Tree-Based QSAR Model for Drug Repurposing in the Discovery of New Antibacterial Compounds against Escherichia coli

 ,
,  ,
,  ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Results
2.1. Tree-Based QSAR Model
2.1.1. Compound Selection and Index Calculation
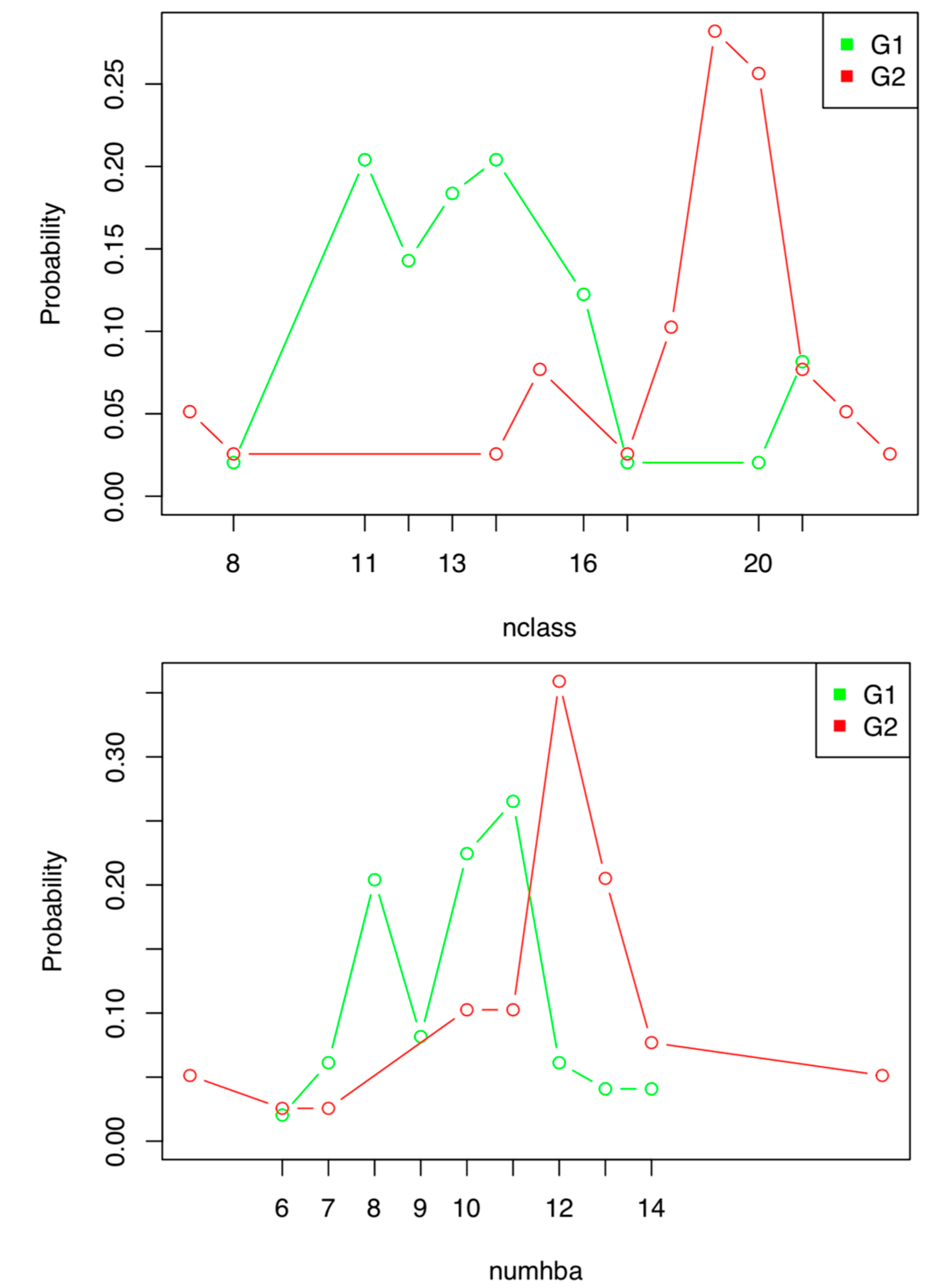
2.1.2. Discrete Index Analysis
2.1.3. Probability Space Construction
- Numhba = 7–11
- Numhba 7–11
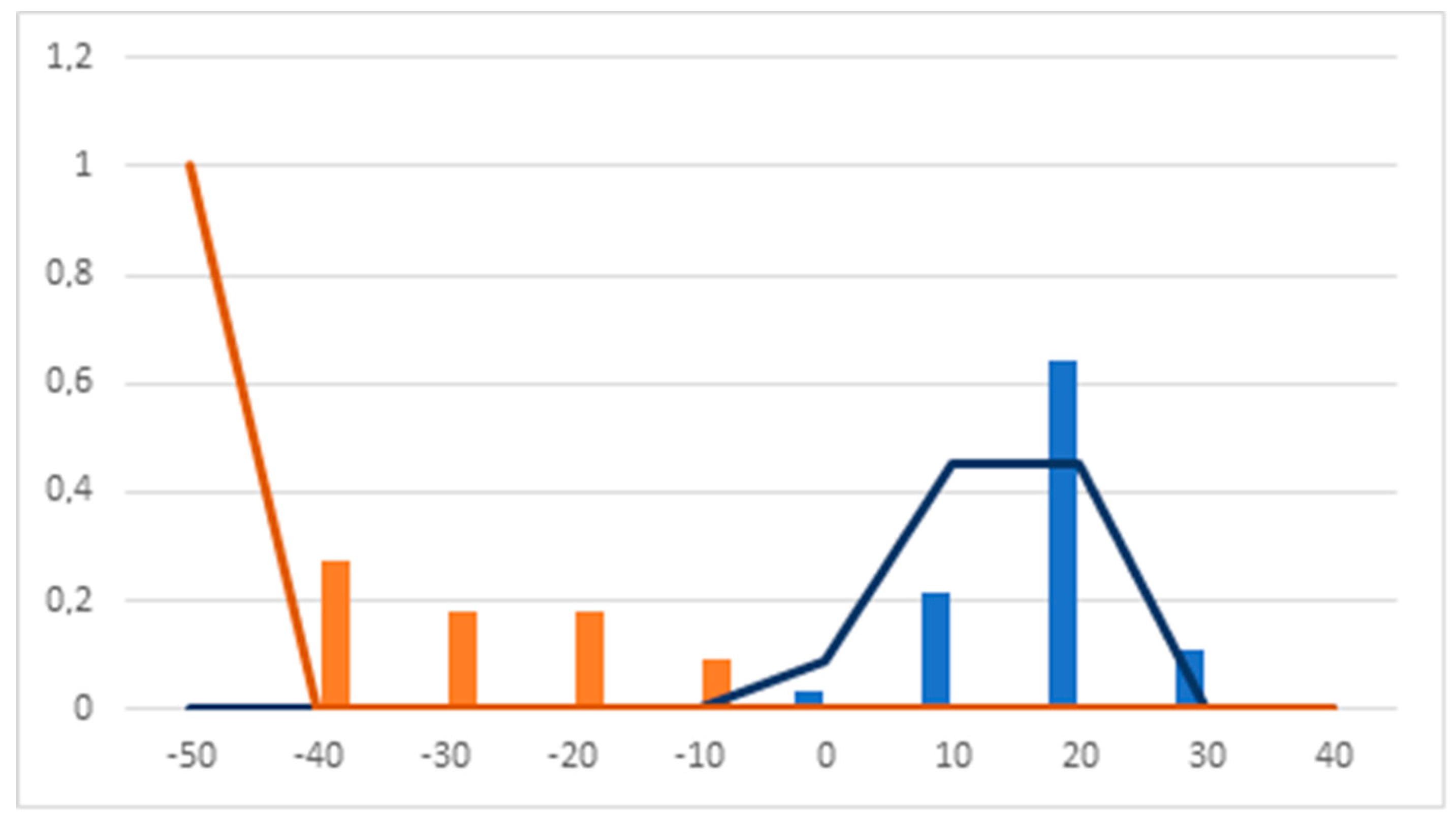
2.1.4. Linear Discriminant Analysis
