Selective Degradation of Target Proteins by Chimeric Small-Molecular Drugs, PROTACs and SNIPERs


Abstract
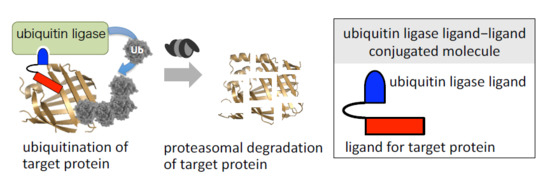
1. Introduction
2. Physiological Degradation of Proteins via the Ubiquitin-Proteasome System
3. Peptidic PROTACs
4. IAP-Mediated Small-Molecular Protein Degraders
5. Selective Knockdown of Target Protein
6. Scope of Protein Knockdown
7. IAPs Pan Antagonist-Mediated Protein Knockdown
8. Cereblon and VHL as Components of Small-Molecular PROTACs
9. Conclusions
Funding
Conflicts of Interest
References
- Brown, D.; Superti-Furga, G. Rediscovering the Sweet Spot in Drug Discovery. Drug Discov. Today 2003, 8, 1067–1077. [Google Scholar] [CrossRef]
- Hopkins, A.L.; Groom, C.R. The Druggable Genome. Nat. Rev. Drug Discov. 2002, 1, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and Specific Genetic Interference by Double-Stranded RNA in Caenorhabditis Elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Brouns, S.J.J.; Jore, M.M.; Lundgren, M.; Westra, E.R.; Slijkhuis, R.J.H.; Snijders, A.P.L.; Dickman, M.J.; Makarova, K.S.; Koonin, E.V.; van der Oost, J.; et al. Small CRISPR RNAs Guide Antiviral Defense in Prokaryotes. Science 2008, 321, 960–964. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Ciechanover, A. THE UBIQUITIN SYSTEM. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef]
- Glickman, M.H.; Ciechanover, A. The Ubiquitin-Proteasome Proteolytic Pathway: Destruction for the Sake of Construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef]
- Ciehanover, A.; Hod, Y.; Hershko, A. A Heat-Stable Polypeptide Component of an ATP-Dependent Proteolytic System from Reticulocytes. Biochem. Biophys. Res. Commun. 1978, 81, 1100–1105. [Google Scholar] [CrossRef]
- Finley, D.; Ciechanover, A.; Varshavsky, A. Thermolability of Ubiquitin-Activating Enzyme from the Mammalian Cell Cycle Mutant Ts85. Cell 1984, 37, 43–55. [Google Scholar] [CrossRef]
- Hershko, A.; Heller, H.; Elias, S.; Ciechanover, A. Components of Ubiquitin-Protein Ligase System. J. Biol. Chem. 1983, 258, 8206–8214. [Google Scholar]
- Li, W.; Bengtson, M.H.; Ulbrich, A.; Matsuda, A.; Reddy, V.A.; Orth, A.; Chanda, S.K.; Batalov, S.; Joazeiro, C.A.P. Genome-Wide and Functional Annotation of Human E3 Ubiquitin Ligases Identifies MULAN, a Mitochondrial E3 That Regulates the Organelle’s Dynamics and Signaling. PLoS ONE 2008, 3, e1487. [Google Scholar] [CrossRef]
- Chau, V.; Tobias, J.W.; Bachmair, A.; Marriott, D.; Ecker, D.J.; Gonda, D.K.; Varshavsky, A. Multiubiquitin Chain Is Confined to Specific Lysine in a Targeted Short-Lived Protein. Science 1989, 243, 1576–1583. [Google Scholar] [CrossRef] [PubMed]
- Arrigo, A.-P.; Tanaka, K.; Goldberg, A.L.; Welch, W.J. Identity of the 19S “prosome” Particle with the Large Multifunctional Protease Complex of Mammalian Cells (the Proteasome). Nature 1988, 331, 192–194. [Google Scholar] [CrossRef] [PubMed]
