Current Evidence on the Protective Effects of Recombinant Human Erythropoietin and Its Molecular Variants against Pathological Hallmarks of Alzheimer’s Disease

 ,
,
Abstract
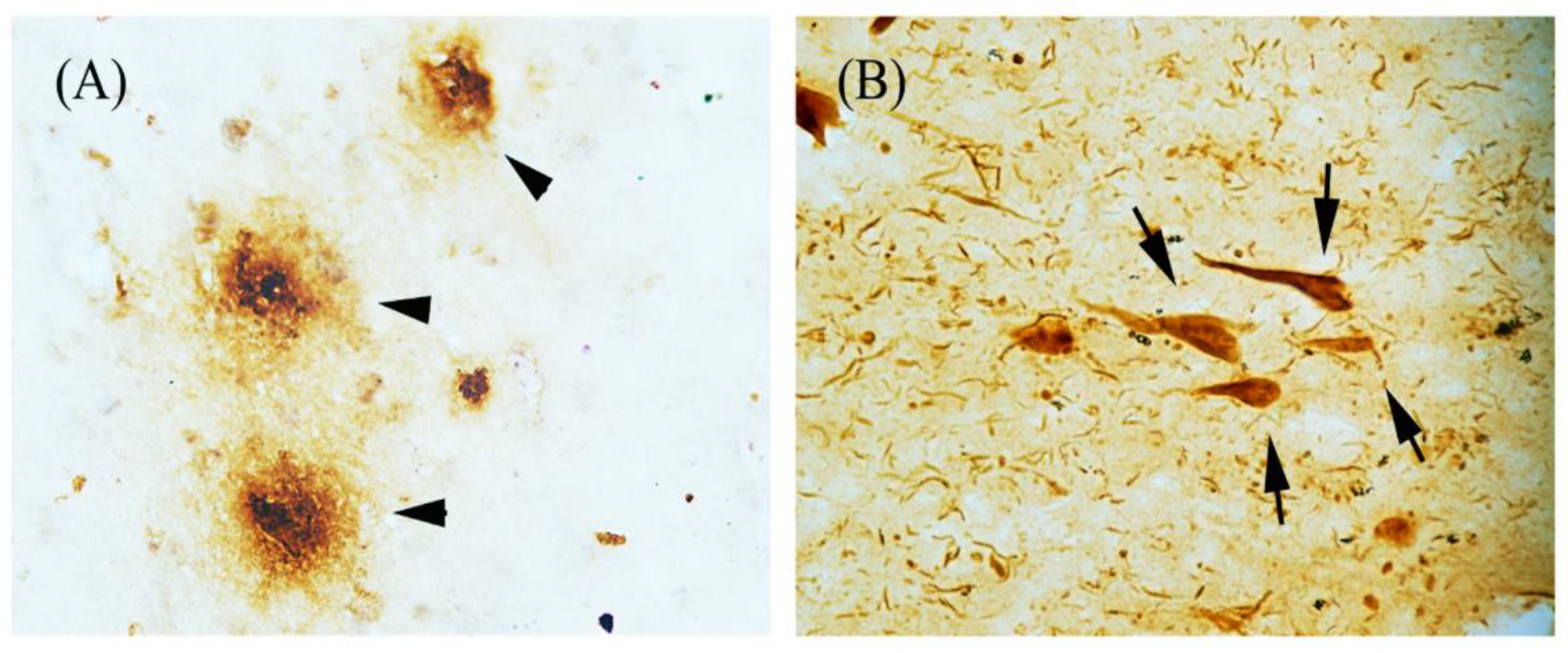
1. Introduction
2. Search Method and Criteria for Literature Inclusion
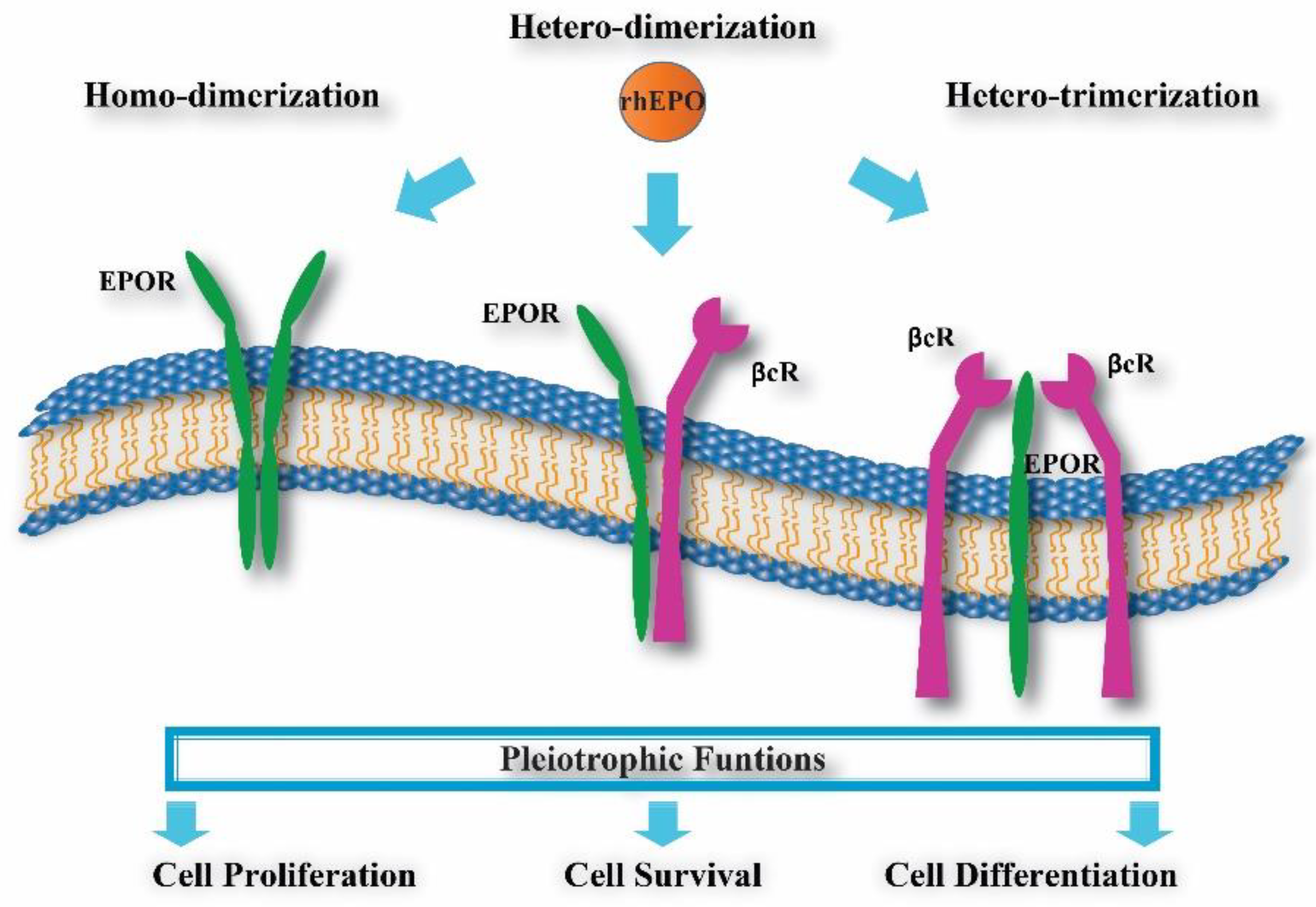
3. rhEPO and Molecular Variants
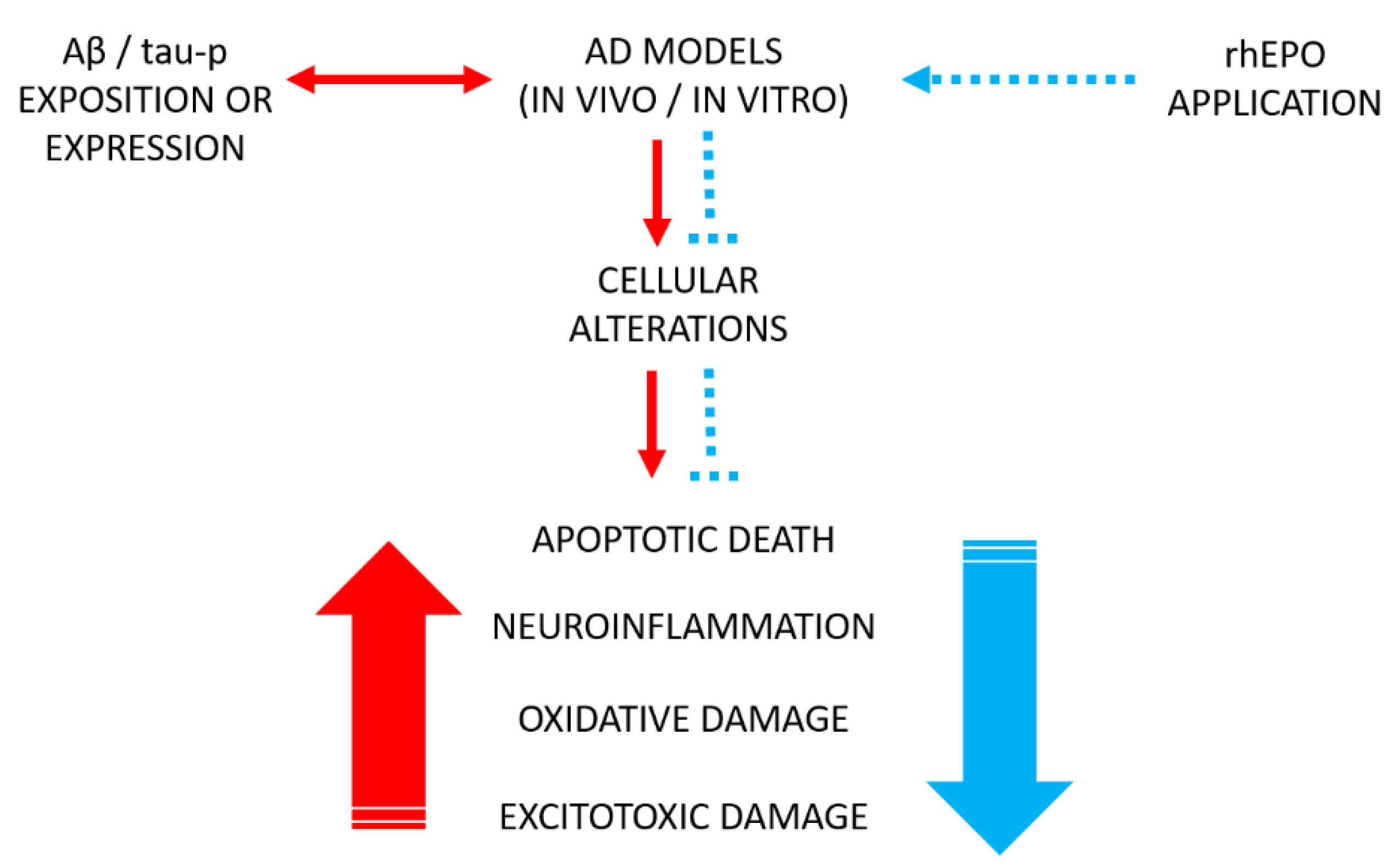
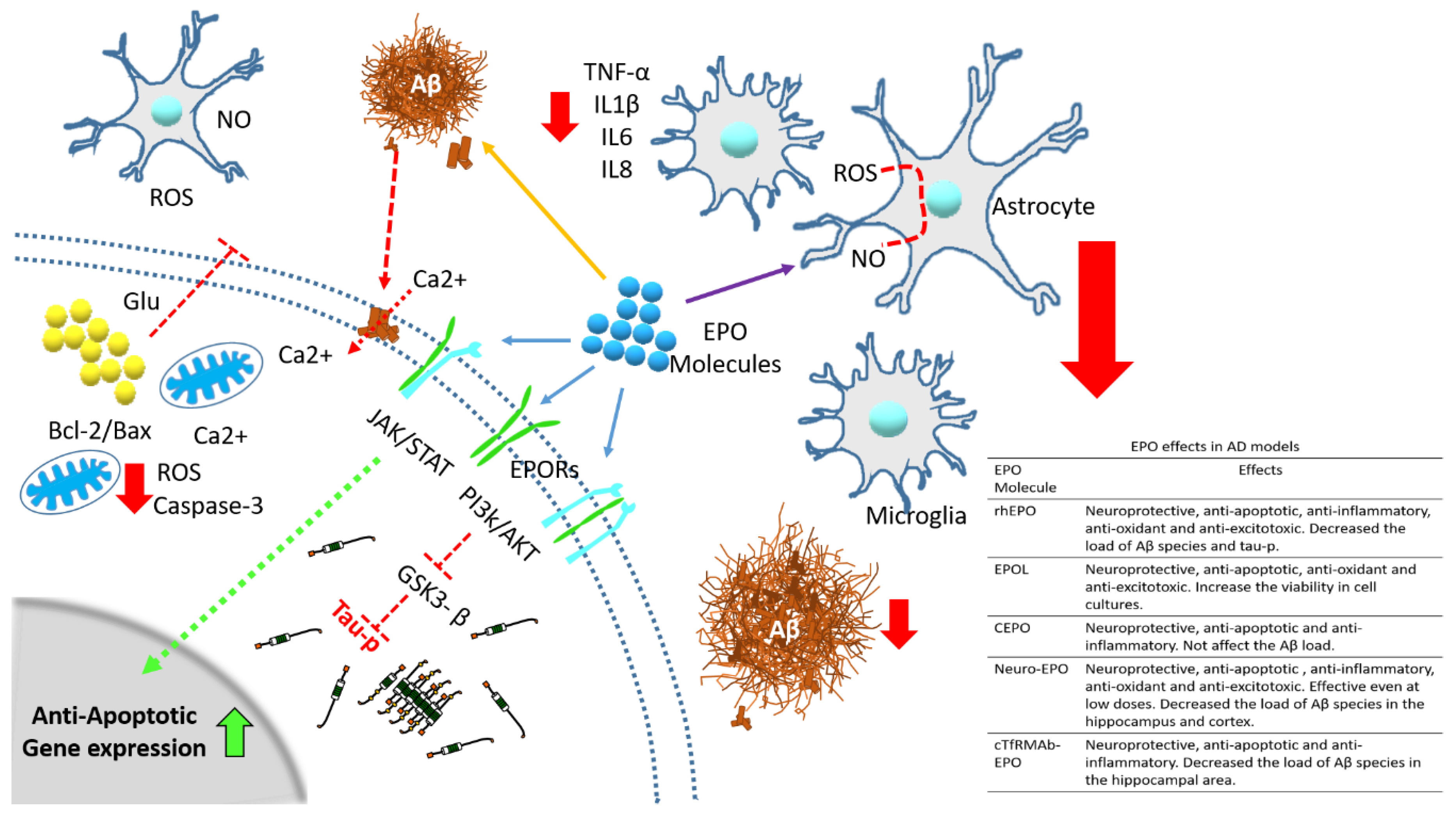
4. Effects of rhEPO and Some Its Molecular Variants in Experimental Models of Alzheimer’s Disease
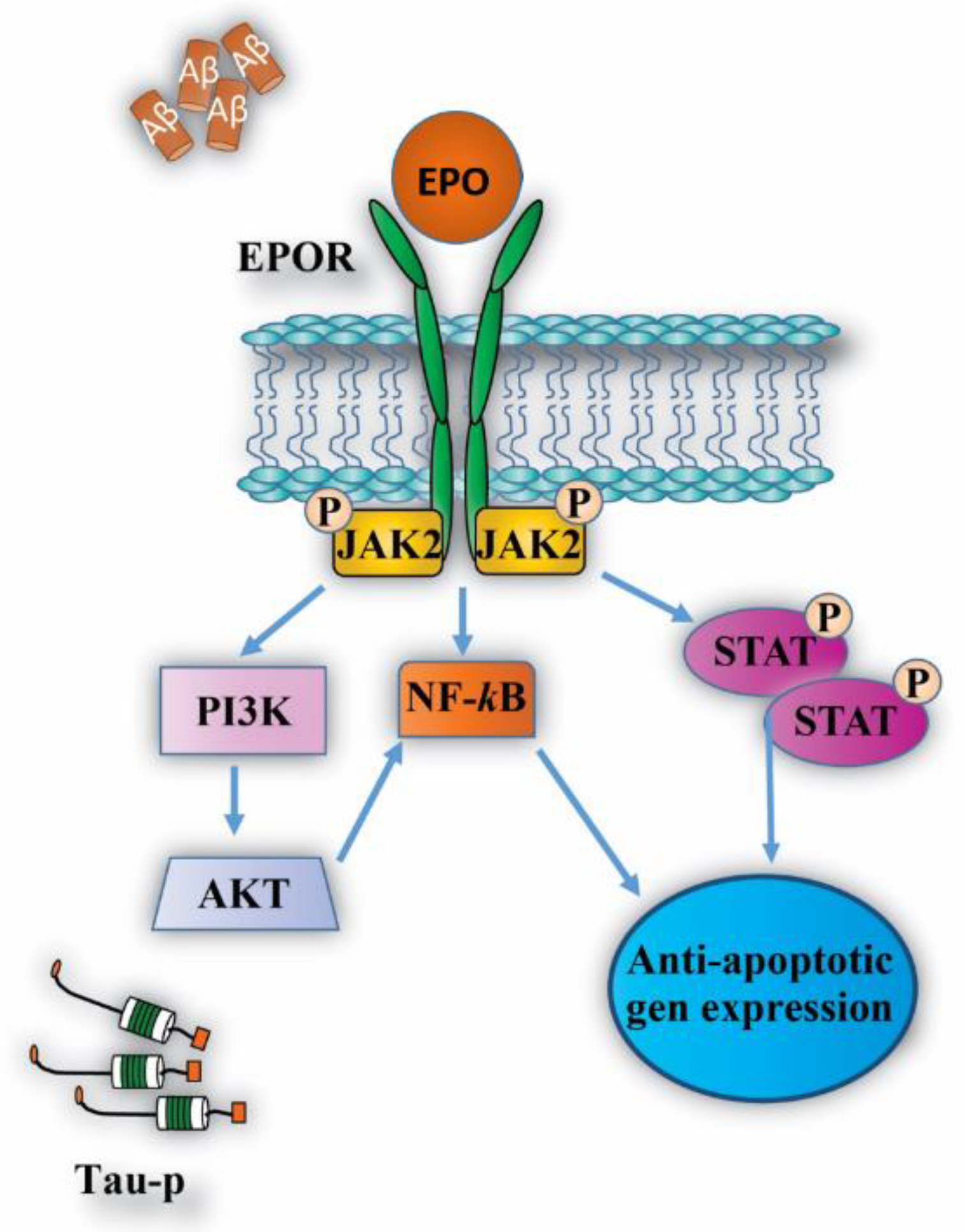
4.1. Neuroprotective Effects
4.2. Anti-Inflammatory Effects
4.3. Anti-Oxidant and Anti-Excitotoxic Effects
5. rhEPO Evidence in Human Trials with Alzheimer’s Disease
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cummings, J.; Zhong, K. Biomarker-driven therapeutic management of alzheimer’s disease: Establishing the foundations. Clin. Pharmacol. Ther. 2014, 95, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Pathways towards and away from alzheimer’s disease. Nature 2004, 430, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer’s disease results from the cerebral accumulation and cytotoxicity of amyloid beta-protein. J. Alzheimers Dis. 2001, 3, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. The molecular pathology of alzheimer’s disease. Neuron 1991, 6, 487–498. [Google Scholar] [CrossRef]
- Hyman, B.T.; Gomez-Isla, T. The natural history of alzheimer neurofibrillary tangles and amyloid deposits. Neurobiol. Aging 1997, 18, 386–387. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Rowan, M.J.; Selkoe, D.J. Amyloid-beta oligomers: Their production, toxicity and therapeutic inhibition. Biochem. Soc. Trans. 2002, 30, 552–557. [Google Scholar] [CrossRef]
- Trojanowski, J.Q.; Lee, V.M. Phosphorylation of paired helical filament tau in alzheimer’s disease neurofibrillary lesions: Focusing on phosphatases. FASEB J. 1995, 9, 1570–1576. [Google Scholar] [CrossRef]
- Gomez-Isla, T.; Hollister, R.; West, H.; Mui, S.; Growdon, J.H.; Petersen, R.C.; Parisi, J.E.; Hyman, B.T. Neuronal loss correlates with but exceeds neurofibrillary tangles in alzheimer’s disease. Ann. Neurol. 1997, 41, 17–24. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Staging of alzheimer’s disease-related neurofibrillary changes. Neurobiol. Aging 1995, 16, 271–278; discussion 278–284. [Google Scholar] [CrossRef]
- Basurto-Islas, G.; Luna-Munoz, J.; Guillozet-Bongaarts, A.L.; Binder, L.I.; Mena, R.; Garcia-Sierra, F. Accumulation of aspartic acid421- and glutamic acid391-cleaved tau in neurofibrillary tangles correlates with progression in alzheimer disease. J. Neuropathol. Exp. Neurol. 2008, 67, 470–483. [Google Scholar] [CrossRef] [PubMed]
- Fisher, J.W. Erythropoietin: Physiology and pharmacology update. Exp. Biol. Med. 2003, 228, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ehrenreich, H.; Aust, C.; Krampe, H.; Jahn, H.; Jacob, S.; Herrmann, M.; Siren, A.L. Erythropoietin: Novel approaches to neuroprotection in human brain disease. Metab. Brain Dis. 2004, 19, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Konishi, Y.; Chui, D.H.; Hirose, H.; Kunishita, T.; Tabira, T. Trophic effect of erythropoietin and other hematopoietic factors on central cholinergic neurons in vitro and in vivo. Brain Res. 1993, 609, 29–35. [Google Scholar] [CrossRef]
- Assaraf, M.I.; Diaz, Z.; Liberman, A.; Miller, W.H., Jr.; Arvanitakis, Z.; Li, Y.; Bennett, D.A.; Schipper, H.M. Brain erythropoietin receptor expression in alzheimer disease and mild cognitive impairment. J. Neuropathol. Exp. Neurol. 2007, 66, 389–398. [Google Scholar] [CrossRef]
