Marine Natural Products, Multitarget Therapy and Repurposed Agents in Alzheimer’s Disease




Abstract
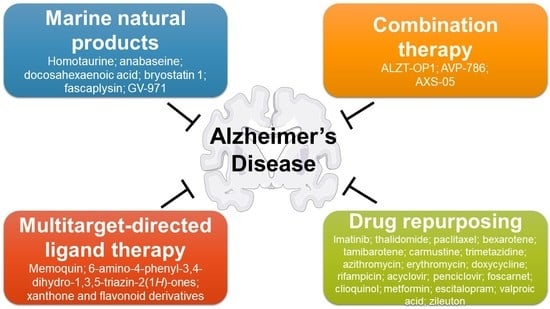
1. Alzheimer’s Disease
1.1. Facts and Characteristics
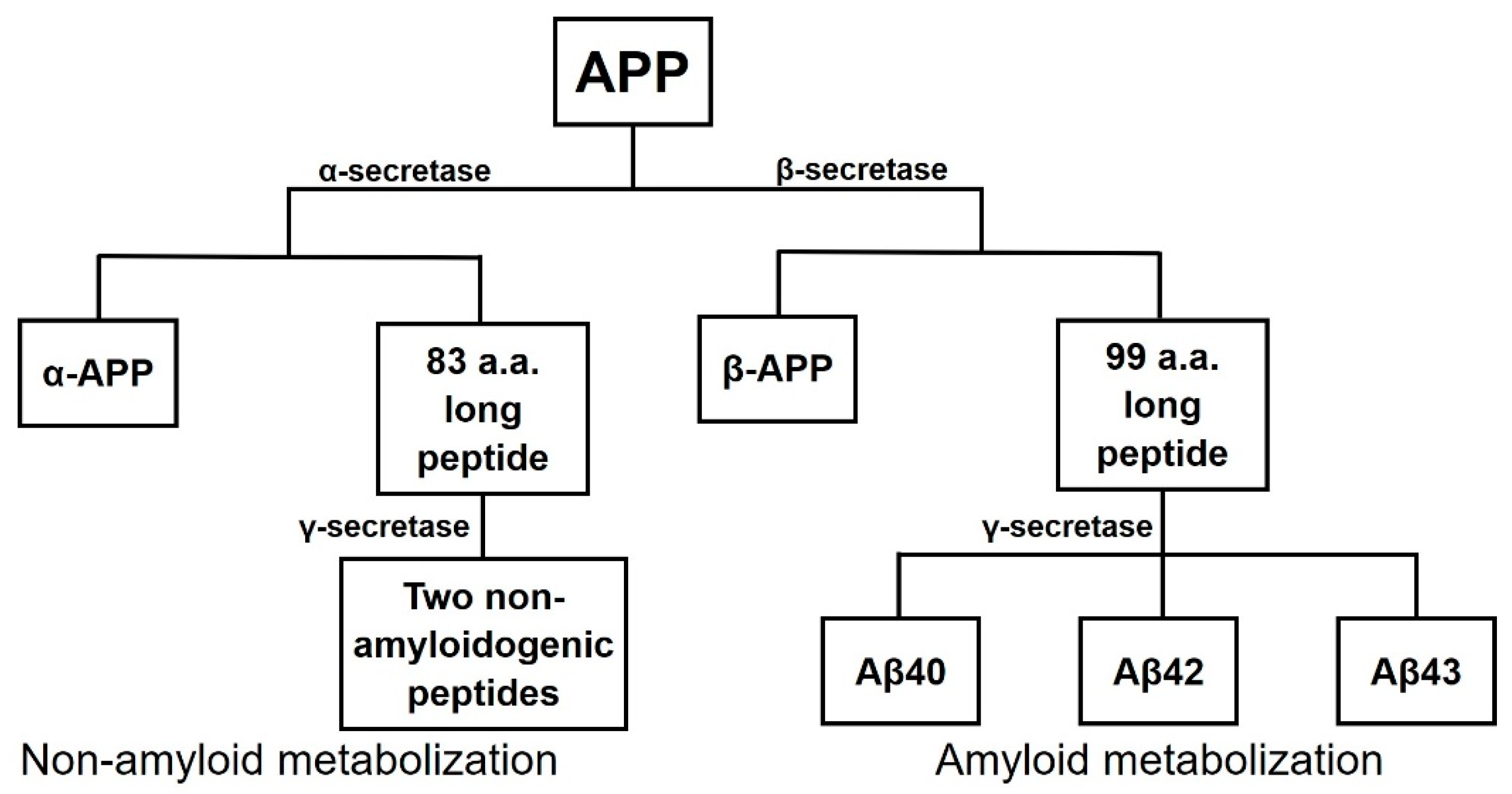
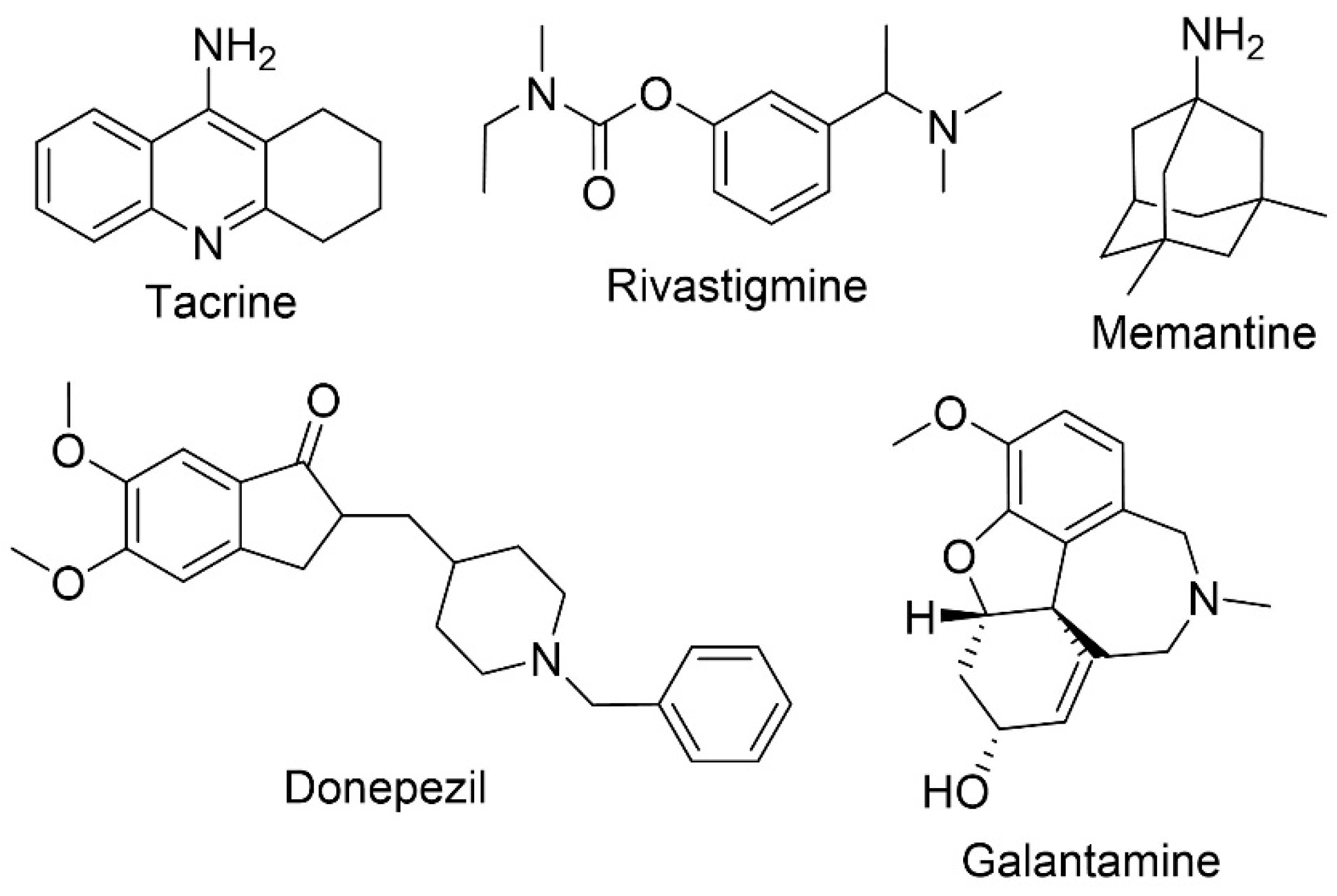
1.2. Approved Therapies
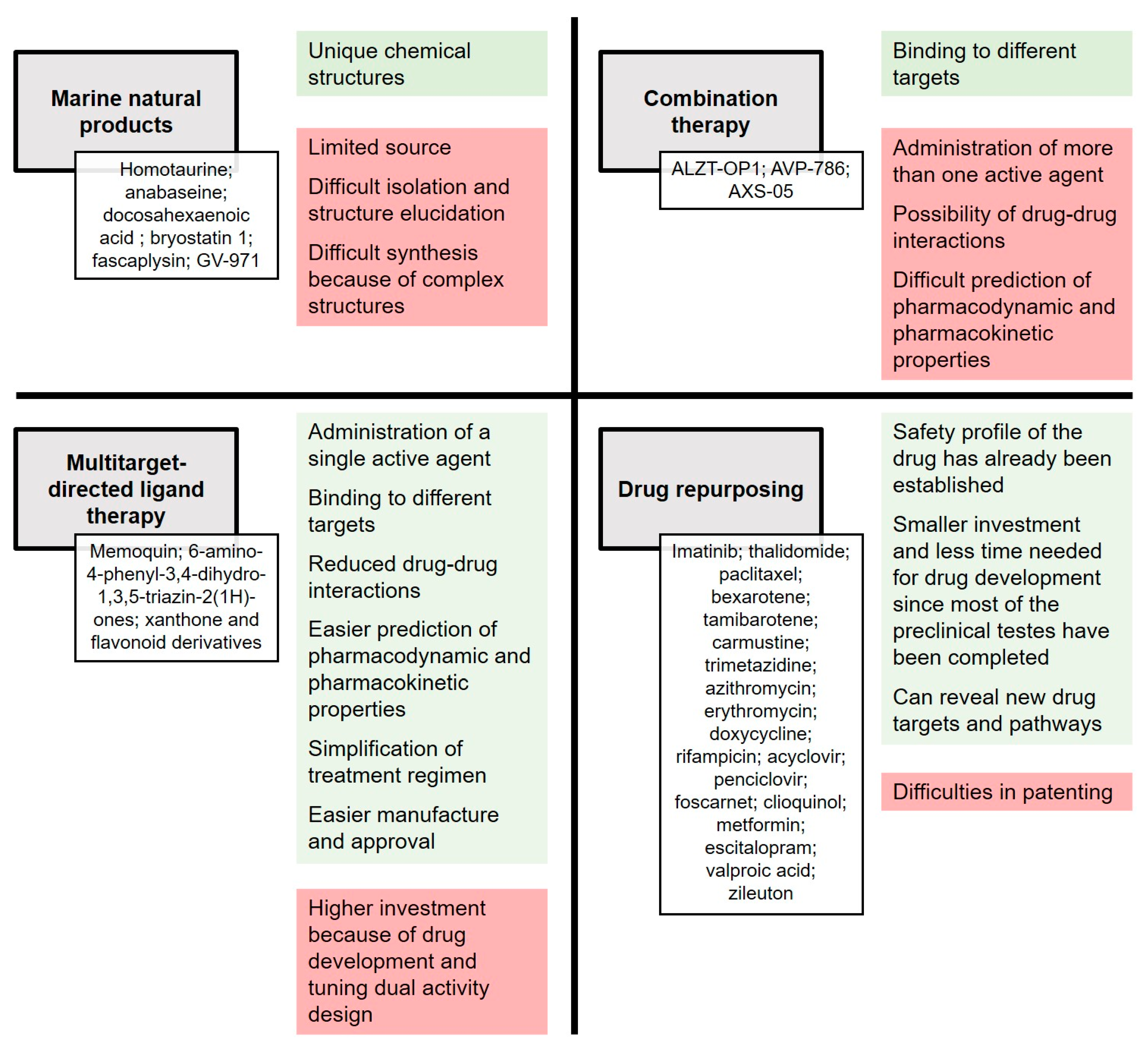
2. Current Approaches and Objectives
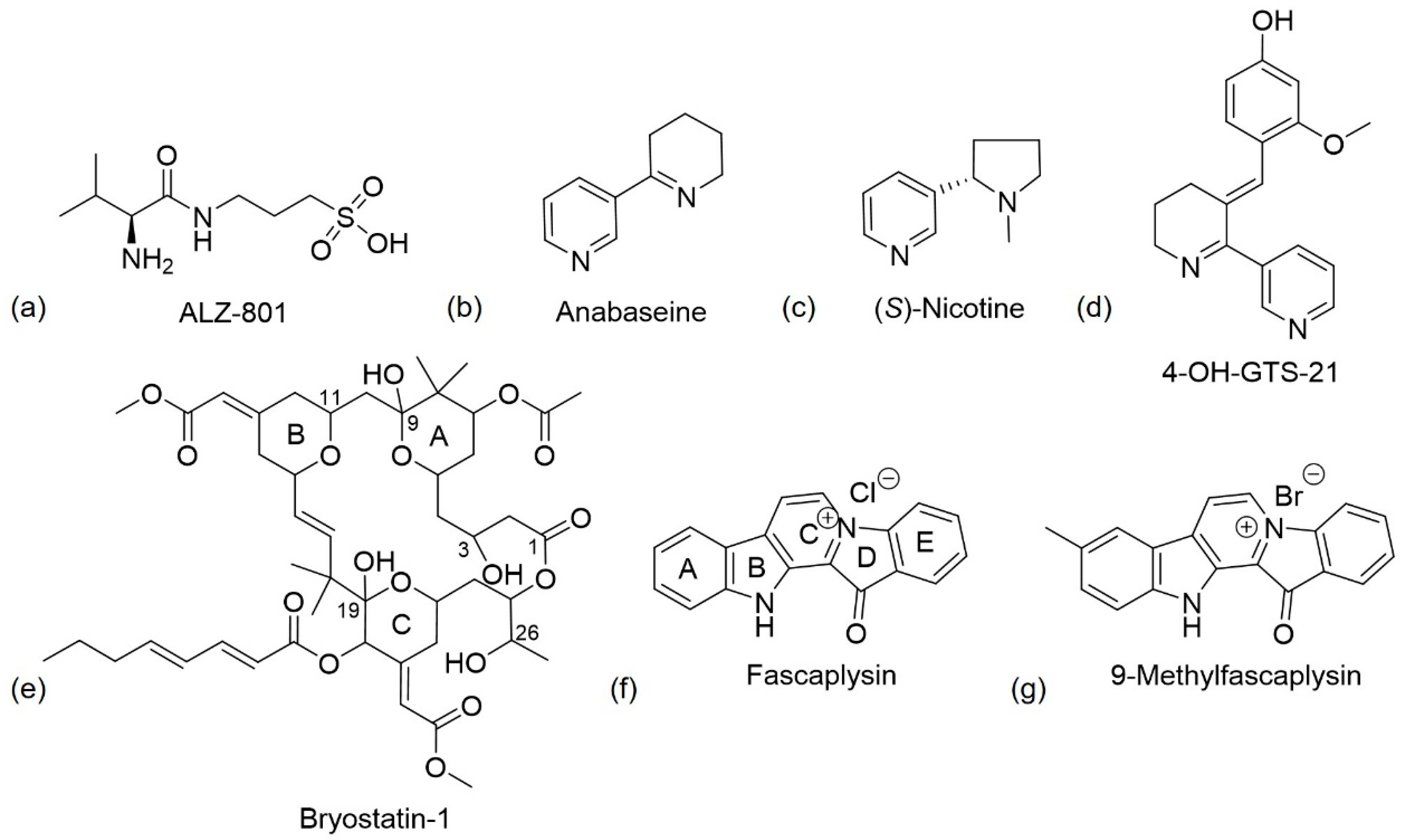
3. Marine Natural Products
3.1. Homotaurine
3.2. Anabaseine
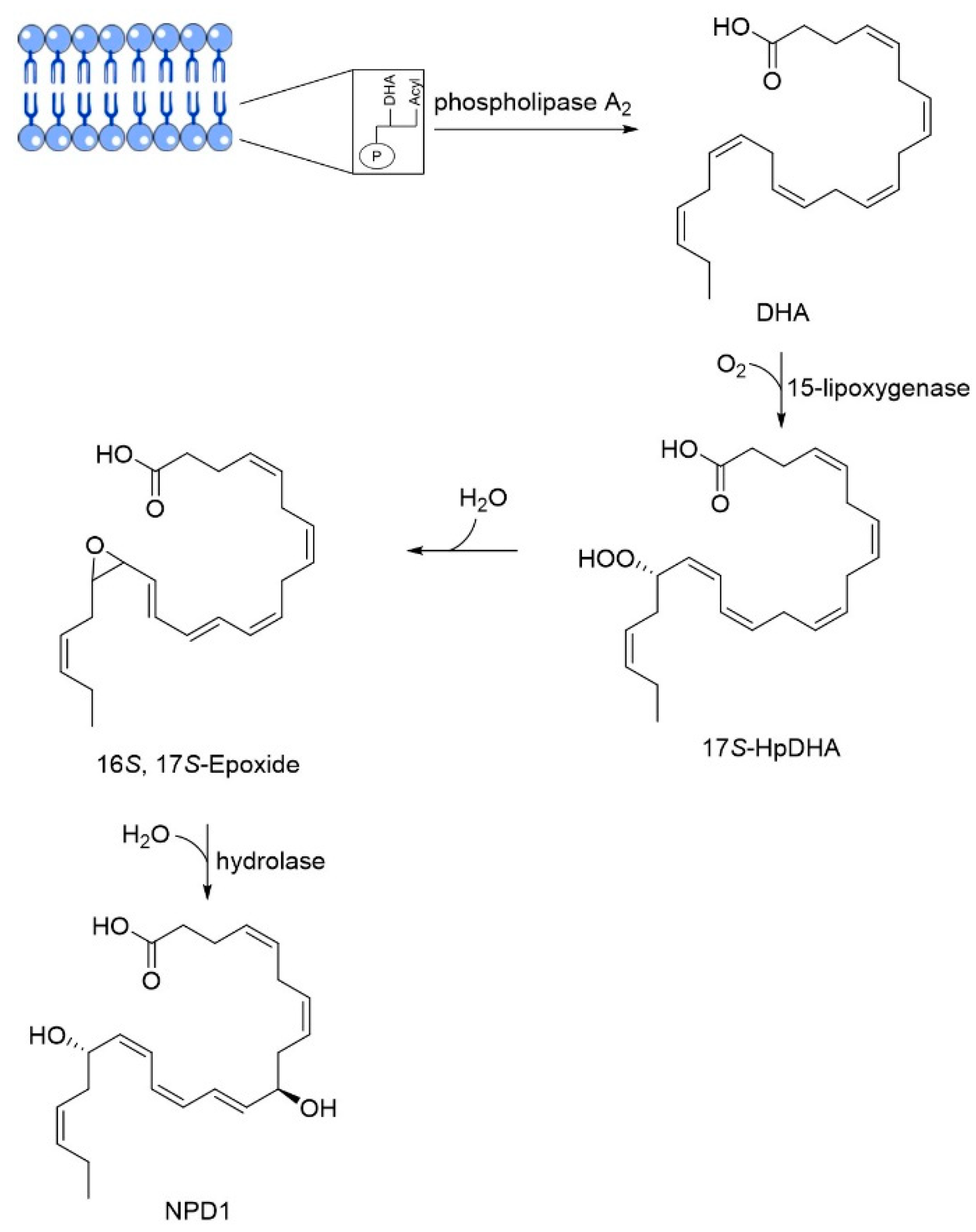
3.3. DHA
3.4. Bryostatin
3.5. Fascaplysin
3.6. GV-971
4. Multitarget Therapy
4.1. Combination Therapy
4.1.1. ALZT-OPT1
4.1.2. AVP-786
4.1.3. AXS-05
4.2. Multitarget-Directed Ligand Therapy
4.2.1. Memoquin
4.2.2. Dual BACE-1/GSK-3β Inhibitors
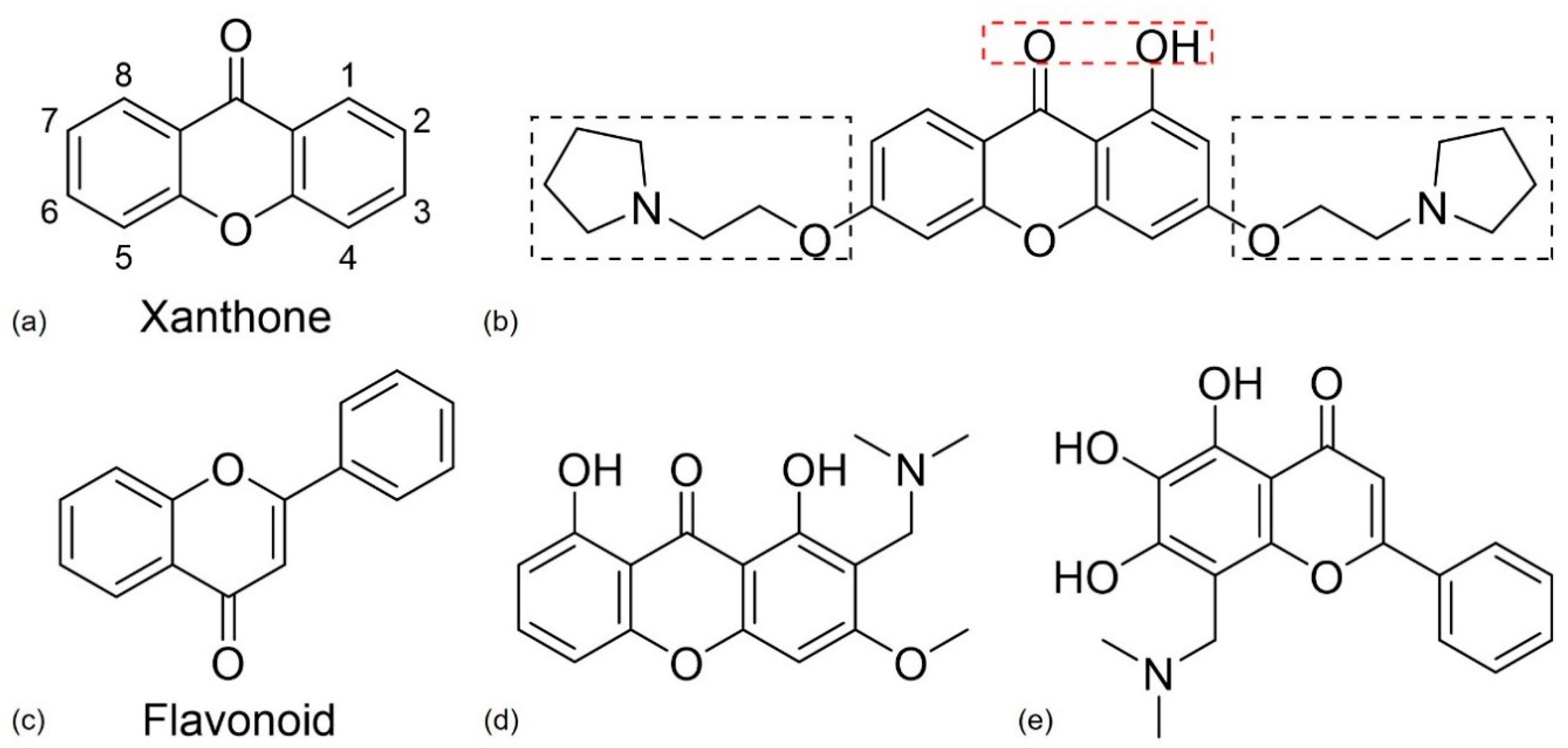
4.2.3. Xanthone and Flavonoid Derivatives
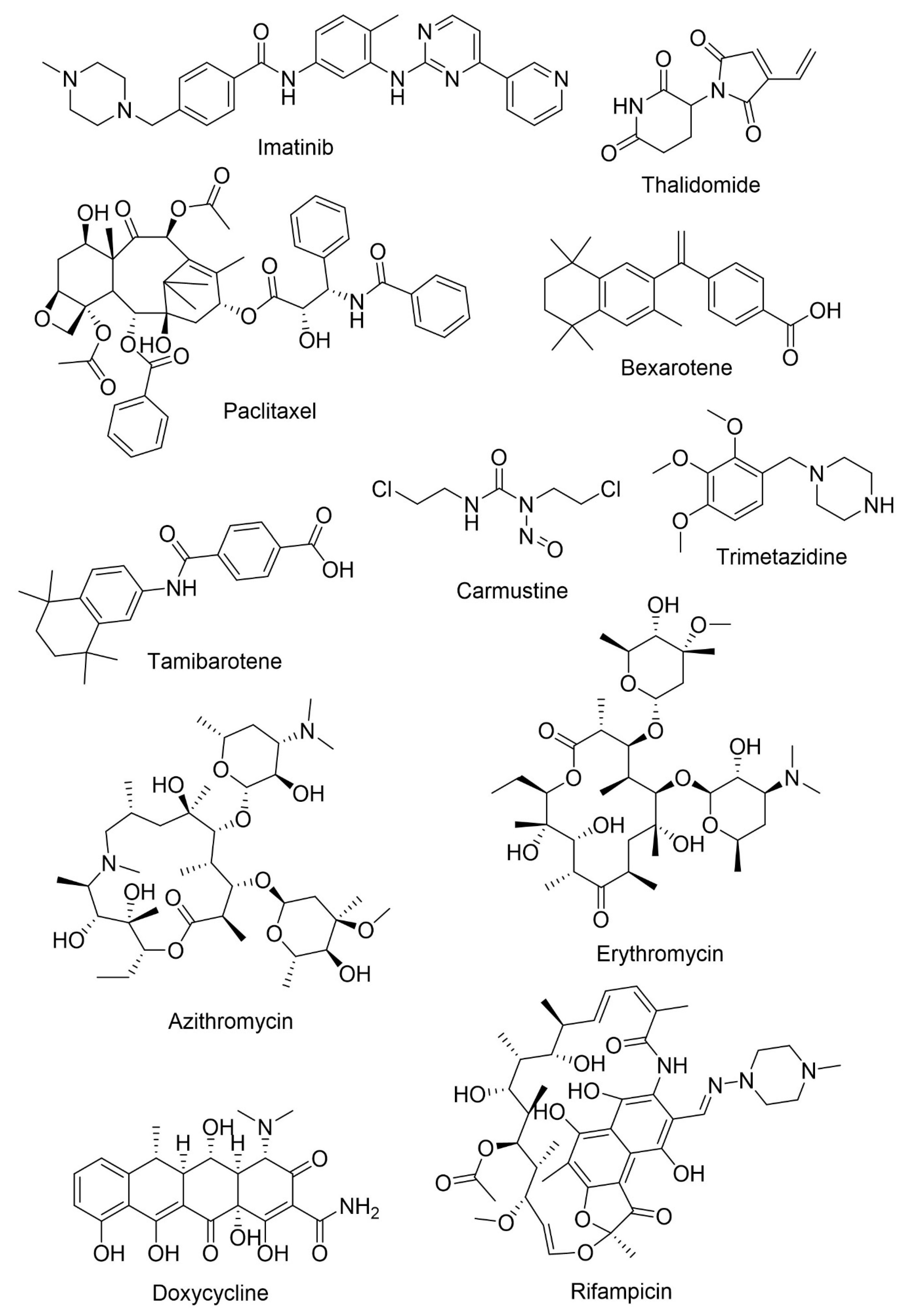
5. Drug Repurposing
6. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, A.; Ekavali. A review on Alzheimer’s disease pathophysiology and its management: An update. Pharm. Rep. 2015, 67, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. 2019 Alzheimer’s disease facts and figures. Alzheimer Dement. 2019, 15, 321–387. [Google Scholar] [CrossRef]
- Cummings, J.L.; Tong, G.; Ballard, C. Treatment Combinations for Alzheimer’s Disease: Current and Future Pharmacotherapy Options. J. Alzheimer Dis. Jad. 2019, 67, 779–794. [Google Scholar] [CrossRef] [PubMed]
- Coman, H.; Nemeş, B. New Therapeutic Targets in Alzheimer’s Disease. Int. J. Gerontol. 2017, 11, 2–6. [Google Scholar] [CrossRef]
- Fish, P.V.; Steadman, D.; Bayle, E.D.; Whiting, P. New approaches for the treatment of Alzheimer’s disease. Bioorgan. Med. Chem. Lett. 2019, 29, 125–133. [Google Scholar] [CrossRef]
- Islam, M.A.; Khandker, S.; Alam, F.; Khalil, D.; Kamal, M.; Gan, S. Alzheimer’s Disease and Natural Products: Future Regimens Emerging From Nature. Curr. Top. Med. Chem. 2017, 17, 1408–1428. [Google Scholar] [CrossRef]
- Honarpisheh, P.; Reynolds, C.R.; Blasco Conesa, M.P.; Moruno Manchon, J.F.; Putluri, N.; Bhattacharjee, M.B.; Urayama, A.; McCullough, L.D.; Ganesh, B.P. Dysregulated Gut Homeostasis Observed Prior to the Accumulation of the Brain Amyloid-β in Tg2576 Mice. Int. J. Mol. Sci. 2020, 21, 1711. [Google Scholar] [CrossRef]
- Dos Santos Guilherme, M.; Todorov, H.; Osterhof, C.; Möllerke, A.; Cub, K.; Hankeln, T.; Gerber, S.; Endres, K. Impact of Acute and Chronic Amyloid-β Peptide Exposure on Gut Microbial Commensals in the Mouse. Front. Microbiol. 2020, 11, 1008. [Google Scholar] [CrossRef]
- Andrade, S.; Ramalho, M.J.; Loureiro, J.A.; Pereira, M.d.C. Natural Compounds for Alzheimer’s Disease Therapy: A Systematic Review of Preclinical and Clinical Studies. Int. J. Mol. Sci. 2019, 20, 2313. [Google Scholar] [CrossRef]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimers Res. 2014, 6, 37. [Google Scholar] [CrossRef] [PubMed]
- Maia, M.A.; Sousa, E. BACE-1 and gamma-Secretase as Therapeutic Targets for Alzheimer’s Disease. Pharmaceuticals 2019, 12. [Google Scholar] [CrossRef]
- Tundis, R.; Loizzo, M.R.; Nabavi, S.M.; Orhan, I.E.; Skalicka-Woźniak, K.; D’Onofrio, G.; Aiello, F. Chapter 3—Natural Compounds and Their Derivatives as Multifunctional Agents for the Treatment of Alzheimer Disease. In Discovery and Development of Neuroprotective Agents from Natural Products; Brahmachari, G., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 63–102. [Google Scholar] [CrossRef]
- Fan, L.Y.; Chiu, M.J. Combotherapy and current concepts as well as future strategies for the treatment of Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2014, 10, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Birks, J. Cholinesterase inhibitors for Alzheimer’s disease. Cochrane Database Syst. Rev. 2006, Cd005593. [Google Scholar] [CrossRef]
- Rogawski, M.A.; Wenk, G.L. The neuropharmacological basis for the use of memantine in the treatment of Alzheimer’s disease. CNS Drug Rev. 2003, 9, 275–308. [Google Scholar] [CrossRef]
