Impact of Sampling Period on Population Pharmacokinetic Analysis of Antibiotics: Why do You Take Blood Samples Following the Fourth Dose?


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
- Recording all doses and times accurately until steady state is achieved can be cumbersome. If there is an error in the record, model development is adversely affected.
- It is difficult to estimate PK parameters accurately and find a significant covariate that affects PK when a patient’s pathophysiological state continues to change until the drug’s steady state is reached.
- The enrolled patient is likely to drop out before steady state is achieved.
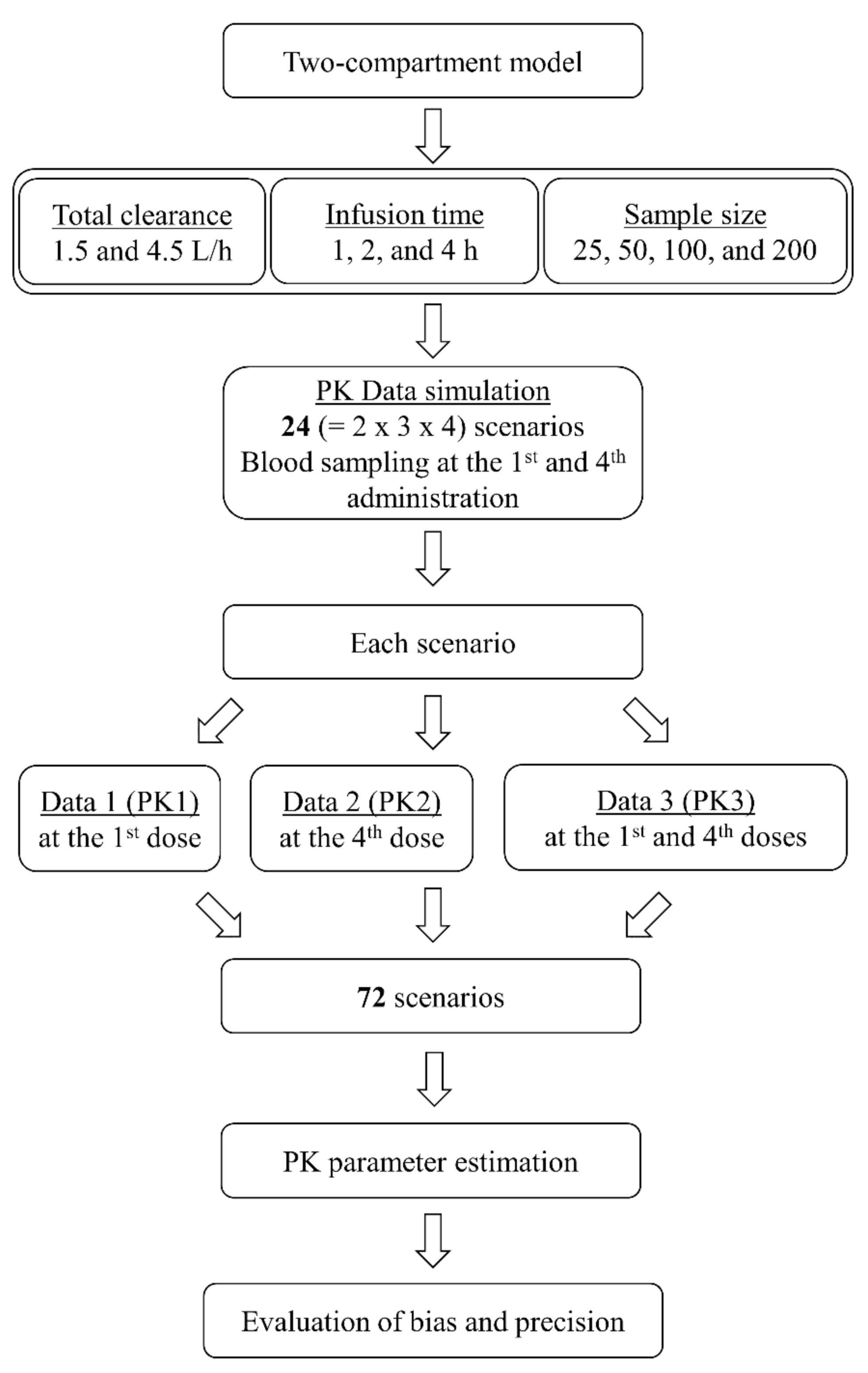
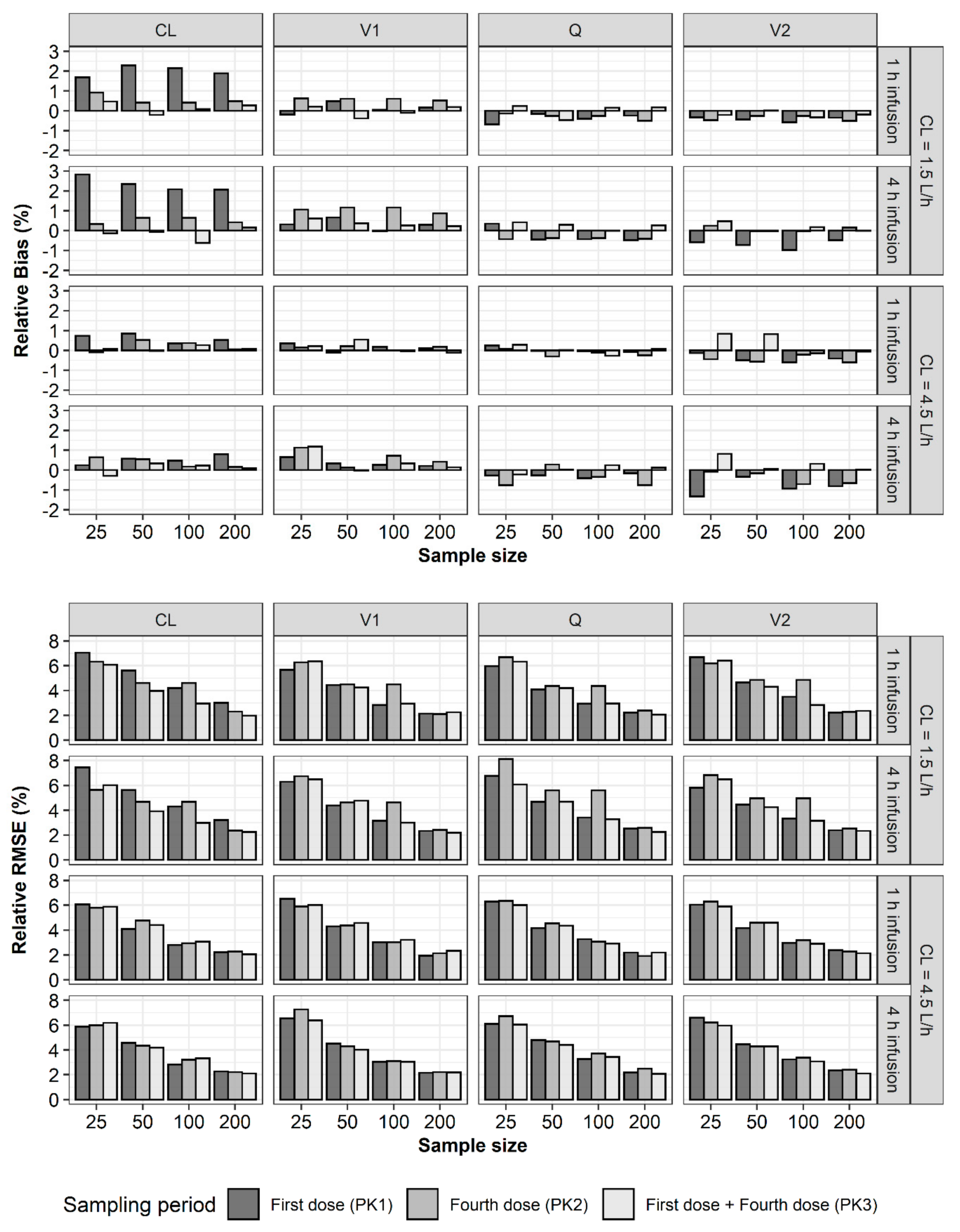
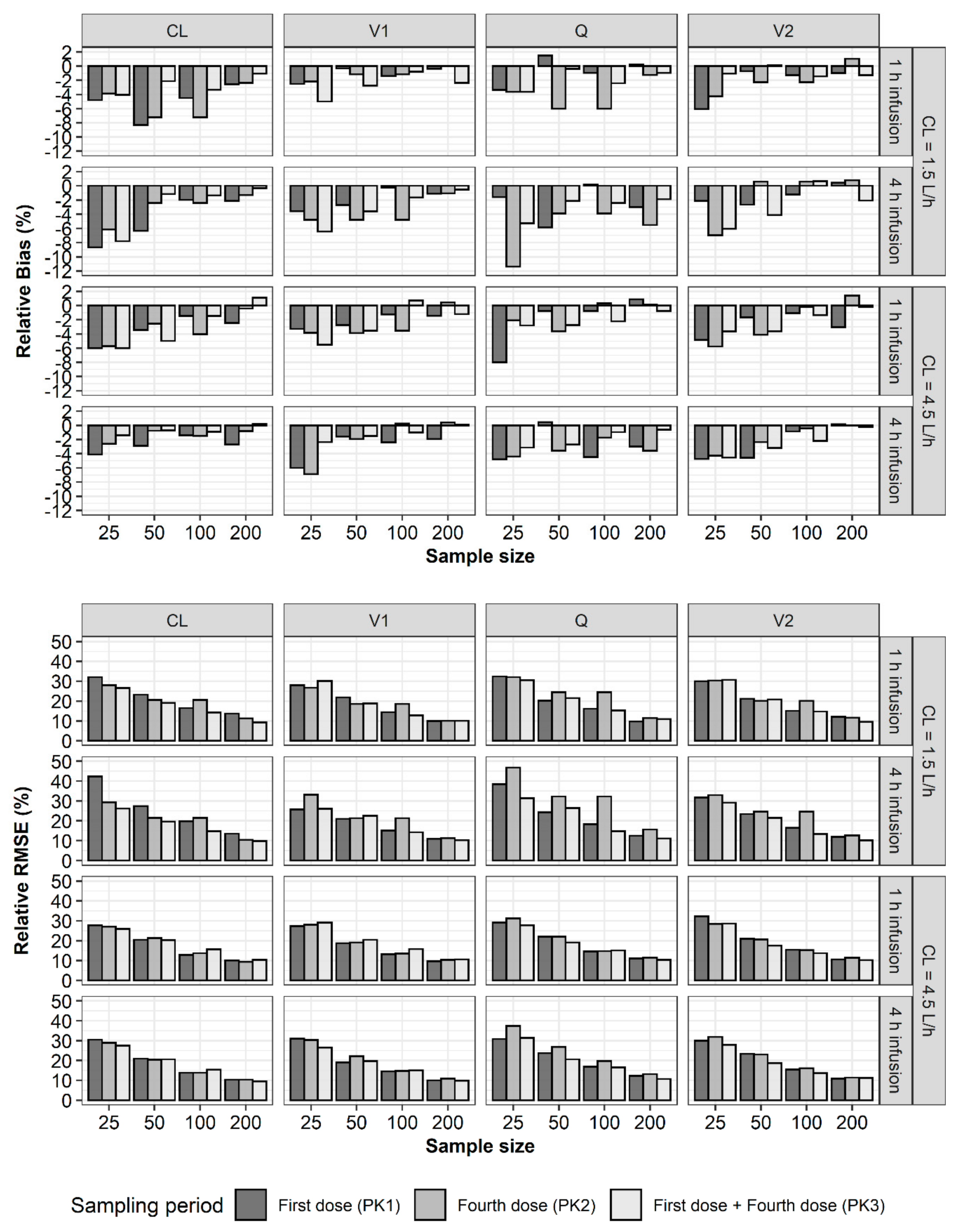
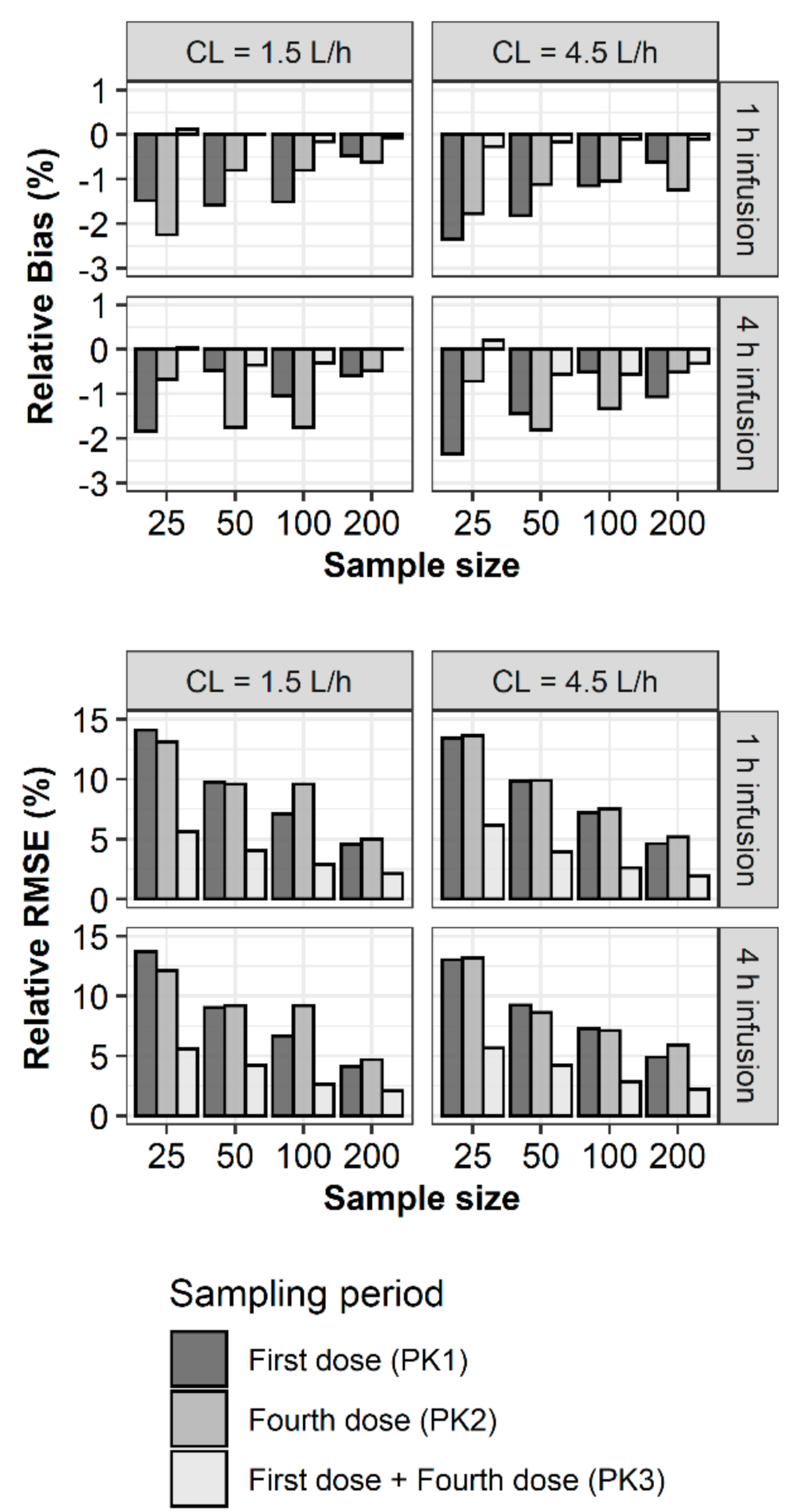
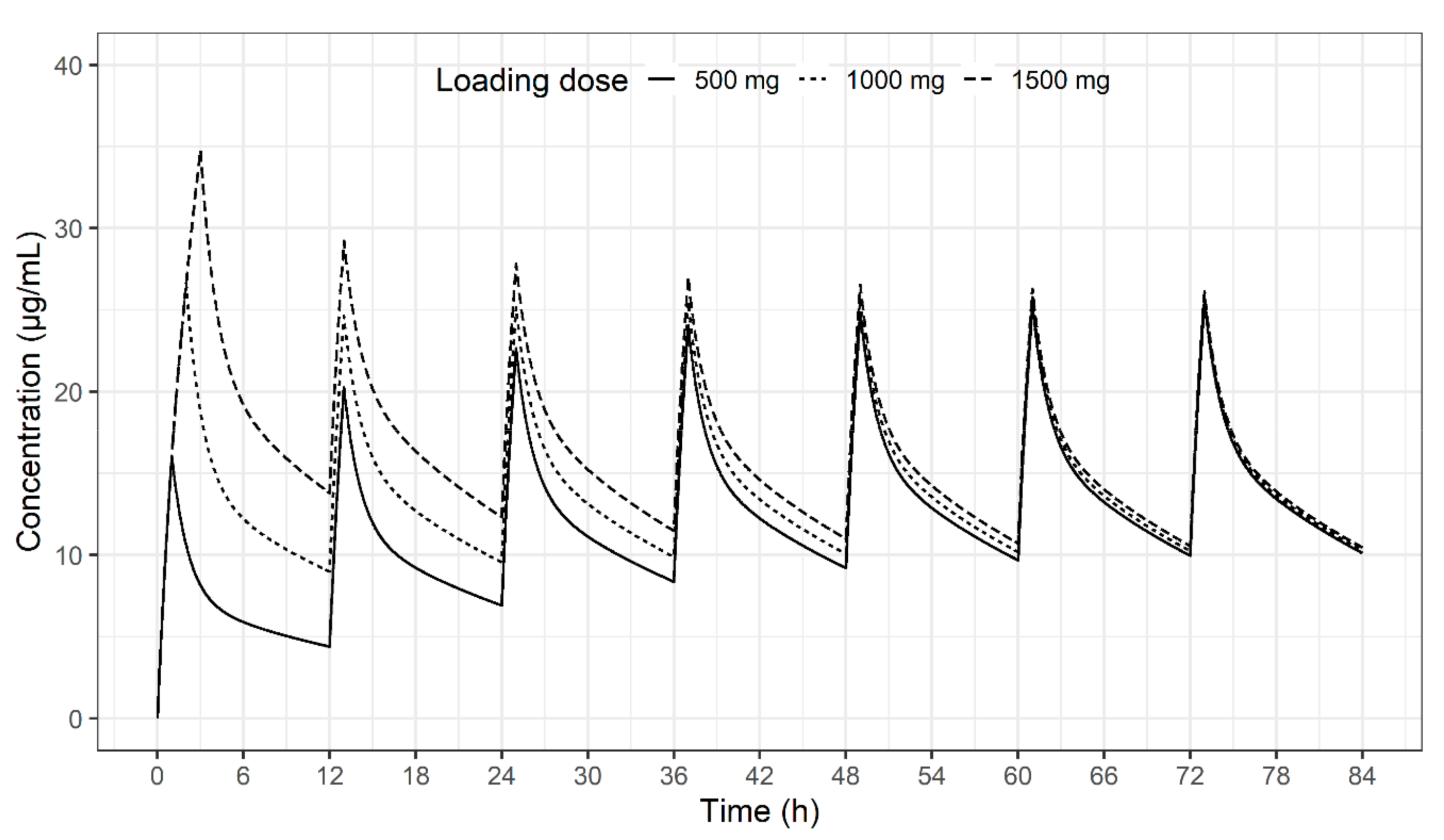
2. Results
3. Discussion
4. Materials and Methods
4.1. Pharmacokinetic Simulation and Estimation
4.2. Bias and Precision of Parameter Estimates
Author Contributions
Funding
Conflicts of Interest
References
- Collins, F.S.; Varmus, H. A new initiative on precision medicine. N. Engl. J. Med. 2015, 372, 793–795. [Google Scholar] [CrossRef]
- Daly, A.K. Pharmacogenomics of adverse drug reactions. Genome Med. 2013, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Workgroup, E.M.; Marshall, S.F.; Burghaus, R.; Cosson, V.; Cheung, S.Y.; Chenel, M.; DellaPasqua, O.; Frey, N.; Hamren, B.; Harnisch, L.; et al. Good Practices in Model-Informed Drug Discovery and Development: Practice, Application, and Documentation. CPT Pharmacomet. Syst. Pharmacol. 2016, 5, 93–122. [Google Scholar] [CrossRef] [PubMed]
