Crosslinked Fibroin Nanoparticles: Investigations on Biostability, Cytotoxicity, and Cellular Internalization

 , and
, and
Abstract
1. Introduction
2. Results and Discussion
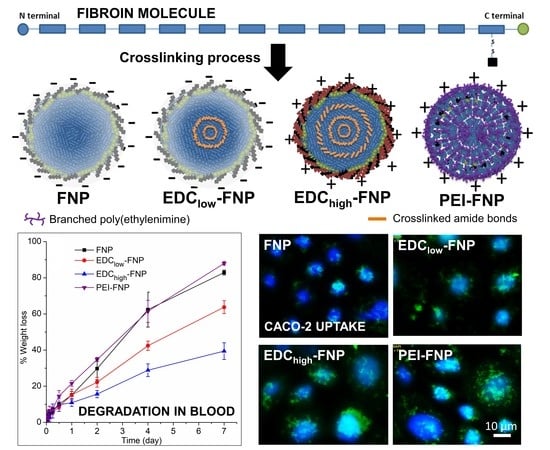
2.1. FNP Characterization
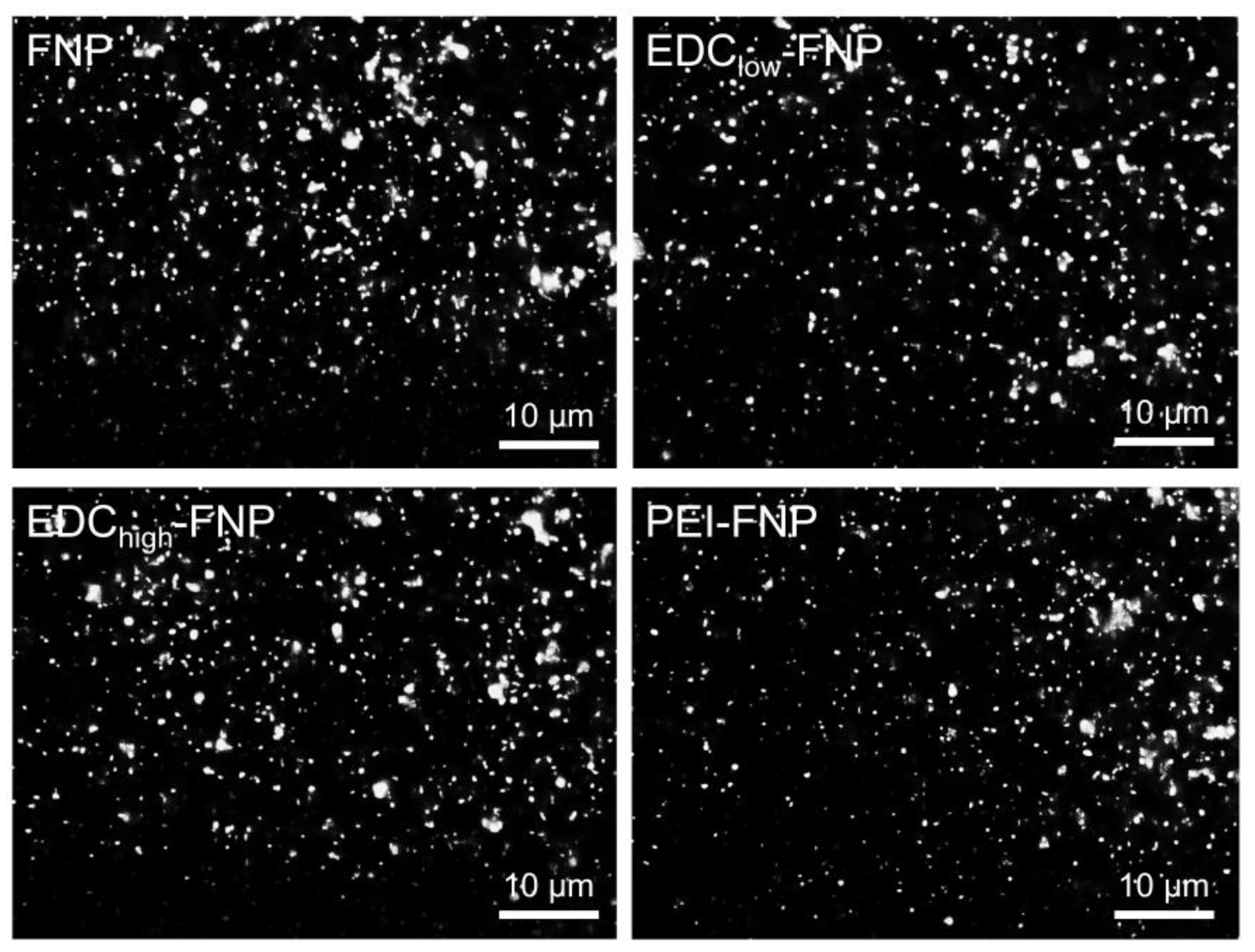
2.2. Biostability and Hemolysis Study
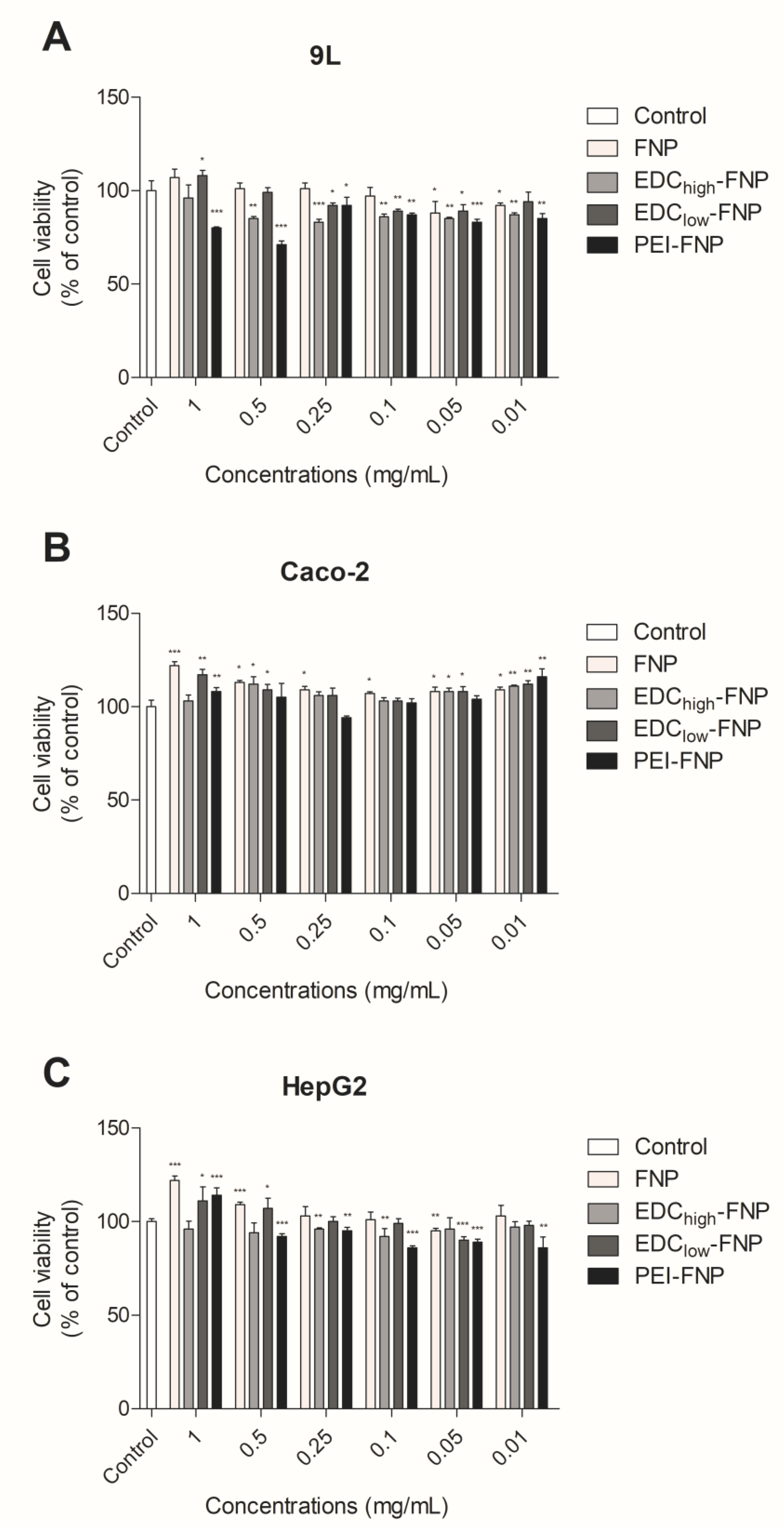
2.3. In Vitro Cytotoxicity
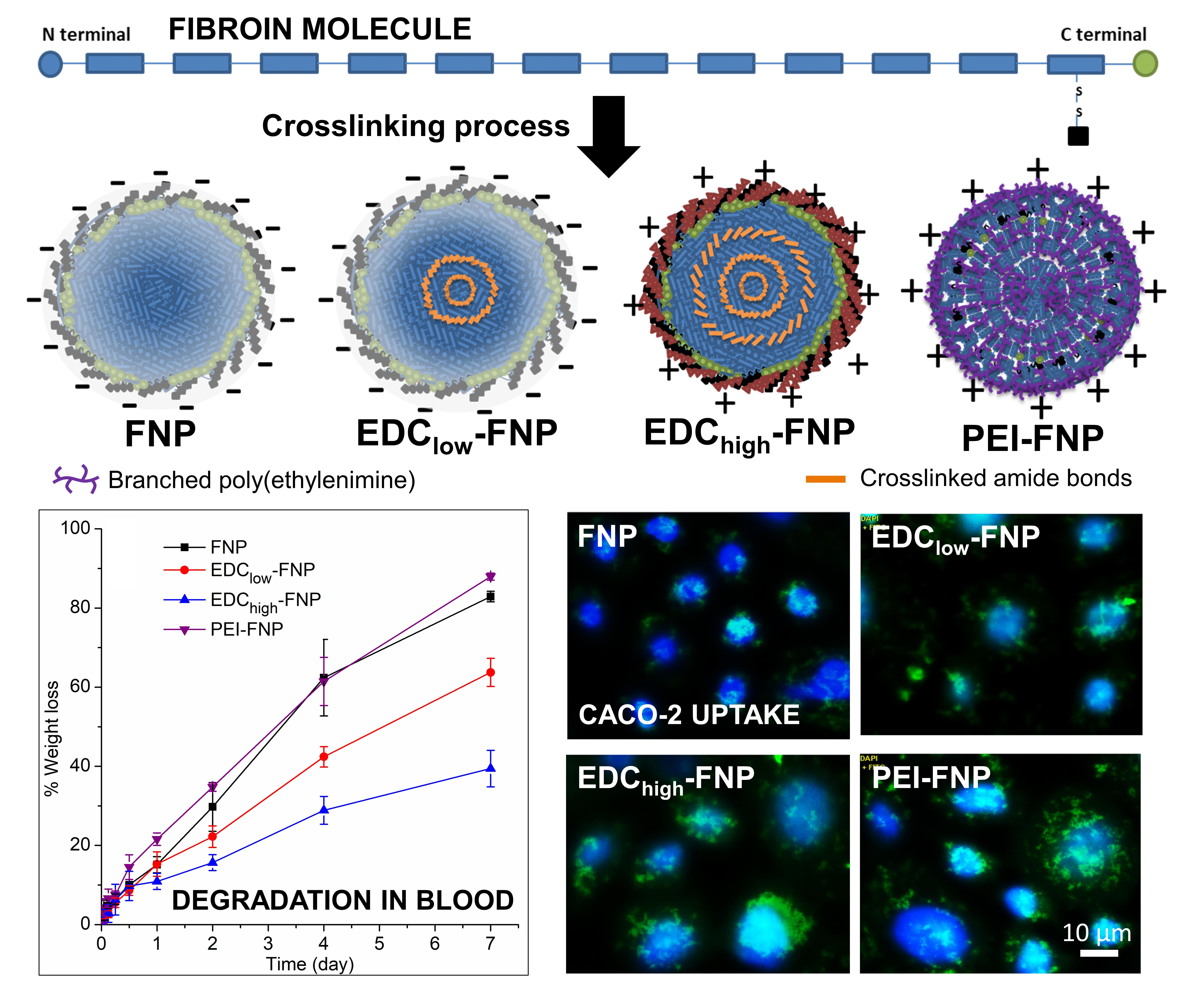
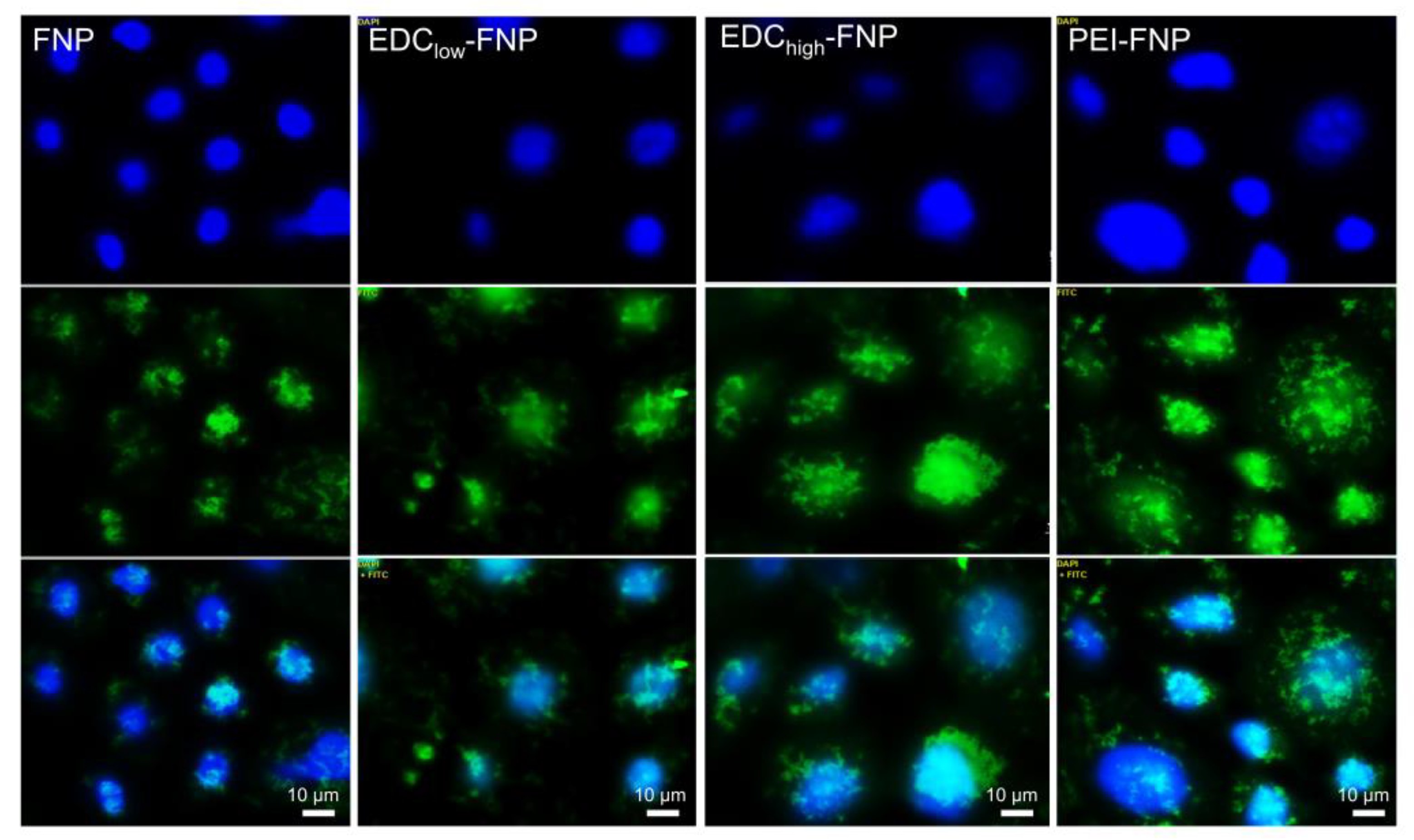
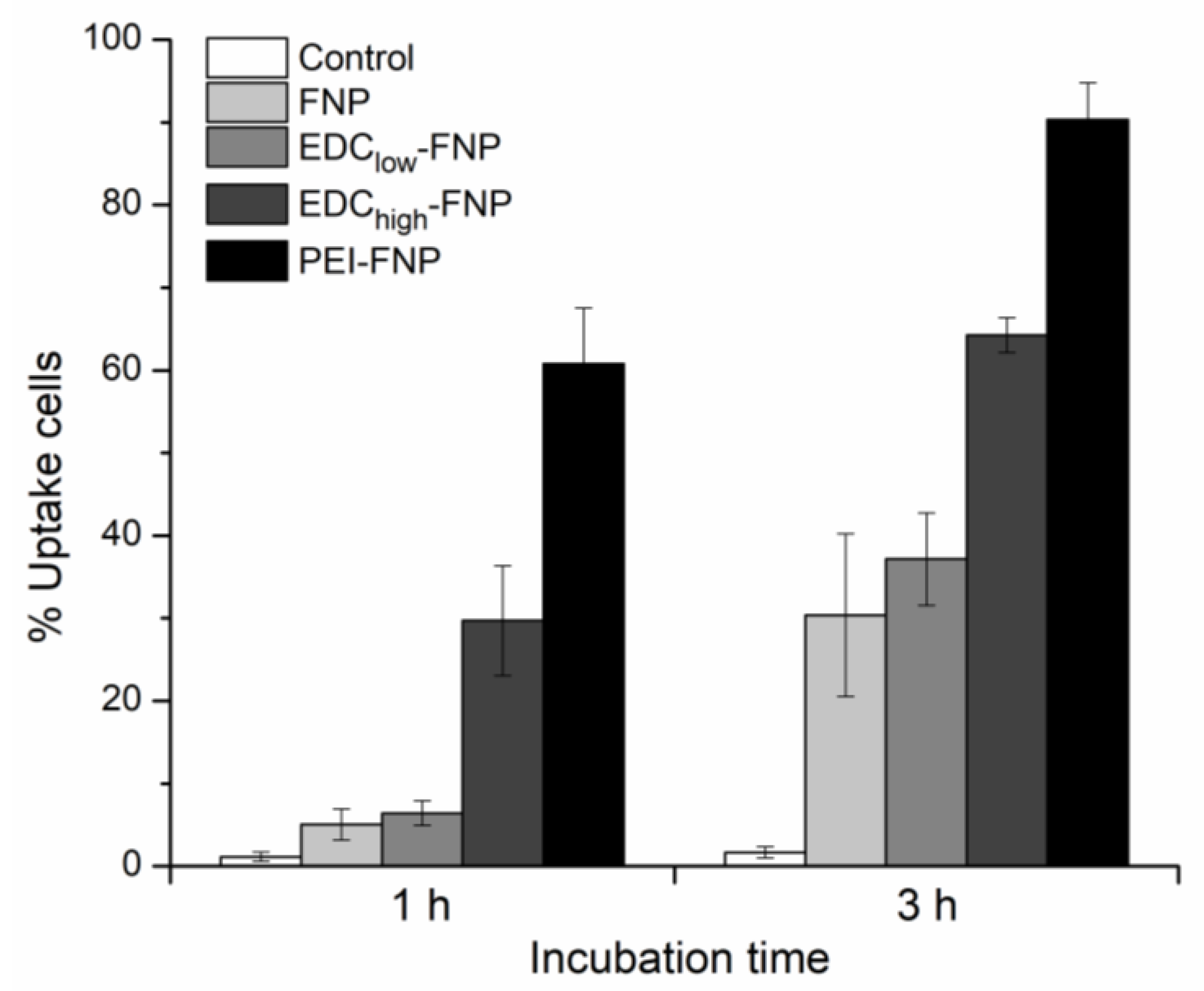
2.4. Cellular Uptake and Flow Cytometry Study
3. Materials and Methods
3.1. Materials
3.2. Fibroin Extraction
3.3. FNP Formulation
3.4. FNP Characterization
3.4.1. Particle Size and Zeta Potential
3.4.2. FITC-Binding Efficiency
3.4.3. FITC Dissolution Profile
3.4.4. Physical Stability
3.5. Biostability Study
3.6. Hemolysis
3.7. Cell Culture
3.8. In Vitro Cytotoxicity
3.9. Cellular Uptake and Flow Cytometry Study
3.10. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Holland, C.; Numata, K.; Rnjak-Kovacina, J.; Seib, F.P. The biomedical use of silk: Past, present, future. Adv. Healthc. Mater. 2019, 8, 1800465. [Google Scholar] [CrossRef]
- Altman, G.H.; Diaz, F.; Jakuba, C.; Calabro, T.; Horan, R.L.; Chen, J.; Lu, H.; Richmond, J.; Kaplan, D.L. Silk-based biomaterials. Biomaterials 2003, 24, 401–416. [Google Scholar] [CrossRef]
- Aramwit, P. Silk: Properties, Production and Uses; Silk materials for drug delivery devices; Nova Science Publishers: Hauppauge, NY, USA, 2012; ISBN 9781621006923. [Google Scholar]