- Riverol, M.; Slachevsky, A.; López, O.L. Efficacy and Tolerability of a Combination Treatment of Memantine and Donepezil for Alzheimer’s Disease: A Literature Review Evidence. Eur. Neurol. J. 2011, 3, 15–19. [Google Scholar]
- Chen, R.; Chan, P.-T.; Chu, H.; Lin, Y.-C.; Chang, P.-C.; Chen, C.-Y.; Chou, K.-R. Treatment effects between monotherapy of donepezil versus combination with memantine for Alzheimer disease: A meta-analysis. PLoS ONE 2017, 12, e0183586. [Google Scholar] [CrossRef]
- Caban, A.; Pisarczyk, K.; Kopacz, K.; Kapusniak, A.; Toumi, M.; Remuzat, C.; Kornfeld, A. Filling the gap in CNS drug development: Evaluation of the role of drug repurposing. J. Mark. Access Health Policy 2017, 5, 1299833. [Google Scholar] [CrossRef]
- Morsy, A.; Trippier, P.C. Current and Emerging Pharmacological Targets for the Treatment of Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 72, S145–S176. [Google Scholar] [CrossRef]
- Ghosh, A.; Cárdenas, E.; Osswald, H. The Design, Development, and Evaluation of BACE1 Inhibitors for the Treatment of Alzheimer’s Disease. In Alzheimer’s Disease II; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar] [CrossRef]
- Cruz, M.I.; Cidade, H.; Pinto, M. Dual/multitargeted xanthone derivatives for Alzheimer’s disease: Where do we stand? Future Med. Chem. 2017, 9, 1611–1630. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s disease drug development pipeline: 2019. Alzheimers Dement. 2019, 5, 272–293. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Aisen, P.S.; DuBois, B.; Frölich, L.; Jack, C.R.; Jones, R.W.; Morris, J.C.; Raskin, J.; Dowsett, S.A.; Scheltens, P. Drug development in Alzheimer’s disease: The path to 2025. Alzheimer Res. Ther. 2016, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Schubert, D.; Maher, P. An alternative approach to drug discovery for Alzheimer’s disease dementia. Future Med. Chem. 2012, 4, 1681–1688. [Google Scholar] [CrossRef]
- Patil, P.; Thakur, A.; Sharma, A.; Flora, S.J.S. Natural products and their derivatives as multifunctional ligands against Alzheimer’s disease. Drug Dev. Res. 2020, 81, 165–183. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, A. The impact of natural products upon modern drug discovery. Curr. Opin. Chem. Biol. 2008, 12, 306–317. [Google Scholar] [CrossRef]
- Alonso, D.; Castro, A.; Martinez, A. Marine compounds for the therapeutic treatment of neurological disorders. Patents 2005, 15, 1377–1386. [Google Scholar] [CrossRef]
- Russo, P.; Kisialiou, A.; Lamonaca, P.; Moroni, R.; Prinzi, G.; Fini, M. New Drugs from Marine Organisms in Alzheimer’s Disease. Mar. Drugs 2015, 14, 5. [Google Scholar] [CrossRef]
- Rubiolo, J.; Alonso, E.; Cagide, E. Marine Compounds as a Starting Point to Drugs. In Seafood and Freshwater Toxins; Routledge: Abingdon, UK, 2014; pp. 1141–1178. [Google Scholar] [CrossRef]
- Blunt, J.W.; Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2018, 35, 8–53. [Google Scholar] [CrossRef]
- Choi, D.Y.; Choi, H. Natural products from marine organisms with neuroprotective activity in the experimental models of Alzheimer’s disease, Parkinson’s disease and ischemic brain stroke: Their molecular targets and action mechanisms. Arch. Pharmacal Res. 2015, 38, 139–170. [Google Scholar] [CrossRef]
- Gervais, F.; Paquette, J.; Morissette, C.; Krzywkowski, P.; Yu, M.; Azzi, M.; Lacombe, D.; Kong, X.; Aman, A.; Laurin, J.; et al. Targeting soluble Abeta peptide with Tramiprosate for the treatment of brain amyloidosis. Neurobiol. Aging 2007, 28, 537–547. [Google Scholar] [CrossRef]
- Aisen, P.S.; Gauthier, S.; Vellas, B.; Briand, R.; Saumier, D.; Laurin, J.; Garceau, D. Alzhemed: A potential treatment for Alzheimer’s disease. Curr. Alzheimer Res. 2007, 4, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Caltagirone, C.; Ferrannini, L.; Marchionni, N.; Nappi, G.; Scapagnini, G.; Trabucchi, M. The potential protective effect of tramiprosate (homotaurine) against Alzheimer’s disease: A review. Aging Clin. Exp. Res. 2012, 24, 580–587. [Google Scholar] [CrossRef]