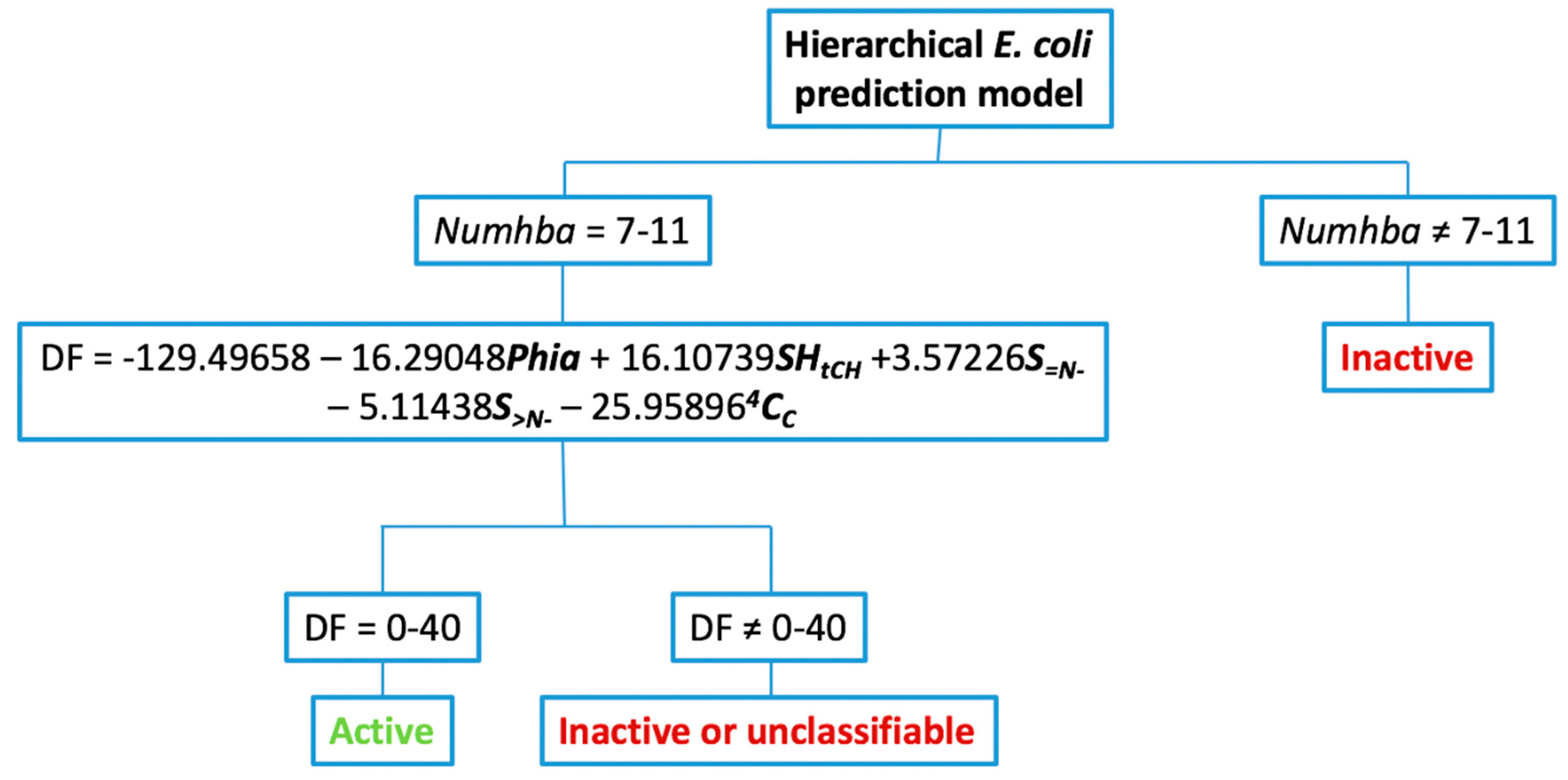
2.1.5. Hierarchical Tree Construction
2.2. Virtual Screening of Adapted DrugBank Database
3. Discussion
4. Materials and Methods
4.1. Tree-Based QSAR Model Construction
- 1.
