Antibody–Drug Conjugates: The Last Decade




Abstract
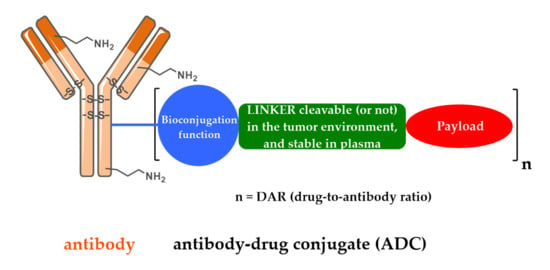
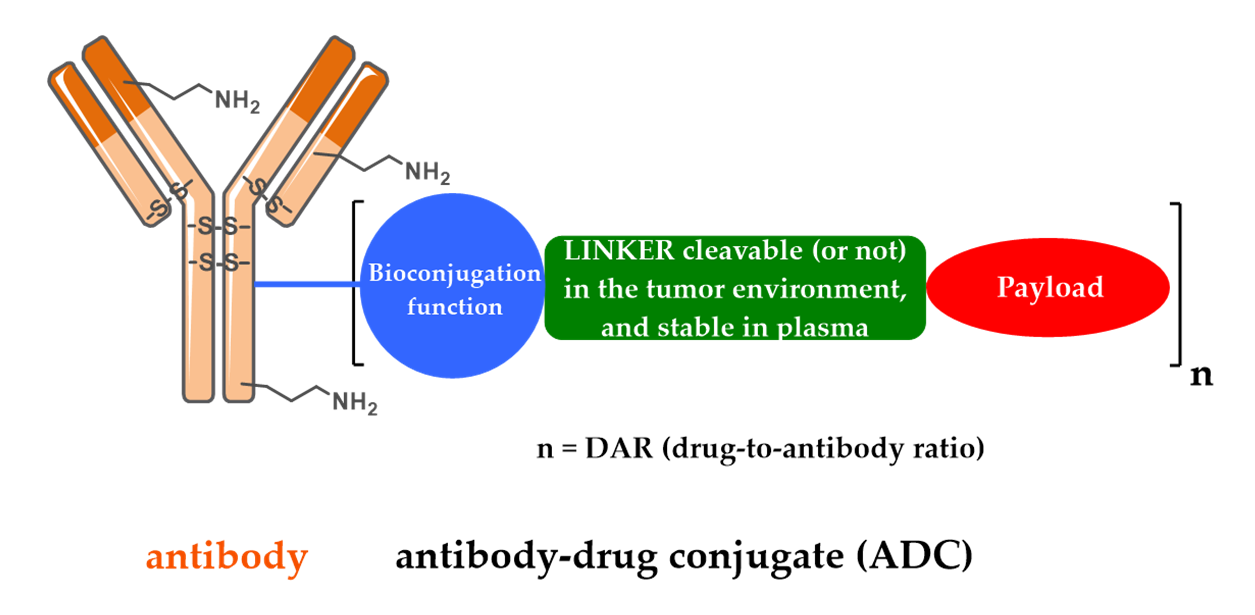
1. Introduction/History
2. Design, Mechanism of Action and Therapeutic Indications of FDA-Approved First- and Second-Generation ADCs
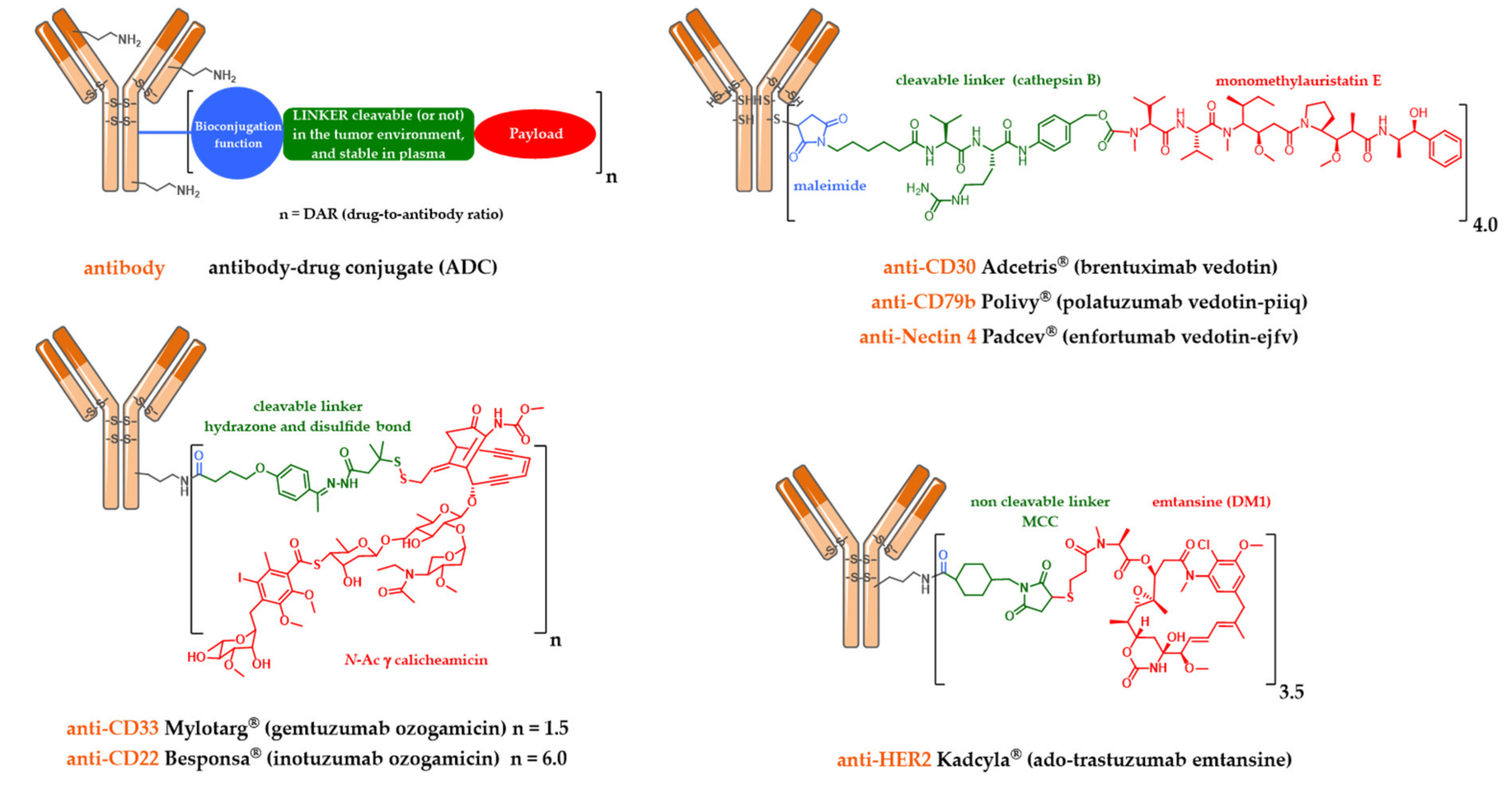
2.1. Mylotarg®, Besponsa® and the First-Generation Cleavable Linker
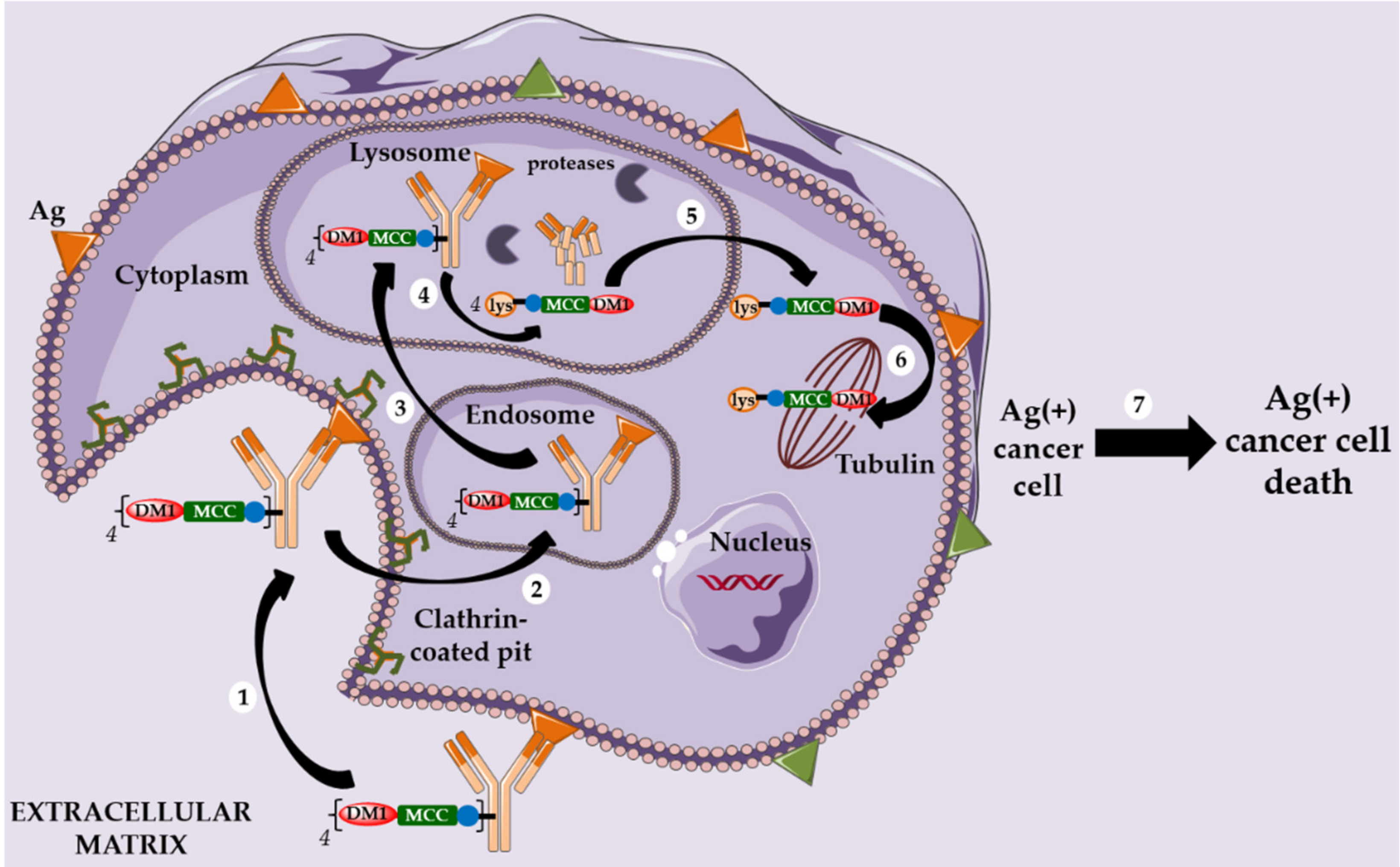
2.2. Kadcyla® and the Notion of Second Generation Non-Cleavable Linkers
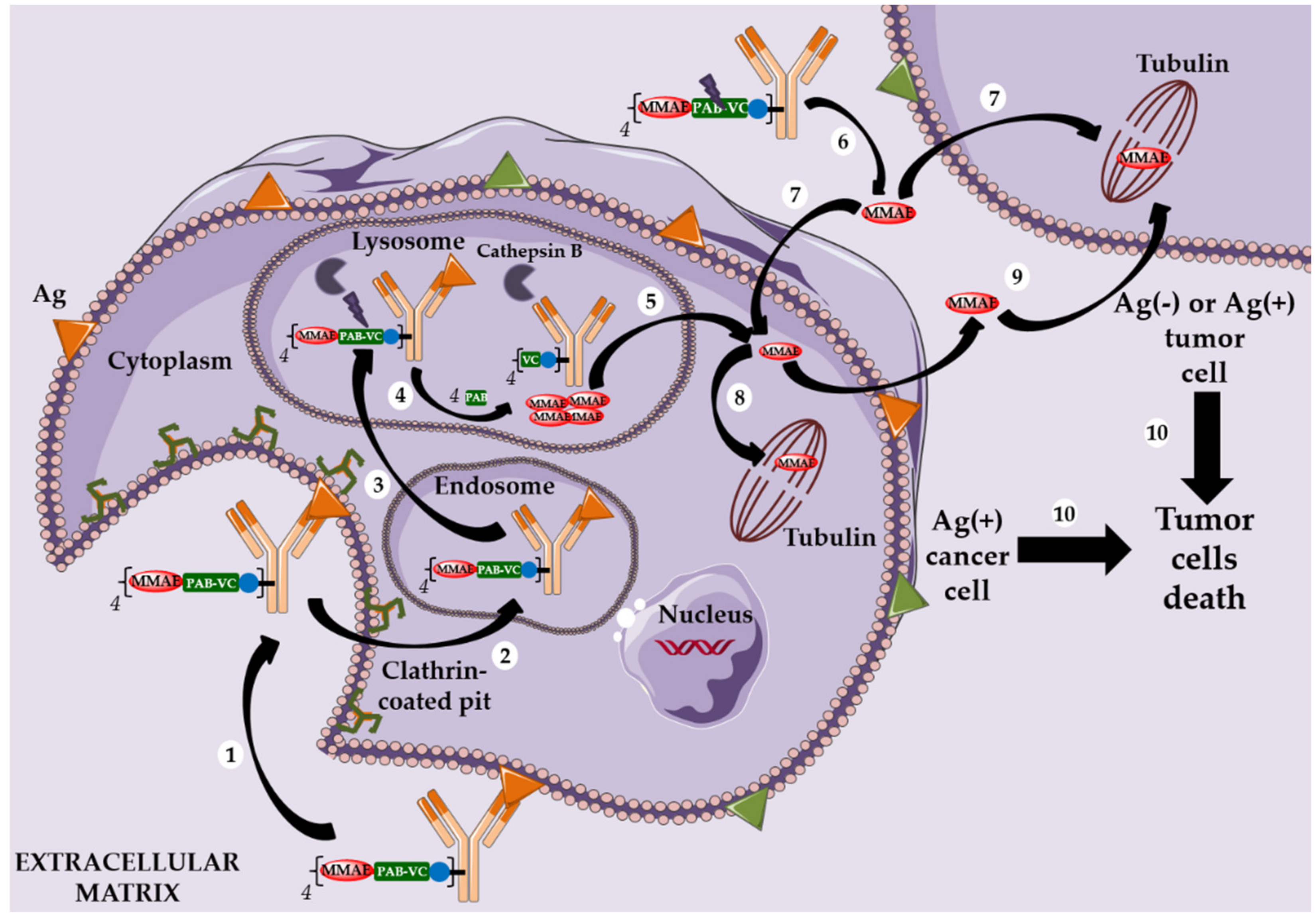
2.3. Adcetris®, Polivy® and the Second-Generation Cleavable Linker
3. Toxicity
3.1. Gemtuzumab Ozogamicin or the Roller Coaster of the First ADC Approved in Humans
3.2. Brentuximab Vedotin for the Treatment of Hodgkin’s Disease
3.3. The Classic or Unexpected Toxicities of ADC
4. Mechanisms of Resistance to ADCs
5. New Strategies in Development: Third Generation ADC
5.1. Limits of Current Approaches
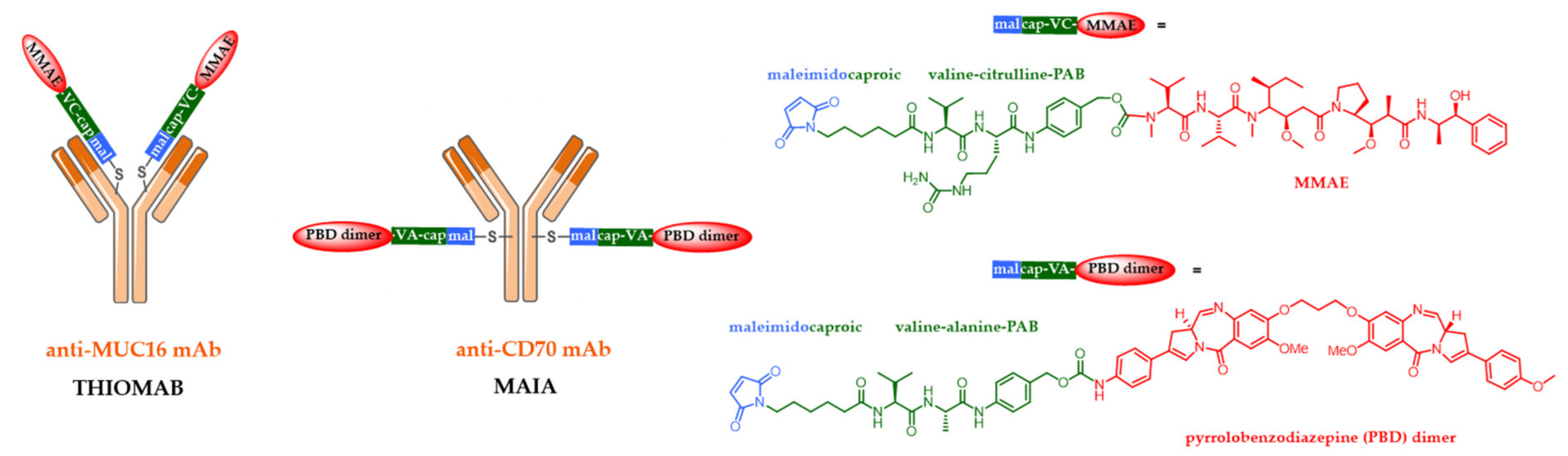
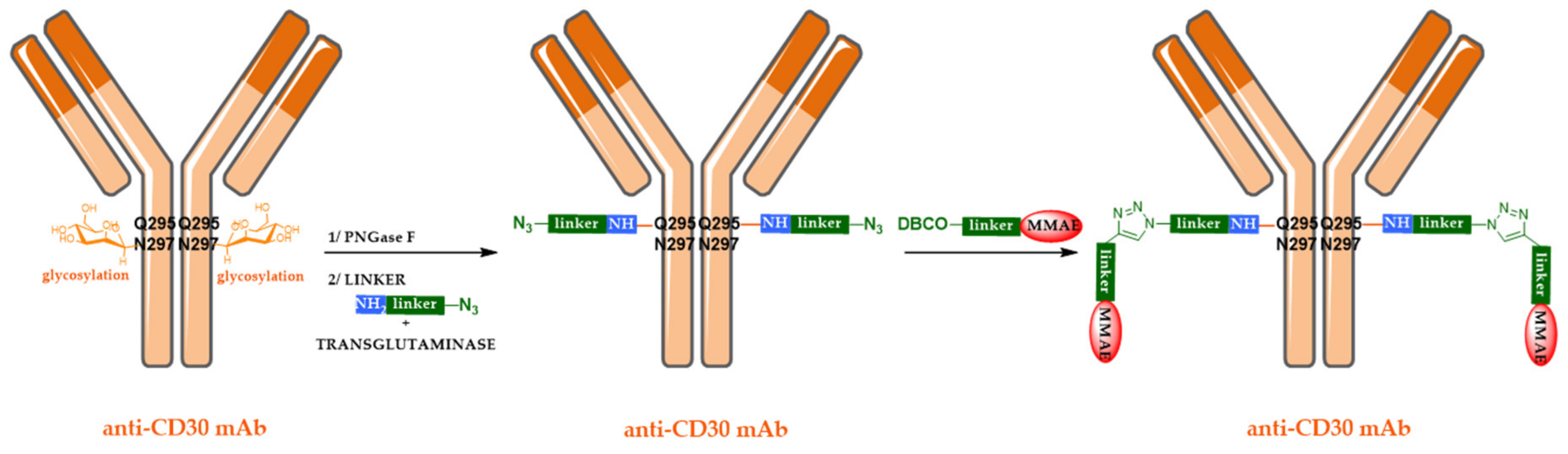
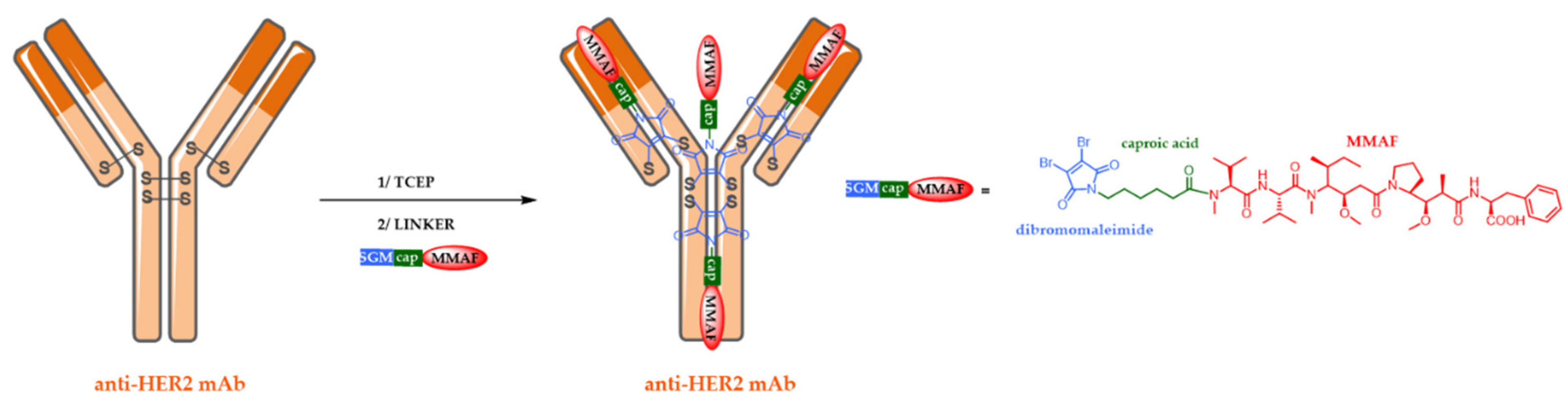
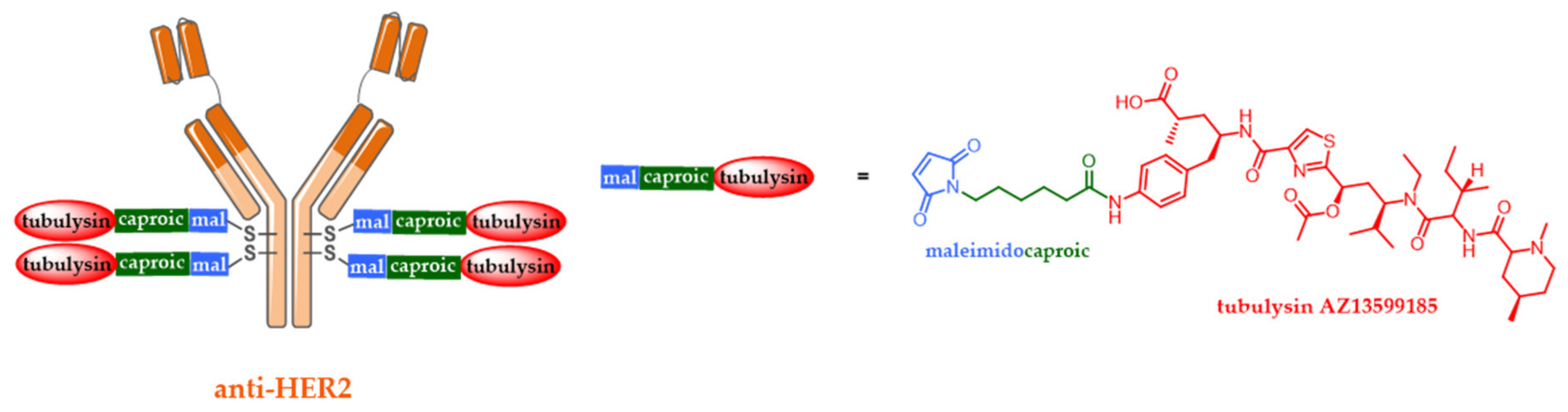
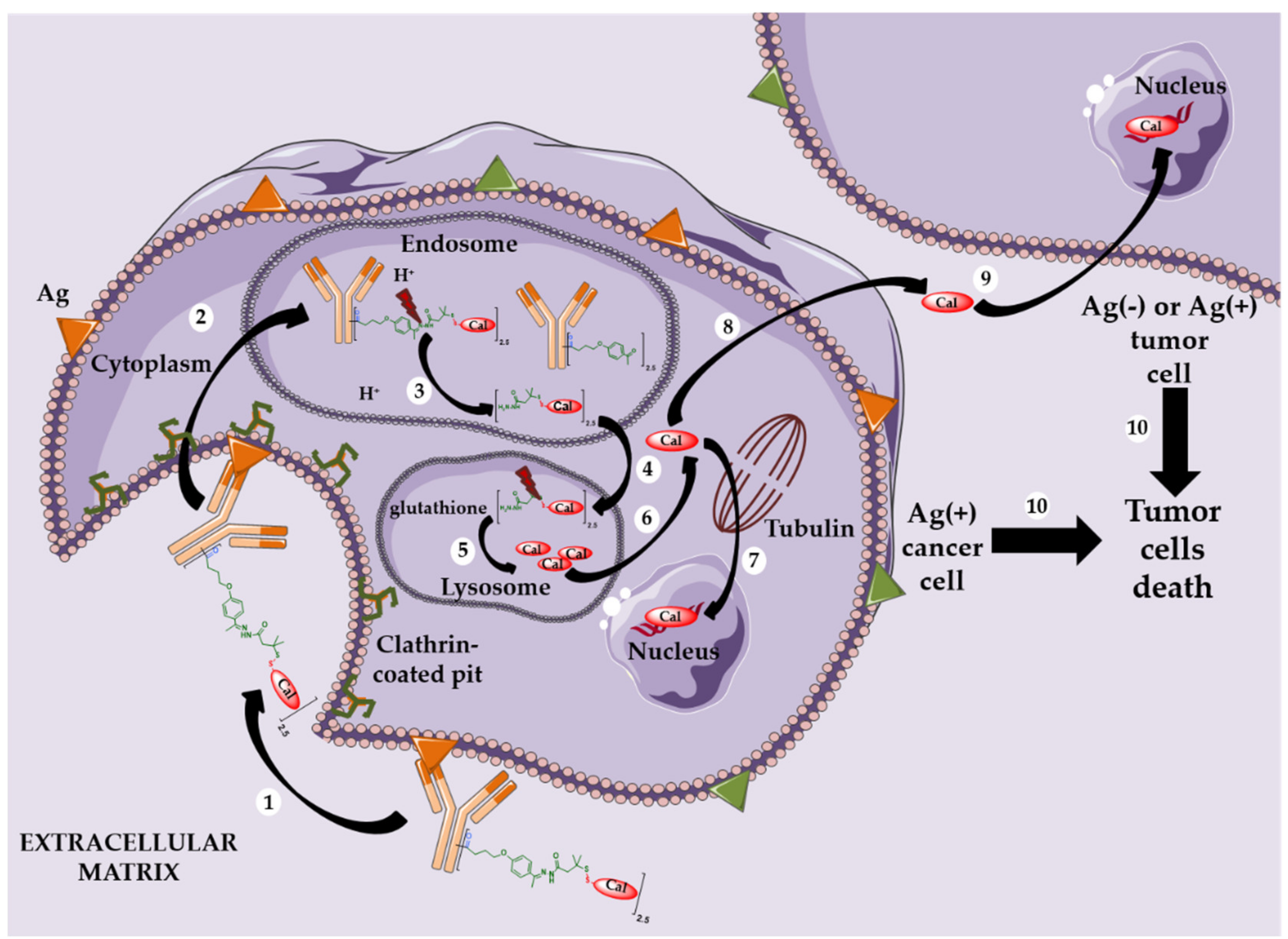
5.2. Site-Specific ADCs
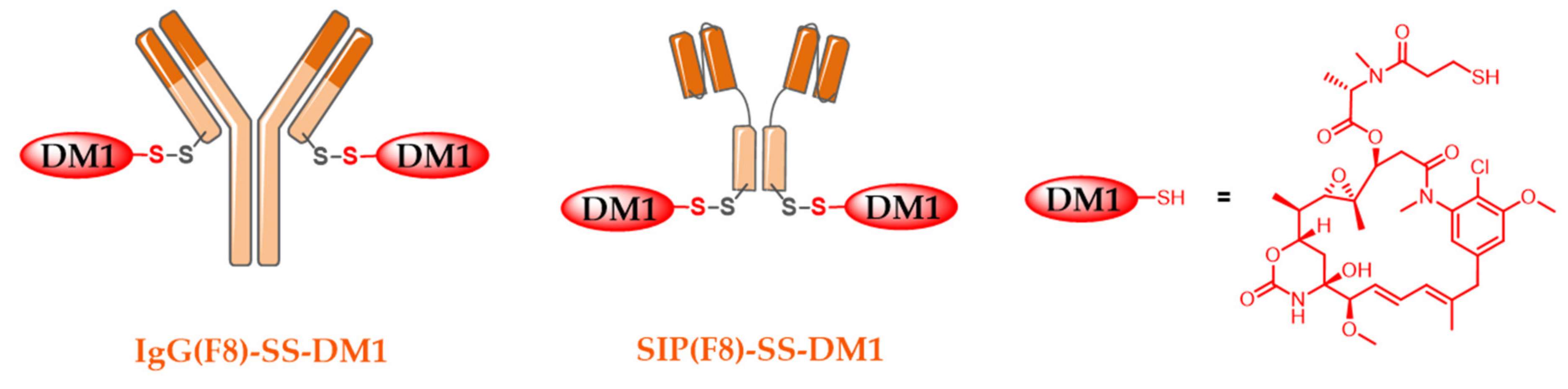
5.3. Alternative ADC Formats
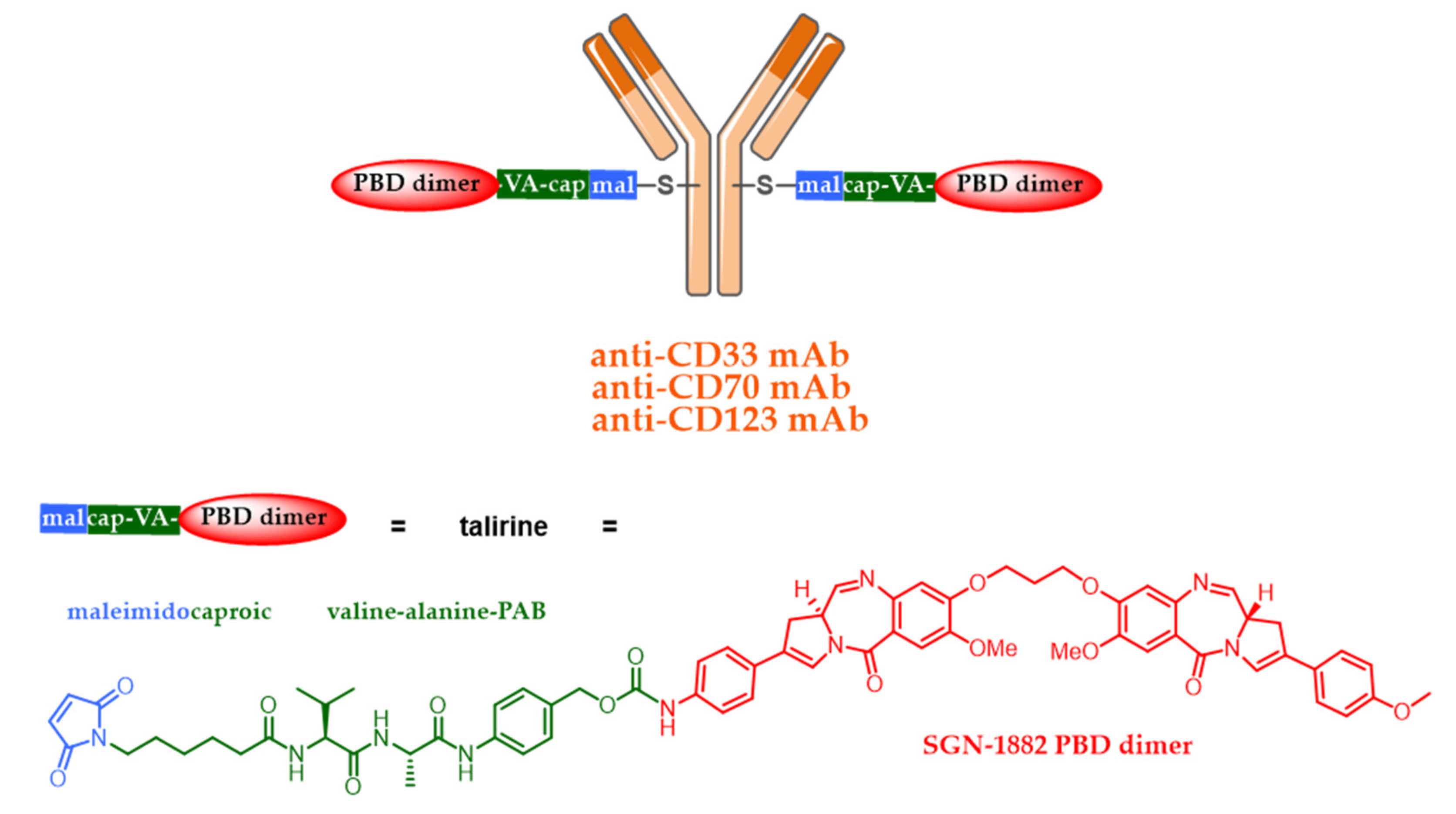
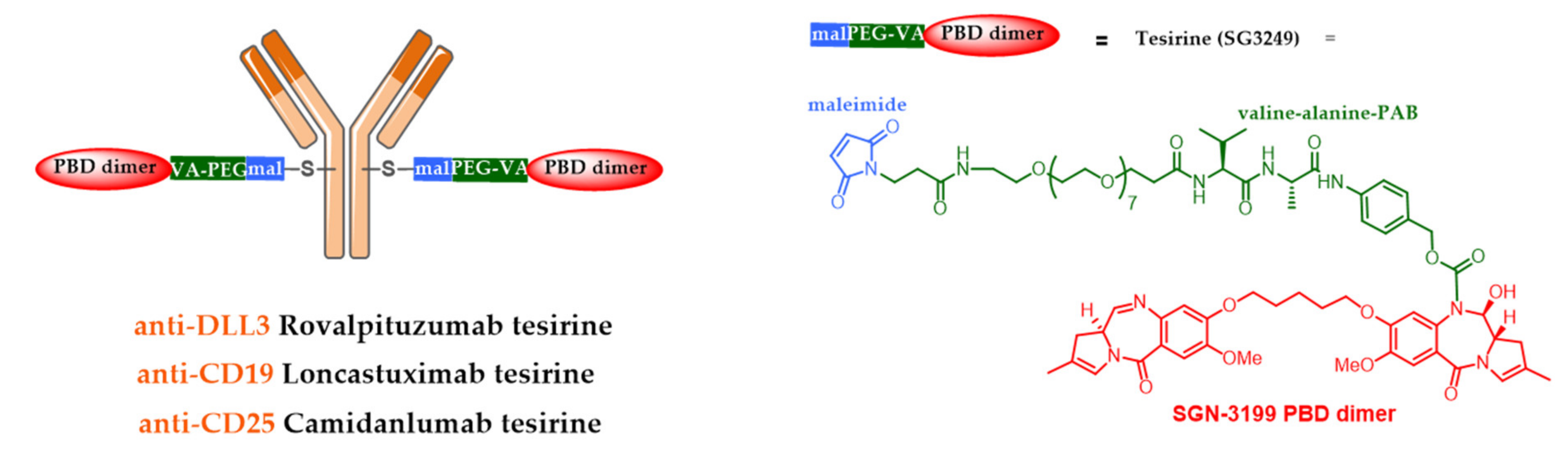
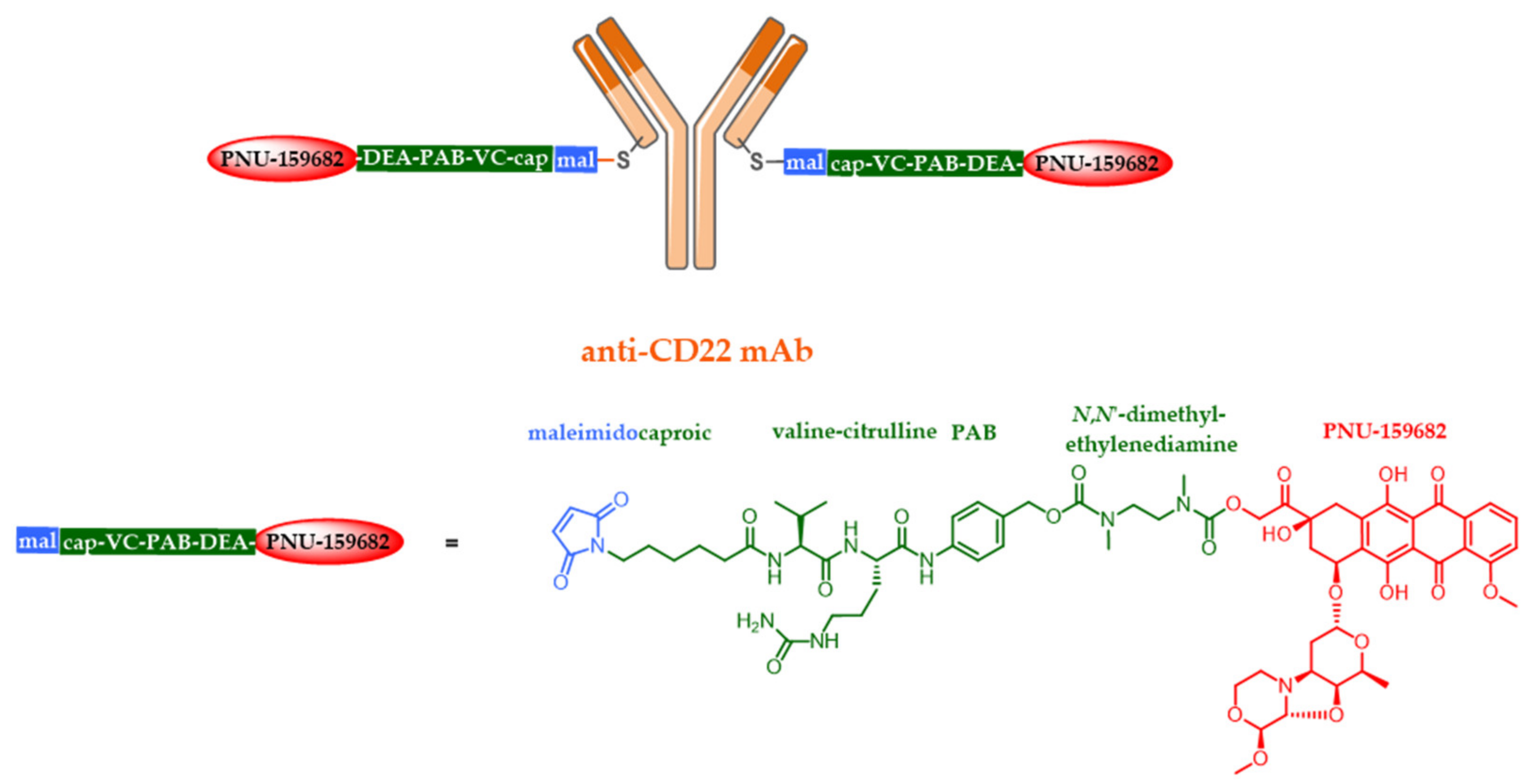
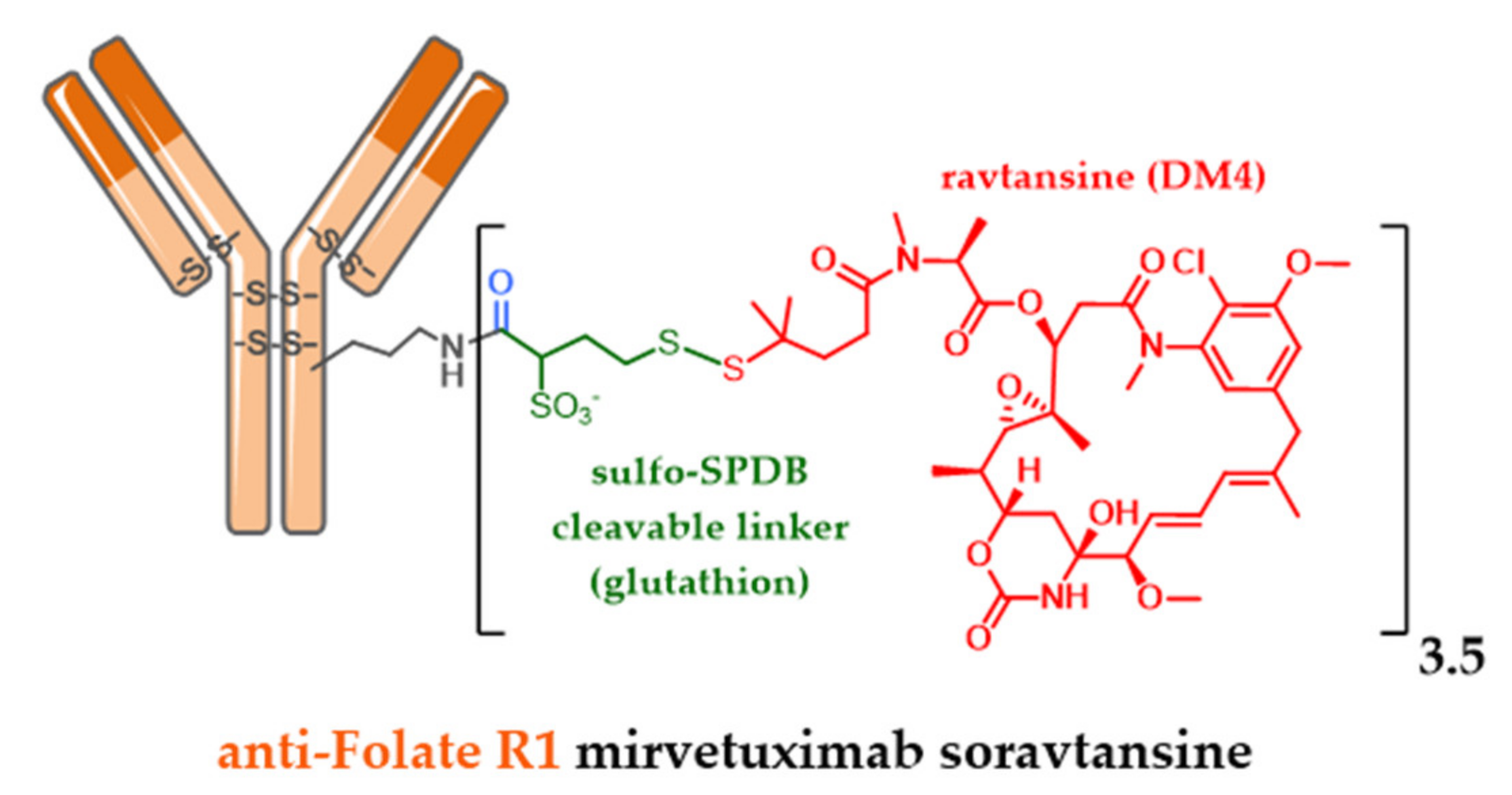
5.4. New Targets and Associated Release Systems
5.5. New Cytotoxic Agents
5.6. Combined Strategies beyond Dogmas: Pivotal Phase II or Phase III ADC
5.7. Successful Iteration beyond Dogmas: Recent Approval of Third Generation Enhertu® and Trodelvy®
6. Indications of ADCs
6.1. Combinations with Conventional Chemotherapy
6.2. Adjuvant, Maintenance or Consolidation Treatments
6.3. Combinations of ADC and Immune Checkpoint Inhibitors
7. Quick Overview of ADC beyond Oncology
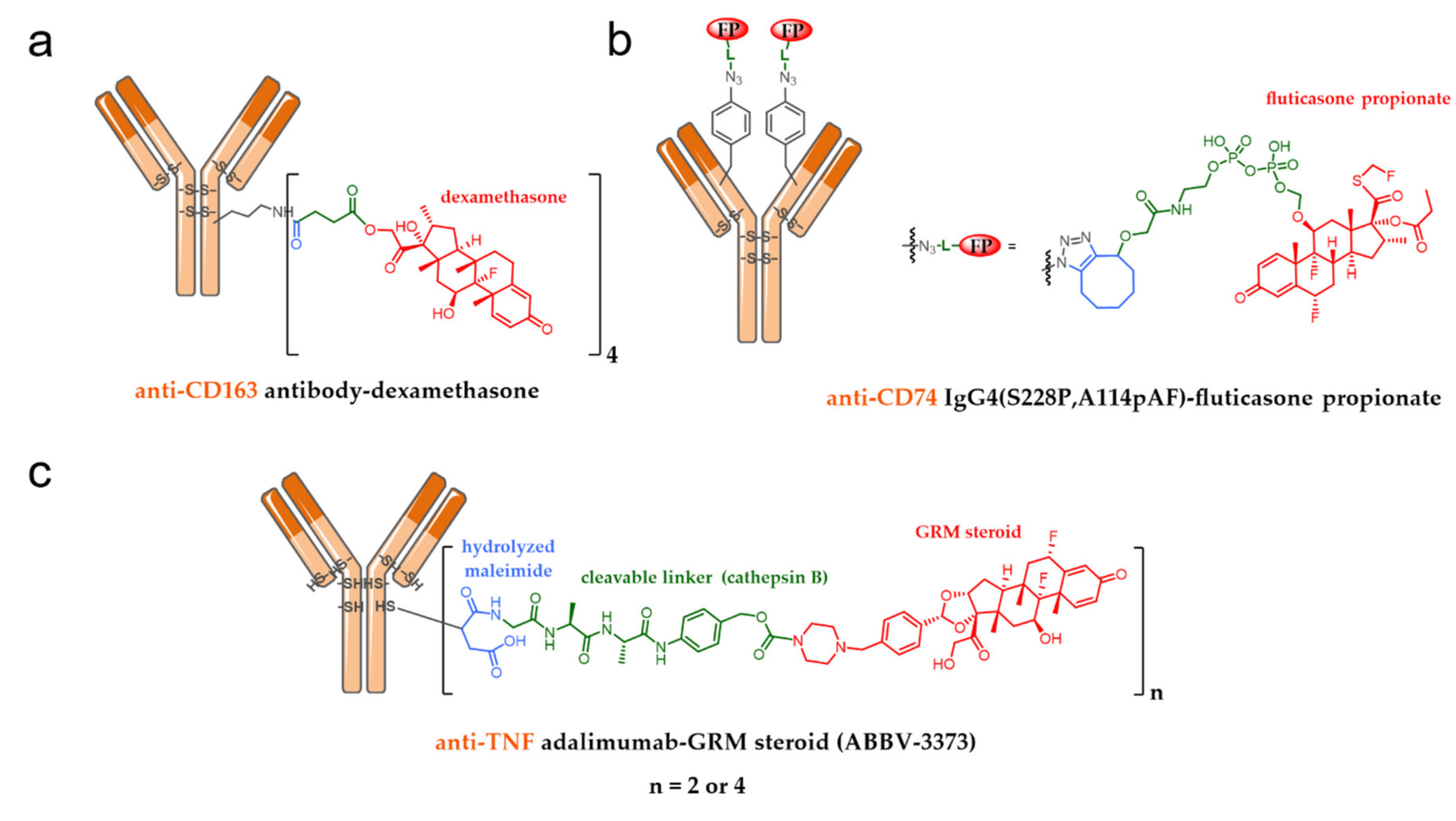
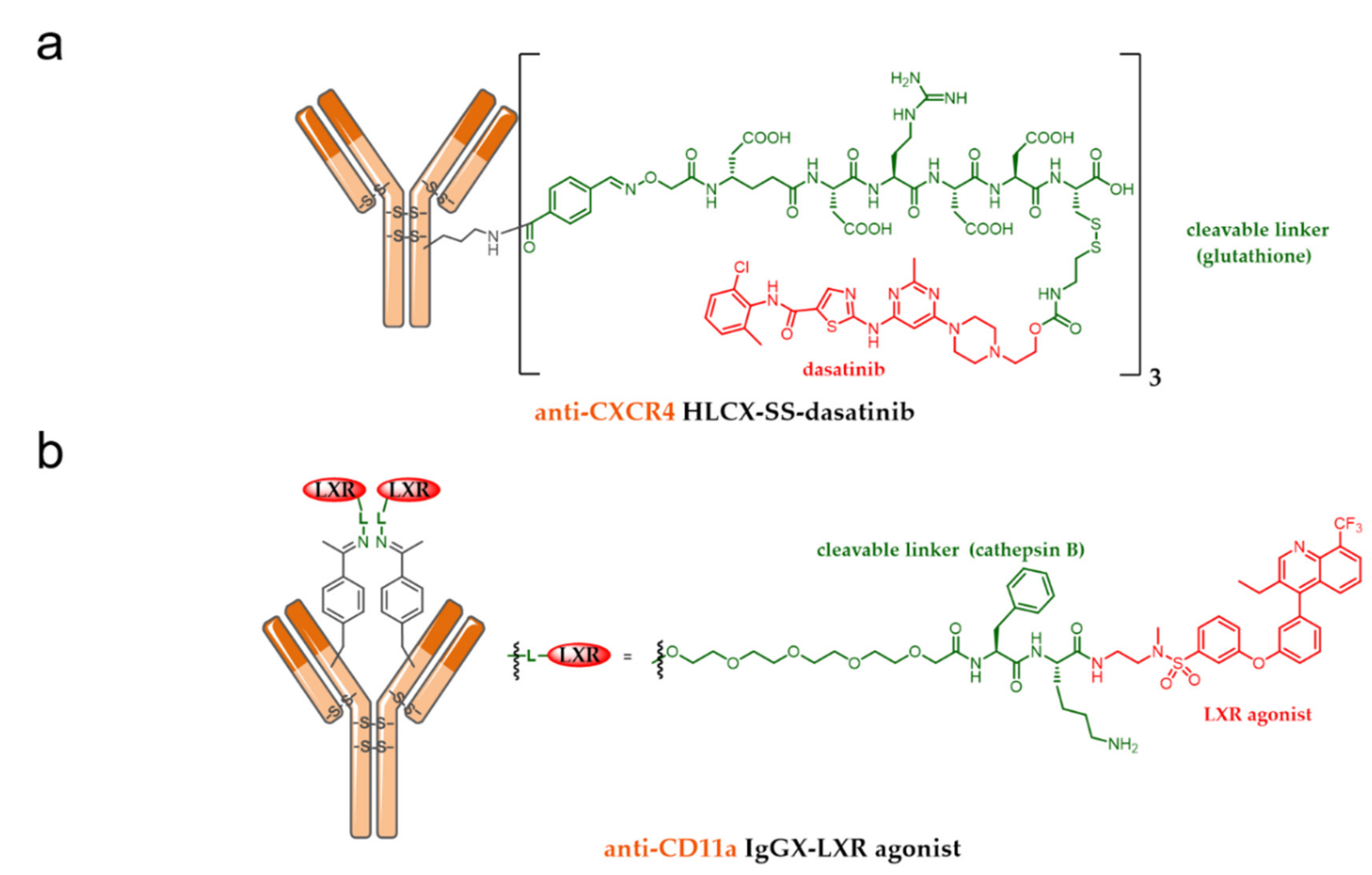
7.1. ADC as an Immunomodulatory Agent
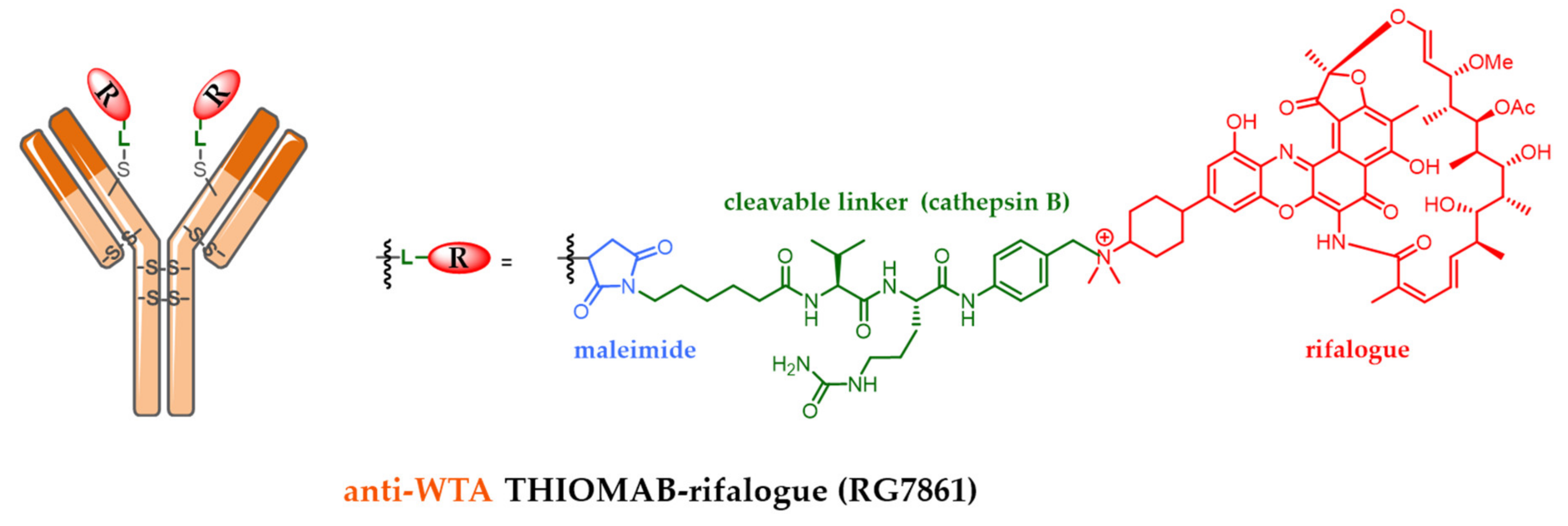
7.2. Antibody-Antibiotic Conjugate
8. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody–drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef] [PubMed]
- Joubert, N.; Denevault-Sabourin, C.; Bryden, F.; Viaud-Massuard, M.C. Towards antibody-drug conjugates and prodrug strategies with extracellular stimuli-responsive drug delivery in the tumor microenvironment for cancer therapy. Eur. J. Med. Chem. 2017, 142, 393–415. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.; Terral, G.; Debaene, F.; Wagner-Rousset, E.; Marcoux, J.; Janin-Bussat, M.-C.; Colas, O.; Van Dorsselaer, A.; Cianférani, S. Cutting-edge mass spectrometry methods for the multi-level structural characterization of antibody-drug conjugates. Expert Rev. Proteom. 2016, 13, 157–183. [Google Scholar] [CrossRef] [PubMed]
- Haeuw, J.F.; Caussanel, V.; Beck, A. Les immunoconjugués, anticorps «armés» pour combattre le cancer. Medecine/Sciences 2009, 25, 1046–1052. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Beck, A.; Dumontet, C.; Joubert, N. Les immuno-conjugués en oncologie, les raisons du succès récent d’une approche ancienne. Médecine/Sciences 2019, 35, 1034–1042. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Bertozzi, C.R. Site-Specific Antibody–Drug Conjugates: The Nexus of Bioorthogonal Chemistry, Protein Engineering, and Drug Development. Bioconjug. Chem. 2015, 26, 176–192. [Google Scholar] [CrossRef]
- Martin, C.; Kizlik-Masson, C.; Pèlegrin, A.; Watier, H.; Viaud-Massuard, M.-C.; Joubert, N. Antibody-drug conjugates: Design and development for therapy and imaging in and beyond cancer, LabEx MAbImprove industrial workshop, July 27–28, 2017, Tours, France. MAbs 2017, 10, 210–221. [Google Scholar] [CrossRef]
- Denevault-Sabourin, C.; Bryden, F.; Viaud-Massuard, M.; Joubert, N. Antibody–Drug Conjugates: Empowering Antibodies for the Fight against Cancer. In Successful Drug Discovery; Wiley: Hoboken, NJ, USA, 2019; pp. 55–82. [Google Scholar]
- Beck, A.; Dumontet, C.; Joubert, N. Les immunoconjugués en oncologie, les nouvelles stratégies en développement. Médecine/Sciences 2019, 35, 1043–1053. [Google Scholar] [CrossRef]
- Joubert, N.; Viaud-Massuard, M.-C. Antibody-Drug conjugates: Historical developments and mechanisms of action. In Optimizing Antibody-Drug Conjugates for Targeted Delivery of Therapeutics; Future Science Ltd.: Bielefeld, Germany, 2015; Volume 51, pp. 6–21. ISBN 9780124071919. [Google Scholar]
- Linenberger, M.L.; Hong, T.; Flowers, D.; Sievers, E.L.; Gooley, T.A.; Bennett, J.M.; Mark, S.; Leopold, L.H.; Appelbaum, F.R.; Bernstein, I.D.; et al. Multidrug-resistance phenotype and clinical responses to gemtuzumab ozogamicin Multidrug-resistance phenotype and clinical responses to gemtuzumab ozogamicin. Blood 2013, 98, 988–994. [Google Scholar] [CrossRef]