- Sakata, E.; Stengel, F.; Fukunaga, K.; Zhou, M.; Saeki, Y.; Förster, F.; Baumeister, W.; Tanaka, K.; Robinson, C.V. The Catalytic Activity of Ubp6 Enhances Maturation of the Proteasomal Regulatory Particle. Mol. Cell 2011, 42, 637–649. [Google Scholar] [CrossRef]
- Yau, R.; Rape, M. The Increasing Complexity of the Ubiquitin Code. Nat. Cell Biol. 2016, 18, 579–586. [Google Scholar] [CrossRef]
- Deng, L.; Wang, C.; Spencer, E.; Yang, L.; Braun, A.; You, J.; Slaughter, C.; Pickart, C.; Chen, Z.J. Activation of the IκB Kinase Complex by TRAF6 Requires a Dimeric Ubiquitin-Conjugating Enzyme Complex and a Unique Polyubiquitin Chain. Cell 2000, 103, 351–361. [Google Scholar] [CrossRef]
- Spence, J.; Sadis, S.; Haas, A.L.; Finley, D. A Ubiquitin Mutant with Specific Defects in DNA Repair and Multiubiquitination. Mol. Cell. Biol. 1995, 15, 1265–1273. [Google Scholar] [CrossRef]
- Iwai, K.; Fujita, H.; Sasaki, Y. Linear Ubiquitin Chains: NF-ΚB Signalling, Cell Death and Beyond. Nat. Rev. Mol. Cell Biol. 2014, 15, 503–508. [Google Scholar] [CrossRef]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric Molecules That Target Proteins to the Skp1-Cullin-F Box Complex for Ubiquitination and Degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef]
- Schneekloth, J.S.; Fonseca, F.N.; Koldobskiy, M.; Mandal, A.; Deshaies, R.; Sakamoto, K.; Crews, C.M. Chemical Genetic Control of Protein Levels: Selective in Vivo Targeted Degradation. J. Am. Chem. Soc. 2004, 126, 3748–3754. [Google Scholar] [CrossRef]
- Schneekloth, A.R.; Pucheault, M.; Tae, H.S.; Crews, C.M. Targeted Intracellular Protein Degradation Induced by a Small Molecule: En Route to Chemical Proteomics. Bioorg. Med. Chem. Lett. 2008, 18, 5904–5908. [Google Scholar] [CrossRef]
- Lin, H.-K.; Wang, L.; Hu, Y.; Altuwaijri, S.; Chang, C. Phosphorylation-Dependent Ubiquitylation and Degradation of Androgen Receptor by Akt Require Mdm2 E3 Ligase. EMBO J. 2002, 21, 4037–4048. [Google Scholar] [CrossRef] [PubMed]
- Logan, I.R.; McNeill, H.V.; Cook, S.; Lu, X.; Lunec, J.; Robson, C.N. Analysis of the MDM2 Antagonist Nutlin-3 in Human Prostate Cancer Cells. Prostate 2007, 67, 900–906. [Google Scholar] [CrossRef] [PubMed]
- Scheepstra, M.; Hekking, K.F.W.; van Hijfte, L.; Folmer, R.H.A. Bivalent Ligands for Protein Degradation in Drug Discovery. Comput. Struct. Biotechnol. J. 2019, 17, 160–176. [Google Scholar] [CrossRef] [PubMed]
- Collins, I.; Wang, H.; Caldwell, J.J.; Chopra, R. Chemical Approaches to Targeted Protein Degradation through Modulation of the Ubiquitin-Proteasome Pathway. Biochem. J. 2017, 474, 1127–1147. [Google Scholar] [CrossRef]
- Itoh, Y.; Ishikawa, M.; Naito, M.; Hashimoto, Y. Protein Knockdown Using Methyl Bestatin-Ligand Hybrid Molecules: Design and Synthesis of Inducers of Ubiquitination-Mediated Degradation of Cellular Retinoic Acid-Binding Proteins. J. Am. Chem. Soc. 2010, 132, 5820–5826. [Google Scholar] [CrossRef]
