Upregulated and Hyperactivated Thalamic Connexin 43 Plays Important Roles in Pathomechanisms of Cognitive Impairment and Seizure of Autosomal Dominant Sleep-Related Hypermotor Epilepsy with S284L-Mutant α4 Subunit of Nicotinic ACh Receptor


Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
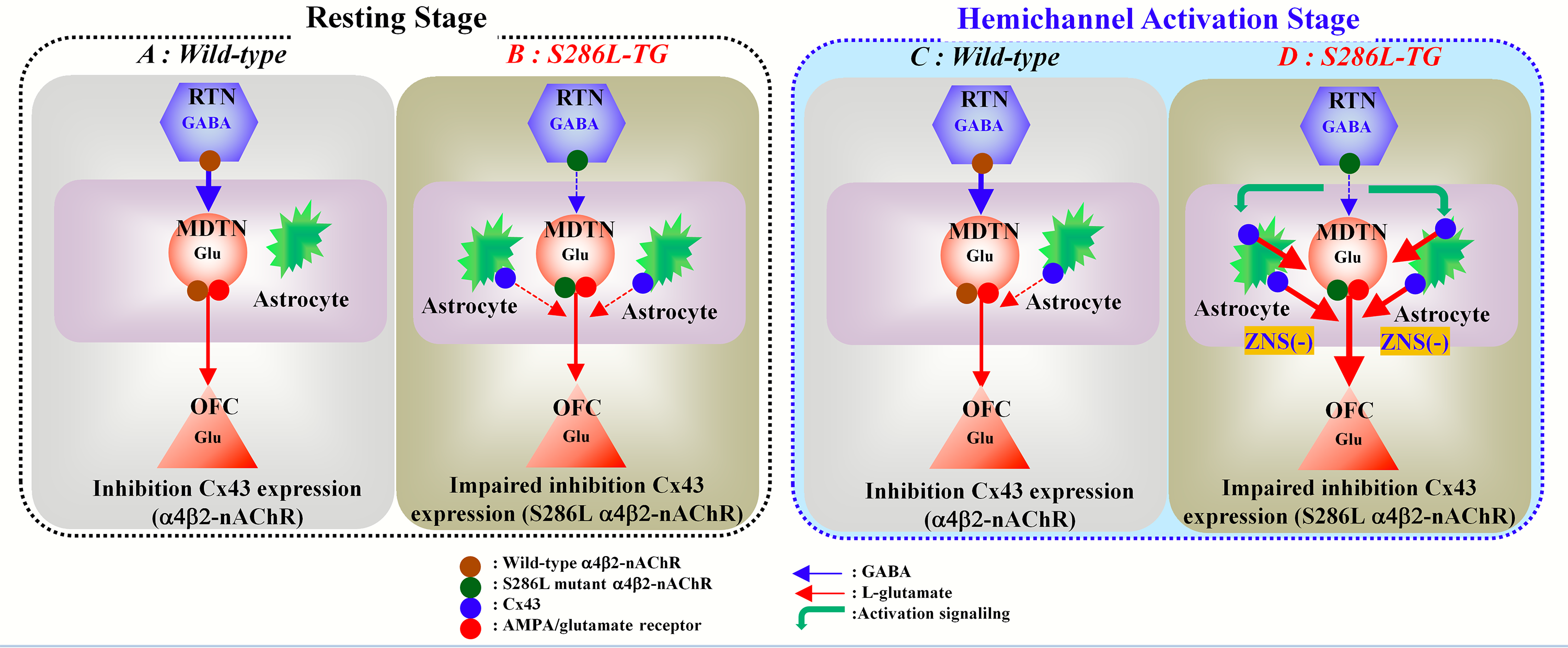
2.1. Effects of Subchronic Administrations of Therapeutic-Relevant Doses of CBZ and ZNS on Cx43 Expression in the Thalamic Cytosol and Plasma Membrane Fractions of S286L-TG
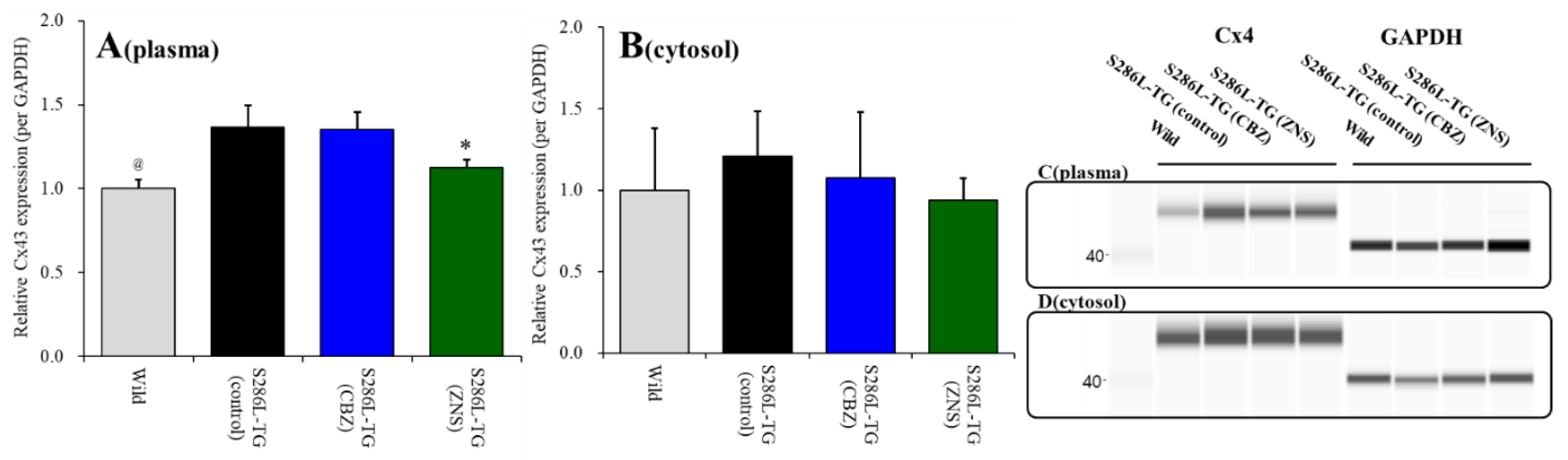
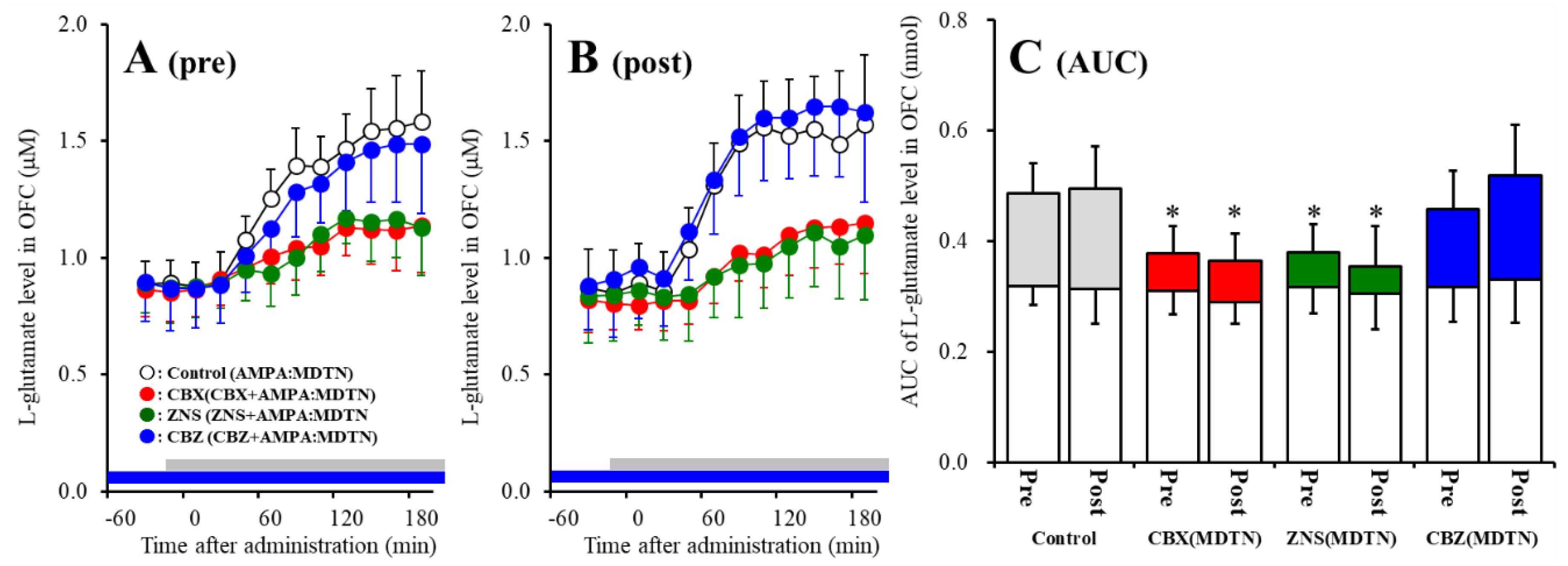
2.2. Effects of Local Administrations of Carbenoxolone (CBX), CBZ, and ZNS into the MDTN on Amino-3-(3-Hydroxy-5-Methyl-Isoxazol-4-yl)propanoic Acid (AMPA)-Evoked l-Glutamate Release in the OFC (Study 1)
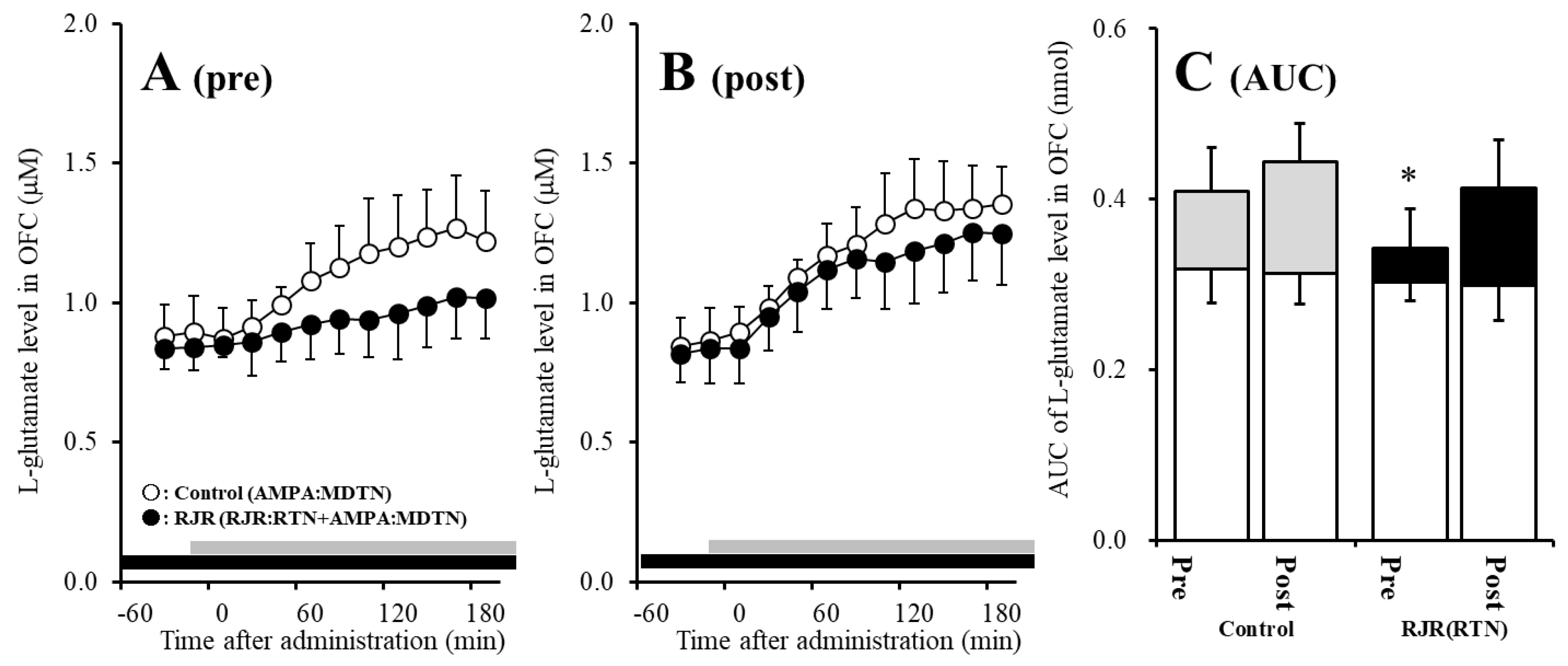
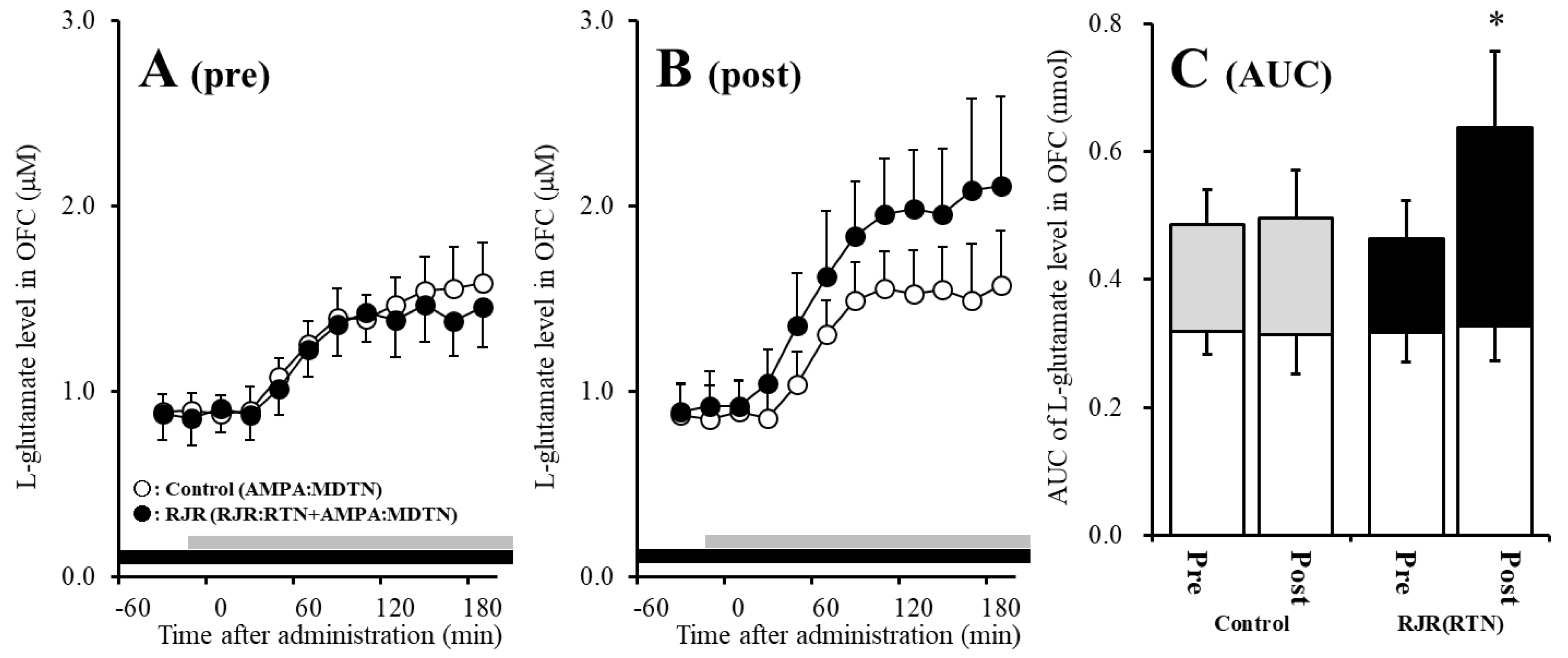
2.3. Effects of Local Administrations of (E)-N-Methyl-4-(3-Pyridinyl)-3-Buten-1-Amine Oxalate (RJR2403) into the RTN on AMPA-Evoked l-Glutamate Release in the OFC (Study 2)
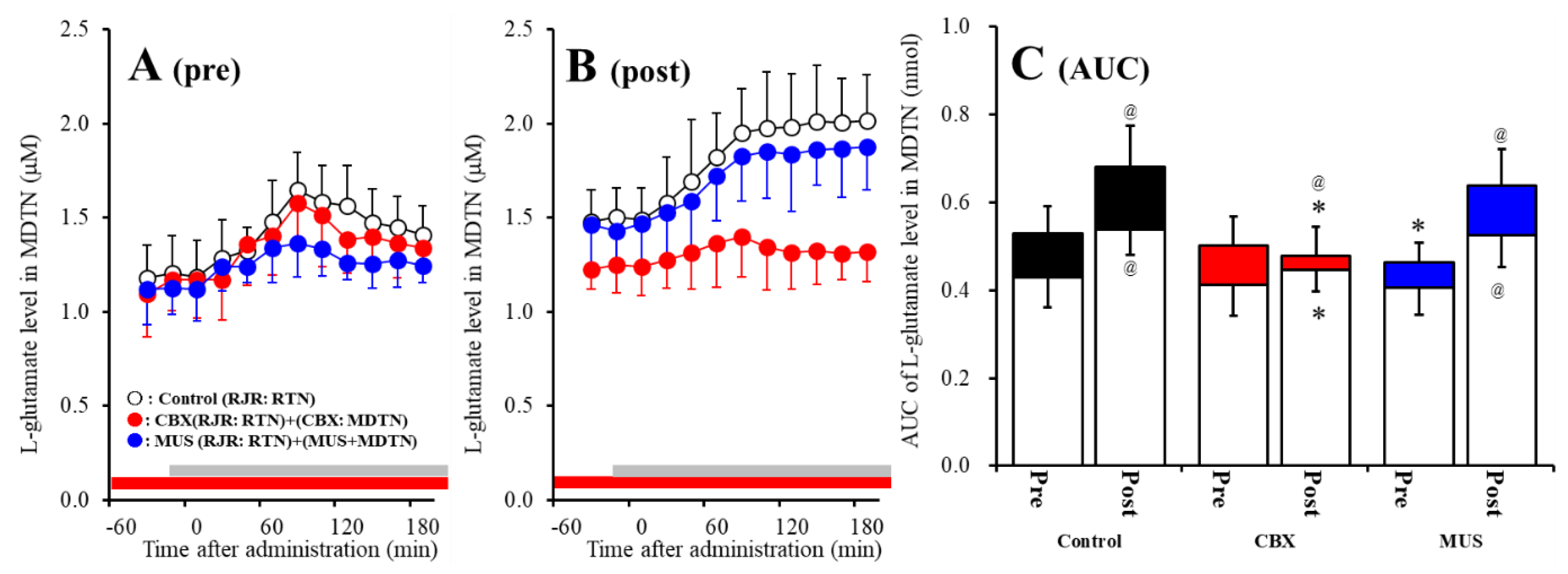
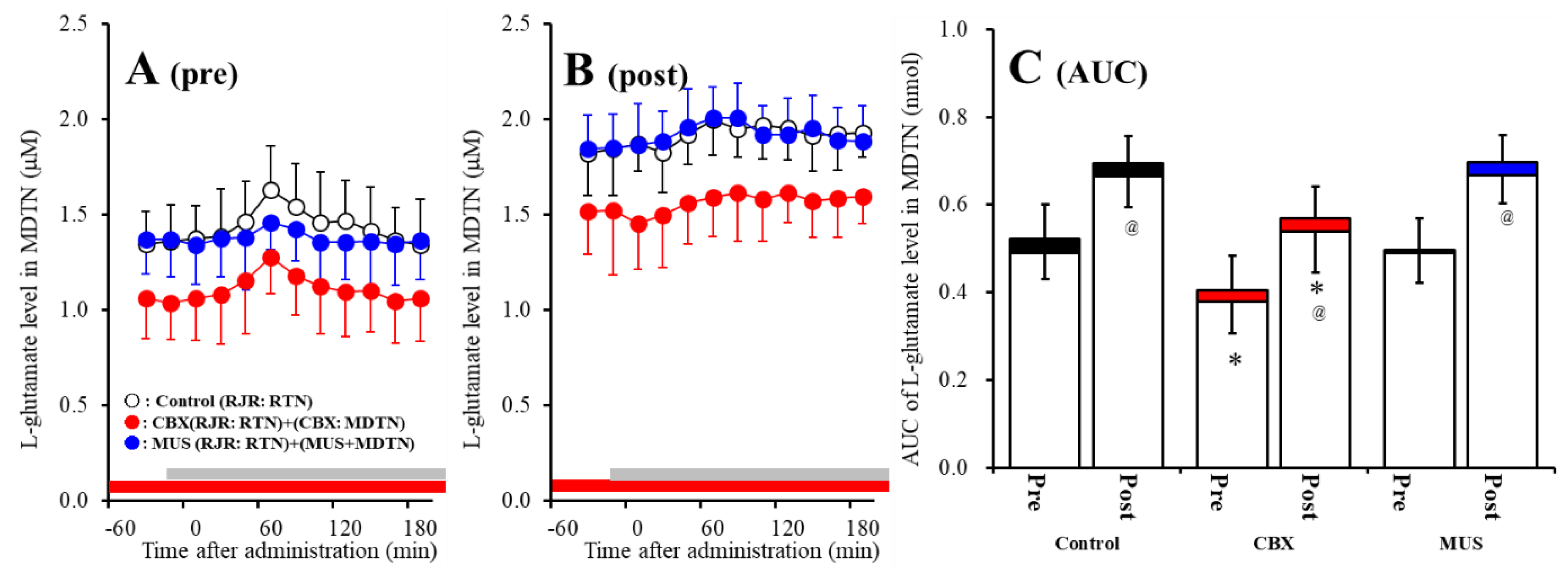
2.4. Effects of Local Administrations of CBX and Muscimol (MUS) into the MDTN on RJR-Evoked l-Glutamate Release in the MDTN (Study 3)
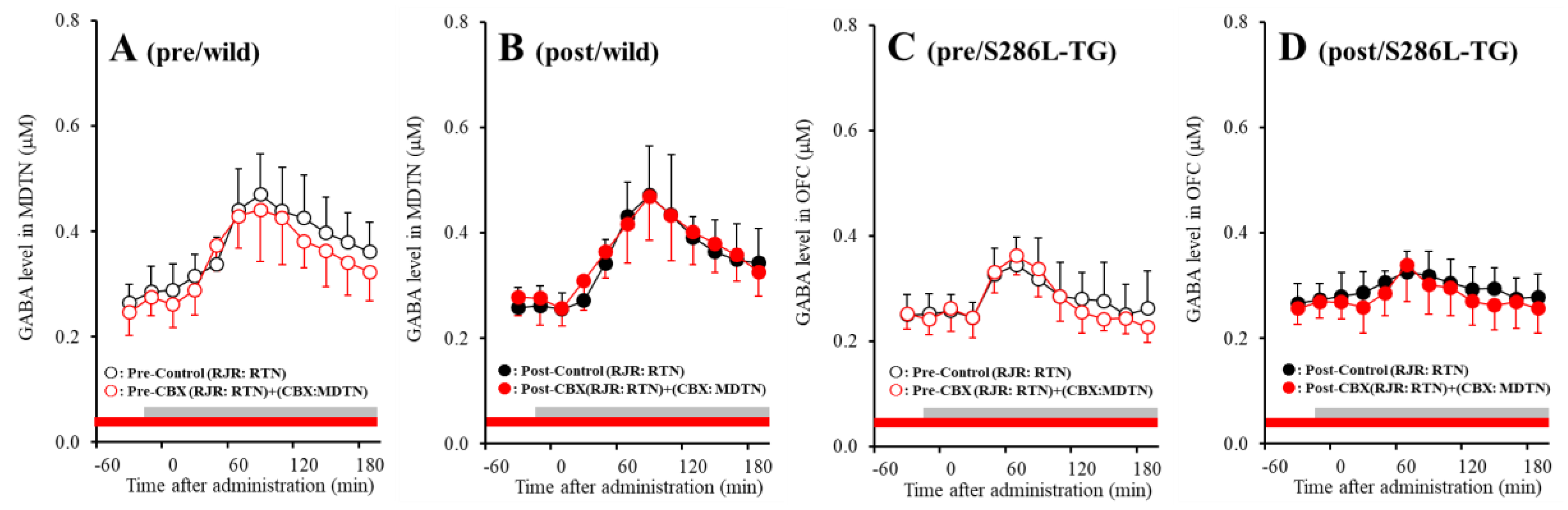
2.5. Effects of Local Administration of CBX into the MDTN on RJR-Evoked GABA Release in the MDTN (Study 3)
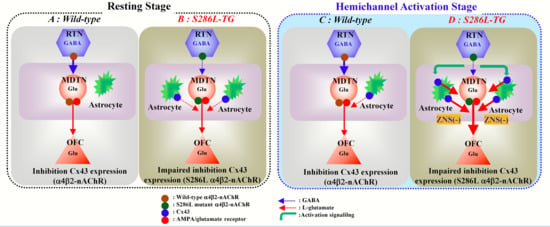
3. Discussion
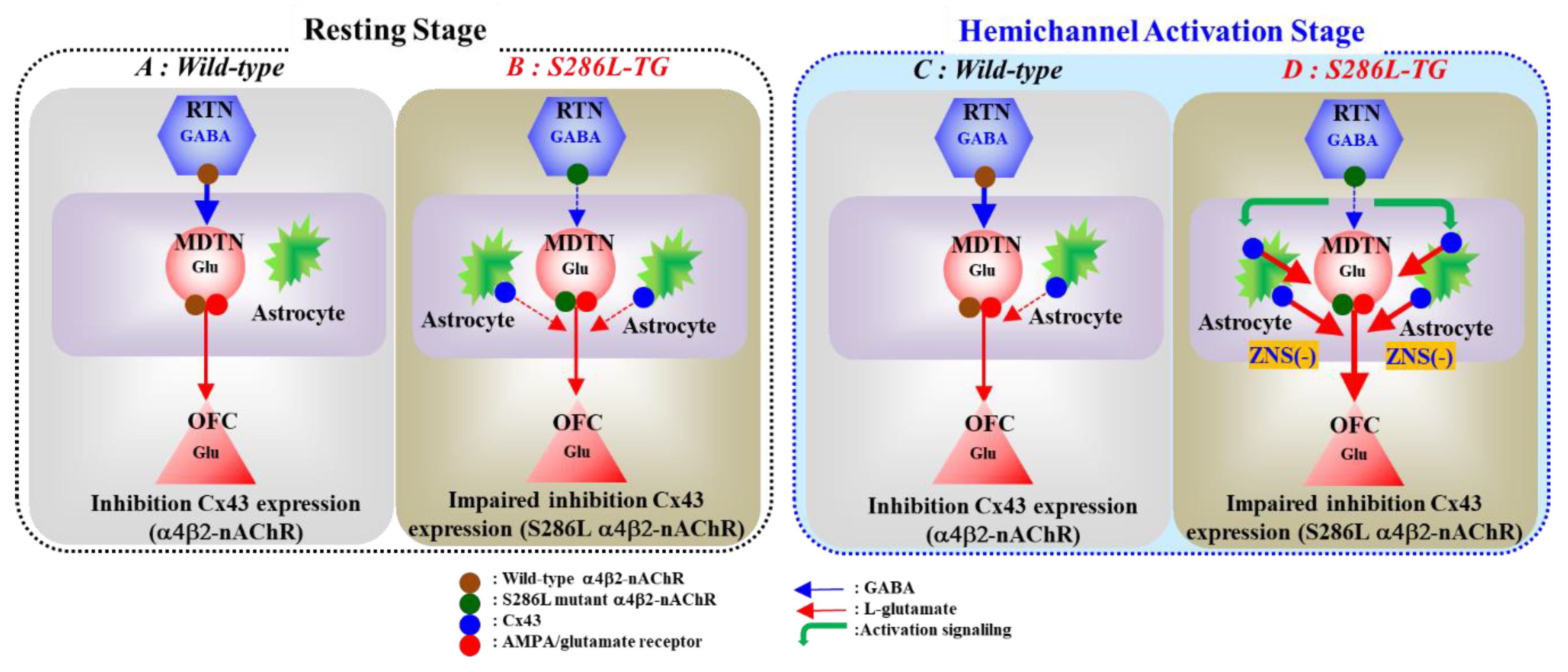
3.1. Pathomechanism of ADSHE Seizures Associated with Cx43
3.2. Pathomechanism of Cognitive Dificit Comorbidity of ADSHE with S284L-Mutation
4. Materials and Methods
4.1. Chemical Agents
4.2. Preparation of the Microdialysis System
4.3. Experimental Designs of Microdialysis Study
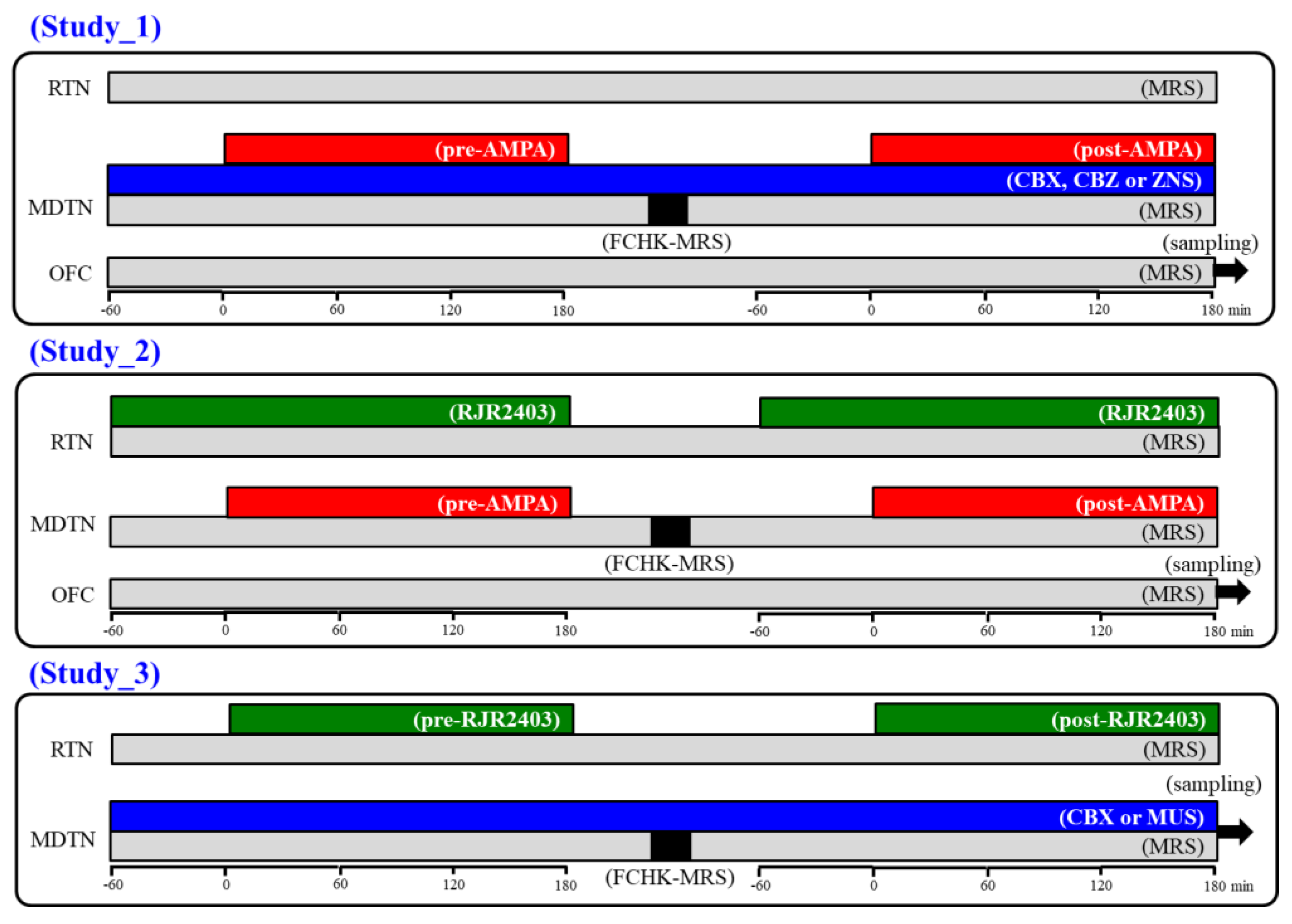
4.3.1. Study 1: Effects of CBX and Therapeutic-Relevant Concentrations of CBZ and ZNS on Glutamatergic Transmission in Thalamocortical Pathway
4.3.2. Study 2: Effects of RJR2403 on Glutamatergic Transmission in Thalamocortical Pathway
4.3.3. Study 3: Effects of α4β2-nAChR in the RTN on Intrathalamic Transmission
4.4. Ultra-High-Performance Liquid-Chromatography (UHPLC)
4.5. Simple Western Analysis
4.6. Data Analysis
4.7. Nomenclature of Targets and Ligands
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Steinlein, O.K. Animal models for autosomal dominant frontal lobe epilepsy: On the origin of seizures. Expert Rev. Neurother. 2010, 10, 1859–1867. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, I.E.; Bhatia, K.P.; Lopes-Cendes, I.; Fish, D.R.; Marsden, C.D.; Andermann, F.; Andermann, E.; Desbiens, R.; Cendes, F.; Manson, J.I.; et al. Autosomal dominant frontal epilepsy misdiagnosed as sleep disorder. Lancet 1994, 343, 515–517. [Google Scholar] [CrossRef]