- Brines, M.L.; Ghezzi, P.; Keenan, S.; Agnello, D.; de Lanerolle, N.C.; Cerami, C.; Itri, L.M.; Cerami, A. Erythropoietin crosses the blood-brain barrier to protect against experimental brain injury. Proc. Natl. Acad. Sci. USA 2000, 97, 10526–10531. [Google Scholar] [CrossRef] [PubMed]
- Juul, S.E.; Anderson, D.K.; Li, Y.; Christensen, R.D. Erythropoietin and erythropoietin receptor in the developing human central nervous system. Pediatr. Res. 1998, 43, 40–49. [Google Scholar] [CrossRef]
- Marti, H.H. Erythropoietin and the hypoxic brain. J. Exp. Biol. 2004, 207, 3233–3242. [Google Scholar] [CrossRef]
- Morishita, E.; Masuda, S.; Nagao, M.; Yasuda, Y.; Sasaki, R. Erythropoietin receptor is expressed in rat hippocampal and cerebral cortical neurons, and erythropoietin prevents in vitro glutamate-induced neuronal death. Neuroscience 1997, 76, 105–116. [Google Scholar] [CrossRef]
- Maiese, K.; Li, F.; Chong, Z.Z. New avenues of exploration for erythropoietin. JAMA 2005, 293, 90–95. [Google Scholar] [CrossRef]
- Sugawa, M.; Sakurai, Y.; Ishikawa-Ieda, Y.; Suzuki, H.; Asou, H. Effects of erythropoietin on glial cell development; oligodendrocyte maturation and astrocyte proliferation. Neurosci. Res. 2002, 44, 391–403. [Google Scholar] [CrossRef]
- Eid, T.; Brines, M. Recombinant human erythropoietin for neuroprotection: What is the evidence? Clin. Breast. Cancer 2002, 3 (Suppl. 3), S109–S115. [Google Scholar] [CrossRef]
- Maiese, K.; Li, F.; Chong, Z.Z. Erythropoietin in the brain: Can the promise to protect be fulfilled? Trends. Pharmacol. Sci. 2004, 25, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Obara, N.; Imagawa, S.; Nakano, Y.; Suzuki, N.; Yamamoto, M.; Nagasawa, T. Suppression of erythropoietin gene expression by cadmium depends on inhibition of hif-1, not stimulation of gata-2. Arch. Toxicol. 2003, 77, 267–273. [Google Scholar] [CrossRef]
- van der Kooij, M.A.; Groenendaal, F.; Kavelaars, A.; Heijnen, C.J.; van Bel, F. Combination of deferoxamine and erythropoietin: Therapy for hypoxia-ischemia-induced brain injury in the neonatal rat? Neurosci. Lett. 2009, 451, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Genc, S.; Koroglu, T.F.; Genc, K. Erythropoietin as a novel neuroprotectant. Restor. Neurol. Neurosci. 2004, 22, 105–119. [Google Scholar]
- Jelkmann, W. Biology of erythropoietin. Clin. Investig. 1994, 72, S3–S10. [Google Scholar]
- Brettschneider, J.; Widl, K.; Ehrenreich, H.; Riepe, M.; Tumani, H. Erythropoietin in the cerebrospinal fluid in neurodegenerative diseases. Neurosci. Lett. 2006, 404, 347–351. [Google Scholar] [CrossRef]
- Chung, Y.H.; Joo, K.M.; Kim, Y.S.; Lee, K.H.; Lee, W.B.; Cha, C.I. Enhanced expression of erythropoietin in the central nervous system of sod1(g93a) transgenic mice. Brain Res. 2004, 1016, 272–280. [Google Scholar] [CrossRef]
- Juul, S.E.; Stallings, S.A.; Christensen, R.D. Erythropoietin in the cerebrospinal fluid of neonates who sustained cns injury. Pediatr. Res. 1999, 46, 543–547. [Google Scholar] [CrossRef]
- Siren, A.L.; Knerlich, F.; Poser, W.; Gleiter, C.H.; Bruck, W.; Ehrenreich, H. Erythropoietin and erythropoietin receptor in human ischemic/hypoxic brain. Acta. Neuropathol. 2001, 101, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Widl, K.; Brettschneider, J.; Schattauer, D.; Sussmuth, S.; Huber, R.; Ludolph, A.C.; Tumani, H. Erythropoietin in cerebrospinal fluid: Age-related reference values and relevance in neurological disease. Neurochem. Res. 2007, 32, 1163–1168. [Google Scholar] [CrossRef] [PubMed]