- Itoh, Y.; Ishikawa, M.; Kitaguchi, R.; Sato, S.; Naito, M.; Hashimoto, Y. Development of Target Protein-Selective Degradation Inducer for Protein Knockdown. Bioorg. Med. Chem. 2011, 19, 3229–3241. [Google Scholar] [CrossRef]
- Itoh, Y.; Kitaguchi, R.; Ishikawa, M.; Naito, M.; Hashimoto, Y. Design, Synthesis and Biological Evaluation of Nuclear Receptor-Degradation Inducers. Bioorg. Med. Chem. 2011, 19, 6768–6778. [Google Scholar] [CrossRef]
- Itoh, Y.; Ishikawa, M.; Kitaguchi, R.; Okuhira, K.; Naito, M.; Hashimoto, Y. Double Protein Knockdown of CIAP1 and CRABP-II Using a Hybrid Molecule Consisting of ATRA and IAPs Antagonist. Bioorg. Med. Chem. Lett. 2012, 22, 4453–4457. [Google Scholar] [CrossRef]
- Okuhira, K.; Demizu, Y.; Hattori, T.; Ohoka, N.; Shibata, N.; Nishimaki-Mogami, T.; Okuda, H.; Kurihara, M.; Naito, M. Development of Hybrid Small Molecules That Induce Degradation of Estrogen Receptor-Alpha and Necrotic Cell Death in Breast Cancer Cells. Cancer Sci. 2013, 104, 1492–1498. [Google Scholar] [CrossRef]
- Tomoshige, S.; Naito, M.; Hashimoto, Y.; Ishikawa, M. Degradation of HaloTag-Fused Nuclear Proteins Using Bestatin-HaloTag Ligand Hybrid Molecules. Org. Biomol. Chem. 2015, 13, 9746–9750. [Google Scholar] [CrossRef]
- Winter, G.E.; Buckley, D.L.; Paulk, J.; Roberts, J.M.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. Phthalimide Conjugation as a Strategy for in Vivo Target Protein Degradation. Science 2015, 348, 1376–1381. [Google Scholar] [CrossRef] [PubMed]
- Zengerle, M.; Chan, K.; Ciulli, A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol. 2015, 10, 1770–1777. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Qian, Y.; Altieri, M.; Dong, H.; Wang, J.; Raina, K.; Hines, J.; Winkler, J.D.; Crew, A.P.; Coleman, K.; et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Article Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol. 2015, 22, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Bondeson, D.P.; Mares, A.; Smith, I.E.D.; Ko, E.; Campos, S.; Miah, A.H.; Mulholland, K.E.; Routly, N.; Buckley, D.L.; Gustafson, J.L.; et al. Catalytic in Vivo Protein Knockdown by Small-Molecule PROTACs. Nat. Chem. Biol. 2015, 11, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Demizu, Y.; Ohoka, N.; Nagakubo, T.; Yamashita, H.; Misawa, T.; Okuhira, K.; Naito, M.; Kurihara, M. Development of a Peptide-Based Inducer of Nuclear Receptors Degradation. Bioorg. Med. Chem. Lett. 2016, 26, 2655–2658. [Google Scholar] [CrossRef] [PubMed]
- Tomoshige, S.; Hashimoto, Y.; Ishikawa, M. Efficient Protein Knockdown of HaloTag-Fused Proteins Using Hybrid Molecules Consisting of IAP Antagonist and HaloTag Ligand. Bioorg. Med. Chem. 2016, 24, 3144–3148. [Google Scholar] [CrossRef]