- Compound selection and index calculation: the compounds selected to build the prediction model must belong to the same structural family. These compounds were divided into two groups, active and inactive. Once the group of compounds had been selected, the molecular descriptors or indexes of each of the molecules were calculated using MOLCONN-Z [54] and DESMOL13 [55] software.
- 2.
- Discrete index analysis: the discrete indexes were analyzed to determine if there were any with a value range that grouped the active compounds. The condition of a compound having the value of that discrete index within the range of maximum probability acted as the first step in the decision tree. Furthermore, this analysis could also act as a fast way to determine new structure activity relationships. It must be noted that, for indexes in which not enough inactive compounds are found in the higher probability space, this methodology cannot be applied for two reasons. Firstly, the lack of inactive compounds makes it impossible to obtain a discriminant function. Moreover, this type of index would lead to overfitting.
- 3.
- Probability space construction: the compounds used for the construction of the model were separated in groups according to their value for the chosen discrete indexes.
- 4.
- Linear Discriminant Analysis: a discriminant function was calculated for the selected group of molecules using only continuous indexes. This DF was calculated using the BMDP (BioMedicine Department Program) module 7M [56]. Compounds used to calculate the DF were randomly split into training and test groups by the BMDP software.
- 5.
- Hierarchical decision tree construction: once the probability spaces of the discrete indexes had been determined and the corresponding discriminant functions (DFs) calculated, a hierarchical tree was built. In the first level of this decision tree, compounds were grouped according to their value for the selected discrete index. Those that lie outside of the maximum probability space were directly classified as inactive. The remaining compounds went on to the second level of the tree. In this level, the DF is applied. Compounds within the established highest activity expectancy range are classified as active, while the rest are classified as inactive or unclassifiable.
4.2. DrugBank Database Construction
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Li, J.W.; Vederas, J.C. Drug discovery and natural products: End of an era or endless frontier? Science 2009, 325, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Karelson, M. Molcular Descriptors in QSAR/QSPR; John Wiley & Sons: New York, NY, USA, 2000. [Google Scholar]
- López-Vallejo, F.; Caulfield, T.; Martínez-Mayorga, K.; Giulianotti, M.A.; Nefzi, A.; Houghten, R.A.; Medina-Franco, J.L. Integrating virtual screening and combinatorial chemistry for accelerated drug discovery. Comb. Chem. High Throughput Screen. 2011, 14, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Engkvist, O.; Wang, Y.; Olivecrona, M.; Blaschke, T. The rise of deep learning in drug discovery. Drug Discov. Today 2018, 23, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- Fox, T.; Kriegl, J.M. Machine learning techniques for in silico modeling of drug metabolism. Curr. Top. Med. Chem. 2006, 6, 1579–1591. [Google Scholar] [CrossRef]
- Maltarrollo, V.G.; Gertrudes, J.C.; Oliveira, P.R.; Honorio, K.M. Applying machine learning techniques for ADME-Tox prediction: A review. Expert Opin. Drug Metab. Toxicol. 2015, 11, 259–271. [Google Scholar] [CrossRef]