- Ginsburg, G.S.; Phillips, K.A. Precision Medicine: From Science to Value. Health Aff. 2018, 37, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Darwich, A.S.; Ogungbenro, K.; Vinks, A.A.; Powell, J.R.; Reny, J.L.; Marsousi, N.; Daali, Y.; Fairman, D.; Cook, J.; Lesko, L.J.; et al. Why has model-informed precision dosing not yet become common clinical reality? lessons from the past and a roadmap for the future. Clin. Pharmacol. Ther. 2017, 101, 646–656. [Google Scholar] [CrossRef] [PubMed]
- Polasek, T.M.; Rostami-Hodjegan, A.; Yim, D.S.; Jamei, M.; Lee, H.; Kimko, H.; Kim, J.K.; Nguyen, P.T.T.; Darwich, A.S.; Shin, J.G. What Does It Take to Make Model-Informed Precision Dosing Common Practice? Report from the 1st Asian Symposium on Precision Dosing. AAPS J. 2019, 21, 17. [Google Scholar] [CrossRef] [PubMed]
- Mould, D.R.; Upton, R.N. Basic concepts in population modeling, simulation, and model-based drug development. CPT Pharmacomet. Syst. Pharmacol. 2012, 1, e6. [Google Scholar] [CrossRef]
- Frye, R.L.; Brooks, M.M.; Nesto, R.W.; Bypass Angioplasty Revascularization, I. Gap between clinical trials and clinical practice: Lessons from the Bypass Angioplasty Revascularization Investigation (BARI). Circulation 2003, 107, 1837–1839. [Google Scholar] [CrossRef]
- Kibbelaar, R.E.; Oortgiesen, B.E.; van der Wal-Oost, A.M.; Boslooper, K.; Coebergh, J.W.; Veeger, N.; Joosten, P.; Storm, H.; van Roon, E.N.; Hoogendoorn, M. Bridging the gap between the randomised clinical trial world and the real world by combination of population-based registry and electronic health record data: A case study in haemato-oncology. Eur. J. Cancer 2017, 86, 178–185. [Google Scholar] [CrossRef]