- Aisen, P.S.; Gauthier, S.; Ferris, S.H.; Saumier, D.; Haine, D.; Garceau, D.; Duong, A.; Suhy, J.; Oh, J.; Lau, W.C.; et al. Tramiprosate in mild-to-moderate Alzheimer’s disease—A randomized, double-blind, placebo-controlled, multi-centre study (the Alphase Study). Arch. Med. Sci. 2011, 7, 102–111. [Google Scholar] [CrossRef]
- Kem, W.R.; Mahnir, V.M.; Prokai, L.; Papke, R.L.; Cao, X.; LeFrancois, S.; Wildeboer, K.; Prokai-Tatrai, K.; Porter-Papke, J.; Soti, F. Hydroxy metabolites of the Alzheimer’s drug candidate 3-[(2, 4-dimethoxy) benzylidene]-anabaseine dihydrochloride (GTS-21): Their molecular properties, interactions with brain nicotinic receptors, and brain penetration. Mol. Pharmacol. 2004, 65, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Kem, W.; Soti, F.; Wildeboer, K.; LeFrancois, S.; MacDougall, K.; Wei, D.-Q.; Chou, K.-C.; Arias, H.R. The Nemertine Toxin Anabaseine and Its Derivative DMXBA (GTS-21): Chemical and Pharmacological Properties. Mar. Drugs 2006, 4, 255–273. [Google Scholar] [CrossRef]
- Dev, K.; Maurya, R. Marine-Derived Anti-Alzheimer’s Agents of Promise: Clinical Aspects and Mode of Action. Neuroprotect. Nat. Prod. Clin. Aspects Mode Action 2017, 153–184. [Google Scholar] [CrossRef]
- Takata, K.; Amamiya, T.; Mizoguchi, H.; Kawanishi, S.; Kuroda, E.; Kitamura, R.; Ito, A.; Saito, Y.; Tawa, M.; Nagasawa, T.; et al. Alpha7 nicotinic acetylcholine receptor-specific agonist DMXBA (GTS-21) attenuates Aβ accumulation through suppression of neuronal γ-secretase activity and promotion of microglial amyloid-β phagocytosis and ameliorates cognitive impairment in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2018, 62, 197–209. [Google Scholar] [CrossRef]
- Quinn, J.F.; Raman, R.; Thomas, R.G.; Yurko-Mauro, K.; Nelson, E.B.; Van Dyck, C.; Galvin, J.E.; Emond, J.; Jack, C.R., Jr.; Weiner, M.; et al. Docosahexaenoic acid supplementation and cognitive decline in Alzheimer disease: A randomized trial. JAMA 2010, 304, 1903–1911. [Google Scholar] [CrossRef]
- Petyaev, I.; Chalyk, N.; Klochkov, V.; Pristensky, D.; Chernyshova, M.; Kyle, N.; Bashmakov, Y. Pharmacokinetics and Oxidation Parameters in Volunteers Supplemented with Microencapsulated Docosahexaenoic Acid. Int. J. Appl. Basic Med. Res. 2018, 8, 148. [Google Scholar] [CrossRef]
- Thomas, J.; Thomas, C.J.; Radcliffe, J.; Itsiopoulos, C. Omega-3 Fatty Acids in Early Prevention of Inflammatory Neurodegenerative Disease: A Focus on Alzheimer’s Disease. BioMed Res. Int. 2015, 2015, 172801. [Google Scholar] [CrossRef]
- Calon, F.; Lim, G.P.; Yang, F.; Morihara, T.; Teter, B.; Ubeda, O.; Rostaing, P.; Triller, A.; Salem, N., Jr.; Ashe, K.H.; et al. Docosahexaenoic acid protects from dendritic pathology in an Alzheimer’s disease mouse model. Neuron 2004, 43, 633–645. [Google Scholar] [CrossRef] [PubMed]
- Lim, G.P.; Calon, F.; Morihara, T.; Yang, F.; Teter, B.; Ubeda, O.; Salem, N., Jr.; Frautschy, S.A.; Cole, G.M. A diet enriched with the omega-3 fatty acid docosahexaenoic acid reduces amyloid burden in an aged Alzheimer mouse model. J. Neurosci. 2005, 25, 3032–3040. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J.; Cui, J.G.; Marcheselli, V.L.; Bodker, M.; Botkjaer, A.; Gotlinger, K.; Serhan, C.N.; Bazan, N.G. A role for docosahexaenoic acid-derived neuroprotectin D1 in neural cell survival and Alzheimer disease. J. Clin. Investig. 2005, 115, 2774–2783. [Google Scholar] [CrossRef] [PubMed]
- Freund-Levi, Y.; Eriksdotter-Jonhagen, M.; Cederholm, T.; Basun, H.; Faxen-Irving, G.; Garlind, A.; Vedin, I.; Vessby, B.; Wahlund, L.O.; Palmblad, J. Omega-3 fatty acid treatment in 174 patients with mild to moderate Alzheimer disease: OmegAD study: A randomized double-blind trial. Arch. Neurol. 2006, 63, 1402–1408. [Google Scholar] [CrossRef] [PubMed]
- Green, K.N.; Martinez-Coria, H.; Khashwji, H.; Hall, E.B.; Yurko-Mauro, K.A.; Ellis, L.; LaFerla, F.M. Dietary docosahexaenoic acid and docosapentaenoic acid ameliorate amyloid-beta and tau pathology via a mechanism involving presenilin 1 levels. J. Neurosci. 2007, 27, 4385–4395. [Google Scholar] [CrossRef] [PubMed]