- Marti, H.H.; Wenger, R.H.; Rivas, L.A.; Straumann, U.; Digicaylioglu, M.; Henn, V.; Yonekawa, Y.; Bauer, C.; Gassmann, M. Erythropoietin gene expression in human, monkey and murine brain. Eur. J. Neurosci. 1996, 8, 666–676. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Ebihara, I.; Shimada, N.; Koide, H. Elevated levels of erythropoietin in cerebrospinal fluid of depressed patients. Am. J. Med. Sci. 1998, 315, 199–201. [Google Scholar]
- Bernaudin, M.; Nedelec, A.S.; Divoux, D.; MacKenzie, E.T.; Petit, E.; Schumann-Bard, P. Normobaric hypoxia induces tolerance to focal permanent cerebral ischemia in association with an increased expression of hypoxia-inducible factor-1 and its target genes, erythropoietin and vegf, in the adult mouse brain. J. Cereb. Blood Flow Metab. 2002, 22, 393–403. [Google Scholar] [CrossRef]
- Prass, K.; Scharff, A.; Ruscher, K.; Lowl, D.; Muselmann, C.; Victorov, I.; Kapinya, K.; Dirnagl, U.; Meisel, A. Hypoxia-induced stroke tolerance in the mouse is mediated by erythropoietin. Stroke 2003, 34, 1981–1986. [Google Scholar] [CrossRef]
- Urena-Guerrero, M.E.; Castaneda-Cabral, J.L.; Rivera-Cervantes, M.C.; Macias-Velez, R.J.; Jarero-Basulto, J.J.; Gudino-Cabrera, G.; Beas-Zarate, C. Neuroprotective and neurestorative effects of epo and vegf: Perspectives for new therapeutic approaches to neurological diseases. Curr. Pharm. Des. 2020, 26, 1263–1276. [Google Scholar] [CrossRef]
- Genc, S.; Akhisaroglu, M.; Kuralay, F.; Genc, K. Erythropoietin restores glutathione peroxidase activity in 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine-induced neurotoxicity in c57bl mice and stimulates murine astroglial glutathione peroxidase production in vitro. Neurosci. Lett. 2002, 321, 73–76. [Google Scholar] [CrossRef]
- Fessel, W.J. Concordance of several subcellular interactions initiates alzheimer’s dementia: Their reversal requires combination treatment. Am. J. Alzheimers Dis. Demen. 2017, 32, 166–181. [Google Scholar] [CrossRef]
- Maurice, T.; Mustafa, M.H.; Desrumaux, C.; Keller, E.; Naert, G.; Garcia-Barcelo, M.D.; Rodríguez Cruz, Y.; Garcia Rodríguez, J.C. Intranasal formulation of erythropoietin (epo) showed potent protective activity against amyloid toxicity in the abeta(2)(5)(-)(3)(5) non-transgenic mouse model of alzheimer’s disease. J. Psychopharmacol. 2013, 27, 1044–1057. [Google Scholar] [CrossRef]
- Cevik, B.; Solmaz, V.; Yigitturk, G.; Cavusoglu, T.; Peker, G.; Erbas, O. Neuroprotective effects of erythropoietin on alzheimer’s dementia model in rats. Adv. Clin. Exp. Med. 2017, 26, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, G.; Arabpour, Z.; Shabrang, M.; Rashidi, B.; Alaei, H.; Sharifi, M.R.; Salami, M.; Reisi, P. Erythropoietin improves spatial learning and memory in streptozotocin model of dementia. Pathophysiology 2013, 20, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Brines, M.; Grasso, G.; Fiordaliso, F.; Sfacteria, A.; Ghezzi, P.; Fratelli, M.; Latini, R.; Xie, Q.W.; Smart, J.; Su-Rick, C.J.; et al. Erythropoietin mediates tissue protection through an erythropoietin and common beta-subunit heteroreceptor. Proc. Natl. Acad. Sci. USA 2004, 101, 14907–14912. [Google Scholar] [CrossRef] [PubMed]
- Leist, M.; Ghezzi, P.; Grasso, G.; Bianchi, R.; Villa, P.; Fratelli, M.; Savino, C.; Bianchi, M.; Nielsen, J.; Gerwien, J.; et al. Derivatives of erythropoietin that are tissue protective but not erythropoietic. Science 2004, 305, 239–242. [Google Scholar] [CrossRef]
- Um, M.; Gross, A.W.; Lodish, H.F. A “classical” homodimeric erythropoietin receptor is essential for the antiapoptotic effects of erythropoietin on differentiated neuroblastoma sh-sy5y and pheochromocytoma pc-12 cells. Cell Signal. 2007, 19, 634–645. [Google Scholar] [CrossRef]
