Altered Brain Adiponectin Receptor Expression in the 5XFAD Mouse Model of Alzheimer’s Disease



Abstract
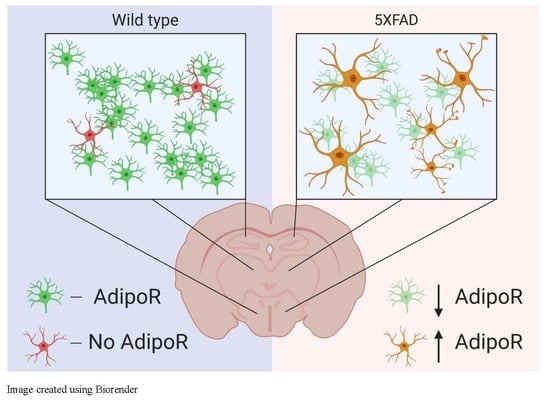
{kind=link}
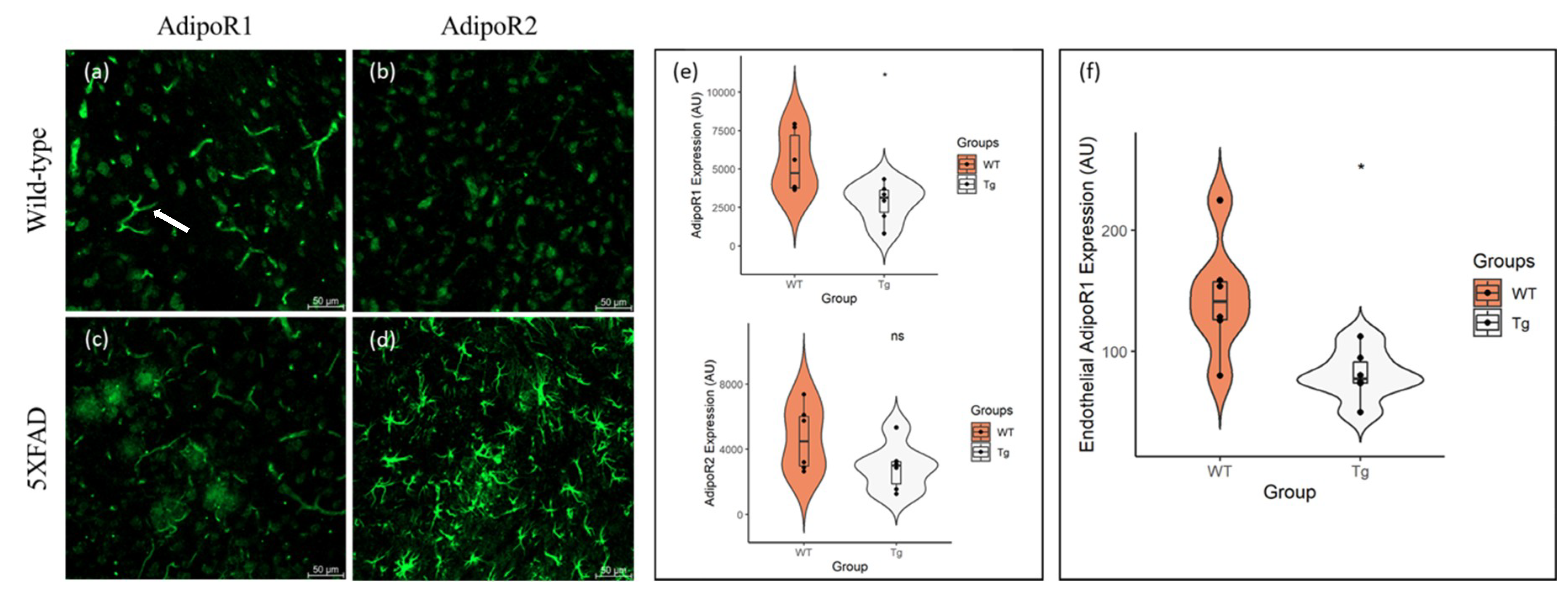{kind=link}
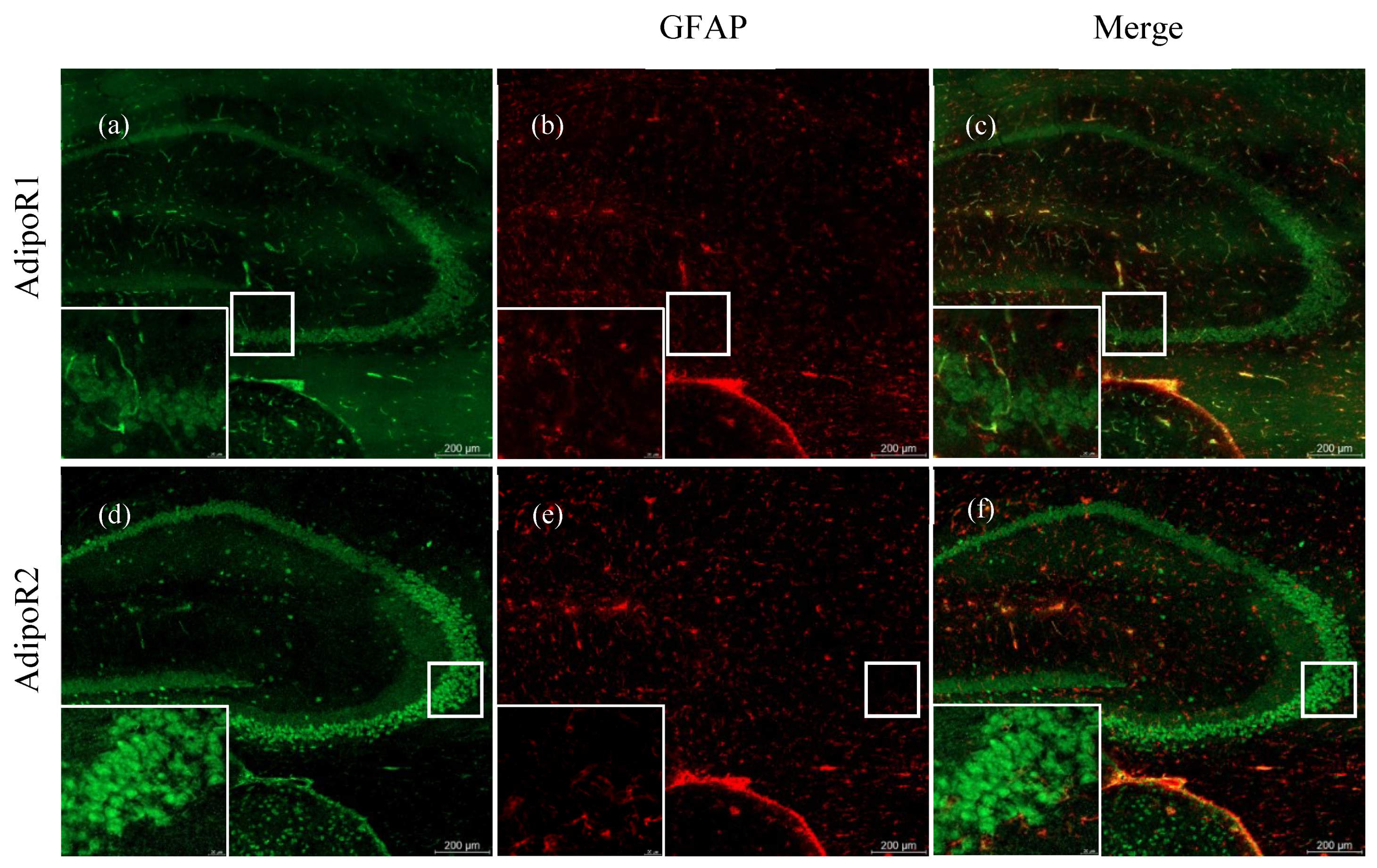{kind=link}
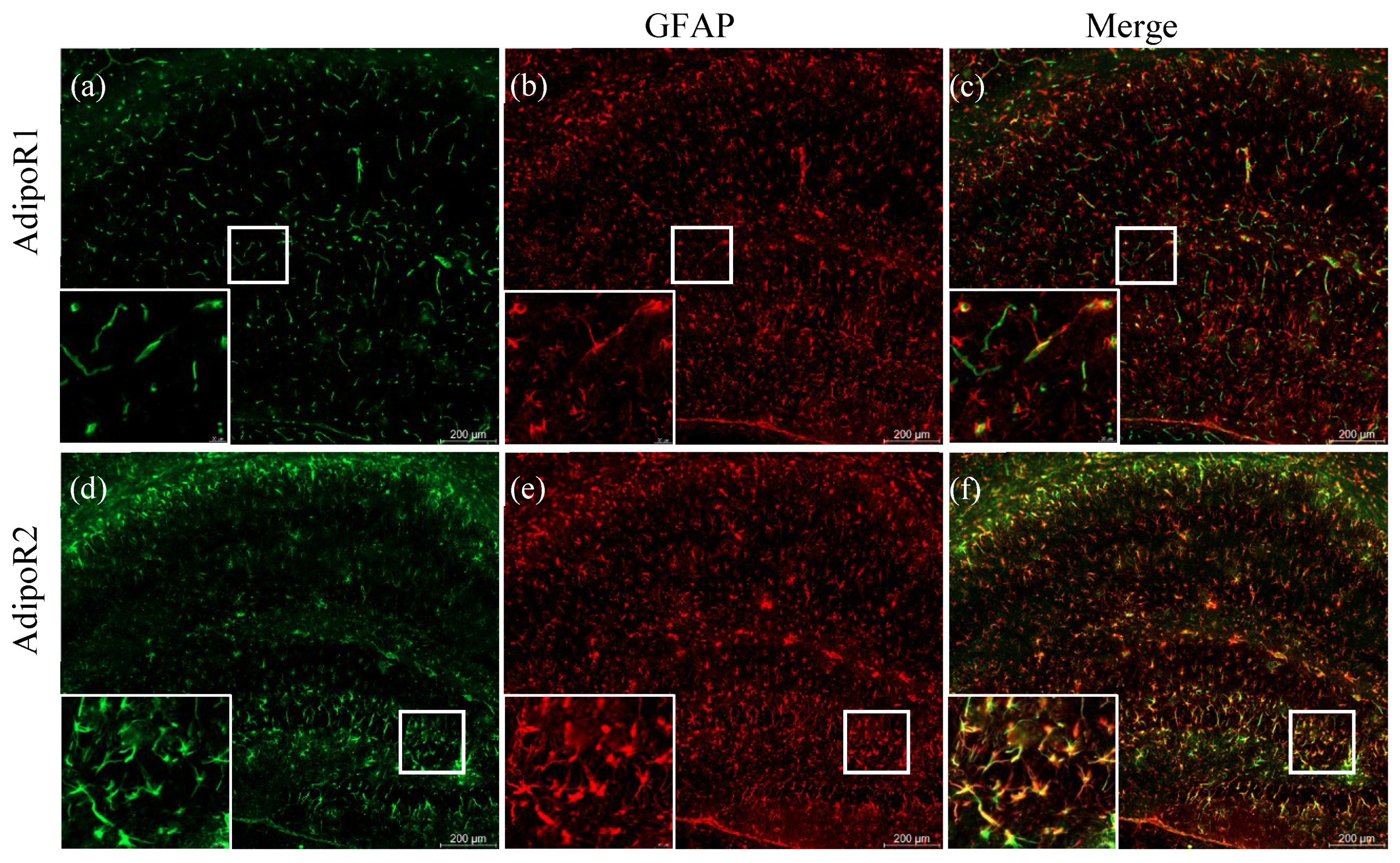{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. AdipoR1 and AdipoR2 Is Expressed throughout the Mouse Cortex and Hippocampus
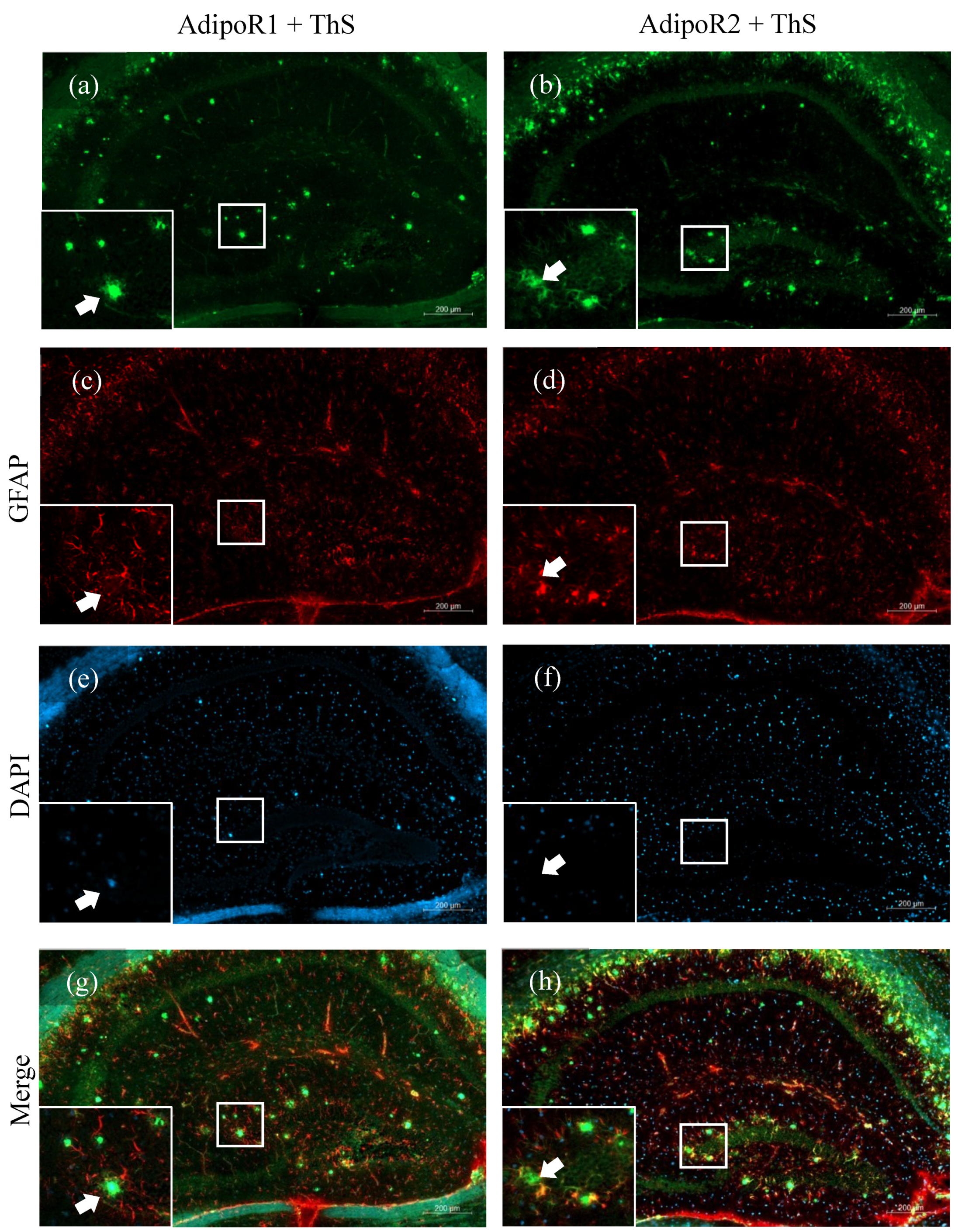
2.2. Astrocytes Express AdipoR2 in the 5XFAD Mouse
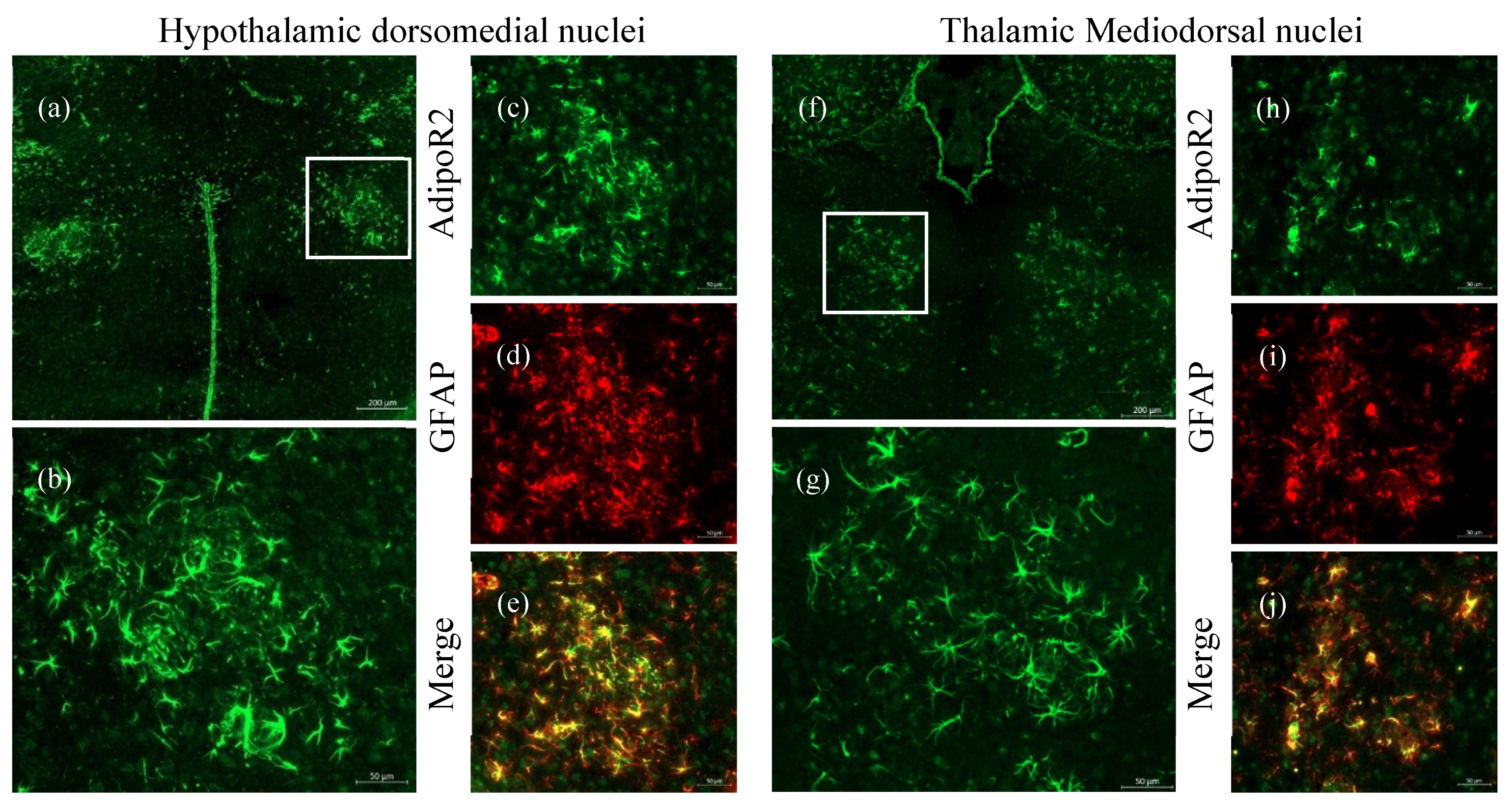
2.3. Increased Expression of AdipoR2 in Thalamic and Hypothalamic Areas
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Animals
4.3. Tissue Collection and Preparation
4.4. Immunofluorescence
4.5. Thioflavin-S Staining
4.6. Quantification Analysis
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Alzheimer’s Association. 2017 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2017, 13, 325–373. [Google Scholar] [CrossRef]
- Arshavsky, Y.I. Alzheimer Disease and Cellular Mechanisms of Memory Storage. J. Neuropathol. Exp. Neurol. 2014, 73, 192–205. [Google Scholar] [CrossRef] [PubMed]
- Murayama, O.; Tomita, T.; Nihonmatsu, N.; Murayama, M.; Sun, X.; Honda, T.; Iwatsubo, T.; Takashima, A. Enhancement of amyloid β 42 secretion by 28 different presenilin 1 mutations of familial Alzheimer’s disease. Neurosci. Lett. 1999, 265, 61–63. [Google Scholar] [CrossRef]
- Wirths, O.; Multhaup, G.; Czech, C.; Blanchard, V.; Moussaoui, S.; Tremp, G.; Pradier, L.; Beyreuther, K.; Bayer, T.A. Intraneuronal Aβ accumulation precedes plaque formation in β-amyloid precursor protein and presenilin-1 double-transgenic mice. Neurosci. Lett. 2001, 306, 116–120. [Google Scholar] [CrossRef]
- Iizuka, T.; Shoji, M.; Kawarabayashi, T.; Sato, M.; Kobayashi, T.; Tada, N.; Kasai, K.; Matsubara, E.; Watanabe, M.; Tomidokoro, Y.; et al. Intracellular generation of amyloid beta-protein from amyloid beta- protein precursor fragment by direct cleavage with beta- and gamma- secretase. Biochem. Biophys. Res. Commun. 1996, 218, 238–242. [Google Scholar] [CrossRef]
- Boissière, F.; Hunot, S.; Faucheux, B.; Duyckaerts, C.; Hauw, J.J.; Agid, Y.; Hirsch, E.C. Nuclear translocation of NF-κB in cholinergic neurons of patients with Alzheimer’s disease. NeuroReport 1997, 8, 2849–2852. [Google Scholar] [CrossRef]