- Fotuhi, M.; Mohassel, P.; Yaffe, K. Fish consumption, long-chain omega-3 fatty acids and risk of cognitive decline or Alzheimer disease: A complex association. Nat. Clin. Pract. Neurol. 2009, 5, 140–152. [Google Scholar] [CrossRef]
- Zhao, Y.; Calon, F.; Julien, C.; Winkler, J.W.; Petasis, N.A.; Lukiw, W.J.; Bazan, N.G. Docosahexaenoic acid-derived neuroprotectin D1 induces neuronal survival via secretase- and PPARgamma-mediated mechanisms in Alzheimer’s disease models. PLoS ONE 2011, 6, e15816. [Google Scholar] [CrossRef]
- Grimm, M.O.W.; Kuchenbecker, J.; Grösgen, S.; Burg, V.K.; Hundsdörfer, B.; Rothhaar, T.L.; Friess, P.; de Wilde, M.C.; Broersen, L.M.; Penke, B.; et al. Docosahexaenoic acid reduces amyloid beta production via multiple pleiotropic mechanisms. J. Biol. Chem. 2011, 286, 14028–14039. [Google Scholar] [CrossRef]
- Nelson, T.J.; Sun, M.-K.; Lim, C.; Sen, A.; Khan, T.; Chirila, F.V.; Alkon, D.L. Bryostatin Effects on Cognitive Function and PKCɛ in Alzheimer’s Disease Phase IIa and Expanded Access Trials. J. Alzheimer’s Dis. Jad. 2017, 58, 521–535. [Google Scholar] [CrossRef]
- Farlow, M.R.; Thompson, R.E.; Wei, L.J.; Tuchman, A.J.; Grenier, E.; Crockford, D.; Wilke, S.; Benison, J.; Alkon, D.L. A Randomized, Double-Blind, Placebo-Controlled, Phase II Study Assessing Safety, Tolerability, and Efficacy of Bryostatin in the Treatment of Moderately Severe to Severe Alzheimer’s Disease. J. Alzheimer’s Dis. Jad. 2019, 67, 555–570. [Google Scholar] [CrossRef]
- Sun, M.-K.; Alkon, D.L. Bryostatin-1: Pharmacology and Therapeutic Potential as a CNS Drug. CNS Drug Rev. 2006, 12, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.K.; Sen, A.; Hongpaisan, J.; Lim, C.S.; Nelson, T.J.; Alkon, D.L. PKCepsilon deficits in Alzheimer’s disease brains and skin fibroblasts. J. Alzheimer’s Dis.Jad. 2015, 43, 491–509. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.R.; Tang, H.F.; Tian, X.L.; Hu, J.J.; Huang, L.L.; Gustafson, K.R. Review of bioactive secondary metabolites from marine bryozoans in the progress of new drugs discovery. Future Med. Chem. 2018, 10, 1497–1514. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Chen, H.; Chang, N.; Xu, Y.; Jiao, J.; Zhang, H. Unlocking the Drug Potential of the Bryostatin Family: Recent Advances in Product Synthesis and Biomedical Applications. Chem. Eur. J. 2020, 26, 1166–1195. [Google Scholar] [CrossRef]
- Bharate, S.; Manda, S.; Joshi, P.; Singh, B.; Vishwakarma, R. Total synthesis and anti-cholinesterase activity of marine-derived bis-indole alkaloid fascaplysin. Rapid Commun. Res. Med. Chem. 2012, 3. [Google Scholar] [CrossRef]
- Manda, S.; Sharma, S.; Wani, A.; Joshi, P.; Kumar, V.; Guru, S.K.; Bharate, S.S.; Bhushan, S.; Vishwakarma, R.A.; Kumar, A.; et al. Discovery of a marine-derived bis-indole alkaloid fascaplysin, as a new class of potent P-glycoprotein inducer and establishment of its structure–activity relationship. Eur. J. Med. Chem. 2016, 107, 1–11. [Google Scholar] [CrossRef]
- Pan, H.B.; Qiu, H.D.; Zhang, K.; Zhang, P.P.; Liang, W.D.; Yang, M.X.; Mou, C.Y.; Lin, M.M.; He, M.; Xiao, X.; et al. Fascaplysin Derivatives Are Potent Multitarget Agents against Alzheimer’s Disease: In Vitro and in Vivo Evidence. ACS Chem. Neurosci. 2019, 10, 4741–4756. [Google Scholar] [CrossRef]
- Sun, Q.; Liu, F.; Sang, J.; Lin, M.; Ma, J.; Xiao, X.; Yan, S.; Naman, C.B.; Wang, N.; He, S.; et al. 9-Methylfascaplysin Is a More Potent Aβ Aggregation Inhibitor than the Marine-Derived Alkaloid, Fascaplysin, and Produces Nanomolar Neuroprotective Effects in SH-SY5Y Cells. Mar. Drugs 2019, 17, 121. [Google Scholar] [CrossRef]
- Alzforum. China Approves Seaweed Sugar as First New Alzheimer’s Drug in 17 Years. Available online: https://www.alzforum.org/news/research-news/china-approves-seaweed-sugar-first-new-alzheimers-drug-17-years (accessed on 9 April 2020).