- Hamann, P.R.; Hinman, L.M.; Hollander, I.; Beyer, C.F.; Lindh, D.; Holcomb, R.; Hallett, W.; Tsou, H.-R.; Upeslacis, J.; Shochat, D.; et al. Gemtuzumab Ozogamicin, A Potent and Selective Anti-CD33 Antibody−Calicheamicin Conjugate for Treatment of Acute Myeloid Leukemia. Bioconjug. Chem. 2002, 13, 47–58. [Google Scholar] [CrossRef]
- Ricart, A.D. Antibody-drug conjugates of calicheamicin derivative: Gemtuzumab ozogamicin and inotuzumab ozogamicin. Clin. Cancer Res. 2011, 17, 6417–6427. [Google Scholar] [CrossRef]
- Beck, A.; D’Atri, V.; Ehkirch, A.; Fekete, S.; Hernandez-Alba, O.; Gahoual, R.; Leize-Wagner, E.; François, Y.; Guillarme, D.; Cianférani, S. Cutting-edge multi-level analytical and structural characterization of antibody-drug conjugates: Present and future. Expert Rev. Proteom. 2019, 16, 337–362. [Google Scholar] [CrossRef] [PubMed]
- Trail, P.; Willner, D.; Lasch, S.; Henderson, A.; Hofstead, S.; Casazza, A.; Firestone, R.; Hellstrom, I.; Hellstrom, K. Cure of xenografted human carcinomas by BR96-doxorubicin immunoconjugates. Science 1993, 261, 212–215. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; DeAngelo, D.J.; Stelljes, M.; Martinelli, G.; Liedtke, M.; Stock, W.; Gökbuget, N.; O’Brien, S.; Wang, K.; Wang, T.; et al. Inotuzumab Ozogamicin versus Standard Therapy for Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2016, 375, 740–753. [Google Scholar] [CrossRef]
- Chari, R.V.J. Targeted cancer therapy: Conferring specificity to cytotoxic drugs. Acc. Chem. Res. 2008, 41, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Erickson, H.K.; Widdison, W.C.; Mayo, M.F.; Whiteman, K.; Audette, C.; Wilhelm, S.D.; Singh, R. Tumor delivery and in vivo processing of disulfide-linked and thioether-linked antibody-maytansinoid conjugates. Bioconjug. Chem. 2010, 21, 84–92. [Google Scholar] [CrossRef]
- Erickson, H.K.; Lewis Phillips, G.D.; Leipold, D.D.; Provenzano, C.A.; Mai, E.; Johnson, H.A.; Gunter, B.; Audette, C.A.; Gupta, M.; Pinkas, J.; et al. The Effect of Different Linkers on Target Cell Catabolism and Pharmacokinetics/Pharmacodynamics of Trastuzumab Maytansinoid Conjugates. Mol. Cancer Ther. 2012, 11, 1133–1142. [Google Scholar] [CrossRef]
- Sun, M.M.C.; Beam, K.S.; Cerveny, C.G.; Hamblett, K.J.; Blackmore, R.S.; Torgov, M.Y.; Handley, F.G.M.; Ihle, N.C.; Senter, P.D.; Alley, S.C. Reduction-alkylation strategies for the modification of specific monoclonal antibody bisulfides. Bioconjug. Chem. 2005, 16, 1282–1290. [Google Scholar] [CrossRef]
- Doronina, S.O.; Toki, B.E.; Torgov, M.Y.; Mendelsohn, B.; Cerveny, C.G.; Chace, D.F.; DeBlanc, R.L.; Gearing, R.P.; Bovee, T.D.; Siegall, C.B.; et al. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat. Biotechnol. 2003, 21, 778–784. [Google Scholar] [CrossRef]
- Katz, J.; Janik, J.E.; Younes, A. Brentuximab vedotin (SGN-35). Clin. Cancer Res. 2011, 17, 6428–6436. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Emmerton, K.K.; Jonas, M.; Zhang, X.; Miyamoto, J.B.; Setter, J.R.; Nicholas, N.D.; Okeley, N.M.; Lyon, R.P.; Benjamin, D.R.; et al. Intracellular released payload influences potency and bystander-killing effects of antibody-drug conjugates in preclinical models. Cancer Res. 2016, 76, 2710–2719. [Google Scholar] [CrossRef] [PubMed]
- Kovtun, Y.V.; Audette, C.A.; Ye, Y.; Xie, H.; Ruberti, M.F.; Phinney, S.J.; Leece, B.A.; Chittenden, T.; Blattler, W.A.; Goldmacher, V.S. Antibody-Drug Conjugates Designed to Eradicate Tumors with Homogeneous and Heterogeneous Expression of the Target Antigen. Cancer Res. 2006, 66, 3214–3221. [Google Scholar] [CrossRef] [PubMed]
- Ogitani, Y.; Hagihara, K.; Oitate, M.; Naito, H.; Agatsuma, T. Bystander killing effect of DS-8201a, a novel anti-human epidermal growth factor receptor 2 antibody-drug conjugate, in tumors with human epidermal growth factor receptor 2 heterogeneity. Cancer Sci. 2016, 107, 1039–1046. [Google Scholar] [CrossRef]
- Dal Corso, A.; Cazzamalli, S.; Gébleux, R.; Mattarella, M.; Neri, D. Protease-Cleavable Linkers Modulate the Anticancer Activity of Noninternalizing Antibody-Drug Conjugates. Bioconjug. Chem. 2017, 28, 1826–1833. [Google Scholar] [CrossRef]
- Teicher, B. Antibody-Drug Conjugate Targets. Curr. Cancer Drug Targets 2009, 9, 982–1004. [Google Scholar] [CrossRef]
- Pfeifer, M.; Zheng, B.; Erdmann, T.; Koeppen, H.; McCord, R.; Grau, M.; Staiger, A.; Chai, A.; Sandmann, T.; Madle, H.; et al. Anti-CD22 and anti-CD79B antibody drug conjugates are active in different molecular diffuse large B-cell lymphoma subtypes. Leukemia 2015, 29, 1578–1586. [Google Scholar] [CrossRef] [PubMed]
- An Anti-CD79B Antibody-Drug Conjugate Is Active in Non-Hodgkin Lymphoma. Cancer Discov. 2015, 5, 576. [CrossRef]
- Challita-Eid, P.M.; Satpayev, D.; Yang, P.; An, Z.; Morrison, K.; Shostak, Y.; Raitano, A.; Nadell, R.; Liu, W.; Lortie, D.R.; et al. Enfortumab vedotin antibody-drug conjugate targeting nectin-4 is a highly potent therapeutic agent in multiple preclinical cancer models. Cancer Res. 2016, 76, 3003–3013. [Google Scholar] [CrossRef]