- Demizu, Y.; Shibata, N.; Hattori, T.; Ohoka, N.; Motoi, H.; Misawa, T.; Shoda, T.; Naito, M.; Kurihara, M. Development of BCR-ABL Degradation Inducers via the Conjugation of an Imatinib Derivative and a CIAP1 Ligand. Bioorg. Med. Chem. Lett. 2016, 26, 4865–4869. [Google Scholar] [CrossRef]
- Ohoka, N.; Misawa, T.; Kurihara, M.; Demizu, Y.; Naito, M. Development of a Peptide-Based Inducer of Protein Degradation Targeting NOTCH1. Bioorg. Med. Chem. Lett. 2017, 27, 4985–4988. [Google Scholar] [CrossRef]
- Ohoka, N.; Shibata, N.; Okuhira, K.; Ito, M.; Nagai, K.; Hattori, T.; Ujikawa, O.; Shimokawa, K.; Sano, O.; Koyama, R.; et al. In Vivo Knockdown of Pathogenic Proteins via Specific and Nongenetic Inhibitor of Apoptosis Protein (IAP)-Dependent Protein Erasers (SNIPERs). J. Biol. Chem. 2017, 292, 4556–4570. [Google Scholar] [CrossRef]
- Tomoshige, S.; Nomura, S.; Ohgane, K.; Hashimoto, Y.; Ishikawa, M. Discovery of Small Molecules That Induce Degradation of Huntingtin. Angew. Chemie Int. Ed. 2017, 56, 11530–11533. [Google Scholar] [CrossRef]
- Shimokawa, K.; Shibata, N.; Sameshima, T.; Miyamoto, N.; Ujikawa, O.; Nara, H.; Ohoka, N.; Hattori, T.; Cho, N.; Naito, M. Targeting the Allosteric Site of Oncoprotein BCR-ABL as an Alternative Strategy for Effective Target Protein Degradation. ACS Med. Chem. Lett. 2017, 8, 1042–1047. [Google Scholar] [CrossRef] [PubMed]
- Tomoshige, S.; Nomura, S.; Ohgane, K.; Hashimoto, Y.; Ishikawa, M. Degradation of Huntingtin Mediated by a Hybrid Molecule Composed of IAP Antagonist Linked to Phenyldiazenyl Benzothiazole Derivative. Bioorg. Med. Chem. Lett. 2018, 28, 707–710. [Google Scholar] [CrossRef] [PubMed]
- Ohoka, N.; Morita, Y.; Nagai, K.; Shimokawa, K.; Ujikawa, O.; Fujimori, I.; Ito, M.; Hayase, Y.; Okuhira, K.; Shibata, N.; et al. Derivatization of Inhibitor of Apoptosis Protein (IAP) Ligands Yields Improved Inducers of Estrogen Receptor α Degradation. J. Biol. Chem. 2018, 293, 6776–6790. [Google Scholar] [CrossRef] [PubMed]
- Okitsu, K.; Hattori, T.; Misawa, T.; Shoda, T.; Kurihara, M.; Naito, M.; Demizu, Y. Development of a Small Hybrid Molecule That Mediates Degradation of His-Tag Fused Proteins. J. Med. Chem. 2018, 61, 576–582. [Google Scholar] [CrossRef]
- Shibata, N.; Nagai, K.; Morita, Y.; Ujikawa, O.; Ohoka, N.; Hattori, T.; Koyama, R.; Sano, O.; Imaeda, Y.; Nara, H.; et al. Development of Protein Degradation Inducers of Androgen Receptor by Conjugation of Androgen Receptor Ligands and Inhibitor of Apoptosis Protein Ligands. J. Med. Chem. 2018, 61, 543–575. [Google Scholar] [CrossRef]
- Yamashita, H.; Tomoshige, S.; Nomura, S.; Ohgane, K.; Hashimoto, Y.; Ishikawa, M. Application of Protein Knockdown Strategy Targeting β-Sheet Structure to Multiple Disease-Associated Polyglutamine Proteins. Bioorg. Med. Chem. 2020, 28, 115175. [Google Scholar] [CrossRef]
- Deveraux, Q.L.; Reed, J.C. IAP Family Proteins--Suppressors of Apoptosis. Genes Dev. 1999, 13, 239–252. [Google Scholar] [CrossRef]