- Zhao, Z.; Li, Y.; Xie, M. Silk fibroin-based nanoparticles for drug delivery. Int. J. Mol. Sci. 2015, 16, 4880–4903. [Google Scholar] [CrossRef] [PubMed]
- Numata, K.; Kaplan, D.L. Silk-based delivery systems of bioactive molecules. Adv. Drug Deliv. Rev. 2010, 62, 1497–1508. [Google Scholar] [CrossRef] [PubMed]
- Din Wani, S.U.; Veerabhadrappa, G.H. Silk fibroin based drug delivery applications: Promises and challenges. Curr. Drug Targets 2018, 19, 1177–1190. [Google Scholar] [CrossRef] [PubMed]
- Wenk, E.; Merkle, H.P.; Meinel, L. Silk fibroin as a vehicle for drug delivery applications. J. Control. Release 2011, 150, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Jeencham, R.; Sutheerawattananonda, M.; Tiyaboonchai, W. Preparation and characterization of chitosan/regenerated silk fibroin (CS/RSF) films as a biomaterial for contact lenses-based ophthalmic drug delivery system. Int. J. Appl. Pharm. 2019, 11, 275–284. [Google Scholar] [CrossRef]
- De Moraes, M.A.; Nogueira, G.M.; Weska, R.F.; Beppu, M.M. Preparation and characterization of insoluble silk fibroin/chitosan blend films. Polymers 2010, 2, 719–727. [Google Scholar] [CrossRef]
- Ribeiro, V.P.; Silva-Correia, J.; Gonçalves, C.; Pina, S.; Radhouani, H.; Montonen, T.; Hyttinen, J.; Roy, A.; Oliveira, A.L.; Reis, R.L.; et al. Rapidly responsive silk fibroin hydrogels as an artificial matrix for the programmed tumor cells death. PLoS One 2018, 13, e0194441. [Google Scholar] [CrossRef]