- Alley, S.C.; Benjamin, D.R.; Jeffrey, S.C.; Okeley, N.M.; Meyer, D.L.; Sanderson, R.J.; Senter, P.D. Contribution of linker stability to the activities of anticancer immunoconjugates. Bioconjug. Chem. 2008, 19, 759–765. [Google Scholar] [CrossRef]
- Donaghy, H. Effects of antibody, drug and linker on the preclinical and clinical toxicities of antibody-drug conjugates. MAbs 2016, 8, 659–671. [Google Scholar] [CrossRef]
- Dorywalska, M.; Dushin, R.; Moine, L.; Farias, S.E.; Zhou, D.; Navaratnam, T.; Lui, V.; Hasa-Moreno, A.; Casas, M.G.; Tran, T.-T.; et al. Molecular Basis of Valine-Citrulline-PABC Linker Instability in Site-Specific ADCs and Its Mitigation by Linker Design. Mol. Cancer Ther. 2016, 15, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Gulesserian, S.; Malinao, M.C.; Kumar-Ganesan, S.; Song, J.; Chang, M.S.; Williams, M.M.; Zeng, Z.; Mattie, M.; Mendelsohn, B.A.; et al. A Potential Mechanism for ADC-Induced Neutropenia: Role of Neutrophils in Their Own Demise. Mol. Cancer Ther. 2017, 16, 1866–1876. [Google Scholar] [CrossRef] [PubMed]
- Castaigne, S.; Pautas, C.; Terré, C.; Raffoux, E.; Bordessoule, D.; Bastie, J.-N.; Legrand, O.; Thomas, X.; Turlure, P.; Reman, O.; et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): A randomised, open-label, phase 3 study. Lancet 2012, 379, 1508–1516. [Google Scholar] [CrossRef]
- A phase 2 study of brentuximab vedotin in patients with relapsed or refractory CD30-positive non-Hodgkin lymphomas: Interim results in patients with DLBCL and other B-cell lymphomas. Clin. Adv. Hematol. Oncol. 2014, 12, 3–4.
- Connors, J.M.; Jurczak, W.; Straus, D.J.; Ansell, S.M.; Kim, W.S.; Gallamini, A.; Younes, A.; Alekseev, S.; Illés, Á.; Picardi, M.; et al. Brentuximab Vedotin with Chemotherapy for Stage III or IV Hodgkin’s Lymphoma. N. Engl. J. Med. 2018, 378, 331–344. [Google Scholar] [CrossRef]
- Younes, A.; Connors, J.M.; Park, S.I.; Fanale, M.; O’Meara, M.M.; Hunder, N.N.; Huebner, D.; Ansell, S.M. Brentuximab vedotin combined with ABVD or AVD for patients with newly diagnosed Hodgkin’s lymphoma: A phase 1, open-label, dose-escalation study. Lancet Oncol. 2013, 14, 1348–1356. [Google Scholar] [CrossRef]
- Carlson, J.A.; Nooruddin, Z.; Rusthoven, C.; Elias, A.; Borges, V.F.; Diamond, J.R.; Kavanagh, B.; Kabos, P. Trastuzumab emtansine and stereotactic radiosurgery: An unexpected increase in clinically significant brain edema. Neurol. Oncol. 2014, 16, 1006–1009. [Google Scholar] [CrossRef]
- Loganzo, F.; Tan, X.; Sung, M.; Jin, G.; Myers, J.S.; Melamud, E.; Wang, F.; Diesl, V.; Follettie, M.T.; Musto, S.; et al. Tumor Cells Chronically Treated with a Trastuzumab-Maytansinoid Antibody-Drug Conjugate Develop Varied Resistance Mechanisms but Respond to Alternate Treatments. Mol. Cancer Ther. 2015, 14, 952–963. [Google Scholar] [CrossRef]
- Chen, R.; Hou, J.; Newman, E.; Kim, Y.; Donohue, C.; Liu, X.; Thomas, S.H.; Forman, S.J.; Kane, S.E. CD30 Downregulation, MMAE Resistance, and MDR1 Upregulation Are All Associated with Resistance to Brentuximab Vedotin. Mol. Cancer Ther. 2015, 14, 1376–1384. [Google Scholar] [CrossRef]
- Hamblett, K.J.; Jacob, A.P.; Gurgel, J.L.; Tometsko, M.E.; Rock, B.M.; Patel, S.K.; Milburn, R.R.; Siu, S.; Ragan, S.P.; Rock, D.A.; et al. SLC46A3 Is Required to Transport Catabolites of Noncleavable Antibody Maytansine Conjugates from the Lysosome to the Cytoplasm. Cancer Res. 2015, 75, 5329–5340. [Google Scholar] [CrossRef]
- Sauveur, J.; Matera, E.-L.; Chettab, K.; Valet, P.; Guitton, J.; Savina, A.; Dumontet, C. Esophageal cancer cells resistant to T-DM1 display alterations in cell adhesion and the prostaglandin pathway. Oncotarget 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.F.; Zheng, B.; Go, M.; Lau, J.; Spencer, S.; Raab, H.; Soriano, R.; Jhunjhunwala, S.; Cohen, R.; Caruso, M.; et al. A novel anti-CD22 anthracycline-based antibody-drug conjugate (ADC) that overcomes resistance to auristatin-based ADCs. Clin. Cancer Res. 2015, 21, 3298–3306. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-H.; Wang, Y.; Zalath, M.; Liu, D.; Cardillo, T.M.; Goldenberg, D.M. Combining ABCG2 Inhibitors with IMMU-132, an Anti-Trop-2 Antibody Conjugate of SN-38, Overcomes Resistance to SN-38 in Breast and Gastric Cancers. Mol. Cancer Ther. 2016, 15, 1910–1919. [Google Scholar] [CrossRef] [PubMed]
- Yasunaga, M.; Manabe, S.; Matsumura, Y. New concept of cytotoxic immunoconjugate therapy targeting cancer-induced fibrin clots. Cancer Sci. 2011, 102, 1396–1402. [Google Scholar] [CrossRef]
- Yasunaga, M.; Manabe, S.; Tarin, D.; Matsumura, Y. Cancer-Stroma Targeting Therapy by Cytotoxic Immunoconjugate Bound to the Collagen 4 Network in the Tumor Tissue. Bioconjug. Chem. 2011, 22, 1776–1783. [Google Scholar] [CrossRef]