- Chami, L.; Buggia-Prévot, V.; Duplan, E.; Delprete, D.; Chami, M.; Peyron, J.F.; Checler, F. Nuclear factor-κB regulates βAPP and β- and γ-secretases differently at physiological and supraphysiological Aβ concentrations. J. Biol. Chem. 2012, 287, 24573–24584. [Google Scholar] [CrossRef]
- Finder, V.H.; Glockshuber, R. Amyloid-β aggregation. Neurodegener. Dis. 2007, 4, 13–27. [Google Scholar] [CrossRef]
- Holsinger, R.M.D.; McLean, C.A.; Beyreuther, K.; Masters, C.L.; Evin, G. Increased expression of the amyloid precursor β-secretase in Alzheimer’s disease. Ann. Neurol. 2002, 51, 783–786. [Google Scholar] [CrossRef]
- Irvine, G.; El-Agnaf, O. Protein Aggregation in the Brain: The Molecular Basis for Alzheimer’s and Parkinson’s Diseases. Mol. Med. 2008, 14, 451–464. [Google Scholar] [CrossRef]
- Mandelkow, E.M.; Stamer, K.; Vogel, R.; Thies, E.; Mandelkow, E. Clogging of axons by tau, inhibition of axonal traffic and starvation of synapses. Neurobiol. Aging 2003, 24, 1079–1085. [Google Scholar] [CrossRef] [PubMed]
- Blasko, I.; Veerhuis, R.; Stampfer-Kountchev, M.; Saurwein-Teissl, M.; Eikelenboom, P.; Grubeck-Loebenstein, B. Costimulatory effects of interferon-β and interleukin-1β or tumor necrosis factor α on the synthesis of Aβ1-40 and Aβ1-42 by human astrocytes. Neurobiol. Dis. 2000, 7, 682–689. [Google Scholar] [CrossRef] [PubMed]
- Garwood, C.J.; Pooler, A.M.; Atherton, J.; Hanger, D.P.; Noble, W. Astrocytes are important mediators of Aβ-induced neurotoxicity and tau phosphorylation in primary culture. Cell Death Dis. 2011, 2, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Grolla, A.A.; Fakhfouri, G.; Balzaretti, G.; Marcello, E.; Gardoni, F.; Canonico, P.L.; DiLuca, M.; Genazzani, A.A.; Lim, D. Aβ leads to Ca2+ signaling alterations and transcriptional changes in glial cells. Neurobiol. Aging 2013, 34, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.M.; Cho, H.J.; Kim, Y.W.; Hwang, J.Y.; Mook-Jung, I. Aβ-induced Ca2+ influx regulates astrocytic BACE1 expression via calcineurin/NFAT4 signals. Biochem. Biophys. Res. Commun. 2012, 425, 649–655. [Google Scholar] [CrossRef]
- Pike, C.J. Sex and the development of Alzheimer’s disease. J. Neurosci. Res. 2017, 95, 671–680. [Google Scholar] [CrossRef]
- Wang, R.; Holsinger, R.M.D. Exercise-induced brain-derived neurotrophic factor expression: Therapeutic implications for Alzheimer’s dementia. Ageing Res. Rev. 2018, 48, 109–121. [Google Scholar] [CrossRef]