- Breiman, L.; Friedman, J.; Stone, C. Classification and Regression Trees (Wadsworth Statistics/Probability); Chapman and Hall/CRC: New York, NY, USA, 1984. [Google Scholar]
- Hdoufane, I.; Bjij, I.; Soliman, M.; Tadjer, A.; Villemin, D.; Bogdanov, J.; Cherqaoui, D. In Silico SAR Studies of HIV-1 Inhibitors. Pharmaceuticals 2018, 11, 69. [Google Scholar] [CrossRef]
- Hu, Y.; Lu, Y.; Wang, S.; Zhang, M.; Qu, X.; Niu, B. Application of Machine Learning Approaches for the design and study of anticancer drugs. Curr. Drug Targets 2019, 20, 488–500. [Google Scholar] [CrossRef]
- Carpenter, K.A.; Huang, X. Machine Learning-based Virtual Screening and Its Applications to Alzheimer’s Drug Discovery: A Review. Curr. Pharm. Des. 2018, 24, 3347–3358. [Google Scholar] [CrossRef]
- James, G.; Witten, D.; Hastie, T.; Tibshirani, R. An Introduction to Statistical Learning with Applications in R; Springer: New York, NY, USA, 2013. [Google Scholar]
- Bueso-Bordils, J.I.; Alemán-López, P.A.; Suay-Garcia, B.; Martín-Algarra, R.; Duart, M.J.; Falcó, A.; Antón-Fos, G.M. Molecular Topology for the Discovery of New Broad-Spectrum Antibacterial Drugs. Biomolecules 2020, 10, 1343. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Kleandrova, V.V.; Cordeiro, M.N.D.S. Chemoinformatics for rational discovery of safe antibacterial drugs: Simultaneous predictions of biological activity against streptococci and toxicological profiles in laboratory animals. Bioorganic Med. Chem. 2013, 21, 2727–2732. [Google Scholar] [CrossRef]
- Ambure, P.; Halder, A.K.; González-Diaz, H.; Cordeiro, M.N.D.S. QSAR-Co: An Open Source Software for Developing Robust Multitasking or Multitarget Classification-Based QSAR Models. J. Chem. Inf. Model. 2019, 59, 2538–2544. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Monteagudo, M.; Borges, F.; Cordeiro, M.N.D.S. Jointly handling potency and toxicity of antimicrobial peptidomimetics by sumple rules from desirability theory and chemoinformatics. J. Chem. Inf. Model. 2011, 51, 3060–3077. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, C.A.; Brown, N. Multi-objective optimization methods in drug design. Drug Discov. Today Technol. 2013, 10, e427–e435. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Sun, W.; Simeonov, A. Drug repurposing screens and synergistic drug- combinations for infectious diseases. Br. J. Pharmacol. 2017, 175, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Aubé, J. Drug Repurposing and the Medicinal Chemist. ACS Med. Chem. Lett. 2012, 3, 442–444. [Google Scholar] [CrossRef]
- Ghofrani, H.A.; Osterloh, I.H.; Grimminger, F. Sildenafil: From angina to erectile dysfunction to pulmonary hypertension and beyon. Nat. Rev. Drug Discov. 2006, 5, 689–702. [Google Scholar] [CrossRef]
- Broder, S. The development of antiretroviral therapy and its impact on the HIV-1/AIDS pandemic. Antivir. Res. 2010, 85, 1. [Google Scholar] [CrossRef]
- Scannell, J.W.; Blanckley, A.; Boldon, H.; Warrington, B. Diagnosing the decline in pharmaceutical R&D efficiency. Nat. Rev. Drug Discov. 2012, 11, 191–200. [Google Scholar] [CrossRef]
- Piddock, L.J. The crisis of no new antibiotics—What is the way forward? Lancet Infect. Dis. 2012, 12, 249–253. [Google Scholar] [CrossRef]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef]
- DrugBank. Available online: https://www.drugbank.ca (accessed on 1 August 2020).