- Deshaies, R.J.; Joazeiro, C.A.P. RING Domain E3 Ubiquitin Ligases. Annu. Rev. Biochem. 2009, 78, 399–434. [Google Scholar] [CrossRef]
- Zheng, N.; Shabek, N. Ubiquitin Ligases: Structure, Function, and Regulation. Annu. Rev. Biochem. 2017, 86, 129–157. [Google Scholar] [CrossRef]
- Mahoney, D.J.; Cheung, H.H.; Mrad, R.L.; Plenchette, S.; Simard, C.; Enwere, E.; Arora, V.; Mak, T.W.; Lacasse, E.C.; Waring, J.; et al. Both CIAP1 and CIAP2 Regulate TNFα-Mediated NF-ΚB Activation. Proc. Natl. Acad. Sci. USA 2008, 105, 11778–11783. [Google Scholar] [CrossRef]
- Yang, Y.; Fang, S.; Jensen, J.P.; Weissman, A.M.; Ashwell, J.D. Ubiquitin Protein Ligase Activity of IAPs and Their Degradation in Proteasomes in Response to Apoptotic Stimuli. Science 2000, 288, 874–877. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.-C. Non-Canonical NF-ΚB Signaling Pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Tamm, I.; Kornblau, S.M.; Segall, H.; Cancers, H.; Leukemias, M.; Krajewski, S.; Welsh, K.; Kitada, S.; Scudiero, D.A.; Tudor, G.; et al. Expression and Prognostic Significance of IAP-Family Genes in Human Cancers and Myeloid Leukemias. Clin. Cancer Res. 2000, 6, 1796–1803. [Google Scholar] [PubMed]
- Umezawa, H.; Aoyagi, T.; Suda, H.; Hamada, M.; Takeuchi, T. Bestatin, an Inhibitor of Aminopeptidase B, Produced by Actinomycetes. J. Antibiot. (Tokyo) 1976, 29, 97–99. [Google Scholar] [CrossRef] [PubMed]
- Ichinose, Y.; Genka, K.; Koike, T.; Kato, H.; Watanabe, Y.; Mori, T.; Iioka, S.; Sakuma, A.; Ohta, M. NK421 Lung Cancer Surgery Group: Randomized Double-Blind Placebo-Controlled Trial of Bestatin in Patients with Resected Stage I Squamous-Cell Lung Carcinoma. J. Natl. Cancer Inst. 2003, 95, 605–610. [Google Scholar] [CrossRef]
- Sekine, K.; Takubo, K.; Kikuchi, R.; Nishimoto, M.; Kitagawa, M.; Abe, F.; Nishikawa, K.; Tsuruo, T.; Naito, M. Small Molecules Destabilize CIAP1 by Activating Auto-Ubiquitylation. J. Biol. Chem. 2008, 283, 8961–8968. [Google Scholar] [CrossRef]
- Sato, S.; Aoyama, H.; Miyachi, H.; Naito, M.; Hashimoto, Y. Demonstration of Direct Binding of CIAP1 Degradation-Promoting Bestatin Analogs to BIR3 Domain: Synthesis and Application of Fluorescent Bestatin Ester Analogs. Bioorg. Med. Chem. Lett. 2008, 18, 3354–3358. [Google Scholar] [CrossRef]
- Gordon, J.J.; Kelly, B.K.; Miller, G.A. Actinonin: An Antibiotic Substance Produced by an Actinomycete. Nature 1962, 195, 701–702. [Google Scholar] [CrossRef]
- Sato, S.; Tetsuhashi, M.; Sekine, K.; Miyachi, H.; Naito, M.; Hashimoto, Y.; Aoyama, H. Degradation-Promoters of Cellular Inhibitor of Apoptosis Protein 1 Based on Bestatin and Actinonin. Bioorg. Med. Chem. 2008, 16, 4685–4698. [Google Scholar] [CrossRef]