- Wang, S.; Li, J.; Xia, W.; Geng, M. A marine-derived acidic oligosaccharide sugar chain specifically inhibits neuronal cell injury mediated by beta-amyloid-induced astrocyte activation in vitro. Neurol Res. 2007, 29, 96–102. [Google Scholar] [CrossRef]
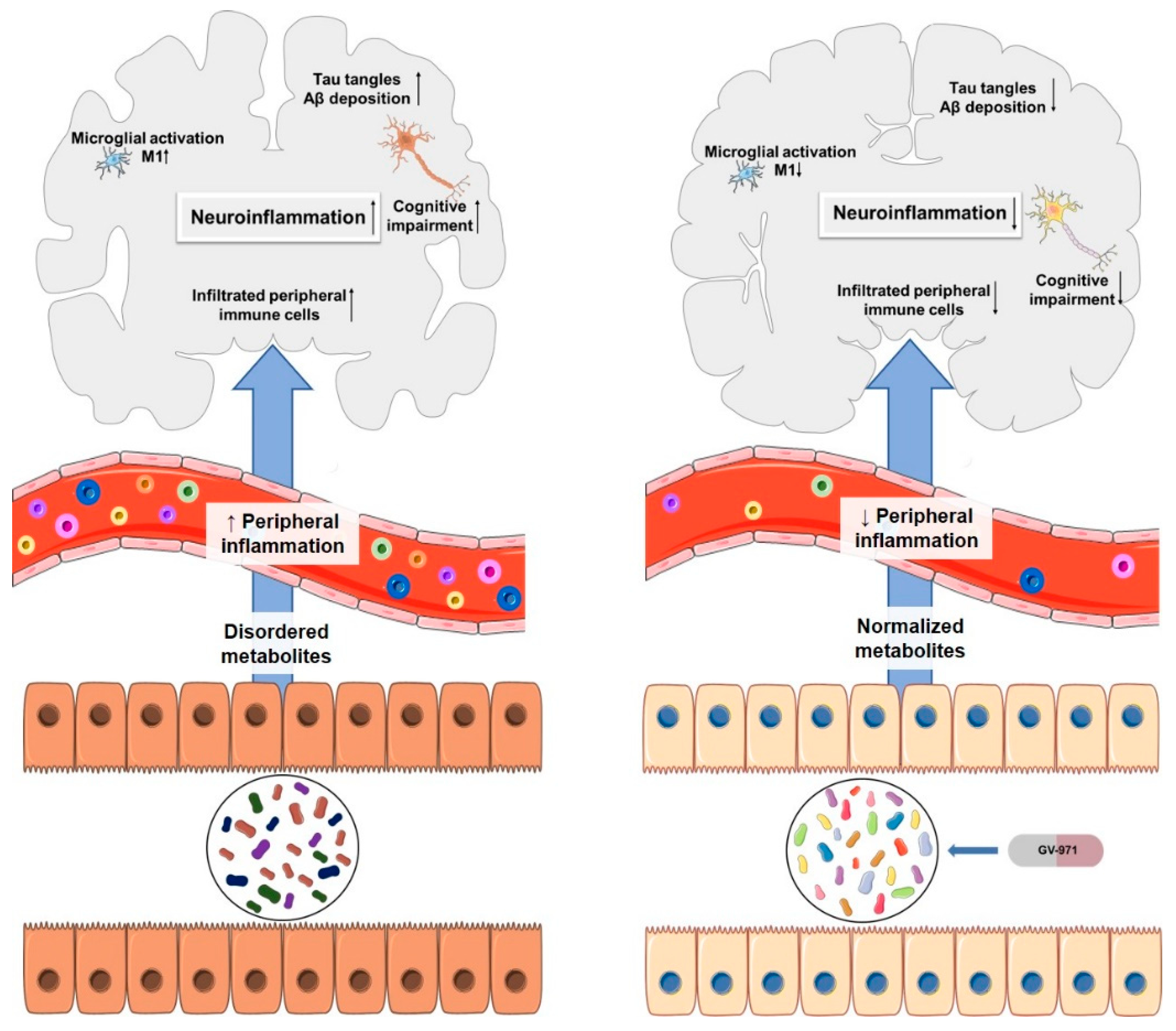
- Wang, X.; Sun, G.; Feng, T.; Zhang, J.; Huang, X.; Wang, T.; Xie, Z.; Chu, X.; Yang, J.; Wang, H.; et al. Sodium oligomannate therapeutically remodels gut microbiota and suppresses gut bacterial amino acids-shaped neuroinflammation to inhibit Alzheimer’s disease progression. Cell Res. 2019, 29, 787–803. [Google Scholar] [CrossRef]
- Syed, Y.Y. Sodium Oligomannate: First Approval. Drugs 2020, 80, 441–444. [Google Scholar] [CrossRef] [PubMed]
- Hey, J.A.; Yu, J.Y.; Versavel, M.; Abushakra, S.; Kocis, P.; Power, A.; Kaplan, P.L.; Amedio, J.; Tolar, M. Clinical Pharmacokinetics and Safety of ALZ-801, a Novel Prodrug of Tramiprosate in Development for the Treatment of Alzheimer’s Disease. Clin. Pharm. 2018, 57, 315–333. [Google Scholar] [CrossRef] [PubMed]
- Alzheon. ALZ-801—Phase 3-Ready Oral Small Molecule Program Designed to Inhibit Formation of Neurotoxic Soluble Amyloid Oligomers Represents Optimized and Differentiated Treatment Paradigm for Alzheimer’s Disease. Available online: https://alzheon.com/alz-801-program-white-paper/ (accessed on 18 April 2020).
- Zawieja, P.; Kornprobst, J.M.; Metais, P. 3-(2,4-dimethoxybenzylidene)-anabaseine: A promising candidate drug for Alzheimer’s disease? Geriatr. Gerontol. Int. 2012, 12, 365–371. [Google Scholar] [CrossRef]
- Slavov, S.H.; Radzvilovits, M.; LeFrancois, S.; Stoyanova-Slavova, I.B.; Soti, F.; Kem, W.R.; Katritzky, A.R. A computational study of the binding of 3-(arylidene) anabaseines to two major brain nicotinic acetylcholine receptors and to the acetylcholine binding protein. Eur. J. Med. Chem. 2010, 45, 2433–2446. [Google Scholar] [CrossRef] [PubMed]
- Oddo, S.; LaFerla, F.M. The role of nicotinic acetylcholine receptors in Alzheimer’s disease. J. Physiol. 2006, 99, 172–179. [Google Scholar] [CrossRef]
- Kombo, D.C.; Mazurov, A.A.; Chewning, J.; Hammond, P.S.; Tallapragada, K.; Hauser, T.A.; Speake, J.; Yohannes, D.; Caldwell, W.S. Discovery of novel α7 nicotinic acetylcholine receptor ligands via pharmacophoric and docking studies of benzylidene anabaseine analogs. Bioorg. Med. Chem. Lett. 2012, 22, 1179–1186. [Google Scholar] [CrossRef]
- Kem, W.R.; Abbott, B.C.; Coates, R.M. Isolation and structure of a hoplonemertine toxin. Toxicon 1971, 9, 15–22. [Google Scholar] [CrossRef]
- Hibbs, R.E.; Sulzenbacher, G.; Shi, J.; Talley, T.T.; Conrod, S.; Kem, W.R.; Taylor, P.; Marchot, P.; Bourne, Y. Structural determinants for interaction of partial agonists with acetylcholine binding protein and neuronal alpha7 nicotinic acetylcholine receptor. EMBO J. 2009, 28, 3040–3051. [Google Scholar] [CrossRef]
- Martin, E.J.; Panickar, K.S.; King, M.A.; Deyrup, M.; Hunter, B.E.; Wang, G.; Meyer, E.M. Cytoprotective actions of 2,4-dimethoxybenzylidene anabaseine in differentiated PC12 cells and septal cholinergic neurons. Drug Dev. Res. 1994, 31, 135–141. [Google Scholar] [CrossRef]
- Gao, F.; Kiesewetter, D.; Chang, L.; Ma, K.; Bell, J.M.; Rapoport, S.I.; Igarashi, M. Whole-body synthesis-secretion rates of long-chain n-3 PUFAs from circulating unesterified alpha-linolenic acid in unanesthetized rats. J. Lipid Res. 2009, 50, 749–758. [Google Scholar] [CrossRef]
- Watters, C.; Edmonds, C.; Rosner, L.; Sloss, K.; Leung, P. A cost analysis of EPA and DHA in fish, supplements and foods. J. Nutr. Food Sci. 2012, 2. [Google Scholar] [CrossRef]
- Bazan, N.G.; Stark, D.T.; Petasis, N.A. Chapter 36—Lipid Mediators: Eicosanoids, Docosanoids and Platelet-Activating Factor. In Basic Neurochemistry, 8th ed.; Brady, S.T., Siegel, G.J., Albers, R.W., Price, D.L., Eds.; Academic Press: New York, NY, USA, 2012; pp. 643–662. [Google Scholar] [CrossRef]
- Pettit, G.R.; Herald, C.L.; Doubek, D.L.; Herald, D.L.; Arnold, E.; Clardy, J. Isolation and structure of bryostatin 1. J. Am. Chem. Soc. 1982, 104, 6846–6848. [Google Scholar] [CrossRef]
- Hale, K.J.; Hummersone, M.G.; Manaviazar, S.; Frigerio, M. The chemistry and biology of the bryostatin antitumour macrolides. Nat. Prod. Rep. 2002, 19, 413–453. [Google Scholar] [CrossRef] [PubMed]
- Naveen kumar, S.; Rajivgandhi, G.; Ramachandran, G.; Manoharan, N. A marine sponge Fascaplysinopsis sp. derived alkaloid fascaplysin inhibits the HepG2 hepatocellular carcinoma cell. Front. Lab. Med. 2018, 2, 41–48. [Google Scholar] [CrossRef]
- Gil-Martins, E.; Barbosa, D.J.; Silva, V.; Remião, F.; Silva, R. Dysfunction of ABC transporters at the blood-brain barrier: Role in neurological disorders. Pharmacol. Ther. 2020, 107554. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, A.; Bolognesi, M.L.; Minarini, A.; Rosini, M.; Tumiatti, V.; Recanatini, M.; Melchiorre, C. Multi-target-directed ligands to combat neurodegenerative diseases. J. Med. Chem. 2008, 51, 347–372. [Google Scholar] [CrossRef]
- Zhou, J.; Jiang, X.; He, S.; Jiang, H.; Feng, F.; Liu, W.; Qu, W.; Sun, H. Rational Design of Multitarget-Directed Ligands: Strategies and Emerging Paradigms. J. Med. Chem. 2019, 62, 8881–8914. [Google Scholar] [CrossRef]
- Prati, F.; Uliassi, E.; Bolognesi, M. Two diseases, one approach: Multitarget drug discovery in Alzheimer’s and neglected tropical diseases. MedChemComm 2014, 5, 853–861. [Google Scholar] [CrossRef]
- Panza, F.; Seripa, D.; Solfrizzi, V.; Imbimbo, B.P.; Lozupone, M.; Leo, A.; Sardone, R.; Gagliardi, G.; Lofano, L.; Creanza, B.C.; et al. Emerging drugs to reduce abnormal β-amyloid protein in Alzheimer’s disease patients. Expert Opin. Emerg. Drugs 2016, 21, 377–391. [Google Scholar] [CrossRef]
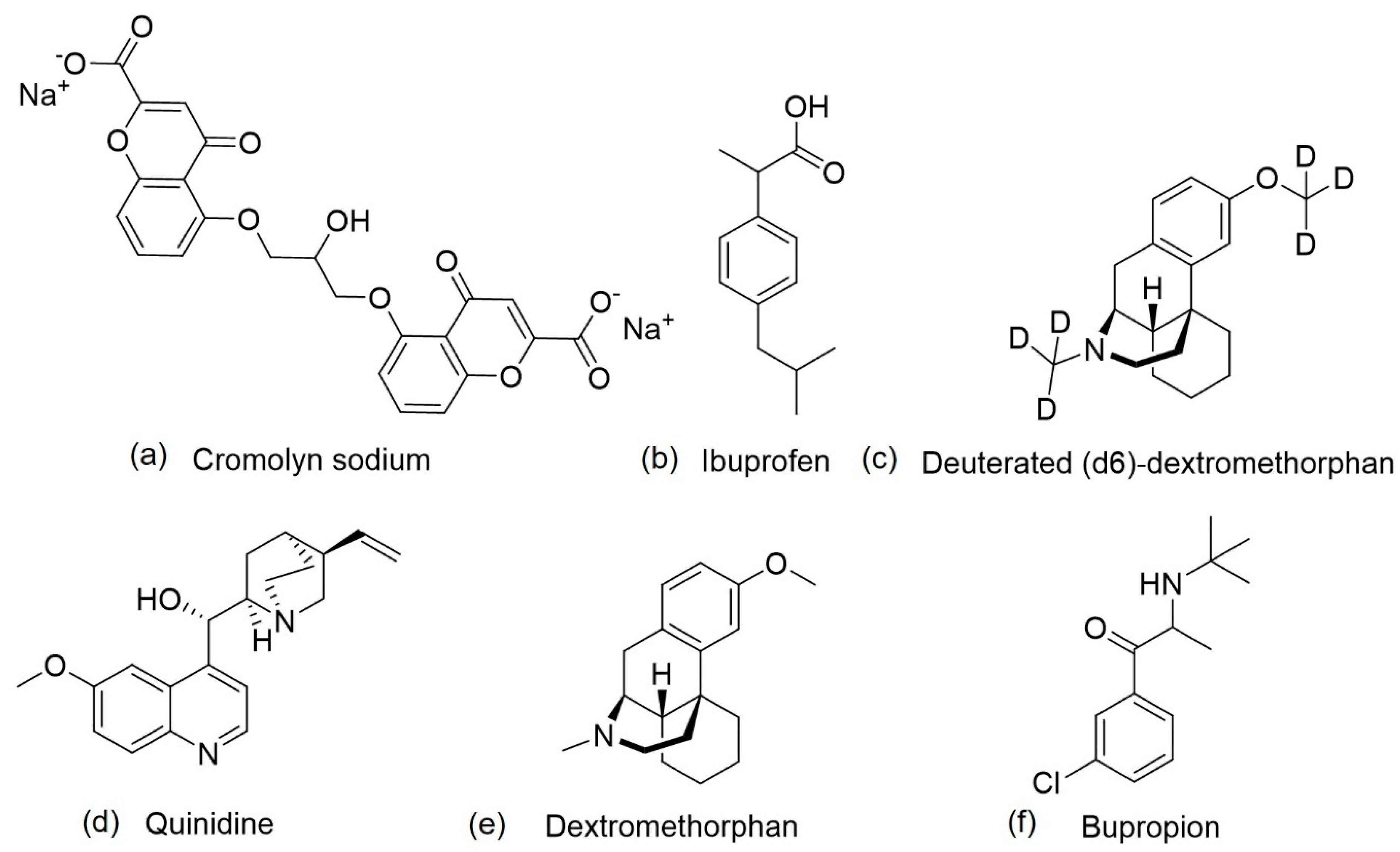
- Hori, Y.; Takeda, S.; Cho, H.; Wegmann, S.; Shoup, T.M.; Takahashi, K.; Irimia, D.; Elmaleh, D.R.; Hyman, B.T.; Hudry, E. A Food and Drug Administration-approved asthma therapeutic agent impacts amyloid β in the brain in a transgenic model of Alzheimer disease. J. Biol. Chem. 2015, 290, 1966–1978. [Google Scholar] [CrossRef]
- Brazier, D.; Perry, R.; Keane, J.; Barrett, K.; Elmaleh, D.R. Pharmacokinetics of Cromolyn and Ibuprofen in Healthy Elderly Volunteers. Clin. Drug Investig. 2017, 37, 1025–1034. [Google Scholar] [CrossRef] [PubMed]
- Pasqualetti, P.; Bonomini, C.; Dal Forno, G.; Paulon, L.; Sinforiani, E.; Marra, C.; Zanetti, O.; Rossini, P.M. A randomized controlled study on effects of ibuprofen on cognitive progression of Alzheimer’s disease. Aging Clin. Exp. Res. 2009, 21, 102–110. [Google Scholar] [CrossRef]
- Weggen, S.; Eriksen, J.L.; Das, P.; Sagi, S.A.; Wang, R.; Pietrzik, C.U.; Findlay, K.A.; Smith, T.E.; Murphy, M.P.; Bulter, T.; et al. A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature 2001, 414, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Griciuc, A.; Hudry, E.; Wan, Y.; Quinti, L.; Ward, J.; Forte, A.; Shen, X.; Ran, C.; Elmaleh, D.; et al. Cromolyn Reduces Levels of the Alzheimer’s Disease-Associated Amyloid β-Protein by Promoting Microglial Phagocytosis. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Garay, R.P.; Grossberg, G.T. AVP-786 for the treatment of agitation in dementia of the Alzheimer’s type. Expert Opin. Investig. Drugs 2017, 26, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, S.T.; Sanacora, G. A new generation of antidepressants: An update on the pharmaceutical pipeline for novel and rapid-acting therapeutics in mood disorders based on glutamate/GABA neurotransmitter systems. Drug Discov. Today 2019, 24, 606–615. [Google Scholar] [CrossRef] [PubMed]
- O’Gorman, C.; Jones, A.; Tabuteau, H. AXS-05 (Dextromethorphan/Bupropion): Psychopharmacology and the potential for therapeutic application in the treatment of neuropsychiatric symptoms. Eur. Neuropsychopharmacol. 2019, 29, S458. [Google Scholar] [CrossRef]