- Thomson, A.H.; Whiting, B. Bayesian parameter estimation and population pharmacokinetics. Clin. Pharm. 1992, 22, 447–467. [Google Scholar] [CrossRef]
- Sheiner, L.B.; Beal, S.; Rosenberg, B.; Marathe, V.V. Forecasting individual pharmacokinetics. Clin. Pharmacol. Ther. 1979, 26, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Standing, J.F. Understanding and applying pharmacometric modelling and simulation in clinical practice and research. Br. J. Clin. Pharmacol. 2017, 83, 247–254. [Google Scholar] [CrossRef] [PubMed]
- De Velde, F.; Mouton, J.W.; de Winter, B.C.M.; van Gelder, T.; Koch, B.C.P. Clinical applications of population pharmacokinetic models of antibiotics: Challenges and perspectives. Pharmacol. Res. 2018, 134, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.A.; Kirkpatrick, C.M.; Lipman, J. Monte Carlo simulations: Maximizing antibiotic pharmacokinetic data to optimize clinical practice for critically ill patients. J. Antimicrob. Chemother. 2011, 66, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Asin-Prieto, E.; Rodriguez-Gascon, A.; Isla, A. Applications of the pharmacokinetic/pharmacodynamic (PK/PD) analysis of antimicrobial agents. J. Infect Chemother. 2015, 21, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.I. Design and power of a population pharmacokinetic study. Pharm. Res. 2001, 18, 75–82. [Google Scholar] [CrossRef]
- Kim, S.W.; Kim, D.J.; Zang, D.Y.; Lee, D.H. Effect of pharmacokinetic model misspecification on antibiotic probability of target attainment predicted by Monte Carlo simulation. Int. J. Clin. Pharmacol. Ther. 2019, 57, 362–374. [Google Scholar] [CrossRef]
