacquot, S.; Masuoka, H.C.; Schinke, T.; Li, L.; Brancorsini, S.; Sassone-Corsi, P.; Townes, T.M.; et al. ATF4 is a substrate of RSK2 and an essential regulator of osteoblast biology; implication for Coffin-Lowry Syndrome. Cell 2004, 117, 387–398. [Google Scholar] [CrossRef]
- Han, J.; Kaufman, R.J. Physiological/pathological ramifications of transcription factors in the unfolded protein response. Genes Dev. 2017, 31, 1417–1438. [Google Scholar] [CrossRef]
- D’Osualdo, A.; Anania, V.G.; Yu, K.; Lill, J.R.; Kaufman, R.J.; Matsuzawa, S.; Reed, J.C. Transcription factor ATF4 induces NLRP1 inflammasome expression during endoplasmic reticulum stress. PLoS ONE 2015, 10, e0130635. [Google Scholar] [CrossRef]
- Ciazynska, M.; Bednarski, I.A.; Wodz, K.; Narbutt, J.; Lesiak, A. NLRP1 and NLRP3 inflammasomes as a new approach to skin carcinogenesis. Oncol. Lett. 2020, 19, 1649–1656. [Google Scholar]
- Muller, J.; Krijgsman, O.; Tsoi, J.; Robert, L.; Hugo, W.; Song, C.; Kong, X.; Possik, P.A.; Cornelissen-Steijger, P.D.; Geukes Foppen, M.H.; et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat. Commun. 2014, 5, 5712. [Google Scholar] [CrossRef]
- Falletta, P.; Sanchez-Del-Campo, L.; Chauhan, J.; Effern, M.; Kenyon, A.; Kershaw, C.J.; Siddaway, R.; Lisle, R.; Freter, R.; Daniels, M.J.; et al. Translation reprogramming is an evolutionarily conserved driver of phenotypic plasticity and therapeutic resistance in melanoma. Genes Dev. 2017, 31, 18–33. [Google Scholar] [CrossRef] [PubMed]
- Singleton, D.C.; Harris, A.L. Targeting the ATF4 pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 1189–1202. [Google Scholar] [CrossRef] [PubMed]
- Matsuguchi, T.; Chiba, N.; Bandow, K.; Kakimoto, K.; Masuda, A.; Ohnishi, T. JNK activity is essential for Atf4 expression and late-stage osteoblast differentiation. J. Bone Miner Res. 2009, 24, 398–410. [Google Scholar] [CrossRef] [PubMed]
- Godwin, P.; Baird, A.M.; Heavey, S.; Barr, M.P.; O’Byrne, K.J.; Gately, K. Targeting nuclear factor-kappa B to overcome resistance to chemotherapy. Front. Oncol. 2013, 3, 120. [Google Scholar] [CrossRef] [PubMed]
- Caporali, S.; Levati, L.; Graziani, G.; Muzi, A.; Atzori, M.G.; Bonmassar, E.; Palmieri, G.; Ascierto, P.A.; D’Atri, S. NF-kappaB is activated in response to temozolomide in an AKT-dependent manner and confers protection against the growth suppressive effect of the drug. J. Transl. Med. 2012, 10, 252. [Google Scholar] [CrossRef] [PubMed]
- Zeligs, K.P.; Neuman, M.K.; Annunziata, C.M. Molecular pathways: The balance between cancer and the immune system challenges the therapeutic specificity of targeting nuclear factor-kappaB signaling for cancer treatment. Clin. Cancer Res. 2016, 22, 4302–4308. [Google Scholar] [CrossRef]
- Peng, C.; Cho, Y.Y.; Zhu, F.; Xu, Y.M.; Wen, W.; Ma, W.Y.; Bode, A.M.; Dong, Z. RSK2 mediates NF-{kappa}B activity through the phosphorylation of IkappaBalpha in the TNF-R1 pathway. FASEB J. 2010, 24, 3490–3499. [Google Scholar] [CrossRef]
- Janostiak, R.; Rauniyar, N.; Lam, T.T.; Ou, J.; Zhu, L.J.; Green, M.R.; Wajapeyee, N. MELK promotes melanoma growth by stimulating the NF-kappaB pathway. Cell Rep. 2017, 21, 2829–2841. [Google Scholar] [CrossRef]
- Xu, C.; Bailly-Maitre, B.; Reed, J.C. Endoplasmic reticulum stress: Cell life and death decisions. J. Clin. Investig. 2005, 115, 2656–2664. [Google Scholar] [CrossRef]
- Cubillos-Ruiz, J.R.; Bettigole, S.E.; Glimcher, L.H. Tumorigenic and immunosuppressive effects of endoplasmic reticulum stress in cancer. Cell 2017, 168, 692–706. [Google Scholar] [CrossRef]