- Lambert, J.M.; Morris, C.Q. Antibody–Drug Conjugates (ADCs) for Personalized Treatment of Solid Tumors: A Review. Adv. Ther. 2017, 34, 1015–1035. [Google Scholar] [CrossRef]
- Barok, M.; Joensuu, H.; Isola, J. Trastuzumab emtansine: Mechanisms of action and drug resistance. Breast Cancer Res. 2014, 16, 3378. [Google Scholar] [CrossRef]
- Panowski, S.; Bhakta, S.; Raab, H.; Polakis, P.; Junutula, J.R. Site-specific antibody drug conjugates for cancer therapy. MAbs 2014, 6, 34–45. [Google Scholar] [CrossRef]
- Jackson, D.Y. Processes for Constructing Homogeneous Antibody Drug Conjugates. Org. Process Res. Dev. 2016, 20, 852–866. [Google Scholar] [CrossRef]
- Junutula, J.R.; Raab, H.; Clark, S.; Bhakta, S.; Leipold, D.D.; Weir, S.; Chen, Y.; Simpson, M.; Tsai, S.P.; Dennis, M.S.; et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol. 2008, 26, 925–932. [Google Scholar] [CrossRef]
- Sutherland, M.S.K.; Walter, R.B.; Jeffrey, S.C.; Burke, P.J.; Yu, C.; Kostner, H.; Stone, I.; Ryan, M.C.; Sussman, D.; Lyon, R.P.; et al. SGN-CD33A: A novel CD33-targeting antibody–drug conjugate using a pyrrolobenzodiazepine dimer is active in models of drug-resistant AML. Blood 2013, 122, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Sandall, S.L.; McCormick, R.; Miyamoto, J.; Biechele, T.; Law, C.-L.; Lewis, T.S. Abstract 946: SGN-CD70A, a pyrrolobenzodiazepine (PBD) dimer linked ADC, mediates DNA damage pathway activation and G2 cell cycle arrest leading to cell death. Cancer Res. 2015, 75, 946. [Google Scholar]
- Strop, P. Versatility of Microbial Transglutaminase Versatility of Microbial Transglutaminase Pavel Strop. Bioconj. Chem. 2014, 25, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Behrens, C.R.; Ha, E.H.; Chinn, L.L.; Bowers, S.; Probst, G.; Fitch-Bruhns, M.; Monteon, J.; Valdiosera, A.; Bermudez, A.; Liao-Chan, S.; et al. Antibody-Drug Conjugates (ADCs) Derived from Interchain Cysteine Cross-Linking Demonstrate Improved Homogeneity and Other Pharmacological Properties over Conventional Heterogeneous ADCs. Mol. Pharm. 2015, 12, 3986–3998. [Google Scholar] [CrossRef]
- Joubert, N.; Viaud-Massuard, M.C.; Respaud, R. Novel Antibody-Drug Conjugates and the Use of Same in Therapy. WIPO Patent WO2015004400, 15 January 2015. [Google Scholar]
- Schumacher, F.F.; Nunes, J.P.M.; Maruani, A.; Chudasama, V.; Smith, M.E.B.; Chester, K.A.; Baker, J.R.; Caddick, S. Next generation maleimides enable the controlled assembly of antibody–drug conjugates via native disulfide bond bridging. Org. Biomol. Chem. 2014, 12, 7261. [Google Scholar] [CrossRef]
- Nunes, J.P.M.; Morais, M.; Vassileva, V.; Robinson, E.; Rajkumar, V.S.; Smith, M.E.B.; Pedley, R.B.; Caddick, S.; Baker, J.R.; Chudasama, V. Functional native disulfide bridging enables delivery of a potent, stable and targeted antibody–drug conjugate (ADC). Chem. Commun. 2015, 51, 10624–10627. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.W.; Strickland, R.A.; Schumacher, F.F.; Caddick, S.; Baker, J.R.; Gibson, M.I.; Haddleton, D.M. Highly efficient disulfide bridging polymers for bioconjugates from radical-compatible dithiophenol maleimides. Chem. Commun. 2012, 48, 4064. [Google Scholar] [CrossRef]
- Govindan, S.V.; Sharkey, R.M.; Goldenberg, D.M. Prospects and progress of antibody-drug conjugates in solid tumor therapies. Expert Opin. Biol. Ther. 2016, 16, 883–893. [Google Scholar] [CrossRef]
- Nagayama, A.; Ellisen, L.W.; Chabner, B.; Bardia, A. Antibody–Drug Conjugates for the Treatment of Solid Tumors: Clinical Experience and Latest Developments. Target. Oncol. 2017, 12, 719–739. [Google Scholar] [CrossRef]
- Jain, R.K. Physiological barriers to delivery of monoclonal antibodies and other macromolecules in tumors. Cancer Res. 1990, 50, 814s–819s. [Google Scholar]
- Deonarain, M.P.; Yahioglu, G.; Stamati, I.; Marklew, J. Emerging formats for next-generation antibody drug conjugates. Expert Opin. Drug Discov. 2015, 10, 463–481. [Google Scholar] [CrossRef]
- Brachet, G.; Respaud, R.; Arnoult, C.; Henriquet, C.; Dhommée, C.; Viaud-Massuard, M.C.; Heuze-Vourc’h, N.; Joubert, N.; Pugnière, M.; Gouilleux-Gruart, V. Increment in Drug Loading on an Antibody-Drug Conjugate Increases Its Binding to the Human Neonatal Fc Receptor in Vitro. Mol. Pharm. 2016, 13, 1405–1412. [Google Scholar] [CrossRef] [PubMed]
- Litvak-Greenfeld, D.; Benhar, I. Risks and untoward toxicities of antibody-based immunoconjugates. Adv. Drug Deliv. Rev. 2012, 64, 1782–1799. [Google Scholar] [CrossRef]
- De Goeij, B.E.C.G.; Lambert, J.M. New developments for antibody-drug conjugate-based therapeutic approaches. Curr. Opin. Immunol. 2016, 40, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Casi, G.; Neri, D. Antibody-Drug Conjugates and Small Molecule-Drug Conjugates: Opportunities and Challenges for the Development of Selective Anticancer Cytotoxic Agents. J. Med. Chem. 2015, 58, 8751–8761. [Google Scholar] [CrossRef]