- Dueber, E.C.; Schoeffler, A.J.; Lingel, A.; Elliott, J.M.; Fedorova, A.V.; Giannetti, A.M.; Zobel, K.; Maurer, B.; Varfolomeev, E.; Wu, P.; et al. Antagonists Induce a Conformational Change in CIAP1 That Promotes Autoubiquitination. Science 2011, 334, 376–380. [Google Scholar] [CrossRef]
- Wermuth, C.G. The Practice of Medicinal Chemistry, 2nd ed.; Academic Press: San Diego, CA, USA, 2003. [Google Scholar]
- Gupta, A.; Williams, B.R.G.; Hanash, S.M.; Rawwas, J. Cellular Retinoic Acid–Binding Protein II Is a Direct Transcriptional Target of MycN in Neuroblastoma. Cancer Res. 2006, 66, 8100–8108. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.S.; Siu, C.H. Purification and Partial Characterization of a Novel Binding Protein for Retinoic Acid from Neonatal Rat. J. Biol. Chem. 1988, 263, 9326–9332. [Google Scholar] [PubMed]
- Breitman, T.R.; Collins, S.J.; Keene, B.R. Terminal Differentiation of Human Promyelocytic Leukemic Cells in Primary Culture in Response to Retinoic Acid. Blood 1981, 57, 1000–1004. [Google Scholar] [CrossRef] [PubMed]
- Kleywegt, G.J.; Bergfors, T.; Senn, H.; le Motte, P.; Gsell, B.; Shud, K.; Jones, T.A. Crystal Structures of Cellular Retinoic Acid Binding Proteins I and II in Complex with All-Trans-Retinoic Acid and a Synthetic Retinoid. Structure 1994, 2, 1241–1258. [Google Scholar] [CrossRef]
- Renaud, J.-P.; Rochel, N.; Ruff, M.; Vivat, V.; Chambon, P.; Gronemeyer, H.; Moras, D. Crystal Structure of the RAR-γ Ligand-Binding Domain Bound to All-Trans Retinoic Acid. Nature 1995, 378, 681–689. [Google Scholar] [CrossRef]
- Shimazawa, R.; Sanda, R.; Mizoguchi, H.; Hashimoto, Y.; Iwasaki, S.; Tanaka, H.; Kagechika, H.; Shudo, K. Fluorescent and Photoaffinity Labeling Probes for Retinoic Acid Receptors. Biochem. Biophys. Res. Commun. 1991, 179, 259–265. [Google Scholar] [CrossRef]
- Okuhira, K.; Ohoka, N.; Sai, K.; Nishimaki-Mogami, T.; Itoh, Y.; Ishikawa, M.; Hashimoto, Y.; Naito, M. Specific Degradation of CRABP-II via CIAP1-Mediated Ubiquitylation Induced by Hybrid Molecules That Crosslink CIAP1 and the Target Protein. FEBS Lett. 2011, 585, 1147–1152. [Google Scholar] [CrossRef]
- Demizu, Y.; Okuhira, K.; Motoi, H.; Ohno, A.; Shoda, T.; Fukuhara, K.; Okuda, H.; Naito, M.; Kurihara, M. Design and Synthesis of Estrogen Receptor Degradation Inducer Based on a Protein Knockdown Strategy. Bioorg. Med. Chem. Lett. 2012, 22, 1793–1796. [Google Scholar] [CrossRef]
- Los, G.V.; Encell, L.P.; McDougall, M.G.; Hartzell, D.D.; Karassina, N.; Zimprich, C.; Wood, M.G.; Learish, R.; Ohana, R.F.; Urh, M.; et al. HaloTag: A Novel Protein Labeling Technology for Cell Imaging and Protein Analysis. ACS Chem. Biol. 2008, 3, 373–382. [Google Scholar] [CrossRef]
- Okuhira, K.; Shoda, T.; Omura, R.; Ohoka, N.; Hattori, T.; Shibata, N.; Demizu, Y.; Sugihara, R.; Ichino, A.; Kawahara, H.; et al. Targeted Degradation of Proteins Localized in Subcellular Compartments by Hybrid Small Molecules. Mol. Pharmacol. 2017, 91, 159–166. [Google Scholar] [CrossRef]