- Ahmed, M.; Malik, M.; Teselink, J.; Lanctot, K.L.; Herrmann, N. Current Agents in Development for Treating Behavioral and Psychological Symptoms Associated with Dementia. Drugs Aging 2019, 36, 589–605. [Google Scholar] [CrossRef]
- Stahl, S.M. Dextromethorphan/Bupropion: A Novel Oral NMDA (N-methyl-d-aspartate) Receptor Antagonist with Multimodal Activity. CNS Spectr. 2019, 24, 461–466. [Google Scholar] [CrossRef]
- Porsteinsson, A.P.; Drye, L.T.; Pollock, B.G.; Devanand, D.P.; Frangakis, C.; Ismail, Z.; Marano, C.; Meinert, C.L.; Mintzer, J.E.; Munro, C.A.; et al. Effect of Citalopram on Agitation in Alzheimer Disease: The CitAD Randomized Clinical Trial. JAMA 2014, 311, 682–691. [Google Scholar] [CrossRef]
- Nguyen, L.; Thomas, K.L.; Lucke-Wold, B.P.; Cavendish, J.Z.; Crowe, M.S.; Matsumoto, R.R. Dextromethorphan: An update on its utility for neurological and neuropsychiatric disorders. Pharmacol. Ther. 2016, 159, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.P.; Traynelis, S.F.; Siffert, J.; Pope, L.E.; Matsumoto, R.R. Pharmacology of dextromethorphan: Relevance to dextromethorphan/quinidine (Nuedexta®) clinical use. Pharmacol. Ther. 2016, 164, 170–182. [Google Scholar] [CrossRef]
- Capon, D.A.; Bochner, F.; Kerry, N.; Mikus, G.; Danz, C.; Somogyi, A.A. The influence of CYP2D6 polymorphism and quinidine on the disposition and antitussive effect of dextromethorphan in humans. Clin. Pharm. 1996, 60, 295–307. [Google Scholar] [CrossRef]
- Patel, K.; Allen, S.; Haque, M.N.; Angelescu, I.; Baumeister, D.; Tracy, D.K. Bupropion: A systematic review and meta-analysis of effectiveness as an antidepressant. Adv. Psychopharmacol. 2016, 6, 99–144. [Google Scholar] [CrossRef] [PubMed]
- Cruz, I.; Puthongking, P.; Cravo, S.; Palmeira, A.; Cidade, H.; Pinto, M.; Sousa, E. Xanthone and Flavone Derivatives as Dual Agents with Acetylcholinesterase Inhibition and Antioxidant Activity as Potential Anti-Alzheimer Agents. J. Chem. 2017, 2017, 1–16. [Google Scholar] [CrossRef]
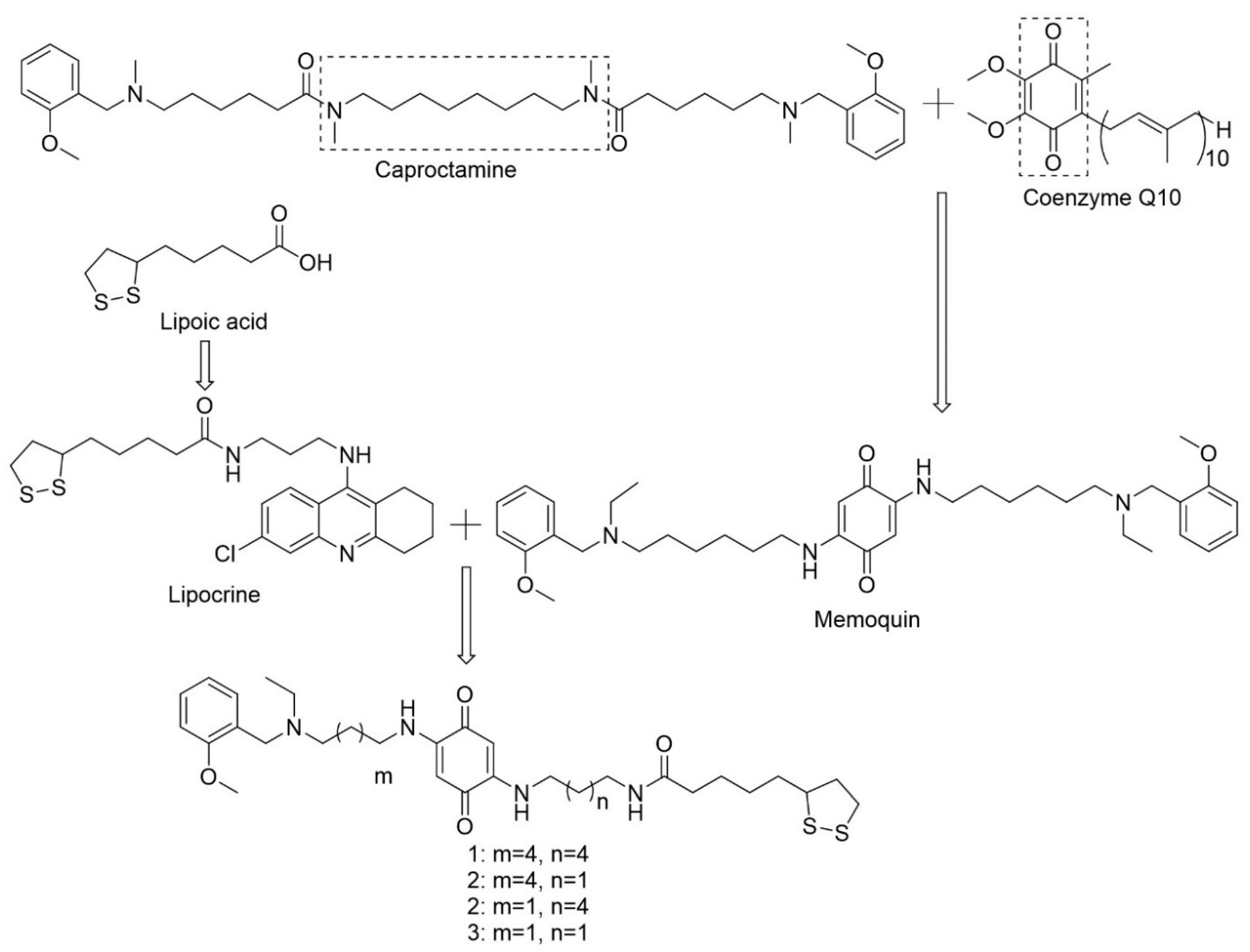
- Cavalli, A.; Bolognesi, M.L.; Capsoni, S.; Andrisano, V.; Bartolini, M.; Margotti, E.; Cattaneo, A.; Recanatini, M.; Melchiorre, C. A small molecule targeting the multifactorial nature of Alzheimer’s disease. Angew. Chem. Int. Ed. Engl. 2007, 46, 3689–3692. [Google Scholar] [CrossRef]
- Bolognesi, M.L.; Cavalli, A.; Melchiorre, C. Memoquin: A multi-target-directed ligand as an innovative therapeutic opportunity for Alzheimer’s disease. Neurother. J. Am. Soc. Exp. Neurother. 2009, 6, 152–162. [Google Scholar] [CrossRef]
- Ortiz, C.J.C.; de Freitas Silva, M.; Gontijo, V.S.; Viegas, F.P.D.; Dias, K.S.T.; Viegas, C., Jr. Design of multi-target directed ligands as a modern approach for the development of innovative drug candidates for Alzheimer’s disease. In Methods in Pharmacology and Toxicology; Humana Press Inc.: Totowa, NJ, USA, 2019; pp. 255–351. [Google Scholar] [CrossRef]
- Bolognesi, M.L.; Cavalli, A.; Bergamini, C.; Fato, R.; Lenaz, G.; Rosini, M.; Bartolini, M.; Andrisano, V.; Melchiorre, C. Toward a Rational Design of Multitarget-Directed Antioxidants: Merging Memoquin and Lipoic Acid Molecular Frameworks. J. Med. Chem. 2009, 52, 7883–7886. [Google Scholar] [CrossRef]
- Capurro, V.; Busquet, P.; Lopes, J.P.; Bertorelli, R.; Tarozzo, G.; Bolognesi, M.L.; Piomelli, D.; Reggiani, A.; Cavalli, A. Pharmacological characterization of memoquin, a multi-target compound for the treatment of Alzheimer’s disease. PLoS ONE 2013, 8, e56870. [Google Scholar] [CrossRef]
- Prati, F.; De Simone, A.; Bisignano, P.; Armirotti, A.; Summa, M.; Pizzirani, D.; Scarpelli, R.; Perez, D.I.; Andrisano, V.; Perez-Castillo, A.; et al. Multitarget drug discovery for Alzheimer’s disease: Triazinones as BACE-1 and GSK-3β inhibitors. Angew. Chem. Int. Ed. Engl. 2015, 54, 1578–1582. [Google Scholar] [CrossRef]
- Prati, F.; Cavalli, A.; Bolognesi, M.L. Navigating the Chemical Space of Multitarget-Directed Ligands: From Hybrids to Fragments in Alzheimer’s Disease. Molecules 2016, 21, 466. [Google Scholar] [CrossRef] [PubMed]
- Prati, F.; De Simone, A.; Armirotti, A.; Summa, M.; Pizzirani, D.; Scarpelli, R.; Bertozzi, S.M.; Perez, D.I.; Andrisano, V.; Perez-Castillo, A.; et al. 3,4-Dihydro-1,3,5-triazin-2(1H)-ones as the First Dual BACE-1/GSK-3β Fragment Hits against Alzheimer’s Disease. Acs Chem. Neurosci. 2015, 6, 1665–1682. [Google Scholar] [CrossRef] [PubMed]
- Alawi, M.S.; Awad, T.A.; Mohamed, M.A.; Khalid, A.; Ismail, E.M.O.; Alfatih, F.; Naz, S.; Ul-Haq, Z. Insights into the molecular basis of acetylcholinesterase inhibition by xanthones: An integrative in silico and in vitro approach. Mol. Simul. 2020, 46, 253–261. [Google Scholar] [CrossRef]
- Kou, X.; Song, L.; Wang, Y.; Yu, Q.; Ju, H.; Yang, A.; Shen, R. Design, synthesis and anti-Alzheimer’s disease activity study of xanthone derivatives based on multi-target strategy. Bioorg. Med. Chem. Lett. 2020, 30. [Google Scholar] [CrossRef]
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An overview. J. Nutr. Sci. 2016, 5, e47. [Google Scholar] [CrossRef]
- Jalili-Baleh, L.; Babaei, E.; Abdpour, S.; Nasir Abbas Bukhari, S.; Foroumadi, A.; Ramazani, A.; Sharifzadeh, M.; Abdollahi, M.; Khoobi, M. A review on flavonoid-based scaffolds as multi-target-directed ligands (MTDLs) for Alzheimer’s disease. Eur. J. Med. Chem. 2018, 152, 570–589. [Google Scholar] [CrossRef]
- Duraes, F.; Pinto, M.; Sousa, E. Old Drugs as New Treatments for Neurodegenerative Diseases. Pharmaceuticals 2018, 11, 44. [Google Scholar] [CrossRef]
- Doan, T.L.; Pollastri, M.; Walters, M.A.; Georg, G.I. Chapter 23—The Future of Drug Repositioning: Old Drugs, New Opportunities. In Annual Reports in Medicinal Chemistry; Macor, J.E., Ed.; Academic Press: Cambridge, MA, USA, 2011; Volume 46, pp. 385–401. [Google Scholar]
- Mucke, H.A. The case of galantamine: Repurposing and late blooming of a cholinergic drug. Future Sci. Oa 2015, 1, FSO73. [Google Scholar] [CrossRef]
- Vezenkov, L.T.; Tsekova, D.S.; Kostadinova, I.; Mihaylova, R.; Vassilev, N.G.; Danchev, N.D. Synthesis of New Galanthamine-Peptide Derivatives Designed for Prevention and Treatment of Alzheimer’s Disease. Curr. Alzheimer Res. 2019, 16, 183–192. [Google Scholar] [CrossRef]
- Kumar, M.; Kulshrestha, R.; Singh, N.; Jaggi, A.S. Expanding spectrum of anticancer drug, imatinib, in the disorders affecting brain and spinal cord. Pharmacol. Res. 2019, 143, 86–96. [Google Scholar] [CrossRef]
- Netzer, W.J.; Dou, F.; Cai, D.; Veach, D.; Jean, S.; Li, Y.; Bornmann, W.G.; Clarkson, B.; Xu, H.; Greengard, P. Gleevec inhibits beta-amyloid production but not Notch cleavage. Proc. Natl. Acad. Sci. USA 2003, 100, 12444–12449. [Google Scholar] [CrossRef] [PubMed]
- Monacelli, F.; Cea, M.; Borghi, R.; Odetti, P.; Nencioni, A. Do Cancer Drugs Counteract Neurodegeneration? Repurposing for Alzheimer’s Disease. J. Alzheimer’s Dis. Jad. 2017, 55, 1295–1306. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Cheng, X.; Staufenbiel, M.; Li, R.; Shen, Y. Long-term treatment of thalidomide ameliorates amyloid-like pathology through inhibition of β-secretase in a mouse model of Alzheimer’s disease. PLoS ONE 2013, 8, e55091. [Google Scholar] [CrossRef]
- Ryu, J.K.; McLarnon, J.G. Thalidomide inhibition of perturbed vasculature and glial-derived tumor necrosis factor-α in an animal model of inflamed Alzheimer’s disease brain. Neurobiol. Dis. 2008, 29, 254–266. [Google Scholar] [CrossRef]
- Brunden, K.R.; Yao, Y.; Potuzak, J.S.; Ferrer, N.I.; Ballatore, C.; James, M.J.; Hogan, A.M.; Trojanowski, J.Q.; Smith, A.B., 3rd; Lee, V.M. The characterization of microtubule-stabilizing drugs as possible therapeutic agents for Alzheimer’s disease and related tauopathies. Pharm. Res. 2011, 63, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Maiti, A.; Shively, S.; Lakhani, F.; McDonald-Jones, G.; Bruce, J.; Lee, E.B.; Xie, S.X.; Joyce, S.; Li, C.; et al. Microtubule-binding drugs offset tau sequestration by stabilizing microtubules and reversing fast axonal transport deficits in a tauopathy model. Proc. Natl. Acad. Sci. USA 2005, 102, 227–231. [Google Scholar] [CrossRef]