- Kim, Y.K.; Lee, D.H.; Jeon, J.; Jang, H.J.; Kim, H.K.; Jin, K.; Lim, S.N.; Lee, S.S.; Park, B.S.; Kim, Y.W.; et al. Population Pharmacokinetic Analysis of Meropenem After Intravenous Infusion in Korean Patients with Acute Infections. Clin. Ther. 2018, 40, 1384–1395. [Google Scholar] [CrossRef]
- Chen, Y.; Wu, D.; Dong, M.; Zhu, Y.; Lu, J.; Li, X.; Chen, C.; Li, Z. Population pharmacokinetics of vancomycin and AUC-guided dosing in Chinese neonates and young infants. Eur. J. Clin. Pharmacol. 2018, 74, 921–930. [Google Scholar] [CrossRef]
- Rhodes, N.J.; Grove, M.E.; Kiel, P.J.; O’Donnell, J.N.; Whited, L.K.; Rose, D.T.; Jones, D.R.; Scheetz, M.H. Population pharmacokinetics of cefepime in febrile neutropenia: Implications for dose-dependent susceptibility and contemporary dosing regimens. Int. J. Antimicrob. Agents 2017, 50, 482–486. [Google Scholar] [CrossRef]
- Lee, D.H.; Kim, Y.K.; Jin, K.; Kang, M.J.; Joo, Y.D.; Kim, Y.W.; Moon, Y.S.; Shin, J.G.; Kiem, S. Population Pharmacokinetic Analysis of Doripenem after Intravenous Infusion in Korean Patients with Acute Infections. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Zhang, Y.; Chen, M.; Zhong, P.; Chen, Y.; Yu, J.; Wu, X.; Wu, J.; Zhang, J. Population Pharmacokinetics and Dosing Regimen Optimization of Meropenem in Cerebrospinal Fluid and Plasma in Patients with Meningitis after Neurosurgery. Antimicrob. Agents Chemother. 2016, 60, 6619–6625. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Qian, Q.; Sun, M.R.; Qian, C.Y.; Zou, S.L.; Wang, M.L.; Wang, L.Y. Population Pharmacokinetics and Pharmacodynamics of Piperacillin/Tazobactam in Patients with Nosocomial Infections. Eur. J. Drug Metab. Pharm. 