- Ortega, Z.; Lucas, J.J. Ubiquitin-Proteasome System Involvement in Huntington’s Disease. Front. Mol. Neurosci. 2014, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Vucic, D. Targeting IAP Proteins for Therapeutic Intervention in Cancer. Nat. Rev. Drug Discov. 2012, 11, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Ndubaku, C.; Varfolomeev, E.; Wang, L.; Zobel, K.; Lau, K.; Elliott, L.O.; Maurer, B.; Fedorova, A.V.; Dynek, J.N.; Koehler, M.; et al. Antagonism of C-IAP and XIAP Proteins Is Required for Efficient Induction of Cell Death by Small-Molecule IAP Antagonists. ACS Chem. Biol. 2009, 4, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Douglass, E.F.; Miller, C.J.; Sparer, G.; Shapiro, H.; Spiegel, D.A. A Comprehensive Mathematical Model for Three-Body Binding Equilibria. J. Am. Chem. Soc. 2013, 135, 6092–6099. [Google Scholar] [CrossRef]
- Shibata, N.; Shimokawa, K.; Nagai, K.; Ohoka, N.; Hattori, T.; Miyamoto, N.; Ujikawa, O.; Sameshima, T.; Nara, H.; Cho, N.; et al. Pharmacological Difference between Degrader and Inhibitor against Oncogenic BCR-ABL Kinase. Sci. Rep. 2018, 8, 13549. [Google Scholar] [CrossRef]
- Lai, A.C.; Crews, C.M. Induced Protein Degradation: An Emerging Drug Discovery Paradigm. Nat. Rev. Drug Discov. 2017, 16, 101–114. [Google Scholar] [CrossRef]
- Cermakova, K.; Hodges, H. Next-Generation Drugs and Probes for Chromatin Biology: From Targeted Protein Degradation to Phase Separation. Molecules 2018, 23, 1958. [Google Scholar] [CrossRef]
- Delport, A.; Hewer, R. Inducing the Degradation of Disease-Related Proteins Using Heterobifunctional Molecules. Molecules 2019, 24, 3272. [Google Scholar] [CrossRef]


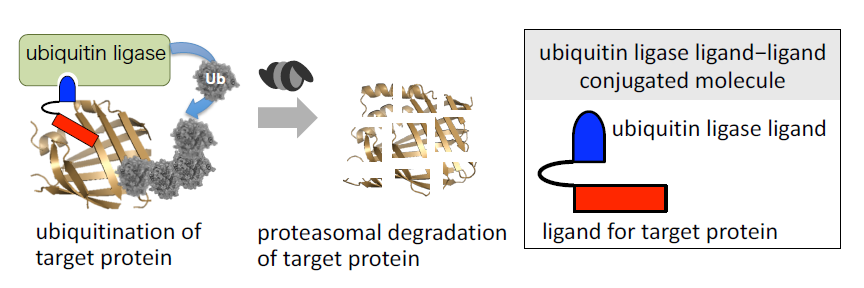{kind=link}
{kind=link}
| Year | E3 | Target Class | Target Protein | Molecular Weight | Compd No. | Chemical Structure | Efficacy in Cells | Efficacy In Vivo | Ref |
|---|---|---|---|---|---|---|---|---|---|
| 2001 | SCFβTRCP | enzyme | MetAP | 1697 | 1 |  | - | - | [18] |
| 2004 | VHL | Protein-protein interaction | FKBP | 2894 | 2 |  | 25 μM | - | [19] |
| 2008 | MDM2 | Endogenous substrate of E3 | AR | 1210 | 3 |  | 10 μM | - | [20] |
| 2010 | cIAP1 | E3/ binding protein (functionally neutral ligand) | cIAP1/ CRABP-II | 809 | 9 |  | 1 μM | - | [25] |
| 2011 | cIAP1 | Binding protein | CRABP-II | 808 | 11 |  | 1 μM | - | [26] |
| 2011 | cIAP1 | receptor | ER | 764 | 12 |  | 30 μM | - | [27] |
| 2011 | cIAP1 | receptor | AR | 770 | 13 |  | 30 μM | - | [27] |
| 2011 | cIAP1 | receptor | RAR | 917 | 14 |  | 10 μM | - | [27] |
| 2012 | IAPs | E3/ binding protein | cIAP1/ CRABP-II | 1062 | 22 |  | 100 nM | - | [28] |
| 2012 | cIAP1 | receptor | ER | 763 | 15 |  | 10 μM | - | [29] |
| 2015 | cIAP1 | Enzyme/tag | HaloTag- fused proteins | 602 | 17 |  | 10 μM | - | [30] |
| 2015 | Celebron | others | BRD4 | 785 | 36 |  | 100 nM | 50 mg/kg, IP | [31] |