- Dong, T.; Mi, R.; Wu, M.; Zhong, N.; Zhao, X.; Chen, X.; Shao, Z. The regenerated silk fibroin hydrogel with designed architecture bioprinted by its microhydrogel. J. Mater. Chem. B. 2019, 7, 4328–4337. [Google Scholar] [CrossRef]
- Martín-Martín, Y.; Fernández-García, L.; Sanchez-Rebato, M.H.; Marí-Buyé, N.; Rojo, F.J.; Pérez-Rigueiro, J.; Ramos, M.; Guinea, G.V.; Panetsos, F.; González-Nieto, D. Evaluation of neurosecretome from mesenchymal stem cells encapsulated in silk fibroin hydrogels. Sci. Rep. 2019, 9, 8801. [Google Scholar] [CrossRef] [PubMed]
- Katayama, H.; Issiki, M.; Yoshitomi, H. Application of fibroin in controlled release tablets containing theophylline. Biol. Pharm. Bull. 2000, 23, 1229–1234. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Melke, J.; Midha, S.; Ghosh, S.; Ito, K.; Hofmann, S. Silk fibroin as biomaterial for bone tissue engineering. Acta Biomater. 2016, 31, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Vepari, C.; Jin, H.-J.; Kim, H.J.; Kaplan, D.L. Electrospun silk-BMP-2 scaffolds for bone tissue engineering. Biomaterials 2006, 27, 3115–3124. [Google Scholar] [CrossRef] [PubMed]
- Chomchalao, P.; Pongcharoen, S.; Sutheerawattananonda, M.; Tiyaboonchai, W. Fibroin and fibroin blended three-dimensional scaffolds for rat chondrocyte culture. Biomed. Eng. Online 2013, 12, 28. [Google Scholar] [CrossRef]
- Wu, J.; Wang, J.; Zhang, J.; Zheng, Z.; Kaplan, D.L.; Li, G.; Wang, X. Oral delivery of curcumin using silk nano- and microparticles. ACS Biomater. Sci. Eng. 2018, 4, 3885–3894. [Google Scholar] [CrossRef]