2016, 41, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Lea-Henry, T.N.; Carland, J.E.; Stocker, S.L.; Sevastos, J.; Roberts, D.M. Clinical Pharmacokinetics in Kidney Disease: Fundamental Principles. Clin. J. Am. Soc. Nephrol. 2018, 13, 1085–1095. [Google Scholar] [CrossRef]
- Wu, C.C.; Shen, L.J.; Hsu, L.F.; Ko, W.J.; Wu, F.L. Pharmacokinetics of vancomycin in adults receiving extracorporeal membrane oxygenation. J. Formos. Med. Assoc. 2016, 115, 560–570. [Google Scholar] [CrossRef]
- Mulla, H.; Pooboni, S. Population pharmacokinetics of vancomycin in patients receiving extracorporeal membrane oxygenation. Br. J. Clin. Pharmacol. 2005, 60, 265–275. [Google Scholar] [CrossRef]
- Donadello, K.; Roberts, J.A.; Cristallini, S.; Beumier, M.; Shekar, K.; Jacobs, F.; Belhaj, A.; Vincent, J.L.; de Backer, D.; Taccone, F.S. Vancomycin population pharmacokinetics during extracorporeal membrane oxygenation therapy: A matched cohort study. Crit. Care 2014, 18, 632. [Google Scholar] [CrossRef]
- Moore, J.N.; Healy, J.R.; Thoma, B.N.; Peahota, M.M.; Ahamadi, M.; Schmidt, L.; Cavarocchi, N.C.; Kraft, W.K. A Population Pharmacokinetic Model for Vancomycin in Adult Patients Receiving Extracorporeal Membrane Oxygenation Therapy. CPT Pharmacomet. Syst. Pharmacol. 2016, 5, 495–502. [Google Scholar] [CrossRef]
- Wu, G.; Cossettini, P.; Furlanut, M. Prediction of blood cyclosporine concentrations in haematological patients with multidrug resistance by one-, two- and three-compartment models using Bayesian and non-linear least squares methods. Pharmacol. Res. 1996, 34, 47–57. [Google Scholar] [CrossRef]