| 2015 | VHL | others | BRD4 | 1002 | 37 |  | 100 nM | - | [32] |
| 2015 | celebron | others | BRD4 | 923 | 38 |  | 0.3 nM | - | [33] |
| 2015 | VHL | enzyme | RIPK | 1060 | 39 |  | 3 nM | - | [34] |
| 2015 | VHL | receptor | ERR | 949 | 40 |  | 100 nM | 100 mg/kg, ip | [34] |
| 2016 | IAPs | Receptor (coactivator binding site) | ER | 3265 | 23 |  | 20 μM | - | [35] |
| 2016 | IAPs | Enzyme/tag | HaloTag- fused protein | 834 | 25 |  | 1 μM | - | [36] |
| 2016 | cIAP1 | enzyme | BCR-ABL | 828 | 18 |  | 30 μM | - | [37] |
| 2017 | IAPs | enzyme (undruggable) | Notch1 | 2989 | 24 |  | 100 μM | - | [38] |
| 2017 | IAPs | E3/ receptor | ER | 1044 | 29 |  | 3 nM | 10 mg/kg, IP | [39] |
| 2017 | IAPs | E3/ enzyme | PDE4 | 1137 | 30 |  | 1 nM | - | [39] |
| 2017 | IAPs | E3/ others | BRD4 | 1056 | 31 |  | 10 nM | - | [39] |
| 2017 | IAPs | E3/ enzyme | BCR-ABL | 1070 | 32 |  | 10 nM | - | [39] |
| 2017 | cIAP1 | aggregation-prone (undruggable) | mHtt | 720 | 19 |  | 10 μM | - | [40] |
| 2017 | cIAP1 | aggregation-prone (undruggable) | mHtt | 662 | 20 |  | 10 μM | - | [40] |
| 2017 | IAPs | enzyme (allosteric site) | BCR-ABL | 1089 | 35 |  | 30 nM | - | [41] |
| 2018 | IAPs | aggregation-prone (undruggable) | mHtt | 974 | 27 |  | 10 μM | - | [42] |
| 2018 | IAPs | E3/ receptor | ER | 1122 | 34 |  | 10 nM | 30 mg/kg, ip | [43] |
| 2018 | IAPs | Tag | HisTag- fused protein | 2116 | 26 |  | 3 μM | - | [44] |
| 2018 | cIAP1 | receptor | AR | 864 | 16 |  | 10 μM | - | [45] |
| 2018 | IAPs | receptor | AR | 984 | 33 |  | 3 μM | - | [45] |
| 2020 | cIAP1 | aggregation-prone (undruggable) | ataxin-3 ataxin-7 atrophin-1 | 19, 20 | (The chemical structures are described above.) | 10 μM | - | [46] |
 | |||||
| Compound | R1 | R2 | Aminopeptidase Inhibition pIC50 | cIAP1 Degradation | |
| Ala | Arg | ||||
| 4 | H | H | 7.0 | 7.5 | ++ |
| 5 | Me | H | 6.9 | 5.7 | +++ |
| 6 | H | OH | 7.1 | 8.7 | + |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ishikawa, M.; Tomoshige, S.; Demizu, Y.; Naito, M. Selective Degradation of Target Proteins by Chimeric Small-Molecular Drugs, PROTACs and SNIPERs. Pharmaceuticals 2020, 13, 74. https://doi.org/10.3390/ph13040074
Ishikawa M, Tomoshige S, Demizu Y, Naito M. Selective Degradation of Target Proteins by Chimeric Small-Molecular Drugs, PROTACs and SNIPERs. Pharmaceuticals. 2020; 13(4):74. https://doi.org/10.3390/ph13040074
Chicago/Turabian StyleIshikawa, Minoru, Shusuke Tomoshige, Yosuke Demizu, and Mikihiko Naito. 2020. "Selective Degradation of Target Proteins by Chimeric Small-Molecular Drugs, PROTACs and SNIPERs" Pharmaceuticals 13, no. 4: 74. https://doi.org/10.3390/ph13040074
APA StyleIshikawa, M., Tomoshige, S., Demizu, Y., & Naito, M. (2020). Selective Degradation of Target Proteins by Chimeric Small-Molecular Drugs, PROTACs and SNIPERs. Pharmaceuticals, 13(4), 74. https://doi.org/10.3390/ph13040074