- Deonarain, M.; Yahioglu, G.; Stamati, I.; Pomowski, A.; Clarke, J.; Edwards, B.; Diez-Posada, S.; Stewart, A. Small-Format Drug Conjugates: A Viable Alternative to ADCs for Solid Tumours? Antibodies 2018, 7, 16. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; Xu, Z.; Chen, Y. Doxorubicin conjugated with a trastuzumab epitope and an MMP-2 sensitive peptide linker for the treatment of HER2-positive breast cancer. Drug Deliv. 2018, 25, 448–460. [Google Scholar] [CrossRef]
- Massa, S.; Xavier, C.; De Vos, J.; Caveliers, V.; Lahoutte, T.; Muyldermans, S.; Devoogdt, N. Site-specific labeling of cysteine-tagged camelid single-domain antibody-fragments for use in molecular imaging. Bioconjug. Chem. 2014, 25, 979–988. [Google Scholar] [CrossRef]
- Albrecht, H.; Burke, P.A.; Natarajan, A.; Xiong, C.; Kalicinsky, M.; Denardo, G.L.; Denardo, S.J. Production of soluble ScFvs with C-terminal-free thiol for site-specific conjugation or stable dimeric ScFvs on demand. Bioconjug. Chem. 2004, 15, 16–26. [Google Scholar] [CrossRef]
- Badescu, G.; Bryant, P.; Bird, M.; Henseleit, K.; Swierkosz, J.; Parekh, V.; Tommasi, R.; Pawlisz, E.; Jurlewicz, K.; Farys, M.; et al. Bridging Disulfides for Stable and Defined Antibody Drug Conjugates. Bioconjug. Chem. 2014, 25, 1124–1136. [Google Scholar] [CrossRef]
- Gebleux, R.; Wulhfard, S.; Casi, G.; Neri, D. Antibody Format and Drug Release Rate Determine the Therapeutic Activity of Noninternalizing Antibody-Drug Conjugates. Mol. Cancer Ther. 2015, 14, 2606–2612. [Google Scholar] [CrossRef] [PubMed]
- Perrino, E.; Steiner, M.; Krall, N.; Bernardes, G.J.L.; Pretto, F.; Casi, G.; Neri, D. Curative properties of noninternalizing antibody-drug conjugates based on maytansinoids. Cancer Res. 2014, 74, 2569–2578. [Google Scholar] [CrossRef]
- Lillo, A.M.; Sun, C.; Gao, C.; Ditzel, H.; Parrish, J.; Gauss, C.-M.; Moss, J.; Felding-Habermann, B.; Wirsching, P.; Boger, D.L.; et al. A Human Single-Chain Antibody Specific for Integrin α3β1 Capable of Cell Internalization and Delivery of Antitumor Agents. Chem. Biol. 2004, 11, 897–906. [Google Scholar] [CrossRef][Green Version]
- Spidel, J.L.; Albone, E.F.; Cheng, X.; Vaessen, B.; Jacob, S.; Milinichik, A.Z.; Verdi, A.; Kline, J.B.; Grasso, L. Engineering humanized antibody framework sequences for optimal site-specific conjugation of cytotoxins. MAbs 2017, 9, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Aubrey, N.; Allard-Vannier, E.; Martin, C.; Bryden, F.; Letast, S.; Colas, C.; Lakhrif, Z.; Collinet, N.; Dimier-Poisson, I.; Chourpa, I.; et al. Site-Specific Conjugation of Auristatins onto Engineered scFv Using Second Generation Maleimide to Target HER2-positive Breast Cancer in Vitro. Bioconjug. Chem. 2018, 29, 3516–3521. [Google Scholar] [CrossRef] [PubMed]
- Bryden, F.; Martin, C.; Letast, S.; Lles, E.; Viéitez-Villemin, I.; Rousseau, A.; Colas, C.; Brachet-Botineau, M.; Allard-Vannier, E.; Larbouret, C.; et al. Impact of cathepsin B-sensitive triggers and hydrophilic linkers on in vitro efficacy of novel site-specific antibody-drug conjugates. Org. Biomol. Chem. 2018, 16, 1882–1889. [Google Scholar] [CrossRef] [PubMed]
- Adams, G.P.; McCartney, J.E.; Tai, M.S.; Oppermann, H.; Huston, J.S.; Stafford, W.F.; Bookman, M.; Fand, I.; Houston, L.L.; Weiner, L.M. Highly specific in vivo tumor targeting by monovalent and divalent forms of 741F8 anti-c-erbB-2 single-chain Fv. Cancer Res. 1993, 53, 4026–4034. [Google Scholar]
- Li, J.Y.; Perry, S.R.; Muniz-Medina, V.; Wang, X.; Wetzel, L.K.; Rebelatto, M.C.; Hinrichs, M.J.M.; Bezabeh, B.Z.; Fleming, R.L.; Dimasi, N.; et al. A Biparatopic HER2-Targeting Antibody-Drug Conjugate Induces Tumor Regression in Primary Models Refractory to or Ineligible for HER2-Targeted Therapy. Cancer Cell 2016, 29, 117–129. [Google Scholar] [CrossRef]
- Uppal, H.; Doudement, E.; Mahapatra, K.; Darbonne, W.C.; Bumbaca, D.; Shen, B.Q.; Du, X.; Saad, O.; Bowles, K.; Olsen, S.; et al. Potential mechanisms for thrombocytopenia development with trastuzumab emtansine (T-DM1). Clin. Cancer Res. 2015, 21, 123–133. [Google Scholar] [CrossRef]
- Matsumura, Y. Cancer stromal targeting (CAST) therapy. Adv. Drug Deliv. Rev. 2012, 64, 710–719. [Google Scholar] [CrossRef]
- Casi, G.; Neri, D. Noninternalizing targeted cytotoxics for cancer therapy. Mol. Pharm. 2015, 12, 1880–1884. [Google Scholar] [CrossRef] [PubMed]
- Casi, G.; Neri, D. Antibody-drug conjugates: Basic concepts, examples and future perspectives. J. Control. Release 2012, 161, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Mantaj, J.; Jackson, P.J.M.; Rahman, K.M.; Thurston, D.E. From Anthramycin to Pyrrolobenzodiazepine (PBD)-Containing Antibody–Drug Conjugates (ADCs). Angew. Chem. Int. Ed. 2017, 56, 462–488. [Google Scholar] [CrossRef] [PubMed]