- Haan, M.N. Therapy insight: Type 2 diabetes mellitus and the risk of late-onset Alzheimer’s disease. Nat. Clin. Pract. Neurol. 2006, 2, 159–166. [Google Scholar] [CrossRef]
- Hazari, M.A.H.; Ram Reddy, B.; Uzma, N.; Santhosh Kumar, B. Cognitive impairment in type 2 diabetes mellitus. Int. J. Diabetes Mellit. 2015, 3, 19–24. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, F.; Inge, G.I.; Iqbal, K.; Gong, C.X. Deficient brain insulin signalling pathway in Alzheimer’s disease and diabetes. J Pathol. 2011, 225, 54–62. [Google Scholar] [CrossRef]
- Plucińska, K.; Dekeryte, R.; Koss, D.; Shearer, K.; Mody, N.; Whitfield, P.D.; Doherty, M.K.; Mingarelli, M.; Welch, A.; Riedel, G.; et al. Neuronal human BACE1 knockin induces systemic diabetes in mice. Diabetologia 2016, 59, 1513–1523. [Google Scholar] [CrossRef] [PubMed]
- Umeda, T.; Tomiyama, T.; Kitajima, E.; Idomoto, T.; Nomura, S.; Lambert, M.P.; Klein, W.L.; Mori, H. Hypercholesterolemia accelerates intraneuronal accumulation of Aβ oligomers resulting in memory impairment in Alzheimer’s disease model mice. Life Sci. 2012, 91, 1169–1176. [Google Scholar] [CrossRef]
- Arnold, S.E.; Arvanitakis, Z.; Macauley-Rambach, S.L.; Koenig, A.M.; Wang, H.Y.; Ahima, R.S.; Craft, S.; Gandy, S.; Buettner, C.; Stoeckel, L.E.; et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nat. Rev. Neurol. 2018, 14, 168–181. [Google Scholar] [CrossRef]
- Heni, M.; Kullmann, S.; Preissl, H.; Fritsche, A.; Häring, H.U. Impaired insulin action in the human brain: Causes and metabolic consequences. Nat. Rev. Endocrinol. 2015, 11, 701–711. [Google Scholar] [CrossRef]
- Jimenez, A.; Pegueroles, J.; Carmona-Iragui, M.; Vilaplana, E.; Montal, V.; Alcolea, D.; Videla, L.; Illán-Gala, I.; Pané, A.; Casajoana, A.; et al. Weight loss in the healthy elderly might be a non-cognitive sign of preclinical Alzheimer’s disease. Oncotarget 2017, 8, 104706–104716. [Google Scholar] [CrossRef]
- Jo, J.; Gavrilova, O.; Pack, S.; Jou, W.; Mullen, S.; Sumner, A.E.; Cushman, S.W.; Periwal, V. Hypertrophy and/or hyperplasia: Dynamics of adipose tissue growth. Plos Comput. Biol. 2009, 5, e1000324. [Google Scholar] [CrossRef]
- Dunmore, S.J.; Brown, J.E.P. The role of adipokines in β-cell failure of type 2 diabetes. J. Endocrinol. 2013, 216, T37–T45. [Google Scholar] [CrossRef] [PubMed]
- Kiliaan, A.J.; Arnoldussen, I.A.C.; Gustafson, D.R. Adipokines: A link between obesity and dementia? Lancet Neurol. 2014, 13, 913–923. [Google Scholar] [CrossRef]