- Ehrenreich, H.; Hasselblatt, M.; Dembowski, C.; Cepek, L.; Lewczuk, P.; Stiefel, M.; Rustenbeck, H.H.; Breiter, N.; Jacob, S.; Knerlich, F.; et al. Erythropoietin therapy for acute stroke is both safe and beneficial. Mol. Med. 2002, 8, 495–505. [Google Scholar] [CrossRef]
- Lewczuk, P.; Hasselblatt, M.; Kamrowski-Kruck, H.; Heyer, A.; Unzicker, C.; Siren, A.L.; Ehrenreich, H. Survival of hippocampal neurons in culture upon hypoxia: Effect of erythropoietin. Neuroreport 2000, 11, 3485–3488. [Google Scholar] [CrossRef]
- Siren, A.L.; Fratelli, M.; Brines, M.; Goemans, C.; Casagrande, S.; Lewczuk, P.; Keenan, S.; Gleiter, C.; Pasquali, C.; Capobianco, A.; et al. Erythropoietin prevents neuronal apoptosis after cerebral ischemia and metabolic stress. Proc. Natl. Acad. Sci. USA 2001, 98, 4044–4049. [Google Scholar] [CrossRef]
- Subiros, N.; Del Barco, D.G.; Coro-Antich, R.M. Erythropoietin: Still on the neuroprotection road. Ther. Adv. Neurol. Disord. 2012, 5, 161–173. [Google Scholar] [CrossRef]
- Osredkar, D.; Sall, J.W.; Bickler, P.E.; Ferriero, D.M. Erythropoietin promotes hippocampal neurogenesis in in vitro models of neonatal stroke. Neurobiol. Dis. 2010, 38, 259–265. [Google Scholar] [CrossRef]
- Erbayraktar, S.; Grasso, G.; Sfacteria, A.; Xie, Q.W.; Coleman, T.; Kreilgaard, M.; Torup, L.; Sager, T.; Erbayraktar, Z.; Gokmen, N.; et al. Asialoerythropoietin is a nonerythropoietic cytokine with broad neuroprotective activity in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 6741–6746. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhu, C.; Wang, X.; Gerwien, J.G.; Schrattenholz, A.; Sandberg, M.; Leist, M.; Blomgren, K. The nonerythropoietic asialoerythropoietin protects against neonatal hypoxia-ischemia as potently as erythropoietin. J. Neurochem. 2004, 91, 900–910. [Google Scholar] [CrossRef] [PubMed]
- Villa, P.; Bigini, P.; Mennini, T.; Agnello, D.; Laragione, T.; Cagnotto, A.; Viviani, B.; Marinovich, M.; Cerami, A.; Coleman, T.R.; et al. Erythropoietin selectively attenuates cytokine production and inflammation in cerebral ischemia by targeting neuronal apoptosis. J. Exp. Med. 2003, 198, 971–975. [Google Scholar] [CrossRef]
- Schmidt, R.E.; Green, K.G.; Feng, D.; Dorsey, D.A.; Parvin, C.A.; Lee, J.M.; Xiao, Q.; Brines, M. Erythropoietin and its carbamylated derivative prevent the development of experimental diabetic autonomic neuropathy in stz-induced diabetic nod-scid mice. Exp. Neurol. 2008, 209, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Castillo, C.; Zaror, S.; Gonzalez, M.; Hidalgo, A.; Burgos, C.F.; Cabezas, O.I.; Hugues, F.; Jimenez, S.P.; Gonzalez-Horta, E.; Gonzalez-Chavarria, I.; et al. Neuroprotective effect of a new variant of epo nonhematopoietic against oxidative stress. Redox. Biol. 2018, 14, 285–294. [Google Scholar] [CrossRef]
- Castillo, C.; Fernandez-Mendivil, C.; Buendia, I.; Saavedra, P.; Meza, C.; Parra, N.C.; Lopez, M.G.; Toledo, J.R.; Fuentealba, J. Neuroprotective effects of epol against oxidative stress induced by soluble oligomers of abeta peptide. Redox. Biol. 2019, 24, 101187. [Google Scholar] [CrossRef]
- Garcia-Rodriguez, J.C.; Sosa-Teste, I. The nasal route as a potential pathway for delivery of erythropoietin in the treatment of acute ischemic stroke in humans. ScientificWorldJournal 2009, 9, 970–981. [Google Scholar] [CrossRef]
- Rodriguez Cruz, Y.; Mengana Tamos, Y.; Munoz Cernuda, A.; Subiros Martines, N.; Gonzalez-Quevedo, A.; Sosa Teste, I.; Garcia Rodriguez, J.C. Treatment with nasal neuro-epo improves the neurological, cognitive, and histological state in a gerbil model of focal ischemia. ScientificWorldJournal 2010, 10, 2288–2300. [Google Scholar] [CrossRef]