- Komarnitsky, P.B.; Lee, H.-J.; Shah, M.; Wong, S.; Gauthier, S.; Dziubinski, J.; Osbaugh, S.; Zhang, F. A phase III study of rovalpituzumab tesirine maintenance therapy following first-line platinum-based chemotherapy in patients with extensive disease small cell lung cancer (ED SCLC). J. Clin. Oncol. 2017, 35, TPS8583. [Google Scholar] [CrossRef]
- Morgensztern, D.; Besse, B.; Greillier, L.; Santana-Davila, R.; Ready, N.; Hann, C.L.; Glisson, B.S.; Farago, A.F.; Dowlati, A.; Rudin, C.M.; et al. Efficacy and safety of rovalpituzumab tesirine in third-line and beyond patients with DLL3-expressing, relapsed/refractory small-cell lung cancer: Results from the phase II TrINITY study. Clin. Cancer Res. 2019, 25, 6958–6966. [Google Scholar] [CrossRef]
- Kahl, B.S.; Hamadani, M.; Radford, J.; Carlo-Stella, C.; Caimi, P.; Reid, E.; Feingold, J.M.; Ardeshna, K.M.; Solh, M.; Heffner, L.T.; et al. A phase I study of ADCT-402 (loncastuximab tesirine), a novel pyrrolobenzodiazepine-based antibody–drug conjugate, in relapsed/refractory B-cell non-Hodgkin lymphoma. Clin. Cancer Res. 2019, 25, 6986–6994. [Google Scholar] [CrossRef]
- Spriano, F.; Tarantelli, C.; Golino, G.; Gaudio, E.; Scalise, L.; Cascione, L.; Zucca, E.; Van Berkel, P.; Stathis, A.; Zammarchi, F.; et al. The anti-CD25 antibody-drug conjugate camidanlumab tesirine (adct-301) presents a strong preclinical activity both as single agent and in combination in lymphoma cell lines. Hematol. Oncol. 2019, 37, 323–324. [Google Scholar] [CrossRef]
- Chari, R.V.J.; Miller, M.L.; Widdison, W.C. Antibody-drug conjugates: An emerging concept in cancer therapy. Angew. Chem. Int. Ed. 2014, 53, 3796–3827. [Google Scholar] [CrossRef]
- Miller, M.; Fishkin, N.; Li, W.; Leece, B.; Mayo, M.; Jones, G.; Reid, E.; Archer, K.; Maloney, E.; Kovtun, Y.; et al. Abstract B126: Potent antigen-specific anti-tumor activity observed with antibody-drug conjugates (ADCs) made using a new class of DNA-crosslinking agents. Mol. Cancer Ther. 2009, 8, B126. [Google Scholar]
- Pahl, A.; Lutz, C.; Hechler, T. Amanitins and their development as a payload for antibody-drug conjugates. Drug Discov. Today Technol. 2018, 30, 85–89. [Google Scholar] [CrossRef]
- Pahl, A.; Lutz, C.; Hechler, T. CHAPTER 19. Amatoxins as RNA Polymerase II Inhibiting Antibody–Drug Conjugate (ADC) Payloads. In Cytotoxic Payloads for Antibody–Drug Conjugates; Royal Society of Chemistry: London, UK, 2019; pp. 398–426. [Google Scholar]
- Tai, Y.-T.; Anderson, K.C. Targeting B-cell maturation antigen in multiple myeloma. Immunotherapy 2015, 7, 1187–1199. [Google Scholar] [CrossRef] [PubMed]
- Hechler, T.; Palfi, A.; Müller, C.; Lutz, C.; Pahl, A.; Kulke, M. Abstract 77: Preclinical evaluation of HDP-101, an anti-BCMA antibody-drug conjugate. In Experimental and Molecular Therapeutics; American Association for Cancer Research: Philadelphia, PA, USA, 2017; p. 77. [Google Scholar]
- Ponte, J.F.; Ab, O.; Lanieri, L.; Lee, J.; Coccia, J.; Bartle, L.M.; Themeles, M.; Zhou, Y.; Pinkas, J.; Ruiz-Soto, R. Mirvetuximab Soravtansine (IMGN853), a Folate Receptor Alpha Targeting Antibody-Drug Conjugate, Potentiates the Activity of Standard of Care Therapeutics in Ovarian Cancer Models. Neoplasia 2016, 18, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Ponte, J.F.; Yoder, N.C.; Laleau, R.; Coccia, J.; Lanieri, L.; Qiu, Q.; Wu, R.; Hong, E.; Bogalhas, M.; et al. Effects of Drug-Antibody Ratio on Pharmacokinetics, Biodistribution, Efficacy, and Tolerability of Antibody-Maytansinoid Conjugates. Bioconjug. Chem. 2017, 28, 1371–1381. [Google Scholar] [CrossRef]
- Widdison, W.C.; Ponte, J.F.; Coccia, J.A.; Lanieri, L.; Setiady, Y.; Dong, L.; Skaletskaya, A.; Hong, E.E.; Wu, R.; Qiu, Q.; et al. Development of Anilino-Maytansinoid ADCs that Efficiently Release Cytotoxic Metabolites in Cancer Cells and Induce High Levels of Bystander Killing. Bioconjug. Chem. 2015, 26, 2261–2278. [Google Scholar] [CrossRef] [PubMed]
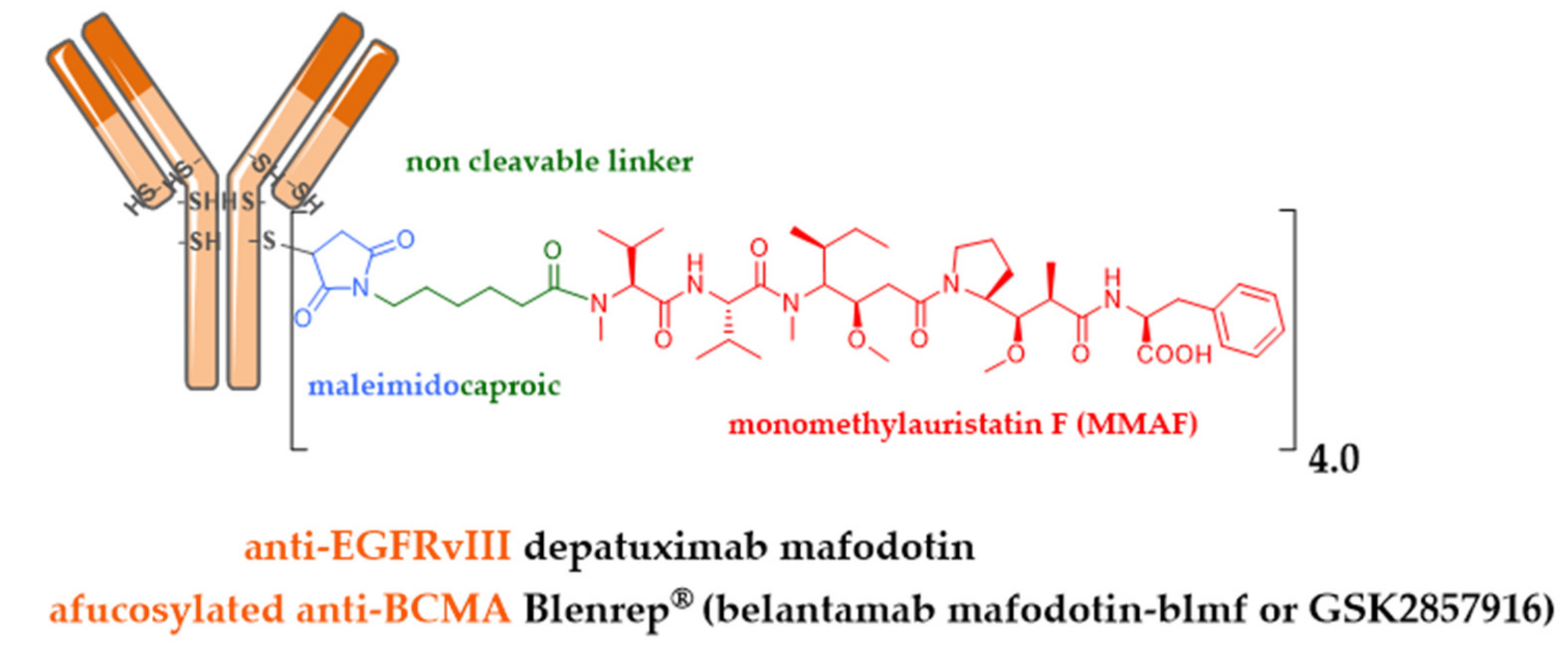
- Goss, G.D.; Vokes, E.E.; Gordon, M.S.; Gandhi, L.; Papadopoulos, K.P.; Rasco, D.W.; Fischer, J.D.S.; Chu, K.L.; Ames, W.W.; Mittapalli, R.K.; et al. Efficacy and safety results of depatuxizumab mafodotin (ABT-414) in patients with advanced solid tumors likely to overexpress epidermal growth factor receptor. Cancer 2018, 124, 2174–2183. [Google Scholar] [CrossRef]
- Van den Bent, M.; Gan, H.K.; Lassman, A.B.; Kumthekar, P.; Merrell, R.; Butowski, N.; Lwin, Z.; Mikkelsen, T.; Nabors, L.B.; Papadopoulos, K.P.; et al. Efficacy of depatuxizumab mafodotin (ABT-414) monotherapy in patients with EGFR-amplified, recurrent glioblastoma: Results from a multi-center, international study. Cancer Chemother. Pharmacol. 2017, 80, 1209–1217. [Google Scholar] [CrossRef]
- Phillips, A.C.; Boghaert, E.R.; Vaidya, K.S.; Mitten, M.J.; Norvell, S.; Falls, H.D.; DeVries, P.J.; Cheng, D.; Meulbroek, J.A.; Buchanan, F.G.; et al. ABT-414, an Antibody-Drug Conjugate Targeting a Tumor-Selective EGFR Epitope. Mol. Cancer Ther. 2016, 15, 661–669. [Google Scholar] [CrossRef]