- Tousi, B. The emerging role of bexarotene in the treatment of Alzheimer’s disease: Current evidence. Neuropsychiatr. Dis. Treat. 2015, 11, 311–315. [Google Scholar] [CrossRef]
- Fukasawa, H.; Nakagomi, M.; Yamagata, N.; Katsuki, H.; Kawahara, K.; Kitaoka, K.; Miki, T.; Shudo, K. Tamibarotene: A candidate retinoid drug for Alzheimer’s disease. Biol. Pharm. Bull. 2012, 35, 1206–1212. [Google Scholar] [CrossRef]
- Hayes, C.; Dey, D.; Palavicini, J.; Wang, H.; Patkar, K.; Minond, D.; Nefzi, A.; Lakshmana, M. Striking reduction of amyloid plaque burden in an Alzheimer’s mouse model after chronic administration of carmustine. BMC Med. 2013, 11, 81. [Google Scholar] [CrossRef]
- Hassanzadeh, G.; Hosseini, A.; Pasbakhsh, P.; Akbari, M.; Ghaffarpour, M.; Takzare, N.; Zahmatkesh, M. Trimetazidine prevents oxidative changes induced in a rat model of sporadic type of Alzheimer’s disease. Acta Med. Iran. 2015, 53, 17–24. [Google Scholar]
- Appleby, B.S.; Nacopoulos, D.; Milano, N.; Zhong, K.; Cummings, J.L. A review: Treatment of Alzheimer’s disease discovered in repurposed agents. Dement. Geriatr. Cogn. Disord. 2013, 35, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Tucker, S.; Ahl, M.; Cho, H.H.; Bandyopadhyay, S.; Cuny, G.D.; Bush, A.I.; Goldstein, L.E.; Westaway, D.; Huang, X.; Rogers, J.T. RNA therapeutics directed to the non coding regions of APP mRNA, in vivo anti-amyloid efficacy of paroxetine, erythromycin, and N-acetyl cysteine. Curr. Alzheimer Res. 2006, 3, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Balducci, C.; Forloni, G. Doxycycline for Alzheimer’s Disease: Fighting β-Amyloid Oligomers and Neuroinflammation. Front. Pharm. 2019, 10, 738. [Google Scholar] [CrossRef]
- Yulug, B.; Hanoglu, L.; Ozansoy, M.; Isık, D.; Kilic, U.; Kilic, E.; Schabitz, W.R. Therapeutic role of rifampicin in Alzheimer’s disease. Psychiatry Clin. Neurosci. 2018, 72, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Panza, F.; Lozupone, M.; Solfrizzi, V.; Watling, M.; Imbimbo, B. Time to test antibacterial therapy in Alzheimer’s disease. Brain 2019, 142, 2905–2929. [Google Scholar] [CrossRef]
- Bareggi, S.R.; Cornelli, U. Clioquinol: Review of its mechanisms of action and clinical uses in neurodegenerative disorders. CNS Neurosci. 2012, 18, 41–46. [Google Scholar] [CrossRef]
- Campbell, J.M.; Stephenson, M.D.; de Courten, B.; Chapman, I.; Bellman, S.M.; Aromataris, E. Metformin Use Associated with Reduced Risk of Dementia in Patients with Diabetes: A Systematic Review and Meta-Analysis. J. Alzheimer’s Dis. Jad. 2018, 65, 1225–1236. [Google Scholar] [CrossRef]
- Ho, T.; Pollock, B.G.; Mulsant, B.H.; Schantz, O.; Devanand, D.P.; Mintzer, J.E.; Porsteinsson, A.P.; Schneider, L.S.; Weintraub, D.; Yesavage, J.; et al. R- and S-citalopram concentrations have differential effects on neuropsychiatric scores in elders with dementia and agitation. Br. J. Clin. Pharmacol. 2016, 82, 784–792. [Google Scholar] [CrossRef]
- Xuan, A.G.; Pan, X.B.; Wei, P.; Ji, W.D.; Zhang, W.J.; Liu, J.H.; Hong, L.P.; Chen, W.L.; Long, D.H. Valproic acid alleviates memory deficits and attenuates amyloid-β deposition in transgenic mouse model of Alzheimer’s disease. Mol. Neurobiol. 2015, 51, 300–312. [Google Scholar] [CrossRef]
- Zhang, X.-Z.; Li, X.-J.; Zhang, H.-Y. Valproic acid as a promising agent to combat Alzheimer’s disease. Brain Res. Bull. 2010, 81, 3–6. [Google Scholar] [CrossRef]
- Di Meco, A.; Lauretti, E.; Vagnozzi, A.N.; Praticò, D. Zileuton restores memory impairments and reverses amyloid and tau pathology in aged Alzheimer’s disease mice. Neurobiol. Aging 2014, 35, 2458–2464. [Google Scholar] [CrossRef] [PubMed]
- Bendlin, B.B. Antidiabetic therapies and Alzheimer disease. Dialogues Clin. Neurosci. 2019, 21, 83–91. [Google Scholar] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Structure | Chemical Family | Relevant Numbers | Therapeutic Purpose | Pharmacokinetic Profile | In the Pipeline | Reference |
|---|---|---|---|---|---|---|
 Homotaurine | Amino sulfonate | NCT0031491 | Anti-amyloid | Safe Oral administration causes gastrointestinal irritation Crosses the blood-brain barrier (BBB) | Failed phase III clinical trials | [33,35,36] |
| Particularities: It binds to Aβ peptide through the sulfonated head, preventing the progression of amyloid cascade and maintaining Aβ in a stable conformation. In vitro, its ability to inhibit Aβ aggregation and fibrillogenesis, decrease the levels of Aβ40 and Aβ42 and interfere with toxic oligomers formation was demonstrated. In clinical trials, it reduced Aβ42 levels in AD patients following three months of treatment, demonstrated benefits in cognitive function and reduction of hippocampus volume loss. The benefit in cognitive function was more prominent in APOE4+ patients. | ||||||
 GTS-21 | Benzylidene | NCT00414622 | Agonist activity against nicotinic acetylcholine receptors (nAChRs) | Lipophilic compound with a high permeation through the BBB. Hydroxy-metabolites of the compound have poorer BBB penetration | Phase II clinical trials | [37,38,39,40] |
| Particularities: It is an anabaseine derivative more potent and selective for α7 nAChRs than the MNP anabaseine. The three rings lie in different planes, unlike the two rings of anabaseine. It possesses some stimulant effect following oral administeration. Effect may be caused by the metabolites. In vitro, GTS-21 reduced Aβ through the suppression of γ-secretase activity and promotion of microglial Aβ phagocytosis. In vivo, neuroprotective effects from amyloid toxicity, apoptosis and necrosis, and improvement in cognitive functions were observed. | ||||||
 DHA | Omega-3 fatty acid | NCT00440050 NCT03613844 | Reducing Aβ formation Improve synaptic function | Low bioavailability and suffers oxidation | Phase III clinical trials | [41,42,43,44,45,46,47,48,49,50,51] |
| Particularities: Docosahexaenoic acid (DHA) is essential for brain health and neurodevelopment and its reduction is associated with AD. In vivo, DHA supplementation reduced tau hyperphosphorylation, neurotoxic damage associated with Aβ plaques, and Aβ deposition through the shifting of APP metabolism via the non-amyloidogenic pathway. The effects are dependent on the stage of AD progression and positive effects were verified in patients in the initial stage of AD. Neuroprotectin D1 (NPD1) is a bioactive DHA-derivative that showed anti-inflammatory, anti-amyloidogenic, and anti-apoptotic activities, also useful for the treatment of the initial stage of AD. | ||||||
 Bryostatin 1 | Macrolide lactone | NCT03560245 | PKC modulator Neuroprotection | Safe and well tolerated | Phase II clinical trials | [52,53,54,55,56,57] |
| Particularities: Bryostatin-1 activates PKC isozyme epsilon (PKCε), causing its downregulation. Consequently, degradation of Aβ, activation of α-secretase generating the synaptogenic non-toxic soluble amyloid-β protein precursor α, reduction of GSK3-β activity that leads to decreasing hyperphosphorylation of tau ensues. Bryostatin-1 elevates synaptic growth factors in the brain possible causing neuroprotective effects. Structure-activity relationship (SAR) studies demonstrated that the 20-membered macrolactone ring is essential, but the elimination of the A-ring and B-ring exocyclic olefin is possible. The C-26 free hydroxyl and C-1 carbonyl group are necessary for interaction and affinity, respectively. The C-19 hydroxyl group might interact with the lipid bilayer and C-3 hydroxyl group is important for the molecules conformation. The C-9 region can be modified to alter pharmacokinetic characteristics and C-20 can be used to form analogues. A phase IIa safety and tolerability test showed that bryostatin-1 increases PKCε and is safe, with a favorable pharmacokinetic and an initial cognitive improvement with a single dose. | ||||||