- Chang, R.; Al Maghribi, A.; Vanderpoel, V.; Vasilevko, V.; Cribbs, D.H.; Boado, R.; Pardridge, W.M.; Sumbria, R.K. Brain penetrating bifunctional erythropoietin-transferrin receptor antibody fusion protein for alzheimer’s disease. Mol. Pharm. 2018, 15, 4963–4973. [Google Scholar] [CrossRef]
- Biebl, M.; Cooper, C.M.; Winkler, J.; Kuhn, H.G. Analysis of neurogenesis and programmed cell death reveals a self-renewing capacity in the adult rat brain. Neurosci. Lett. 2000, 291, 17–20. [Google Scholar] [CrossRef]
- Cras, P.; Smith, M.A.; Richey, P.L.; Siedlak, S.L.; Mulvihill, P.; Perry, G. Extracellular neurofibrillary tangles reflect neuronal loss and provide further evidence of extensive protein cross-linking in alzheimer disease. Acta. Neuropathol. 1995, 89, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Mosconi, L.; Herholz, K.; Prohovnik, I.; Nacmias, B.; De Cristofaro, M.T.; Fayyaz, M.; Bracco, L.; Sorbi, S.; Pupi, A. Metabolic interaction between apoe genotype and onset age in alzheimer’s disease: Implications for brain reserve. J. Neurol. Neurosurg. Psychiatry. 2005, 76, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Ma, R.; Huang, C.; Tang, Q.; Fu, Q.; Liu, H.; Hu, B.; Xiang, J. Protective effect of erythropoietin on beta-amyloid-induced pc12 cell death through antioxidant mechanisms. Neurosci. Lett. 2008, 442, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.C.; Chong, Z.Z.; Hou, J.; Maiese, K. The forkhead transcription factor foxo3a controls microglial inflammatory activation and eventual apoptotic injury through caspase 3. Curr. Neurovasc. Res. 2009, 6, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda, F.J.; Fierro, H.; Fernandez, E.; Castillo, C.; Peoples, R.W.; Opazo, C.; Aguayo, L.G. Nature of the neurotoxic membrane actions of amyloid-beta on hippocampal neurons in alzheimer’s disease. Neurobiol. Aging 2014, 35, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Saez-Orellana, F.; Godoy, P.A.; Bastidas, C.Y.; Silva-Grecchi, T.; Guzman, L.; Aguayo, L.G.; Fuentealba, J. Atp leakage induces p2xr activation and contributes to acute synaptic excitotoxicity induced by soluble oligomers of beta-amyloid peptide in hippocampal neurons. Neuropharmacology 2016, 100, 116–123. [Google Scholar] [CrossRef]
- Zempel, H.; Thies, E.; Mandelkow, E.; Mandelkow, E.M. Abeta oligomers cause localized ca(2+) elevation, missorting of endogenous tau into dendrites, tau phosphorylation, and destruction of microtubules and spines. J. Neurosci. 2010, 30, 11938–11950. [Google Scholar] [CrossRef]
- Sepulveda, F.J.; Parodi, J.; Peoples, R.W.; Opazo, C.; Aguayo, L.G. Synaptotoxicity of alzheimer beta amyloid can be explained by its membrane perforating property. PLoS ONE 2010, 5, e11820. [Google Scholar] [CrossRef]
- Fuchsberger, T.; Martinez-Bellver, S.; Giraldo, E.; Teruel-Marti, V.; Lloret, A.; Vina, J. Abeta induces excitotoxicity mediated by apc/c-cdh1 depletion that can be prevented by glutaminase inhibition promoting neuronal survival. Sci. Rep. 2016, 6, 31158. [Google Scholar] [CrossRef]
- Kumar, A.; Singh, A. A review on alzheimer’s disease pathophysiology and its management: An update. Pharmacol. Rep. 2015, 67, 195–203. [Google Scholar] [CrossRef]