- Tinuper, P.; Bisulli, F.; Cross, J.H.; Hesdorffer, D.; Kahane, P.; Nobili, L.; Provini, F.; Scheffer, I.E.; Tassi, L.; Vignatelli, L.; et al. Definition and diagnostic criteria of sleep-related hypermotor epilepsy. Neurology 2016, 86, 1834–1842. [Google Scholar] [CrossRef]
- Okada, M.; Zhu, G.; Yoshida, S.; Kaneko, S. Validation criteria for genetic animal models of epilepsy. Epilepsy Seizure 2010, 3, 109–120. [Google Scholar] [CrossRef][Green Version]
- Nobili, L.; Proserpio, P.; Combi, R.; Provini, F.; Plazzi, G.; Bisulli, F.; Tassi, L.; Tinuper, P. Nocturnal frontal lobe epilepsy. Curr. Neurol. Neurosci. Rep. 2014, 14, 424. [Google Scholar] [CrossRef] [PubMed]
- Provini, F.; Plazzi, G.; Tinuper, P.; Vandi, S.; Lugaresi, E.; Montagna, P. Nocturnal frontal lobe epilepsy. A clinical and polygraphic overview of 100 consecutive cases. Brain 1999, 122, 1017–1031. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, I.E.; Bhatia, K.P.; Lopes-Cendes, I.; Fish, D.R.; Marsden, C.D.; Andermann, E.; Andermann, F.; Desbiens, R.; Keene, D.; Cendes, F.; et al. Autosomal dominant nocturnal frontal lobe epilepsy. A distinctive clinical disorder. Brain 1995, 118, 61–73. [Google Scholar] [CrossRef]
- Provini, F.; Plazzi, G.; Montagna, P.; Lugaresi, E. The wide clinical spectrum of nocturnal frontal lobe epilepsy. Sleep Med. Rev. 2000, 4, 375–386. [Google Scholar] [CrossRef]
- Rozycka, A.; Skorupska, E.; Kostyrko, A.; Trzeciak, W.H. Evidence for S284L mutation of the CHRNA4 in a white family with autosomal dominant nocturnal frontal lobe epilepsy. Epilepsia 2003, 44, 1113–1117. [Google Scholar] [CrossRef]
- Ito, M.; Kobayashi, K.; Fujii, T.; Okuno, T.; Hirose, S.; Iwata, H.; Mitsudome, A.; Kaneko, S. Electroclinical picture of autosomal dominant nocturnal frontal lobe epilepsy in a Japanese family. Epilepsia 2000, 41, 52–58. [Google Scholar] [CrossRef]
- Hirose, S.; Iwata, H.; Akiyoshi, H.; Kobayashi, K.; Ito, M.; Wada, K.; Kaneko, S.; Mitsudome, A. A novel mutation of CHRNA4 responsible for autosomal dominant nocturnal frontal lobe epilepsy. Neurology 1999, 53, 1749–1753. [Google Scholar] [CrossRef] [PubMed]
- Miyajima, T.; Kumada, T.; Saito, K.; Fujii, T. Autism in siblings with autosomal dominant nocturnal frontal lobe epilepsy. Brain Dev. 2013, 35, 155–157. [Google Scholar] [CrossRef] [PubMed]
- Steinlein, O.K.; Mulley, J.C.; Propping, P.; Wallace, R.H.; Phillips, H.A.; Sutherland, G.R.; Scheffer, I.E.; Berkovic, S.F. A missense mutation in the neuronal nicotinic acetylcholine receptor alpha 4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy. Nat. Genet. 1995, 11, 201–203. [Google Scholar] [CrossRef] [PubMed]