- Baimark, Y.; Srihanam, P.; Srisuwan, Y.; Phinyocheep, P. Preparation of porous silk fibroin microparticles by a water-in-oil emulsification-diffusion method. J. Appl. Polym. Sci. 2010, 118, 1127–1133. [Google Scholar] [CrossRef]
- Gianak, O.; Kyzas, G.Z.; Samanidou, V.F.; Deliyanni, E.A. A review for the synthesis of silk fibroin nanoparticles with different techniques and their ability to be used for drug delivery. Curr. Anal. Chem. 2019, 15, 339–348. [Google Scholar] [CrossRef]
- Mottaghitalab, F.; Farokhi, M.; Shokrgozar, M.A.; Atyabi, F.; Hosseinkhani, H. Silk fibroin nanoparticle as a novel drug delivery system. J. Control. Release 2015, 206, 161–176. [Google Scholar] [CrossRef]
- Chomchalao, P.; Nimtrakul, P.; Pham, D.T.; Tiyaboonchai, W. Development of amphotericin B-loaded fibroin nanoparticles: A novel approach for topical ocular application. J. Mater. Sci. 2020, 55, 5268–5279. [Google Scholar] [CrossRef]
- Zhang, Y.-Q.; Shen, W.-D.; Xiang, R.-L.; Zhuge, L.-J.; Gao, W.-J.; Wang, W.-B. Formation of silk fibroin nanoparticles in water-miscible organic solvent and their characterization. J. Nanoparticle Res. 2007, 9, 885–900. [Google Scholar] [CrossRef]
- Pham, D.T.; Tiyaboonchai, W. Fibroin nanoparticles: A promising drug delivery system. Drug Deliv. 2020, 27, 431–448. [Google Scholar] [CrossRef] [PubMed]
- Pham, D.T.; Saelim, N.; Tiyaboonchai, W. Crosslinked fibroin nanoparticles using EDC or PEI for drug delivery: Physicochemical properties, crystallinity and structure. J. Mater. Sci. 2018, 53, 14087–14103. [Google Scholar] [CrossRef]