- Amin, S.A.; Adhikari, N.; Bhargava, S.; Jha, T.; Gayen, S. Designing Potential Antitrypanosomal Thiazol-2-ethylamines through Predictive Regression Based and Classification Based QSAR Analyses. Curr. Drug Discov. Technol. 2017, 14, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Jha, T.; Adhikari, N.; Saha, A.; Amin, S.A. Multiple molecular modelling studies on some derivatives and analogues of glutamic acid as matrix metalloproteinase-2 inhibitors. SAR QSAR Environ. Res. 2018, 29, 43–68. [Google Scholar] [CrossRef] [PubMed]
- Bueso-Bordils, J.I.; Pérez-Gracia, M.T.; Suay-Garcia, B.; Duart, M.J.; Algarra, R.V.M.; Lahuerta-Zamora, L.; Antón-Fos, G.M.; Alemán-López, P.A. Topological pattern for the search of new active drugs against methicillin resistant Staphylococcus aureus. Eur. J. Med. Chem. 2017, 138, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Santana, K.D.; Rivera-Borroto, O.M.; Puris, A.; Pham-The, H.; Le-Thi-Thu, H.; Rasulev, B.; Casañola-Martin, G.M. Beyond model interpretability using LDA and decisión tres for a-amylase and a-glucosidase inhibitor classification studies. Chem. Biol. Drug Des. 2019, 94, 1414–1421. [Google Scholar] [CrossRef]
- Roy, K.; Kar, S.; Das, R.N. “QSAR/QSPR Modeling: Introduction”. A Primer on QSAR/QSPR Modeling—Fundamental Concepts; Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar]
- Webb, A.R. “Introduction to Statistical Pattern Recognition”. Statistical Pattern Recognition; Wiley-Blackwell: Oxford, UK, 2011. [Google Scholar]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Kier, L.B. An index of molecular flexibility from kappa shape attributes. Quant. Struct.-Act. Relat. 1989, 8, 221–224. [Google Scholar] [CrossRef]
- Gund, P. Three-dimensional pharmacophoric pattern searching. Prog. Mol. Subcell. Biol. 1977, 5, 117–143. [Google Scholar]
- Kier, L.B.; Hall, L.H. The E-state as an extended free valence. J. Chem. Inf. Comput. Sci. 1997, 37, 548–552. [Google Scholar] [CrossRef]
- Ejim, L.; A Farha, M.; Falconer, S.B.; Wildenhain, J.; Coombes, B.K.; Tyers, M.; Brown, E.D.; Wright, G.D. Combinations of antibiotics and nonantibiotic drugs enhance antimicrobial efficacy. Nat. Chem. Biol. 2011, 6, 348–350. [Google Scholar] [CrossRef]
- Thangamani, S.; Mohammad, H.; Abushahba, M.F.N.; Sobreira, T.J.P.; Seleem, M.N. Repurposing auranofin for the treatment of cutaneous staphylococcal infections. Int. J. Antimicrob. Agents 2016, 47, 195–201. [Google Scholar] [CrossRef]
- Thangamani, S.; Younis, W.; Seleem, M.N. Repurposing Clinical Molecule Ebselen to Combat Drug Resistant Pathogens. PLoS ONE 2015, 10, e0133877. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.E.; Vilcheze, C.; Jacobs, W.R., Jr.; Ramon-García, S.; Thompson, C.J. Anthelmintic avermectins kill Mycobacterium tuberculosis, including multidrug-resistant clinical strains. Antimicrob. Agents Chemother. 2013, 57, 1040–1046. [Google Scholar] [CrossRef] [PubMed]
- Kinnings, S.L.; Liu, N.; Buchmeier, N.; Tonge, P.J.; Xie, L.; Bourne, P.E. Drug discovery using chemical system biology: Repositioning the safe medicine comtan to treat multi- drug and extensively drug resistant tuberculosis. PLoS Comput. Biol. 2009, 5, e1000423. [Google Scholar] [CrossRef] [PubMed]
- Ordway, D.J.; Viveiros, M.; Leandro, C.; Bettencourt, R.; Almeida, J.; Martins, M.; Kristiansen, J.E.; Molnar, J.; Amaral, L. Clinical concentrations of thioridazine kill intracellular multidrug-resistant Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2003, 47, 917–922. [Google Scholar] [CrossRef] [PubMed]