- Corazzari, M.; Gagliardi, M.; Fimia, G.M.; Piacentini, M. Endoplasmic reticulum stress, unfolded protein response, and cancer cell fate. Front. Oncol. 2017, 7, 78. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.J.; Lee, C.C.; Shih, Y.L.; Lin, C.H.; Wang, S.H.; Chen, T.H.; Shih, C.M. Inhibition of mitochondria- and endoplasmic reticulum stress-mediated autophagy augments temozolomide-induced apoptosis in glioma cells. PLoS ONE 2012, 7, e38706. [Google Scholar] [CrossRef] [PubMed]
- Dey, S.; Sayers, C.M.; Verginadis, I.I.; Lehman, S.L.; Cheng, Y.; Cerniglia, G.J.; Tuttle, S.W.; Feldman, M.D.; Zhang, P.J.; Fuchs, S.Y.; et al. ATF4-dependent induction of heme oxygenase 1 prevents anoikis and promotes metastasis. J. Clin. Investig. 2015, 125, 2592–2608. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Tian, C.; Hoffman, T.E.; Jacobsen, N.K.; Spencer, S.L. Rapidly induced drug adaptation mediates escape from BRAF inhibition in single melanoma cells. BioRxiv 2020. [Google Scholar] [CrossRef]
- Ma, X.H.; Piao, S.F.; Dey, S.; McAfee, Q.; Karakousis, G.; Villanueva, J.; Hart, L.S.; Levi, S.; Hu, J.; Zhang, G.; et al. Targeting ER stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma. J. Clin. Investig. 2014, 124, 1406–1417. [Google Scholar] [CrossRef]
- Raskin, L.; Fullen, D.R.; Giordano, T.J.; Thomas, D.G.; Frohm, M.L.; Cha, K.B.; Ahn, J.; Mukherjee, B.; Johnson, T.M.; Gruber, S.B. Transcriptome profiling identifies HMGA2 as a biomarker of melanoma progression and prognosis. J. Investig. Dermatol. 2013, 133, 2585–2592. [Google Scholar] [CrossRef]
- Kabbarah, O.; Nogueira, C.; Feng, B.; Nazarian, R.M.; Bosenberg, M.; Wu, M.; Scott, K.L.; Kwong, L.N.; Xiao, Y.; Cordon-Cardo, C.; et al. Integrative genome comparison of primary and metastatic melanomas. PLoS ONE 2010, 5, e10770. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, H.; Wei, S.; Wang, Z.; Ji, G. Inhibition of ER stress-related IRE1alpha/CREB/NLRP1 pathway promotes the apoptosis of human chronic myelogenous leukemia cell. Mol. Immunol. 2018, 101, 377–385. [Google Scholar] [CrossRef]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef]
- Verma, D.; Bivik, C.; Farahani, E.; Synnerstad, I.; Fredrikson, M.; Enerback, C.; Rosdahl, I.; Soderkvist, P. Inflammasome polymorphisms confer susceptibility to sporadic malignant melanoma. Pigment Cell Melanoma Res. 2012, 25, 506–513. [Google Scholar] [CrossRef]
- Levandowski, C.B.; Mailloux, C.M.; Ferrara, T.M.; Gowan, K.; Ben, S.; Jin, Y.; McFann, K.K.; Holland, P.J.; Fain, P.R.; Dinarello, C.A.; et al. NLRP1 haplotypes associated with vitiligo and autoimmunity increase interleukin-1beta processing via the NLRP1 inflammasome. Proc. Natl. Acad. Sci. USA 2013, 110, 2952–2956. [Google Scholar] [CrossRef] [PubMed]
- Stork, P.J.; Schmitt, J.M. Crosstalk between cAMP and MAP kinase signaling in the regulation of cell proliferation. Trends Cell Biol. 2002, 12, 258–266. [Google Scholar] [CrossRef]
- Dumaz, N.; Hayward, R.; Martin, J.; Ogilvie, L.; Hedley, D.; Curtin, J.A.; Bastian, B.C.; Springer, C.; Marais, R. In melanoma, RAS mutations are accompanied by switching signaling from BRAF to CRAF and disrupted cyclic AMP signaling. Cancer Res. 2006, 66, 9483–9491. [Google Scholar] [CrossRef] [PubMed]