- Lonial, S.; Lee, H.C.; Badros, A.; Trudel, S.; Nooka, A.K.; Chari, A.; Abdallah, A.O.; Callander, N.; Lendvai, N.; Sborov, D.; et al. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM-2): A two-arm, randomised, open-label, phase 2 study. Lancet Oncol. 2020, 21, 207–221. [Google Scholar] [CrossRef]
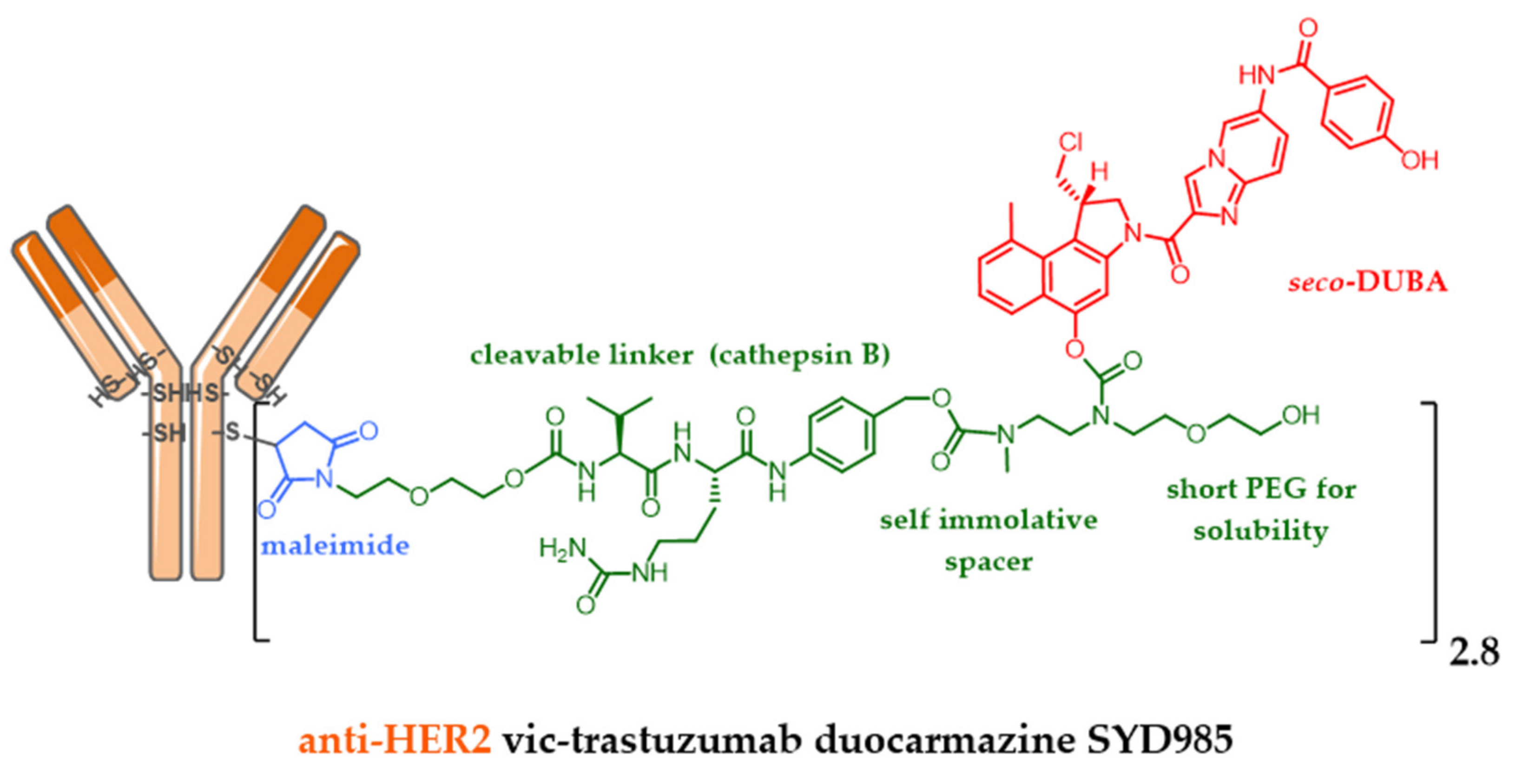
- Elgersma, R.C.; Coumans, R.G.E.; Huijbregts, T.; Menge, W.M.P.B.; Joosten, J.A.F.; Spijker, H.J.; De Groot, F.M.H.; Van Der Lee, M.M.C.; Ubink, R.; Van Den Dobbelsteen, D.J.; et al. Design, synthesis, and evaluation of linker-duocarmycin payloads: Toward selection of HER2-targeting antibody-drug conjugate SYD985. Mol. Pharm. 2015, 12, 1813–1835. [Google Scholar] [CrossRef]
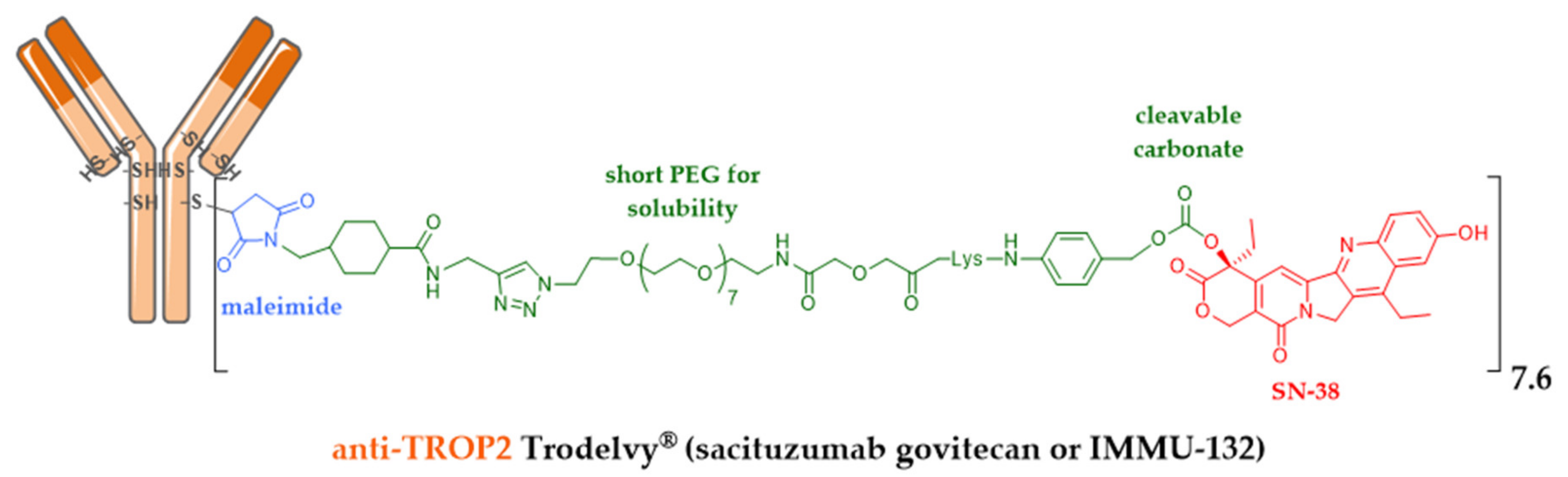
- Goldenberg, D.M.; Cardillo, T.M.; Govindan, S.V.; Rossi, E.A.; Sharkey, R.M. Trop-2 is a novel target for solid cancer therapy with sacituzumab govitecan (IMMU-132), an antibody-drug conjugate (ADC)*. Oncotarget 2015, 6, 22496–22512. [Google Scholar] [CrossRef]
- Cardillo, T.M.; Govindan, S.V.; Sharkey, R.M.; Trisal, P.; Arrojo, R.; Liu, D.; Rossi, E.A.; Chang, C.H.; Goldenberg, D.M. Sacituzumab govitecan (IMMU-132), an Anti-Trop-2/SN-38 antibody-drug conjugate: Characterization and efficacy in pancreatic, gastric, and other cancers. Bioconjug. Chem. 2015, 26, 919–931. [Google Scholar] [CrossRef] [PubMed]
- Govindan, S.V.; Starodub, A.N.; Juric, D.; Abramson, V.; Sharkey, R.M.; Wegener, W.A.; Tolaney, S.M.; Kalinsky, K.; O’Shaughnessy, J.; Maliakal, P.; et al. Efficacy and Safety of Anti-Trop-2 Antibody Drug Conjugate Sacituzumab Govitecan (IMMU-132) in Heavily Pretreated Patients With Metastatic Triple-Negative Breast Cancer. J. Clin. Oncol. 2018, 2017, 2141–2148. [Google Scholar]
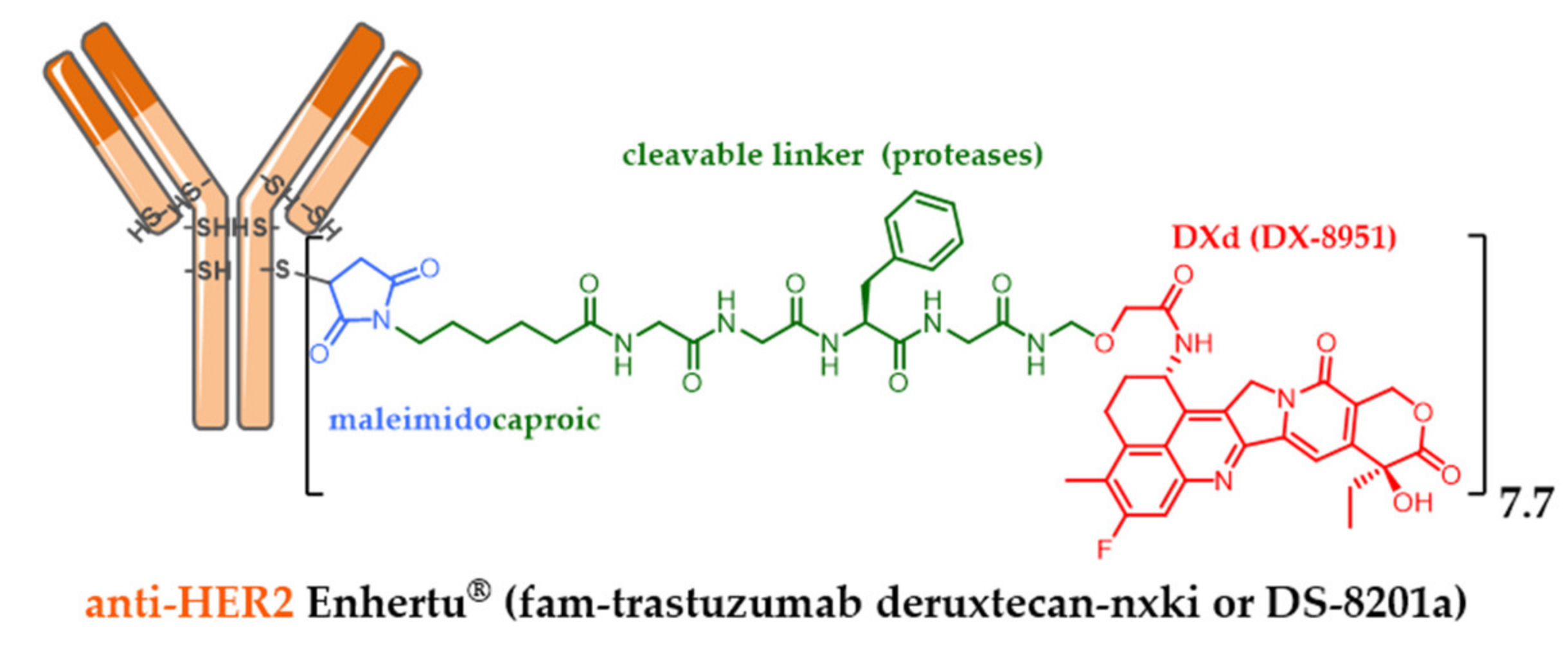
- Nakada, T.; Masuda, T.; Naito, H.; Yoshida, M.; Ashida, S.; Morita, K.; Miyazaki, H.; Kasuya, Y.; Ogitani, Y.; Yamaguchi, J.; et al. Novel antibody drug conjugates containing exatecan derivative-based cytotoxic payloads. Bioorg. Med. Chem. Lett. 2016, 26, 1542–1545. [Google Scholar] [CrossRef] [PubMed]
- Ogitani, Y.; Aida, T.; Hagihara, K.; Yamaguchi, J.; Ishii, C.; Harada, N.; Soma, M.; Okamoto, H.; Oitate, M.; Arakawa, S.; et al. DS-8201a, a novel HER2-targeting ADC with a novel DNA topoisomerase I inhibitor, demonstrates a promising antitumor efficacy with differentiation from T-DM1. Clin. Cancer Res. 2016, 22, 5097–5108. [Google Scholar] [CrossRef]
- Nakada, T.; Sugihara, K.; Jikoh, T.; Abe, Y.; Agatsuma, T. Drug Discovery: Recent Progress and the Future The Latest Research and Development into the Antibody—Drug for HER2 Cancer Therapy. Chem. Pharm. Bull. 2019, 67, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Hagenbeek, A.; Mooij, H.; Zijlstra, J.; Lugtenburg, P.; van Imhoff, G.; Nijland, M.; Tonino, S.; Hutchings, M.; Spiering, M.; Liu, R.; et al. Phase I dose-escalation study of brentuximab-vedotin combined with dexamethasone, high-dose cytarabine and cisplatin, as salvage treatment in relapsed/refractory classical Hodgkin lymphoma: The HOVON/LLPC Transplant BRaVE study. Haematologica 2019, 104, e151–e153. [Google Scholar] [CrossRef] [PubMed]
- Von Minckwitz, G.; Huang, C.-S.; Mano, M.S.; Loibl, S.; Mamounas, E.P.; Untch, M.; Wolmark, N.; Rastogi, P.; Schneeweiss, A.; Redondo, A.; et al. Trastuzumab Emtansine for Residual Invasive HER2-Positive Breast Cancer. N. Engl. J. Med. 2019, 380, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, K.M.; Rennert, P.D.; Freeman, G.J. Combination cancer immunotherapy and new immunomodulatory targets. Nat. Rev. Drug Discov. 2015, 14, 561–584. [Google Scholar] [CrossRef] [PubMed]
- Herrera, A.F.; Moskowitz, A.J.; Bartlett, N.L.; Vose, J.M.; Ramchandren, R.; Feldman, T.A.; LaCasce, A.S.; Ansell, S.M.; Moskowitz, C.H.; Fenton, K.; et al. Interim results of brentuximab vedotin in combination with nivolumab in patients with relapsed or refractory Hodgkin lymphoma. Blood 2018, 131, 1183–1194. [Google Scholar] [CrossRef] [PubMed]
- Armand, P.; Engert, A.; Younes, A.; Fanale, M.; Santoro, A.; Zinzani, P.L.; Timmerman, J.M.; Collins, G.P.; Ramchandren, R.; Cohen, J.B.; et al. Nivolumab for Relapsed/Refractory Classic Hodgkin Lymphoma After Failure of Autologous Hematopoietic Cell Transplantation: Extended Follow-Up of the Multicohort Single-Arm Phase II CheckMate 205 Trial. J. Clin. Oncol. 2018, 36, 1428–1439. [Google Scholar] [CrossRef] [PubMed]
- Faltas, B.; Goldenberg, D.M.; Ocean, A.J.; Govindan, S.V.; Wilhelm, F.; Sharkey, R.M.; Hajdenberg, J.; Hodes, G.; Nanus, D.M.; Tagawa, S.T. Sacituzumab Govitecan, a Novel Antibody–Drug Conjugate, in Patients With Metastatic Platinum-Resistant Urothelial Carcinoma. Clin. Genitourin. Cancer 2016, 14, e75–e79. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Wang, R.E.; Wang, F. Antibody-drug conjugates for non-oncological indications. Expert Opin. Biol. Ther. 2016, 16, 591–593. [Google Scholar] [CrossRef] [PubMed]