- Steinlein, O.K.; Stoodt, J.; Mulley, J.; Berkovic, S.; Scheffer, I.E.; Brodtkorb, E. Independent occurrence of the CHRNA4 Ser248Phe mutation in a Norwegian family with nocturnal frontal lobe epilepsy. Epilepsia 2000, 41, 529–535. [Google Scholar] [CrossRef]
- Magnusson, A.; Stordal, E.; Brodtkorb, E.; Steinlein, O. Schizophrenia, psychotic illness and other psychiatric symptoms in families with autosomal dominant nocturnal frontal lobe epilepsy caused by different mutations. Psychiatr. Genet. 2003, 13, 91–95. [Google Scholar] [CrossRef]
- Saenz, A.; Galan, J.; Caloustian, C.; Lorenzo, F.; Marquez, C.; Rodriguez, N.; Jimenez, M.D.; Poza, J.J.; Cobo, A.M.; Grid, D.; et al. Autosomal dominant nocturnal frontal lobe epilepsy in a Spanish family with a Ser252Phe mutation in the CHRNA4 gene. Arch. Neurol. 1999, 56, 1004–1009. [Google Scholar] [CrossRef]
- McLellan, A.; Phillips, H.A.; Rittey, C.; Kirkpatrick, M.; Mulley, J.C.; Goudie, D.; Stephenson, J.B.; Tolmie, J.; Scheffer, I.E.; Berkovic, S.F.; et al. Phenotypic comparison of two Scottish families with mutations in different genes causing autosomal dominant nocturnal frontal lobe epilepsy. Epilepsia 2003, 44, 613–617. [Google Scholar] [CrossRef]
- Cho, Y.W.; Motamedi, G.K.; Laufenberg, I.; Sohn, S.I.; Lim, J.G.; Lee, H.; Yi, S.D.; Lee, J.H.; Kim, D.K.; Reba, R.; et al. A Korean kindred with autosomal dominant nocturnal frontal lobe epilepsy and mental retardation. Arch. Neurol 2003, 60, 1625–1632. [Google Scholar] [CrossRef]
- Phillips, H.A.; Favre, I.; Kirkpatrick, M.; Zuberi, S.M.; Goudie, D.; Heron, S.E.; Scheffer, I.E.; Sutherland, G.R.; Berkovic, S.F.; Bertrand, D.; et al. CHRNB2 is the second acetylcholine receptor subunit associated with autosomal dominant nocturnal frontal lobe epilepsy. Am. J. Hum. Genet. 2001, 68, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Steinlein, O.K.; Magnusson, A.; Stoodt, J.; Bertrand, S.; Weiland, S.; Berkovic, S.F.; Nakken, K.O.; Propping, P.; Bertrand, D. An insertion mutation of the CHRNA4 gene in a family with autosomal dominant nocturnal frontal lobe epilepsy. Hum. Mol. Genet. 1997, 6, 943–947. [Google Scholar] [CrossRef]
- Fukuyama, K.; Fukuzawa, M.; Shiroyama, T.; Okada, M. Pathogenesis and pathophysiology of autosomal dominant sleep-related hypermotor epilepsy with S284L-mutant alpha4 subunit of nicotinic ACh receptor. Br. J. Pharmacol. 2020, 177, 2143–2162. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, K.; Fukuzawa, M.; Shiroyama, T.; Okada, M. Pathomechanism of nocturnal paroxysmal dystonia in autosomal dominant sleep-related hypermotor epilepsy with S284L-mutant α4 subunit of nicotinic ACh receptor. Biomed. Pharmacother. Biomed. Pharmacother. 2020, 126, 110070. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, K.; Fukuzawa, M.; Ruri, O.; Okada, M. Upregulated Connexin 43 Induced by Loss-of-Functional S284L-Mutant alpha4 Subunit of Nicotinic ACh Receptor Contributes to Pathomechanisms of Autosomal Dominant Sleep-Related Hypermotor Epilepsy. Pharmaceuticals (Basel) 2020, 13, 58. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues-Pinguet, N.; Jia, L.; Li, M.; Figl, A.; Klaassen, A.; Truong, A.; Lester, H.A.; Cohen, B.N. Five ADNFLE mutations reduce the Ca2+ dependence of the mammalian alpha4beta2 acetylcholine response. J. Physiol. 2003, 550, 11–26. [Google Scholar] [CrossRef]
- Rodrigues-Pinguet, N.O.; Pinguet, T.J.; Figl, A.; Lester, H.A.; Cohen, B.N. Mutations linked to autosomal dominant nocturnal frontal lobe epilepsy affect allosteric Ca2+ activation of the alpha 4 beta 2 nicotinic acetylcholine receptor. Mol. Pharmacol. 2005, 68, 487–501. [Google Scholar] [CrossRef]
- Zhu, G.; Okada, M.; Yoshida, S.; Ueno, S.; Mori, F.; Takahara, T.; Saito, R.; Miura, Y.; Kishi, A.; Tomiyama, M.; et al. Rats harboring S284L Chrna4 mutation show attenuation of synaptic and extrasynaptic GABAergic transmission and exhibit the nocturnal frontal lobe epilepsy phenotype. J. Neurosci. 2008, 28, 12465–12476. [Google Scholar] [CrossRef]
- Ernst, C.; Nagy, C.; Kim, S.; Yang, J.P.; Deng, X.; Hellstrom, I.C.; Choi, K.H.; Gershenfeld, H.; Meaney, M.J.; Turecki, G. Dysfunction of astrocyte connexins 30 and 43 in dorsal lateral prefrontal cortex of suicide completers. Biol. Psychiatry 2011, 70, 312–319. [Google Scholar] [CrossRef]
- Miguel-Hidalgo, J.J.; Wilson, B.A.; Hussain, S.; Meshram, A.; Rajkowska, G.; Stockmeier, C.A. Reduced connexin 43 immunolabeling in the orbitofrontal cortex in alcohol dependence and depression. J. Psychiatr. Res. 2014, 55, 101–109. [Google Scholar] [CrossRef]
- Fukuyama, K.; Okubo, R.; Murata, M.; Shiroyama, T.; Okada, M. Activation of Astroglial Connexin is Involved in Concentration-Dependent Double-Edged Sword Clinical Action of Clozapine. Cells 2020, 9, 414. [Google Scholar] [CrossRef]
- Sun, J.-D.; Liu, Y.; Yuan, Y.-H.; Li, J.; Chen, N.-H. Gap junction dysfunction in the prefrontal cortex induces depressive-like behaviors in rats. Neuropsychopharmacology 2012, 37, 1305–1320. [Google Scholar] [CrossRef]
- Irwin, L.N. Gene expression in the hippocampus of behaviorally stimulated rats: Analysis by DNA microarray. Mol. Brain Res. 2001, 96, 163–169. [Google Scholar] [CrossRef]
- Ribeiro-Rodrigues, T.M.; Martins-Marques, T.; Morel, S.; Kwak, B.R.; Girao, H. Role of connexin 43 in different forms of intercellular communication-gap junctions, extracellular vesicles and tunnelling nanotubes. J. Cell Sci. 2017, 130, 3619–3630. [Google Scholar] [CrossRef] [PubMed]
- Dallerac, G.; Rouach, N. Astrocytes as new targets to improve cognitive functions. Prog. Neurobiol. 2016, 144, 48–67. [Google Scholar] [CrossRef] [PubMed]