- Sanz, C.; Calasanz, M.J.; Andreu, E.; Richard, C.; Prosper, F.; Fernandez-Luna, J.L. NALP1 is a transcriptional target for cAMP-response-element-binding protein (CREB) in myeloid leukaemia cells. Biochem. J. 2004, 384, 281–286. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sand, J.; Fenini, G.; Grossi, S.; Hennig, P.; Di Filippo, M.; Levesque, M.; Werner, S.; French, L.E.; Beer, H.D. The NLRP1 inflammasome pathway is silenced in cutaneous squamous cell carcinoma. J. Investig. Dermatol. 2019, 139, 1788–1797 e6. [Google Scholar] [CrossRef]
- Marchetti, C. The NLRP3 inflammasome as a pharmacological target. J. Cardiovasc. Pharmacol. 2019, 74, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Li, X.; Liu, Y.; Xia, Y.; Chang, R.; Zhang, C. Inflammasome inhibitors: Promising therapeutic approaches against cancer. J. Hematol. Oncol. 2019, 12, 64. [Google Scholar] [CrossRef]
- Bleda, S.; De Haro, J.; Varela, C.; Ferruelo, A.; Acin, F. Aspirin therapy inhibits NLRP1 (nucleotide-binding domain-like receptor protein 1) inflammasome gene expression in patients with peripheral artery disease. J. Vasc. Surg. 2015, 61, 1103–1104. [Google Scholar] [CrossRef][Green Version]
- Ellis, L.Z.; Liu, W.; Luo, Y.; Okamoto, M.; Qu, D.; Dunn, J.H.; Fujita, M. Green tea polyphenol epigallocatechin-3-gallate suppresses melanoma growth by inhibiting inflammasome and IL-1beta secretion. Biochem. Biophys. Res. Commun. 2011, 414, 551–556. [Google Scholar] [CrossRef]
- Lau, E.; Sedy, J.; Sander, C.; Shaw, M.A.; Feng, Y.; Scortegagna, M.; Claps, G.; Robinson, S.; Cheng, P.; Srivas, R.; et al. Transcriptional repression of IFNbeta1 by ATF2 confers melanoma resistance to therapy. Oncogene 2015, 34, 5739–5748. [Google Scholar] [CrossRef]
- Ouwens, D.M.; de Ruiter, N.D.; van der Zon, G.C.; Carter, A.P.; Schouten, J.; van der Burgt, C.; Kooistra, K.; Bos, J.L.; Maassen, J.A.; van Dam, H. Growth factors can activate ATF2 via a two-step mechanism: Phosphorylation of Thr71 through the Ras-MEK-ERK pathway and of Thr69 through RalGDS-Src-p38. EMBO J. 2002, 21, 3782–3793. [Google Scholar] [CrossRef] [PubMed]
- McDermott, M.; Eustace, A.J.; Busschots, S.; Breen, L.; Crown, J.; Clynes, M.; O’Donovan, N.; Stordal, B. In vitro development of chemotherapy and targeted therapy drug-resistant cancer cell lines: A practical guide with case studies. Front. Oncol. 2014, 4, 40. [Google Scholar] [CrossRef] [PubMed]
- Rafehi, H.; Orlowski, C.; Georgiadis, G.T.; Ververis, K.; El-Osta, A.; Karagiannis, T.C. Clonogenic assay: Adherent cells. J. Vis. Exp. 2011, 49, e2573. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhai, Z.; Vaddi, P.K.; Samson, J.M.; Takegami, T.; Fujita, M. NLRP1 Functions Downstream of the MAPK/ERK Signaling via ATF4 and Contributes to Acquired Targeted Therapy Resistance in Human Metastatic Melanoma. Pharmaceuticals 2021, 14, 23. https://doi.org/10.3390/ph14010023
Zhai Z, Vaddi PK, Samson JM, Takegami T, Fujita M. NLRP1 Functions Downstream of the MAPK/ERK Signaling via ATF4 and Contributes to Acquired Targeted Therapy Resistance in Human Metastatic Melanoma. Pharmaceuticals. 2021; 14(1):23. https://doi.org/10.3390/ph14010023
Chicago/Turabian StyleZhai, Zili, Prasanna K. Vaddi, Jenny Mae Samson, Tomoya Takegami, and Mayumi Fujita. 2021. "NLRP1 Functions Downstream of the MAPK/ERK Signaling via ATF4 and Contributes to Acquired Targeted Therapy Resistance in Human Metastatic Melanoma" Pharmaceuticals 14, no. 1: 23. https://doi.org/10.3390/ph14010023
APA StyleZhai, Z., Vaddi, P. K., Samson, J. M., Takegami, T., & Fujita, M. (2021). NLRP1 Functions Downstream of the MAPK/ERK Signaling via ATF4 and Contributes to Acquired Targeted Therapy Resistance in Human Metastatic Melanoma. Pharmaceuticals, 14(1), 23. https://doi.org/10.3390/ph14010023