- Song, J.; Lee, W.T.; Park, K.A.; Lee, J.E. Association between risk factors for vascular dementia and adiponectin. Biomed Res Int 2014, 2014, 261672. [Google Scholar] [CrossRef] [PubMed]
- Trayhurn, P. Hypoxia and Adipose Tissue Function and Dysfunction in Obesity. Physiol. Rev. 2013, 93, 1–21. [Google Scholar] [CrossRef]
- Bjursell, M.; Ahnmark, A.; Bohlooly-Y, M.; William-Olsson, L.; Rhedin, M.; Peng, X.R.; Ploj, K.; Gerdin, A.K.; Arnerup, G.; Elmgren, A.; et al. Opposing effects of adiponectin receptors 1 and 2 on energy metabolism. Diabetes 2007, 56, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Kashiwagi, H.; Shiraga, M.; Tadokoro, S.; Kamae, T.; Ujiie, H.; Honda, S.; Miyata, S.; Ijiri, Y.; Yamamoto, J.; et al. Adiponectin acts as an endogenous antithrombotic factor. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 224–230. [Google Scholar] [CrossRef]
- Nicolas, S.; Cazareth, J.; Zarif, H.; Guyon, A.; Heurteaux, C.; Chabry, J.; Petit-Paitel, A. Globular Adiponectin Limits Microglia Pro-Inflammatory Phenotype through an AdipoR1/NF-κB Signaling Pathway. Front. Cell. Neurosci. 2017, 11, 352. [Google Scholar] [CrossRef] [PubMed]
- Di Zazzo, E.; Polito, R.; Bartollino, S.; Nigro, E.; Porcile, C.; Bianco, A.; Daniele, A.; Moncharmont, B. Adiponectin as Link Factor between Adipose Tissue and Cancer. Int. J. Mol. Sci. 2019, 20, 839. [Google Scholar] [CrossRef]
- Coope, A.; Milanski, M.; Araújo, E.P.; Tambascia, M.; Saad, M.J.A.; Geloneze, B.; Velloso, L.A. AdipoR1 mediates the anorexigenic and insulin/leptin-like actions of adiponectin in the hypothalamus. FEBS Lett. 2008, 582, 1471–1476. [Google Scholar] [CrossRef] [PubMed]
- Mojiminiyi, O.A.; Abdella, N.A.; Al Arouj, M.; Ben Nakhi, A. Adiponectin, insulin resistance and clinical expression of the metabolic syndrome in patients with Type 2 diabetes. Int. J. Obes. 2007, 31, 213–220. [Google Scholar] [CrossRef][Green Version]
- Yoon, M.J.; Lee, G.Y.; Chung, J.-J.; Ahn, Y.H.; Hong, S.H.; Kim, J.B. Adiponectin Increases Fatty Acid Oxidation in Skeletal Muscle Cells by Sequential Activation of AMP-Activated Protein Kinase, p38 Mitogen-Activated Protein Kinase, and Peroxisome Proliferator–Activated Receptor α. Diabetes 2006, 55, 2562–2570. [Google Scholar] [CrossRef]
- Gradinaru, D.; Margina, D.; Borsa, C.; Ionescu, C.; Ilie, M.; Costache, M.; Dinischiotu, A.; Prada, G.I. Adiponectin: Possible link between metabolic stress and oxidative stress in the elderly. Aging Clin. Exp. Res. 2017, 29, 621–629. [Google Scholar] [CrossRef]