- Walz, J.M.; Avelar, R.L.; Longtine, K.J.; Carter, K.L.; Mermel, L.A.; Heard, S.O. Anti-infective external coating of central venous catheters: A randomized, noninferiority trial comparing 5-fluorouracil with chlorhexidine/silver sulfadiazine in preventing catheter colonization. Crit. Care Med. 2010, 38, 2095–2102. [Google Scholar] [CrossRef] [PubMed]
- Carlson-Banning, K.M.; Chou, A.; Liu, Z.; Hamill, R.J.; Song, Y.; Zechiedrich, L. Toward repurposing ciclopirox as an antibiotic against drug-resistant Acinetobacter baumannii, Escherichia coli, and Klebsiella pneumoniae. PLoS ONE 2013, 8, e69646. [Google Scholar] [CrossRef]
- Khodaverdian, V.; Pesho, M.; Truitt, B.; Bollinger, L.; Patel, P.; Nithianantham, S.; Yu, G.; Delaney, E.; Jankowsky, E.; Shoham, M. Discovery of antivirulence agents against methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2013, 57, 3645–3652. [Google Scholar] [CrossRef]
- Rosch, J.W.; Boyd, A.R.; Hinojosa, E.; Pestina, T.; Hu, Y.; Persons, D.A.; Orihuela, C.J.; Tuomanen, E.I. Statins protect against fulminant pneumococcal infection and cytolysin toxicity in a mouse model of sickle cell disease. J. Clin. Investig. 2010, 120, 627–635. [Google Scholar] [CrossRef]
- Perlmutter, J.I.; Forbes, L.T.; Krysan, D.J.; Ebsworth-Mojica, K.; Colquhoun, J.M.; Wang, J.L.; Dunman, P.M.; Flaherty, D.P. Repurposing the antihistamine terfandine for antimicrobial activity against Staphylococcus aureus. J. Med. Chem. 2014, 57, 8540–8562. [Google Scholar] [CrossRef]
- Pinault, L.; Han, J.S.; Kang, C.M.; Franco, J.; Ronning, D.R. Zafirlukast inhibits complexation of Lsr2 with DNA and growth of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2013, 57, 2134–2140. [Google Scholar] [CrossRef]
- Bisacchi, G.S.; Hale, M.R. A “Double-Edged” Scaffold: Antitumor Power within the Antibacterial Quinolone. Curr. Med. Chem. 2016, 23, 520–577. [Google Scholar] [CrossRef] [PubMed]
- Soo, V.W.C.; Kwan, B.W.; Quezada, H.; Castillo-Juárez, I.; Pérez-Eretza, B.; García-Contreras, S.J.; Martínez-Vázquez, M.; Wood, T.K.; García-Contreras, R. Repurposing of Anticancer Drugs for the Treatment of Bacterial Infections. Curr. Top. Med. Chem. 2017, 17, 1157–1176. [Google Scholar] [CrossRef] [PubMed]
- Boder, C.A.; Jorgensen, J.H.; Drutz, D.J. Antibacterial activities of antineoplastic agents. Antimicrob. Agents Chemother. 1985, 28, 437–439. [Google Scholar] [CrossRef]
- Shah, Z.; Mahbuba, R.; Turcotte, B. The anticancer drug tirapazamine has antimicrobial activity against Escherichia coli, Staphylococcus aureus and Clostridium difficile. FEMS Microbiol. Lett. 2013, 347, 61–69. [Google Scholar] [CrossRef]
- Schleimer, R.P. Glucocorticoids Supress Inflammation but Spare Innate Immune Responses in Airway Epithelium. Proc. Am. Thorac. Soc. 2004, 1, 222–230. [Google Scholar] [CrossRef]
- Wang, J.; Wang, R.; Wang, H.; Yang, X.; Yang, J.; Xiong, W.; Wen, Q.; Ma, L. Glucocorticoids Supress Antimicrobial Autophagy and Nitric Oxide Production and Facilitate Mycobacterial Survival in Macrophages. Sci. Rep. 2017, 7, 982. [Google Scholar] [CrossRef]
- Lalut, J.; Santoni, G.; Karila, D.; Lecoutey, C.; Davis, A.; Nachon, F.; Silman, I.; Sussman, J.; Weik, M.; Maurice, T.; et al. Novel multitarget-directed ligands targeting acetylcholinesterase and σ1 receptors as lead compounds for treatment of Alzheimer’s disease: Synthesis, evaluation, and structural characterization of their complexes with acetylcholinesterase. Eur. J. Med. Chem. 2018, 162, 234–248. [Google Scholar] [CrossRef]