- Pham, D.T.; Saelim, N.; Tiyaboonchai, W. Alpha mangostin loaded crosslinked silk fibroin-based nanoparticles for cancer chemotherapy. Colloids Surf. B 2019, 181, 705–713. [Google Scholar] [CrossRef]
- Pham, D.T.; Saelim, N.; Tiyaboonchai, W. Paclitaxel loaded EDC-crosslinked fibroin nanoparticles: A potential approach for colon cancer treatment. Drug Deliv. Transl. Res. 2020, 10, 413–424. [Google Scholar] [CrossRef]
- Gupta, V.; Aseh, A.; Ríos, C.N.; Aggarwal, B.B.; Mathur, A.B. Fabrication and characterization of silk fibroin-derived curcumin nanoparticles for cancer therapy. Int. J. Nanomedicine 2009, 4, 115–122. [Google Scholar] [CrossRef]
- Cao, Y.; Liu, F.; Chen, Y.; Yu, T.; Lou, D.; Guo, Y.; Li, P.; Wang, Z.; Ran, H. Drug release from core-shell PVA/silk fibroin nanoparticles fabricated by one-step electrospraying. Sci. Rep. 2017, 7, 11913. [Google Scholar] [CrossRef]
- Pham, D.T.; Saelim, N.; Tiyaboonchai, W. Design of experiments model for the optimization of silk fibroin based nanoparticles. Int. J. Appl. Pharm. 2018, 10, 195–201. [Google Scholar] [CrossRef]
- Pham, D.T.; Tetyczka, C.; Hartl, S.; Absenger-Novak, M.; Fröhlich, E.; Tiyaboonchai, W.; Roblegg, E. Comprehensive investigations of fibroin and poly(ethylenimine) functionalized fibroin nanoparticles for ulcerative colitis treatment. J. Drug Deliv. Sci. Technol. 2019, 11, 101484. [Google Scholar] [CrossRef]
- Jeong, H.; Hwang, J.; Lee, H.; Hammond, P.T.; Choi, J.; Hong, J. In vitro blood cell viability profiling of polymers used in molecular assembly. Sci. Rep. 2017, 7, 9481. [Google Scholar] [CrossRef]