- Wu, G.; Furlanut, M. Prediction of serum vancomycin concentrations using one-, two- and three-compartment models with implemented population pharmacokinetic parameters and with the Bayesian method. J. Pharm. Pharmacol. 1998, 50, 851–856. [Google Scholar] [CrossRef]
- Marsot, A.; Boulamery, A.; Bruguerolle, B.; Simon, N. Vancomycin: A review of population pharmacokinetic analyses. Clin. Pharm. 2012, 51, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Rybak, M.J.; Le, J.; Lodise, T.P.; Levine, D.P.; Bradley, J.S.; Liu, C.; Mueller, B.A.; Pai, M.P.; Wong-Beringer, A.; Rotschafer, J.C.; et al. Therapeutic Monitoring of Vancomycin for Serious Methicillin-resistant Staphylococcus aureus Infections: A Revised Consensus Guideline and Review by the American Society of Health-system Pharmacists, the Infectious Diseases Society of America, the Pediatric Infectious Diseases Society, and the Society of Infectious Diseases Pharmacists. Clin. Infect Dis. 2020. [Google Scholar] [CrossRef]
- Pai, S.M.; Girgis, S.; Batra, V.K.; Girgis, I.G. Population pharmacodynamic parameter estimation from sparse sampling: Effect of sigmoidicity on parameter estimates. AAPS J. 2009, 11, 535–540. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Choe, S.; Lee, D. Parameter estimation for sigmoid Emax models in exposure-response relationship. Transl. Clin. Pharmacol. 2017, 25, 74–84. [Google Scholar] [CrossRef]
- Girgis, S.; Pai, S.M.; Girgis, I.G.; Batra, V.K. Pharmacodynamic parameter estimation: Population size versus number of samples. AAPS J. 2005, 7, 46. [Google Scholar] [CrossRef]