- Yamauchi, T.; Iwabu, M.; Okada-Iwabu, M.; Kadowaki, T. Adiponectin receptors: A review of their structure, function and how they work. Best Pract. Res. Clin. Endocrinol. Metab. 2014, 28, 15–23. [Google Scholar] [CrossRef]
- Yamauchi, T.; Nio, Y.; Maki, T.; Kobayashi, M.; Takazawa, T.; Iwabu, M.; Okada-Iwabu, M.; Kawamoto, S.; Kubota, N.; Kubota, T.; et al. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nat. Med. 2007, 13, 332–339. [Google Scholar] [CrossRef]
- Ng, R.C.L.; Cheng, O.Y.; Jian, M.; Kwan, J.S.C.; Ho, P.W.L.; Cheng, K.K.Y.; Yeung, P.K.K.; Zhou, L.L.; Hoo, R.L.C.; Chung, S.K.; et al. Chronic adiponectin deficiency leads to Alzheimer’s disease-like cognitive impairments and pathologies through AMPK inactivation and cerebral insulin resistance in aged mice. Mol. Neurodegener. 2016, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Waragai, M.; Ho, G.; Takamatsu, Y.; Sekiyama, K.; Sugama, S.; Takenouchi, T.; Masliah, E.; Hashimoto, M. Importance of adiponectin activity in the pathogenesis of Alzheimer’s disease. Ann. Clin. Transl. Neurol. 2017, 4, 591–600. [Google Scholar] [CrossRef]
- Wennberg, A.M.V.; Gustafson, D.; Hagen, C.E.; Roberts, R.O.; Knopman, D.; Jack, C.; Petersen, R.C.; Mielke, M.M. Serum Adiponectin Levels, Neuroimaging, and Cognition in the Mayo Clinic Study of Aging. J. Alzheimer’s Dis. 2016, 53, 573–581. [Google Scholar] [CrossRef]
- Waragai, M.; Adame, A.; Trinh, I.; Sekiyama, K.; Takamatsu, Y.; Une, K.; Masliah, E.; Hashimoto, M. Possible Involvement of Adiponectin, the Anti-Diabetes Molecule, in the Pathogenesis of Alzheimer’s Disease. J. Alzheimer’s Dis. 2016, 52, 1453–1459. [Google Scholar] [CrossRef] [PubMed]
- Ng, R.C.; Jian, M.; Ma, O.K.; Bunting, M.; Kwan, J.S.; Zhou, G.J.; Senthilkumar, K.; Iyaswamy, A.; Chan, P.K.; Li, M.; et al. Chronic oral administration of adipoRon reverses cognitive impairments and ameliorates neuropathology in an Alzheimer’s disease mouse model. Mol. Psychiatry 2020. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.W.; Abid, N.B.; Jo, M.H.; Jo, M.G.; Yoon, G.H.; Kim, M.O. Suppression of adiponectin receptor 1 promotes memory dysfunction and Alzheimer’s disease-like pathologies. Sci. Rep. 2017, 7, 12435. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, F.; Liu, F.; Chen, S.-y. Aging effects on circulating adiponectin levels and the expressions of adiponectin and adiponectin receptor 1 in the brains of male rats. Int. J. Gerontol. 2018, 12, 227–232. [Google Scholar] [CrossRef]