- Fröhlich, E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int. J. Nanomedicine 2012, 7, 5577–5591. [Google Scholar] [CrossRef] [PubMed]
- Florea, B.I.; Meaney, C.; Junginger, H.E.; Borchard, G. Transfection efficiency and toxicity of polyethylenimine in differentiated Calu-3 and nondifferentiated COS-1 cell cultures. AAPS PharmSci. 2002, 4, E12. [Google Scholar] [CrossRef] [PubMed]
- Kafil, V.; Omidi, Y. Cytotoxic impacts of linear and branched polyethylenimine nanostructures in a431 cells. Bioimpacts 2011, 1, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Brelje, T.C.; Wessendorf, M.W.; Sorenson, R.L. Chapter 5 - Multicolor Laser Scanning Confocal Immunofluorescence Microscopy: Practical Application and Limitations. In Cell Biological Applications of Confocal Microscopy; Matsumoto, B., Ed.; Methods in Cell Biology; Academic Press: Cambridge, MA, USA, 2002; Volume 70, pp. 165–249e. [Google Scholar]
- Seib, F.P.; Maitz, M.F.; Hu, X.; Werner, C.; Kaplan, D.L. Impact of processing parameters on the haemocompatibility of Bombyx mori silk films. Biomaterials 2012, 33, 1017–1023. [Google Scholar] [CrossRef] [PubMed]
- Lazzari, S.; Moscatelli, D.; Codari, F.; Salmona, M.; Morbidelli, M.; Diomede, L. Colloidal stability of polymeric nanoparticles in biological fluids. J. Nanopart. Res. 2012, 14, 920. [Google Scholar] [CrossRef]