- Hall, L.H. MOLCONN-Z Software; Eastern Nazarene College: Quincy, MA, USA, 1995. [Google Scholar]
- DESMOL13 Software. Unidad de Investigación de Diseño de Fármacos y Conectividad Molecular; Facultad de Farmacia, Universidad de Valencia: Valencia, Spain, 2000. [Google Scholar]
- Dixon, W.J.; Brown, M.B.; Engelman, L.; Jenrich, R.I. BMDP Statistical Software Manual; University of California Press: Berkeley, CA, USA, 1990. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Group | Active | Inactive | % Hits |
|---|---|---|---|
| Active training | 28 | 0 | 100 |
| Inactive training | 0 | 8 | 100 |
| Active test | 11 | 0 | 100 |
| Inactive test | 0 | 1 | 100 |
| TOTAL | 39 | 9 | 100 |
| Therapeutic Use | Nº of Selected Candidates |
|---|---|
| Antineoplastic | 12 |
| Glucocorticoid | 12 |
| Antiviral | 10 |
| Antibiotic | 8 |
| Anti-inflammatory | 4 |
| Antioxidant | 3 |
| Neuroprotector | 3 |
| Bronchodilator | 2 |
| Diabetic neuropathy | 2 |
| Glutamate receptor | 2 |
| Growth factor | 2 |
| Immunomodulator | 2 |
| Alzheimer | 1 |
| Anti-convulsive | 1 |
| Antifungal | 1 |
| Anti-infective | 1 |
| Anti-rheumatic | 1 |
| Antitussive | 1 |
| Anxiolytic | 1 |
| Benzodiazepine antagonist | 1 |
| Cardiotonic | 1 |
| Diabetes | 1 |
| GABA antagonist | 1 |
| Mannosidase inhibitor | 1 |
| Nucleoside | 1 |
| Phenylketonuria | 1 |
| Protein | 1 |
| Pulmonary arterial hypertension | 1 |
| Vitamin | 1 |
| Experimental drugs | 55 |
| Drug | Therapeutic Use | Antibacterial Activity | Ref. |
|---|---|---|---|
| Loperamide | Antidiarrheal | Salmonella enterica | [35] |
| Auranofin | Rheumatoid arthritis | MRSA | [36] |
| Ebselen | No clinical use | MRSA | [37] |
| Ivermectin | Anthelmintic | M. tuberculosis | [38] |
| Entacapone | Anti-Parkinson | M. tuberculosis | [39] |
| Thioridazine | Antipsychotic | M. tuberculosis | [40] |
| 5-Fluorouracil | Antineoplastic | Broad spectrum | [41] |
| Niclosamide | Anthelmintic | P. aeruginosa | [42] |
| Diflunisal | Anti-inflammatory | MRSA | [43] |
| Statins | Hypolipidemic | P. aeruginosa | [44] |
| Terfenadine | Antihistaminic | S. aureus | [45] |
| M. tuberculosis | |||
| Zafirlukast | Asthma | M. tuberculosis | [46] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suay-Garcia, B.; Falcó, A.; Bueso-Bordils, J.I.; Anton-Fos, G.M.; Pérez-Gracia, M.T.; Alemán-López, P.A. Tree-Based QSAR Model for Drug Repurposing in the Discovery of New Antibacterial Compounds against Escherichia coli. Pharmaceuticals 2020, 13, 431. https://doi.org/10.3390/ph13120431
Suay-Garcia B, Falcó A, Bueso-Bordils JI, Anton-Fos GM, Pérez-Gracia MT, Alemán-López PA. Tree-Based QSAR Model for Drug Repurposing in the Discovery of New Antibacterial Compounds against Escherichia coli. Pharmaceuticals. 2020; 13(12):431. https://doi.org/10.3390/ph13120431
Chicago/Turabian StyleSuay-Garcia, Beatriz, Antonio Falcó, J. Ignacio Bueso-Bordils, Gerardo M. Anton-Fos, M. Teresa Pérez-Gracia, and Pedro A. Alemán-López. 2020. "Tree-Based QSAR Model for Drug Repurposing in the Discovery of New Antibacterial Compounds against Escherichia coli" Pharmaceuticals 13, no. 12: 431. https://doi.org/10.3390/ph13120431
APA StyleSuay-Garcia, B., Falcó, A., Bueso-Bordils, J. I., Anton-Fos, G. M., Pérez-Gracia, M. T., & Alemán-López, P. A. (2020). Tree-Based QSAR Model for Drug Repurposing in the Discovery of New Antibacterial Compounds against Escherichia coli. Pharmaceuticals, 13(12), 431. https://doi.org/10.3390/ph13120431