- Guillod-Maximin, E.; Roy, A.F.; Vacher, C.M.; Aubourg, A.; Bailleux, V.; Lorsignol, A.; Pénicaud, L.; Parquet, M.; Taouis, M. Adiponectin receptors are expressed in hypothalamus and colocalized with proopiomelanocortin and neuropeptide Y in rodent arcuate neurons. J. Endocrinol. 2009, 200, 93–105. [Google Scholar] [CrossRef]
- Song, J.; Choi, S.M.; Whitcomb, D.J.; Kim, B.C. Adiponectin controls the apoptosis and the expression of tight junction proteins in brain endothelial cells through AdipoR1 under beta amyloid toxicity. Cell Death Dis. 2017, 8, e3102. [Google Scholar] [CrossRef]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-Amyloid Aggregates, Neurodegeneration, and Neuron Loss in Transgenic Mice with Five Familial Alzheimer’s Disease Mutations: Potential Factors in Amyloid Plaque Formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef]
- Ishii, M.; Iadecola, C. Metabolic and Non-Cognitive Manifestations of Alzheimers Disease: The Hypothalamus as Both Culprit and Target of Pathology. Cell Metab. 2015, 22, 761–776. [Google Scholar] [CrossRef] [PubMed]
- Douglass, J.D.; Dorfman, M.D.; Fasnacht, R.; Shaffer, L.D.; Thaler, J.P. Astrocyte IKKβ/NF-κB signaling is required for diet-induced obesity and hypothalamic inflammation. Mol. Metab. 2017, 6, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Reichel, J.M.; Han, C.; Zuniga-Hertz, J.P.; Cai, D. Astrocytic Process Plasticity and IKKβ/NF-κB in Central Control of Blood Glucose, Blood Pressure, and Body Weight. Cell Metab. 2017, 25, 1091–1102.e1094. [Google Scholar] [CrossRef] [PubMed]
- Polanco, J.C.; Li, C.; Bodea, L.G.; Martinez-Marmol, R.; Meunier, F.A.; Götz, J. Amyloid-β and tau complexity—Towards improved biomarkers and targeted therapies. Nat. Rev. Neurol. 2018, 14, 22–40. [Google Scholar] [CrossRef]
- Gyoneva, S.; Swanger, S.A.; Zhang, J.; Weinshenker, D.; Traynelis, S.F. Altered motility of plaque-associated microglia in a model of Alzheimer’s disease. Neuroscience 2016, 330, 410–420. [Google Scholar] [CrossRef] [PubMed]
- Henstridge, C.M.; Hyman, B.T.; Spires-Jones, T.L. Beyond the neuron–cellular interactions early in Alzheimer disease pathogenesis. Nat. Rev. Neurosci. 2019, 20, 94–108. [Google Scholar] [CrossRef]