- Moore, T.L.; Rodriguez-Lorenzo, L.; Hirsch, V.; Balog, S.; Urban, D.; Jud, C.; Rothen-Rutishauser, B.; Lattuada, M.; Petri-Fink, A. Nanoparticle colloidal stability in cell culture media and impact on cellular interactions. Chem. Soc. Rev. 2015, 44, 6287–6305. [Google Scholar] [CrossRef]
- Cao, Y.; Wang, B. Biodegradation of silk biomaterials. Int. J. Mol. Sci. 2009, 10, 1514–1524. [Google Scholar] [CrossRef]
- Wongpinyochit, T.; Johnston, B.F.; Seib, F.P. Degradation behavior of silk nanoparticles—Enzyme responsiveness. ACS Biomater. Sci. Eng. 2018, 4, 942–951. [Google Scholar] [CrossRef]
- Xu, H.-L.; ZhuGe, D.-L.; Chen, P.-P.; Tong, M.-Q.; Lin, M.-T.; Jiang, X.; Zheng, Y.-W.; Chen, B.; Li, X.-K.; Zhao, Y.-Z. Silk fibroin nanoparticles dyeing indocyanine green for imaging-guided photo-thermal therapy of glioblastoma. Drug Deliv. 2018, 25, 364–375. [Google Scholar] [CrossRef]
- Shirakura, T.; Ray, A.; Kopelman, R. Polyethylenimine incorporation into hydrogel nanomatrices for enhancing nanoparticle-assisted chemotherapy. RSC Adv. 2016, 6, 48016–48024. [Google Scholar] [CrossRef]
- Tang, H.; Ye, H.; Zhang, H.; Zheng, Y. Wrapping of nanoparticles by the cell membrane: The role of interactions between the nanoparticles. Soft Matter 2015, 11, 8674–8683. [Google Scholar] [CrossRef] [PubMed]
- Lyklema, J. Electrokinetics after Smoluchowski. Colloids Surfaces A Physicochem. Eng. Asp. 2003, 222, 5–14. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Particle Size (nm) | Polydispersity Index | Zeta Potential (mV) |
|---|---|---|---|
| Blank particles | |||
| FNP | 282.1 ± 15.0 | 0.13 ± 0.02 | −17.54 ± 0.63 |