- Duerrschmidt, N.; Hagen, A.; Gaertner, C.; Wermke, A.; Nowicki, M.; Spanel-Borowski, K.; Stepan, H.; Mohr, F.W.; Dhein, S. Nicotine effects on human endothelial intercellular communication via alpha4beta2 and alpha3beta2 nicotinic acetylcholine receptor subtypes. Naunyn. Schmiedebergs Arch. Pharmacol. 2012, 385, 621–632. [Google Scholar] [CrossRef]
- Lapato, A.S.; Tiwari-Woodruff, S.K. Connexins and pannexins: At the junction of neuro-glial homeostasis & disease. J. Neurosci. Res. 2018, 96, 31–44. [Google Scholar]
- Li, Q.; Li, Q.Q.; Jia, J.N.; Liu, Z.Q.; Zhou, H.H.; Mao, X.Y. Targeting gap junction in epilepsy: Perspectives and challenges. Biomed. Pharmacother. Biomed. Pharmacother. 2019, 109, 57–65. [Google Scholar] [CrossRef]
- Wu, X.M.; Wang, G.L.; Miao, J.; Feng, J.C. Effect of connexin 36 blockers on the neuronal cytoskeleton and synaptic plasticity in kainic acid-kindled rats. Transl. Neurosci. 2015, 6, 252–258. [Google Scholar] [CrossRef]
- Jin, M.; Dai, Y.; Xu, C.; Wang, Y.; Wang, S.; Chen, Z. Effects of meclofenamic acid on limbic epileptogenesis in mice kindling models. Neurosci. Lett. 2013, 543, 110–114. [Google Scholar] [CrossRef]
- Mylvaganam, S.; Ramani, M.; Krawczyk, M.; Carlen, P.L. Roles of gap junctions, connexins, and pannexins in epilepsy. Front. Physiol. 2014, 5, 172. [Google Scholar] [CrossRef]
- Fasciani, I.; Temperan, A.; Perez-Atencio, L.F.; Escudero, A.; Martinez-Montero, P.; Molano, J.; Gomez-Hernandez, J.M.; Paino, C.L.; Gonzalez-Nieto, D.; Barrio, L.C. Regulation of connexin hemichannel activity by membrane potential and the extracellular calcium in health and disease. Neuropharmacology 2013, 75, 479–490. [Google Scholar] [CrossRef]
- Kar, R.; Batra, N.; Riquelme, M.A.; Jiang, J.X. Biological role of connexin intercellular channels and hemichannels. Arch. Biochem. Biophys. 2012, 524, 2–15. [Google Scholar] [CrossRef]
- Carmignoto, G.; Haydon, P.G. Astrocyte calcium signaling and epilepsy. Glia 2012, 60, 1227–1233. [Google Scholar] [CrossRef]
- Rusakov, D.; Fine, A. Extracellular Ca2+ Depletion Contributes to Fast Activity-Dependent Modulation of Synaptic Transmission in the Brain. Neuron 2003, 37, 287. [Google Scholar] [CrossRef]
- Okada, M.; Fukuyama, K.; Shiroyama, T.; Ueda, Y. Lurasidone inhibits NMDA antagonist-induced functional abnormality of thalamocortical glutamatergic transmission via 5-HT7 receptor blockade. Br. J. Pharmacol. 2019, 176, 4002–4018. [Google Scholar] [CrossRef]
- Okada, M.; Fukuyama, K.; Kawano, Y.; Shiroyama, T.; Suzuki, D.; Ueda, Y. Effects of acute and sub-chronic administrations of guanfacine on catecholaminergic transmissions in the orbitofrontal cortex. Neuropharmacology 2019, 156, 107547. [Google Scholar] [CrossRef]