- Savic, R.M.; Karlsson, M.O. Importance of shrinkage in empirical bayes estimates for diagnostics: Problems and solutions. AAPS J. 2009, 11, 558–569. [Google Scholar] [CrossRef]
- Xu, X.S.; Yuan, M.; Karlsson, M.O.; Dunne, A.; Nandy, P.; Vermeulen, A. Shrinkage in nonlinear mixed-effects population models: Quantification, influencing factors, and impact. AAPS J. 2012, 14, 927–936. [Google Scholar] [CrossRef]
- Johnson, J.A.; Gong, L.; Whirl-Carrillo, M.; Gage, B.F.; Scott, S.A.; Stein, C.M.; Anderson, J.L.; Kimmel, S.E.; Lee, M.T.; Pirmohamed, M.; et al. Clinical Pharmacogenetics Implementation Consortium Guidelines for CYP2C9 and VKORC1 genotypes and warfarin dosing. Clin. Pharmacol. Ther. 2011, 90, 625–629. [Google Scholar] [CrossRef]
- Chatzizisis, Y.S.; Koskinas, K.C.; Misirli, G.; Vaklavas, C.; Hatzitolios, A.; Giannoglou, G.D. Risk factors and drug interactions predisposing to statin-induced myopathy: Implications for risk assessment, prevention and treatment. Drug Saf. 2010, 33, 171–187. [Google Scholar] [CrossRef]
- Lilja, J.J.; Kivisto, K.T.; Neuvonen, P.J. Grapefruit juice-simvastatin interaction: Effect on serum concentrations of simvastatin, simvastatin acid, and HMG-CoA reductase inhibitors. Clin. Pharmacol. Ther. 1998, 64, 477–483. [Google Scholar] [CrossRef]
- Blot, S.I.; Pea, F.; Lipman, J. The effect of pathophysiology on pharmacokinetics in the critically ill patient--concepts appraised by the example of antimicrobial agents. Adv. Drug Deliv. Rev. 2014, 77, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.A.; Abdul-Aziz, M.H.; Lipman, J.; Mouton, J.W.; Vinks, A.A.; Felton, T.W.; Hope, W.W.; Farkas, A.; Neely, M.N.; Schentag, J.J.; et al. Individualised antibiotic dosing for patients who are critically ill: Challenges and potential solutions. Lancet Infect Dis. 2014, 14, 498–509. [Google Scholar] [CrossRef]