- Graversen, J.H.; Svendsen, P.; Dagnæs-Hansen, F.; Dal, J.; Anton, G.; Etzerodt, A.; Petersen, M.D.; Christensen, P.A.; Møller, H.J.; Moestrup, S.K. Targeting the hemoglobin scavenger receptor CD163 in macrophages highly increases the anti-inflammatory potency of dexamethasone. Mol. Ther. 2012, 20, 1550–1558. [Google Scholar] [CrossRef] [PubMed]
- Graversen, J.H.; Moestrup, S.K. Drug trafficking into macrophages via the endocytotic receptor CD163. Membranes 2015, 5, 228–252. [Google Scholar] [CrossRef]
- Brandish, P.E.; Palmieri, A.; Antonenko, S.; Beaumont, M.; Benso, L.; Cancilla, M.; Cheng, M.; Fayadat-Dilman, L.; Feng, G.; Figueroa, I.; et al. Development of Anti-CD74 Antibody-Drug Conjugates to Target Glucocorticoids to Immune Cells. Bioconjug. Chem. 2018, 29, 2357–2369. [Google Scholar] [CrossRef]
- McPherson, M.J.; Hobson, A.D.; Hayes, M.E.; Marvin, C.C.; Schmidt, D.; Waegell, W.; Goess, C.; Oh, J.Z.; Hernandez, A., Jr.; Randolph, J.T. Glucocorticoid Receptor Agonist and Immunoconjugates Thereof. U.S. Patent No. WO2017210471, 7 December 2017. [Google Scholar]
- Wang, R.E.; Liu, T.; Wang, Y.; Cao, Y.; Du, J.; Luo, X.; Deshmukh, V.; Kim, C.H.; Lawson, B.R.; Tremblay, M.S.; et al. An immunosuppressive antibody-drug conjugate. J. Am. Chem. Soc. 2015, 137, 3229–3232. [Google Scholar] [CrossRef]
- Lim, R.K.V.; Yu, S.; Cheng, B.; Li, S.; Kim, N.J.; Cao, Y.; Chi, V.; Kim, J.Y.; Chatterjee, A.K.; Schultz, P.G.; et al. Targeted Delivery of LXR Agonist Using a Site-Specific Antibody-Drug Conjugate. Bioconjug. Chem. 2015, 26, 2216–2222. [Google Scholar] [CrossRef]
- Hardt, W.D. Antibiotics: Homed to the hideout. Nature 2015, 527, 309–310. [Google Scholar] [CrossRef]
- Lehar, S.M.; Pillow, T.; Xu, M.; Staben, L.; Kajihara, K.K.; Vandlen, R.; DePalatis, L.; Raab, H.; Hazenbos, W.L.; Hiroshi Morisaki, J.; et al. Novel antibody-antibiotic conjugate eliminates intracellular S. aureus. Nature 2015, 527, 323–328. [Google Scholar] [CrossRef]























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Company | ADC (Cytotoxic) | Isotype and Target | Indication/Approval Date (Trade Name)/Clinical Status |
|---|---|---|---|
| Pfizer | gemtuzumab ozogamicin (CAL) | IgG4 CD33 | 2000–2010/2017 AML (Mylotarg®) |
| Seattle Genetics | brentuximab vedotin (AUR) | IgG1 CD30 | 2011 ALCL and Hodgkin lymphoma (Adcetris®) |
| Roche | trastuzumab emtansine (MAY) | IgG1 HER2+ | 2013 metastatic HER2+++ breast cancer (Kadcyla®) ** |
| Pfizer | inotuzumab ozogamicin (CAL) | IgG4 CD22 | 2017 ALL and CLL (Besponsa®) |
| Roche | polatuzumab vedotin (AUR) | IgG1 CD79b | 2019 DLBCL (Polivy®) |
| Seattle Genetics | enfortumab vedotin (AUR) | IgG1 Nectin 4 | 2019 urothelial cancer (Padcev®) ** |
| Daiichi Sankyo | trastuzumab deruxtecan (EXA) | IgG1 HER2+ | 2019 metastatic HER2+++ breast cancer (Enhertu®) ** |
| Immunomedics | sacituzumab govitecan (IRI) | IgG1 TROP-2 | 2020, metastatic TNBC (Trodelvy®) ** |
| GSK | belantamab mafodotin (AUR, MMAF) | IgG1afuc BCMA | 2020, multiple myeloma (Blenrep®) |
| Byondis * | trastuzumab duocarmazine (DUO) | IgG1 HER2+ | Ph 3 metastatic HER2+++ breast cancer (vs T-DM1) ** |
| Bio-Thera | BAT8001 (MAY) | IgG1 HER2+ | Ph 3 breast cancer HER2+++ (vs T-DM1) ** |
| ImmunoGen | mirvetuximab soravtansine (MAY) | IgG1 Folate R1 | Ph 3 epithelial ovarian cancer ** |
| Sanofi (IMG) | SAR408701 (MAY, DM4) | IgG1 CEACAM5 | Ph 3 metastatic small cell lung cancer ** |
| ADC-Therapeutics | loncastuximab tesirine (PBD) | IgG1 CD19 | Piv. Ph2 diffuse large B-Cell lymphoma (BLA 2H2020) |
| ADC-Therapeutics | camidanlumab tesirine (PBD) | IgG1 CD25 | Pivotal Ph2 Hodgkin lymphoma |
| Seattle Genetics | vadastuximab talirine (PBD) | IgG1 CD33 | Ph 3 AML (stopped, 2017) |
| AbbVie | rovalpituzumab tesirine (PBD) | IgG1 DLL3 | Ph 3 small cell lung cancer (stopped, 2019) ** |
| AbbVie | depatuxizumab mafodotin (AUR) | IgG1 EGFRvIII | Ph 3 glioblastome (stopped, 2019) ** |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joubert, N.; Beck, A.; Dumontet, C.; Denevault-Sabourin, C. Antibody–Drug Conjugates: The Last Decade. Pharmaceuticals 2020, 13, 245. https://doi.org/10.3390/ph13090245
Joubert N, Beck A, Dumontet C, Denevault-Sabourin C. Antibody–Drug Conjugates: The Last Decade. Pharmaceuticals. 2020; 13(9):245. https://doi.org/10.3390/ph13090245
Chicago/Turabian StyleJoubert, Nicolas, Alain Beck, Charles Dumontet, and Caroline Denevault-Sabourin. 2020. "Antibody–Drug Conjugates: The Last Decade" Pharmaceuticals 13, no. 9: 245. https://doi.org/10.3390/ph13090245
APA StyleJoubert, N., Beck, A., Dumontet, C., & Denevault-Sabourin, C. (2020). Antibody–Drug Conjugates: The Last Decade. Pharmaceuticals, 13(9), 245. https://doi.org/10.3390/ph13090245