- Maiese, K.; Chong, Z.Z.; Hou, J.; Shang, Y.C. New strategies for alzheimer’s disease and cognitive impairment. Oxid. Med. Cell. Longev. 2009, 2, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.T.; Chu, K.; Park, J.E.; Jung, K.H.; Jeon, D.; Lim, J.Y.; Lee, S.K.; Kim, M.; Roh, J.K. Erythropoietin improves memory function with reducing endothelial dysfunction and amyloid-beta burden in alzheimer’s disease models. J. Neurochem. 2012, 120, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Park, M.H.; Lee, S.M.; Lee, J.W.; Son, D.J.; Moon, D.C.; Yoon, D.Y.; Hong, J.T. Erk-mediated production of neurotrophic factors by astrocytes promotes neuronal stem cell differentiation by erythropoietin. Biochem. Biophys. Res. Commun. 2006, 339, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Vinothkumar, G.; Krishnakumar, S.; Shivashekar, G.; Sreedhar, S.; Dinesh, S.; Sundaram, A.; Balakrishnan, D.; Venkataraman, P. Therapeutic impact of rhuepo on abnormal platelet app, bace 1, presenilin 1, adam 10 and abeta expressions in chronic kidney disease patients with cognitive dysfunction like alzheimer’s disease: A pilot study. Biomed. Pharmacother. 2018, 104, 211–222. [Google Scholar]
- Cerami, A.; Brines, M.; Ghezzi, P.; Cerami, C.; Itri, L.M. Neuroprotective properties of epoetin alfa. Nephrol. Dial. Transplant. 2002, 17 (Suppl. 1), 8–12. [Google Scholar] [CrossRef]
- Agnello, D.; Bigini, P.; Villa, P.; Mennini, T.; Cerami, A.; Brines, M.L.; Ghezzi, P. Erythropoietin exerts an anti-inflammatory effect on the cns in a model of experimental autoimmune encephalomyelitis. Brain Res. 2002, 952, 128–134. [Google Scholar] [CrossRef]
- Pregi, N.; Wenker, S.; Vittori, D.; Leiros, C.P.; Nesse, A. Tnf-alpha-induced apoptosis is prevented by erythropoietin treatment on sh-sy5y cells. Exp. Cell Res. 2009, 315, 419–431. [Google Scholar] [CrossRef]
- Ma, C.; Cheng, F.; Wang, X.; Zhai, C.; Yue, W.; Lian, Y.; Wang, Q. Erythropoietin pathway: A potential target for the treatment of depression. Int. J. Mol. Sci. 2016, 17, 677. [Google Scholar] [CrossRef]
- Ma, R.; Xiong, N.; Huang, C.; Tang, Q.; Hu, B.; Xiang, J.; Li, G. Erythropoietin protects pc12 cells from beta-amyloid(25-35)-induced apoptosis via pi3k/akt signaling pathway. Neuropharmacology 2009, 56, 1027–1034. [Google Scholar] [CrossRef]
- Kilic, E.; Kilic, U.; Soliz, J.; Bassetti, C.L.; Gassmann, M.; Hermann, D.M. Brain-derived erythropoietin protects from focal cerebral ischemia by dual activation of erk-1/-2 and akt pathways. FASEB J. 2005, 19, 2026–2028. [Google Scholar] [CrossRef]
- Chong, Z.Z.; Kang, J.Q.; Maiese, K. Erythropoietin is a novel vascular protectant through activation of akt1 and mitochondrial modulation of cysteine proteases. Circulation 2002, 106, 2973–2979. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Suh, Y.H.; Kim, S.; Kim, Y. Comparison of the structures of beta amyloid peptide (25–35) and substance p in trifluoroethanol/water solution. J. Biomol. Struct. Dyn. 1999, 17, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Kubo, T.; Nishimura, S.; Oda, T. Amyloid beta-peptide alters the distribution of early endosomes and inhibits phosphorylation of akt in the presence of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (mtt). Brain Res. Mol. Brain Res. 2002, 106, 94–100. [Google Scholar] [CrossRef]
- Sun, Z.K.; Yang, H.Q.; Pan, J.; Zhen, H.; Wang, Z.Q.; Chen, S.D.; Ding, J.Q. Protective effects of erythropoietin on tau phosphorylation induced by beta-amyloid. J. Neurosci. Res. 2008, 86, 3018–3027. [Google Scholar] [CrossRef]
- Chong, Z.Z.; Li, F.; Maiese, K. Erythropoietin requires nf-kappab and its nuclear translocation to prevent early and late apoptotic neuronal injury during beta-amyloid toxicity. Curr. Neurovasc. Res. 2005, 2, 387–399. [Google Scholar] [CrossRef]