 Fascaplysin | Bis-indole alkaloid | - | AChE inhibition P-glycoprotein (P-gp) induction | Possibility in crossing BBB | - | [58,59,60,61] |
| Particularities: The principal interaction of fascaplysin with AChE is a π-π interaction. It binds parallelly in AChE active site gorge, with B-ring orientating to the catalytic site and D-ring orientating towards peripheral site. The quaternary status of the C-ring nitrogen is essential for P-gp activity. Fascaplysin increases P-gp activity leading to a higher Aβ clearance. In vitro, 9-methylfascaplysin is more potent than fascaplysin in inhibiting Aβ fibrillation and protection against the neurotoxicity associated with Aβ oligomer. It acts as an antioxidant, can prevent cognitive dysfunction, decreases neuroinflammation and reduces tau hyperphosphorylation. | ||||||
 GV-971 n = 1–9; m = 0,1 or 2; m’ = 0 or 1 | Acidic oligosaccharide | NCT02293915 | Anti-inflammatory | Crosses BBB via Glut-1 transporter Low oral bioavailability | Completed successfully phase III clinical trial | [62,63,64,65] |
| Particularities: Restoration of the normal microbial profile leading to a reduction of neuroinflammation associated with T helper type 1 (Th1) cells. Studies, in vivo, demonstrated its ability to reduce Aβ deposition, tau phosphorylation, improve cognitive function and, reduce Th1 cells, thereby alleviating neuroinflammation. In clinical trials, increased Aβ clearance and improved cognitive function were observed. | ||||||
| Agent | Characteristics | Mechanism of Action | AD Target | Particularities | Relevant Numbers | References |
|---|---|---|---|---|---|---|
| ALZT-OP1 | Cromolyn + ibuprofen | Mast cell stabilizer and anti-inflammatory activity | Amyloid and inflammation | The co-administration of cromolyn and ibuprofen was safe and well tolerated; ibuprofen acts on γ-secretase modulation, instead of inhibiting cyclooxygenase; cromolyn acts on microglia, increasing Aβ phagocytosis, in vivo, and decreases the aggregation of Aβ peptide, in vitro. | NCT02547818 | [85,86,87,88,89,90] |
| AVP-786 | Deuterated (d6)-dextromethorphan + quinidine | Activation of sigma-1 receptors, NDMA receptor antagonist and cytochrome P450 2D6 inhibition | Agitation | Quinidine decreases dextromethorphan metabolization; deuterium also decreases dextromethorphan metabolization, reducing the necessary dose of quinidine. | NCT02442765 NCT02442778 NCT02446132 | [91,92] |
| AXS-05 | Bupropion + dextromethorphan | NMDA receptor antagonist, sigma-1 receptor agonist, a serotonin and noradrenaline reuptake inhibitor, and cytochrome P450 2D6 inhibition | Agitation | Safe and well tolerated; bupropion and dextromethorphan possess synergist effects; increases the amount of dopamine, serotonin glutamate and noradrenaline; increases plasma levels of dextromethorphan. | NCT03226522 | [93,94,95] |
| Agent | Current Treatment | Mechanism in AD | Particularities | References |
|---|---|---|---|---|
| Imatinib | Chronic myeloid leukemia and gastrointestinal stomal tumor | Neuroprotection and reduction of Aβ formation | Inhibits the interaction of γ-secretase activating protein with γ-secretase | [118,119,120] |
| Low BBB permeability and suffers efflux mediated by P-gp | ||||
| Thalidomide | Multiple myeloma and severe erythema nodosum leprosum | Anti-inflammatory, neuroprotection, and anti-angiogenic activities | Decreases glial activation and Aβ neuropathology through the inhibition of tumor necrosis factor-α (TNFα) | [121,122] |
| Poor BBB permeability | ||||
| Paclitaxel | Ovarian and breast cancer and non-small cell lung cancer | Antimitotic agent | Reduces tau phosphorylation | [114,123,124] |
| Poor BBB permeability and P-gp-mediated efflux | ||||
| Bexarotene | Cutaneous T-cell lymphomas | Anti-amyloid | Increases APOE concentration, reduces Aβ levels and amyloid deposition and improves cognition | [125] |
| High BBB permeability | ||||
| Tamibarotene | Acute promyelocytic leukemia | Immunomodulatory activity | Improves cortical acetylcholine decrease, decreases proinflammatory cytokines and chemokines, improves behavioral symptoms | [126] |
| Good BBB permeability | ||||
| Carmustine | Brain cancer | Anti-amyloid | Reduces Aβ production and neuroinflammation | [120,127] |
| Its lipophilic structure confers good BBB permeability | ||||
| Trimetazidine | Angina pectoris | Neuroprotection | Increases the expression of DHCR24 and reduces oxidative stress | [128] |
| Crosses the BBB | ||||
| Azithromycin | Bacterial infections | Anti-amyloid | Alters APP processing leading to a reduction in Aβ levels | [129,130] |
| Erythromycin | Bacterial infections | Anti-amyloid | Alters APP processing leading to a reduction in Aβ levels | [129,130] |
| Possible neuroprotective effect | ||||
| Doxycycline | Bacterial pneumonia, syphilis, cholera, early Lyme disease, acne, and chlamydia infections | Anti-amyloid | Reduces neuroinflammation and reduces Aβ oligomers | [131] |
| Crosses BBB and has a safe clinical profile | ||||
| Rifampicin | Tuberculosis, leprosy, Legionnaires’ disease and Mycobacterium avium complex | Anti-amyloid | Modulates neuroinflammation and Aβ metabolism | [132] |
| Crosses the BBB | ||||
| Acyclovir | Human herpes virus infections | Anti-amyloid and anti-tau | Reduces Aβ accumulation and phosphorylated tau protein in cell models | [114,133] |
| The prodrug, valacyclovir, is hydrolyzed in vivo to acyclovir which has the ability to cross BBB | ||||
| Penciclovir | Human herpes virus infections | Anti-amyloid and anti-tau | Reduces Aβ accumulation and phosphorylated tau protein in cell models | [114,133] |
| Foscarnet | Human herpes virus infections | Anti-amyloid and anti-tau | Reduces Aβ accumulation and phosphorylated tau protein in cell models | [114,133] |
| Clioquinol | Skin infections | Anti-amyloid | Reduces amyloid deposits in vivo by preventing metal-Aβ interactions | [114,129,134] |
| Acts as a zinc, iron and copper chelator and, by reducing the concentration of the ions, it also acts as an antioxidant | ||||
| Metformin | Antihyperglycemic drug | Anti-amyloid | Prevents hyperinsulinemia, reduces inflammation and oxidative stress | [129,135] |
| Observation of mixed results. In vivo, metformin reduced tau hyperphosphorylation but there is evidence that it can increase BACE-1 activity increasing Aβ levels. The co-administration with insulin decreased Aβ levels | ||||
| Escitalopram | Antidepressant | Agitation | Escitalopram is the (S)-enantiomer of citalopram | [96,136] |
| Currently in phase III clinical trials for agitation reduction | ||||
| Valproic acid | Antiepileptic | Anti-inflammatory and neuroprotection | In vivo, improves memory, reduces the accumulation of Aβ deposits and decreases inflammation. | [137,138] |
| Possible modulation of microglia | ||||
| Zileuton | Antiasthma | Anti-amyloid and anti-tau | Specific inhibitor of 5-lipoxygenase | [139] |
| Reduces β-amyloid and tau phosphorylation, and improves cognitive function |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martins, M.; Silva, R.; M. M. Pinto, M.; Sousa, E. Marine Natural Products, Multitarget Therapy and Repurposed Agents in Alzheimer’s Disease. Pharmaceuticals 2020, 13, 242. https://doi.org/10.3390/ph13090242
Martins M, Silva R, M. M. Pinto M, Sousa E. Marine Natural Products, Multitarget Therapy and Repurposed Agents in Alzheimer’s Disease. Pharmaceuticals. 2020; 13(9):242. https://doi.org/10.3390/ph13090242
Chicago/Turabian StyleMartins, Márcia, Renata Silva, Madalena M. M. Pinto, and Emília Sousa. 2020. "Marine Natural Products, Multitarget Therapy and Repurposed Agents in Alzheimer’s Disease" Pharmaceuticals 13, no. 9: 242. https://doi.org/10.3390/ph13090242
APA StyleMartins, M., Silva, R., M. M. Pinto, M., & Sousa, E. (2020). Marine Natural Products, Multitarget Therapy and Repurposed Agents in Alzheimer’s Disease. Pharmaceuticals, 13(9), 242. https://doi.org/10.3390/ph13090242