| EDClow-FNP | 289.4 ± 11.1 | 0.16 ± 0.01 | −18.10 ± 0.93 |
| EDChigh-FNP | 310.1 ± 10.5 | 0.14 ± 0.01 | −26.79 ± 0.88 |
| PEI-FNP | 305.2 ± 12.8 | 0.16 ± 0.02 | −29.32 ± 1.05 |
| FITC-bound particles | |||
| FNP | 300.7 ± 12.6 | 0.12 ± 0.01 | −17.32 ± 0.96 |
| EDClow-FNP | 296.4 ± 15.3 | 0.16 ± 0.02 | −18.21 ± 1.31 |
| EDChigh-FNP | 292.3 ± 13.4 | 0.14 ± 0.02 | +27.21 ± 1.07 |
| PEI-FNP | 311.0 ± 10.9 | 0.15 ± 0.03 | +30.59 ± 1.23 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pham, D.T.; Saelim, N.; Cornu, R.; Béduneau, A.; Tiyaboonchai, W. Crosslinked Fibroin Nanoparticles: Investigations on Biostability, Cytotoxicity, and Cellular Internalization. Pharmaceuticals 2020, 13, 86. https://doi.org/10.3390/ph13050086
Pham DT, Saelim N, Cornu R, Béduneau A, Tiyaboonchai W. Crosslinked Fibroin Nanoparticles: Investigations on Biostability, Cytotoxicity, and Cellular Internalization. Pharmaceuticals. 2020; 13(5):86. https://doi.org/10.3390/ph13050086
Chicago/Turabian StylePham, Duy Toan, Nuttawut Saelim, Raphaël Cornu, Arnaud Béduneau, and Waree Tiyaboonchai. 2020. "Crosslinked Fibroin Nanoparticles: Investigations on Biostability, Cytotoxicity, and Cellular Internalization" Pharmaceuticals 13, no. 5: 86. https://doi.org/10.3390/ph13050086
APA StylePham, D. T., Saelim, N., Cornu, R., Béduneau, A., & Tiyaboonchai, W. (2020). Crosslinked Fibroin Nanoparticles: Investigations on Biostability, Cytotoxicity, and Cellular Internalization. Pharmaceuticals, 13(5), 86. https://doi.org/10.3390/ph13050086