- Schuetze, M.; Park, M.T.; Cho, I.Y.; MacMaster, F.P.; Chakravarty, M.M.; Bray, S.L. Morphological Alterations in the Thalamus, Striatum, and Pallidum in Autism Spectrum Disorder. Neuropsychopharmacology 2016, 41, 2627–2637. [Google Scholar] [CrossRef]
- Karlsen, A.S.; Korbo, S.; Uylings, H.B.; Pakkenberg, B. A stereological study of the mediodorsal thalamic nucleus in Down syndrome. Neuroscience 2014, 279, 253–259. [Google Scholar] [CrossRef]
- Leeman-Markowski, B.A.; Smart, O.L.; Faught, R.E.; Gross, R.E.; Meador, K.J. Cessation of gamma activity in the dorsomedial nucleus associated with loss of consciousness during focal seizures. Epilepsy Behav. 2015, 51, 215–220. [Google Scholar] [CrossRef]
- Porrino, L.J.; Crane, A.M.; Goldman-Rakic, P.S. Direct and indirect pathways from the amygdala to the frontal lobe in rhesus monkeys. J. Comp. Neurol. 1981, 198, 121–136. [Google Scholar] [CrossRef]
- Russchen, F.T.; Amaral, D.G.; Price, J.L. The afferent input to the magnocellular division of the mediodorsal thalamic nucleus in the monkey, Macaca fascicularis. J. Comp. Neurol. 1987, 256, 175–210. [Google Scholar] [CrossRef]
- McCormick, D.A.; Wang, Z. Serotonin and noradrenaline excite GABAergic neurones of the guinea-pig and cat nucleus reticularis thalami. J. Physiol. 1991, 442, 235–255. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, K.; Kato, R.; Murata, M.; Shiroyama, T.; Okada, M. Clozapine Normalizes a Glutamatergic Transmission Abnormality Induced by an Impaired NMDA Receptor in the Thalamocortical Pathway via the Activation of a Group III Metabotropic Glutamate Receptor. Biomolecules 2019, 9, 234. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Hasegawa, T.; Suzuki, D.; Motomura, E.; Okada, M. Amantadine Combines Astroglial System Xc(-) Activation with Glutamate/NMDA Receptor Inhibition. Biomolecules 2019, 9, 191. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Fukuyama, K.; Kawano, Y.; Shiroyama, T.; Ueda, Y. Memantine protects thalamocortical hyper-glutamatergic transmission induced by NMDA receptor antagonism via activation of system xc(). Pharmacol. Res. Perspect. 2019, 7, e00457. [Google Scholar] [CrossRef]
- Fukuyama, K.; Hasegawa, T.; Okada, M. Cystine/Glutamate Antiporter and Aripiprazole Compensate NMDA Antagonist-Induced Dysfunction of Thalamocortical L-Glutamatergic Transmission. Int. J. Mol. Sci. 2018, 19, 3645. [Google Scholar] [CrossRef]
- Alexander, S.P.H.; Mathie, A.; Peters, J.A.; Veale, E.L.; Striessnig, J.; Kelly, E.; Armstrong, J.F.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; et al. THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Ion channels. Br. J. Pharmacol. 2019, 176 (Suppl. S1), S142–S228. [Google Scholar] [CrossRef]