- Zhi-Kun, S.; Hong-Qi, Y.; Zhi-Quan, W.; Jing, P.; Zhen, H.; Sheng-Di, C. Erythropoietin prevents pc12 cells from beta-amyloid-induced apoptosis via pi3kakt pathway. Transl. Neurodegener. 2012, 1, 7. [Google Scholar] [CrossRef]
- Ma, R.; Hu, J.; Huang, C.; Wang, M.; Xiang, J.; Li, G. Jak2/stat5/bcl-xl signalling is essential for erythropoietin-mediated protection against apoptosis induced in pc12 cells by the amyloid beta-peptide abeta25–35. Br. J. Pharmacol. 2014, 171, 3234–3245. [Google Scholar] [CrossRef]
- Pregi, N.; Vittori, D.; Perez, G.; Leiros, C.P.; Nesse, A. Effect of erythropoietin on staurosporine-induced apoptosis and differentiation of sh-sy5y neuroblastoma cells. Biochim. Biophys. Acta 2006, 1763, 238–246. [Google Scholar] [CrossRef]
- Somervaille, T.C.; Linch, D.C.; Khwaja, A. Growth factor withdrawal from primary human erythroid progenitors induces apoptosis through a pathway involving glycogen synthase kinase-3 and bax. Blood 2001, 98, 1374–1381. [Google Scholar] [CrossRef]
- Rossig, L.; Badorff, C.; Holzmann, Y.; Zeiher, A.M.; Dimmeler, S. Glycogen synthase kinase-3 couples akt-dependent signaling to the regulation of p21cip1 degradation. J. Biol. Chem. 2002, 277, 9684–9689. [Google Scholar] [CrossRef]
- Su, B.; Wang, X.; Nunomura, A.; Moreira, P.I.; Lee, H.G.; Perry, G.; Smith, M.A.; Zhu, X. Oxidative stress signaling in alzheimer’s disease. Curr. Alzheimer. Res. 2008, 5, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Balaji, V.; Kaniyappan, S.; Mandelkow, E.; Wang, Y.; Mandelkow, E.M. Pathological missorting of endogenous mapt/tau in neurons caused by failure of protein degradation systems. Autophagy 2018, 14, 2139–2154. [Google Scholar] [CrossRef] [PubMed]
- Peters, C.; Fernandez-Perez, E.J.; Burgos, C.F.; Espinoza, M.P.; Castillo, C.; Urrutia, J.C.; Streltsov, V.A.; Opazo, C.; Aguayo, L.G. Inhibition of amyloid beta-induced synaptotoxicity by a pentapeptide derived from the glycine zipper region of the neurotoxic peptide. Neurobiol. Aging 2013, 34, 2805–2814. [Google Scholar] [CrossRef] [PubMed]
- Garzon, F.; Coimbra, D.; Parcerisas, A.; Rodriguez, Y.; Garcia, J.C.; Soriano, E.; Rama, R. Neuroepo preserves neurons from glutamate-induced excitotoxicity. J. Alzheimers Dis. 2018, 65, 1469–1483. [Google Scholar] [CrossRef] [PubMed]
- Reisi, P.; Alaei, H.; Babri, S.; Sharifi, M.R.; Mohaddes, G. Effects of treadmill running on spatial learning and memory in streptozotocin-induced diabetic rats. Neurosci. Lett. 2009, 455, 79–83. [Google Scholar] [CrossRef]
- Ishrat, T.; Parveen, K.; Hoda, M.N.; Khan, M.B.; Yousuf, S.; Ansari, M.A.; Saleem, S.; Islam, F. Effects of pycnogenol and vitamin e on cognitive deficits and oxidative damage induced by intracerebroventricular streptozotocin in rats. Behav. Pharmacol. 2009, 20, 567–575. [Google Scholar] [CrossRef]
- Arabpoor, Z.; Hamidi, G.; Rashidi, B.; Shabrang, M.; Alaei, H.; Sharifi, M.R.; Salami, M.; Dolatabadi, H.R.; Reisi, P. Erythropoietin improves neuronal proliferation in dentate gyrus of hippocampal formation in an animal model of alzheimer’s disease. Adv. Biomed. Res. 2012, 1, 50. [Google Scholar]
- Mehla, J.; Pahuja, M.; Gupta, Y.K. Streptozotocin-induced sporadic alzheimer’s disease: Selection of appropriate dose. J. Alzheimers Dis. 2013, 33, 17–21. [Google Scholar] [CrossRef]
- Salkovic-Petrisic, M.; Knezovic, A.; Hoyer, S.; Riederer, P. What have we learned from the streptozotocin-induced animal model of sporadic alzheimer’s disease, about the therapeutic strategies in alzheimer’s research. J. Neural. Transm. 2013, 120, 233–252. [Google Scholar] [CrossRef]
- Maurice, T.; Lockhart, B.P.; Privat, A. Amnesia induced in mice by centrally administered beta-amyloid peptides involves cholinergic dysfunction. Brain Res. 1996, 706, 181–193. [Google Scholar] [CrossRef]