- Zhao, L.; Chen, S.; Sherchan, P.; Ding, Y.; Zhao, W.; Guo, Z.; Yu, J.; Tang, J.; Zhang, J.H. Recombinant CTRP9 administration attenuates neuroinflammation via activating adiponectin receptor 1 after intracerebral hemorrhage in mice. J. Neuroinflamm. 2018, 15, 215. [Google Scholar] [CrossRef]
- Baker, L.D.; Cross, D.J.; Minoshima, S.; Belongia, D.; Stennis Watson, G.; Craft, S. Insulin resistance and alzheimer-like reductions in regional cerebral glucose metabolism for cognitively normal adults with prediabetes or early type 2 diabetes. Arch. Neurol. 2011, 68, 51–57. [Google Scholar] [CrossRef]
- Diehl, T.; Mullins, R.; Kapogiannis, D. Insulin resistance in Alzheimer’s disease. Transl. Res. 2017, 183, 26–40. [Google Scholar] [CrossRef]
- Ferreira, L.S.S.; Fernandes, C.S.; Vieira, M.N.N.; De Felice, F.G. Insulin Resistance in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 830. [Google Scholar] [CrossRef]
- Hayashi, K.; Kurioka, S.; Yamaguchi, T.; Morita, M.; Kanazawa, I.; Takase, H.; Wada, A.; Kitagaki, H.; Nagai, A.; Bokura, H.; et al. Association of cognitive dysfunction with hippocampal atrophy in elderly Japanese people with type 2 diabetes. Diabetes Res. Clin. Pract. 2011, 94, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; de la Monte, S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease--is this type 3 diabetes? J. Alzheimer’s Dis. JAD 2005, 7, 63–80. [Google Scholar] [CrossRef]
- Arrieta-Cruz, I.; Gutiérrez-Juárez, R. The Role of Insulin Resistance and Glucose Metabolism Dysregulation in the Development of Alzheimer’s Disease. Rev. Investig. Clin. 2016, 68, 53–58. [Google Scholar] [CrossRef]
- Yamauchi, T.; Kamon, J.; Minokoshi, Y.; Ito, Y.; Waki, H.; Uchida, S.; Yamashita, S.; Noda, M.; Kita, S.; Ueki, K.; et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 2002, 8, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Cova, I.; Clerici, F.; Rossi, A.; Cucumo, V.; Ghiretti, R.; Maggiore, L.; Pomati, S.; Galimberti, D.; Scarpini, E.; Mariani, C.; et al. Weight loss predicts progression of mild cognitive impairment to Alzheimer’s disease. PLoS ONE 2016, 11, e0151710. [Google Scholar] [CrossRef] [PubMed]
- Mander, B.A.; Winer, J.R.; Jagust, W.J.; Walker, M.P. Sleep: A Novel Mechanistic Pathway, Biomarker, and Treatment Target in the Pathology of Alzheimer’s Disease? Trends Neurosci. 2016, 39, 552–566. [Google Scholar] [CrossRef]