- Wang, N.; De Bock, M.; Decrock, E.; Bol, M.; Gadicherla, A.; Bultynck, G.; Leybaert, L. Connexin targeting peptides as inhibitors of voltage- and intracellular Ca2+-triggered Cx43 hemichannel opening. Neuropharmacology 2013, 75, 506–516. [Google Scholar] [CrossRef]
- Yoshida, S.; Okada, M.; Zhu, G.; Kaneko, S. Effects of zonisamide on neurotransmitter exocytosis associated with ryanodine receptors. Epilepsy Res. 2005, 67, 153–162. [Google Scholar] [CrossRef]
- Yamamura, S.; Ohoyama, K.; Nagase, H.; Okada, M. Zonisamide enhances delta receptor-associated neurotransmitter release in striato-pallidal pathway. Neuropharmacology 2009, 57, 322–331. [Google Scholar] [CrossRef]
- Kawata, Y.; Okada, M.; Murakami, T.; Kamata, A.; Zhu, G.; Kaneko, S. Pharmacological discrimination between effects of carbamazepine on hippocampal basal, Ca(2+)- and K(+)-evoked serotonin release. Br. J. Pharmacol. 2001, 133, 557–567. [Google Scholar] [CrossRef]
- Okada, M.; Hirano, T.; Mizuno, K.; Kawata, Y.; Wada, K.; Murakami, T.; Tasaki, H.; Kaneko, S. Effects of carbamazepine on hippocampal serotonergic system. Epilepsy Res. 1998, 31, 187–198. [Google Scholar] [CrossRef]
- Yamamura, S.; Hamaguchi, T.; Ohoyama, K.; Sugiura, Y.; Suzuki, D.; Kanehara, S.; Nakagawa, M.; Motomura, E.; Matsumoto, T.; Tanii, H.; et al. Topiramate and zonisamide prevent paradoxical intoxication induced by carbamazepine and phenytoin. Epilepsy Res. 2009, 84, 172–186. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Zhu, G.; Yoshida, S.; Kanai, K.; Hirose, S.; Kaneko, S. Exocytosis mechanism as a new targeting site for mechanisms of action of antiepileptic drugs. Life Sci. 2002, 72, 465–473. [Google Scholar] [CrossRef]
- McGrath, J.C.; Lilley, E. Implementing guidelines on reporting research using animals (ARRIVE etc.): New requirements for publication in BJP. Br. J. Pharmacol. 2015, 172, 3189–3193. [Google Scholar] [CrossRef]
- Paxinos, G.; Watson, C. The Rat Brain: In Stereotoxic Coordinates, 6th ed.; Academic Press: San Diego, CA, USA, 2007. [Google Scholar]
- Okada, M.; Wada, K.; Kiryu, K.; Kawata, Y.; Mizuno, K.; Kondo, T.; Tasaki, H.; Kaneko, S. Effects of Ca2+ channel antagonists on striatal dopamine and DOPA release, studied by in vivo microdialysis. Br. J. Pharmacol. 1998, 123, 805–814. [Google Scholar] [CrossRef]
- Okada, M.; Kawata, Y.; Mizuno, K.; Wada, K.; Kondo, T.; Kaneko, S. Interaction between Ca2+, K+, carbamazepine and zonisamide on hippocampal extracellular glutamate monitored with a microdialysis electrode. Br. J. Pharmacol. 1998, 124, 1277–1285. [Google Scholar] [CrossRef]
- Mtui, E.; Gruener, G.; Dockery, P. Fitzgerald’s Clinical Neuroanatomy and Neuroscience, 7th ed.; Elsevier: Philadelphia, PA, USA, 2015. [Google Scholar]