- Velenosi, T.J.; Urquhart, B.L. Pharmacokinetic considerations in chronic kidney disease and patients requiring dialysis. Expert Opin. Drug Metab. Toxicol. 2014, 10, 1131–1143. [Google Scholar] [CrossRef] [PubMed]
- Shekar, K.; Fraser, J.F.; Smith, M.T.; Roberts, J.A. Pharmacokinetic changes in patients receiving extracorporeal membrane oxygenation. J. Crit. Care 2012, 27, 741.e9–741.e18. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.W.; Kim, D.J.; Zang, D.Y.; Lee, D.-H. Impact of Sampling Period on Population Pharmacokinetic Analysis of Antibiotics: Why do You Take Blood Samples Following the Fourth Dose? Pharmaceuticals 2020, 13, 249. https://doi.org/10.3390/ph13090249
Kim SW, Kim DJ, Zang DY, Lee D-H. Impact of Sampling Period on Population Pharmacokinetic Analysis of Antibiotics: Why do You Take Blood Samples Following the Fourth Dose? Pharmaceuticals. 2020; 13(9):249. https://doi.org/10.3390/ph13090249
Chicago/Turabian StyleKim, So Won, Dong Jin Kim, Dae Young Zang, and Dong-Hwan Lee. 2020. "Impact of Sampling Period on Population Pharmacokinetic Analysis of Antibiotics: Why do You Take Blood Samples Following the Fourth Dose?" Pharmaceuticals 13, no. 9: 249. https://doi.org/10.3390/ph13090249
APA StyleKim, S. W., Kim, D. J., Zang, D. Y., & Lee, D.-H. (2020). Impact of Sampling Period on Population Pharmacokinetic Analysis of Antibiotics: Why do You Take Blood Samples Following the Fourth Dose? Pharmaceuticals, 13(9), 249. https://doi.org/10.3390/ph13090249