- Harper, D.G.; Stopa, E.G.; Kuo-Leblanc, V.; McKee, A.C.; Asayama, K.; Volicer, L.; Kowall, N.; Satlin, A. Dorsomedial SCN neuronal subpopulations subserve different functions in human dementia. Brain 2008, 131, 1609–1617. [Google Scholar] [CrossRef]
- Vercruysse, P.; Vieau, D.; Blum, D.; Petersén, Å.; Dupuis, L. Hypothalamic alterations in neurodegenerative diseases and their relation to abnormal energy metabolism. Front. Mol. Neurosci. 2018, 11, 2. [Google Scholar] [CrossRef]
- Arnemann, K.L.; Stöber, F.; Narayan, S.; Rabinovici, G.D.; Jagust, W.J. Metabolic brain networks in aging and preclinical Alzheimer’s disease. Neuroimage Clin. 2018, 17, 987–999. [Google Scholar] [CrossRef]
- Landau, S.M.; Mintun, M.A.; Joshi, A.D.; Koeppe, R.A.; Petersen, R.C.; Aisen, P.S.; Weiner, M.W.; Jagust, W.J. Amyloid deposition, hypometabolism, and longitudinal cognitive decline. Ann. Neurol. 2012, 72, 578–586. [Google Scholar] [CrossRef]
- Roher, A.; Maarouf, C.; Kokjohn, T.; Whiteside, C.; Macias, M.; Kalback, W.; Sabbagh, M.; Beach, T.; Vassar, R. Molecular Differences and Similarities Between Alzheimer’s Disease and the 5XFAD Transgenic Mouse Model of Amyloidosis. Biochem. Insights 2013, 6, 1–10. [Google Scholar] [CrossRef]
- Potapova, T.A.; Sivakumar, S.; Flynn, J.N.; Li, R.; Gorbsky, G.J. Mitotic progression becomes irreversible in prometaphase and collapses when Wee1 and Cdc25 are inhibited. Mol. Biol. Cell 2011, 22, 1191–1206. [Google Scholar] [CrossRef] [PubMed]
- McCloy, R.A.; Rogers, S.; Caldon, C.E.; Lorca, T.; Castro, A.; Burgess, A. Partial inhibition of Cdk1 in G 2 phase overrides the SAC and decouples mitotic events. Cell Cycle 2014, 13, 1400–1412. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pratap, A.A.; Holsinger, R.M.D. Altered Brain Adiponectin Receptor Expression in the 5XFAD Mouse Model of Alzheimer’s Disease. Pharmaceuticals 2020, 13, 150. https://doi.org/10.3390/ph13070150
Pratap AA, Holsinger RMD. Altered Brain Adiponectin Receptor Expression in the 5XFAD Mouse Model of Alzheimer’s Disease. Pharmaceuticals. 2020; 13(7):150. https://doi.org/10.3390/ph13070150
Chicago/Turabian StylePratap, Anishchal A., and R. M. Damian Holsinger. 2020. "Altered Brain Adiponectin Receptor Expression in the 5XFAD Mouse Model of Alzheimer’s Disease" Pharmaceuticals 13, no. 7: 150. https://doi.org/10.3390/ph13070150
APA StylePratap, A. A., & Holsinger, R. M. D. (2020). Altered Brain Adiponectin Receptor Expression in the 5XFAD Mouse Model of Alzheimer’s Disease. Pharmaceuticals, 13(7), 150. https://doi.org/10.3390/ph13070150