- Okada, M.; Fukuyama, K.; Shiroyama, T.; Ueda, Y. Carbamazepine Attenuates Astroglial L-Glutamate Release Induced by Pro-Inflammatory Cytokines via Chronically Activation of Adenosine A2A Receptor. Int. J. Mol. Sci. 2019, 20, 3727. [Google Scholar] [CrossRef]
- Okada, M.; Fukuyama, K.; Nakano, T.; Ueda, Y. Pharmacological Discrimination of Effects of MK801 on Thalamocortical, Mesothalamic, and Mesocortical Transmissions. Biomolecules 2019, 9, 746. [Google Scholar] [CrossRef]
- Curtis, M.J.; Alexander, S.; Cirino, G.; Docherty, J.R.; George, C.H.; Giembycz, M.A.; Hoyer, D.; Insel, P.A.; Izzo, A.A.; Ji, Y.; et al. Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. Br. J. Pharmacol. 2018, 175, 987–993. [Google Scholar] [CrossRef]
- Harding, S.D.; Sharman, J.L.; Faccenda, E.; Southan, C.; Pawson, A.J.; Ireland, S.; Gray, A.J.G.; Bruce, L.; Alexander, S.P.H.; Anderton, S.; et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Res. 2018, 46, D1091–D1106. [Google Scholar] [CrossRef]
- Alexander, S.P.H.; Christopoulos, A.; Davenport, A.P.; Kelly, E.; Mathie, A.; Peters, J.A.; Veale, E.L.; Armstrong, J.F.; Faccenda, E.; Harding, S.D.; et al. THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: G protein-coupled receptors. Br. J. Pharmacol. 2019, 176 (Suppl. S1), S21–S141. [Google Scholar] [CrossRef] [PubMed]










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fukuyama, K.; Fukuzawa, M.; Okada, M. Upregulated and Hyperactivated Thalamic Connexin 43 Plays Important Roles in Pathomechanisms of Cognitive Impairment and Seizure of Autosomal Dominant Sleep-Related Hypermotor Epilepsy with S284L-Mutant α4 Subunit of Nicotinic ACh Receptor. Pharmaceuticals 2020, 13, 99. https://doi.org/10.3390/ph13050099
Fukuyama K, Fukuzawa M, Okada M. Upregulated and Hyperactivated Thalamic Connexin 43 Plays Important Roles in Pathomechanisms of Cognitive Impairment and Seizure of Autosomal Dominant Sleep-Related Hypermotor Epilepsy with S284L-Mutant α4 Subunit of Nicotinic ACh Receptor. Pharmaceuticals. 2020; 13(5):99. https://doi.org/10.3390/ph13050099
Chicago/Turabian StyleFukuyama, Kouji, Masashi Fukuzawa, and Motohiro Okada. 2020. "Upregulated and Hyperactivated Thalamic Connexin 43 Plays Important Roles in Pathomechanisms of Cognitive Impairment and Seizure of Autosomal Dominant Sleep-Related Hypermotor Epilepsy with S284L-Mutant α4 Subunit of Nicotinic ACh Receptor" Pharmaceuticals 13, no. 5: 99. https://doi.org/10.3390/ph13050099
APA StyleFukuyama, K., Fukuzawa, M., & Okada, M. (2020). Upregulated and Hyperactivated Thalamic Connexin 43 Plays Important Roles in Pathomechanisms of Cognitive Impairment and Seizure of Autosomal Dominant Sleep-Related Hypermotor Epilepsy with S284L-Mutant α4 Subunit of Nicotinic ACh Receptor. Pharmaceuticals, 13(5), 99. https://doi.org/10.3390/ph13050099