- Rodriguez Cruz, Y.; Strehaiano, M.; Rodriguez Obaya, T.; Garcia Rodriguez, J.C.; Maurice, T. An intranasal formulation of erythropoietin (neuro-epo) prevents memory deficits and amyloid toxicity in the appswe transgenic mouse model of alzheimer’s disease. J. Alzheimers Dis. 2017, 55, 231–248. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative memory deficits, abeta elevation, and amyloid plaques in transgenic mice. Science 1996, 274, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Borchelt, D.R.; Davis, J.; Fischer, M.; Lee, M.K.; Slunt, H.H.; Ratovitsky, T.; Regard, J.; Copeland, N.G.; Jenkins, N.A.; Sisodia, S.S.; et al. A vector for expressing foreign genes in the brains and hearts of transgenic mice. Genet. Anal. 1996, 13, 159–163. [Google Scholar] [CrossRef]
- Armand-Ugon, M.; Aso, E.; Moreno, J.; Riera-Codina, M.; Sanchez, A.; Vegas, E.; Ferrer, I. Memory improvement in the abetapp/ps1 mouse model of familial alzheimer’s disease induced by carbamylated-erythropoietin is accompanied by modulation of synaptic genes. J. Alzheimers Dis. 2015, 45, 407–421. [Google Scholar] [CrossRef] [PubMed]
- Modabbernia, A.; Ashrafi, M.; Modabbernia, M.J. Let’s try erythropoietin in alzheimer’s disease. Med. Hypotheses 2010, 75, 270–271. [Google Scholar] [CrossRef] [PubMed]
- Itagaki, S.; McGeer, P.L.; Akiyama, H.; Zhu, S.; Selkoe, D. Relationship of microglia and astrocytes to amyloid deposits of alzheimer disease. J. Neuroimmunol. 1989, 24, 173–182. [Google Scholar] [CrossRef]
- Campana, W.M.; Li, X.; Shubayev, V.I.; Angert, M.; Cai, K.; Myers, R.R. Erythropoietin reduces schwann cell tnf-alpha, wallerian degeneration and pain-related behaviors after peripheral nerve injury. Eur. J. Neurosci. 2006, 23, 617–626. [Google Scholar] [CrossRef]
- Gunnarson, E.; Song, Y.; Kowalewski, J.M.; Brismar, H.; Brines, M.; Cerami, A.; Andersson, U.; Zelenina, M.; Aperia, A. Erythropoietin modulation of astrocyte water permeability as a component of neuroprotection. Proc. Natl. Acad. Sci. USA 2009, 106, 1602–1607. [Google Scholar] [CrossRef]
- Kim, S.M.; Song, J.; Kim, S.; Han, C.; Park, M.H.; Koh, Y.; Jo, S.A.; Kim, Y.Y. Identification of peripheral inflammatory markers between normal control and alzheimer’s disease. BMC Neurol. 2011, 11, 51. [Google Scholar] [CrossRef]
- Park, J.; Lee, S.Y.; Shon, J.; Kim, K.; Lee, H.J.; Kim, K.A.; Lee, B.Y.; Oh, S.H.; Kim, N.K.; Kim, O.J. Adalimumab improves cognitive impairment, exerts neuroprotective effects and attenuates neuroinflammation in an abeta1-40-injected mouse model of alzheimer’s disease. Cytotherapy 2019, 21, 671–682. [Google Scholar] [CrossRef]
- Schipper, H.M. Astrocytes, brain aging, and neurodegeneration. Neurobiol. Aging 1996, 17, 467–480. [Google Scholar] [CrossRef]
- Chong, Z.Z.; Kang, J.Q.; Maiese, K. Apaf-1, bcl-xl, cytochrome c, and caspase-9 form the critical elements for cerebral vascular protection by erythropoietin. J. Cereb. Blood Flow Metab. 2003, 23, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Chong, Z.Z.; Kang, J.Q.; Maiese, K. Metabotropic glutamate receptors promote neuronal and vascular plasticity through novel intracellular pathways. Histol. Histopathol. 2003, 18, 173–189. [Google Scholar] [PubMed]
- Naert, G.; Rivest, S. Cc chemokine receptor 2 deficiency aggravates cognitive impairments and amyloid pathology in a transgenic mouse model of alzheimer’s disease. J. Neurosci. 2011, 31, 6208–6220. [Google Scholar] [CrossRef] [PubMed]
- Nagai, A.; Nakagawa, E.; Choi, H.B.; Hatori, K.; Kobayashi, S.; Kim, S.U. Erythropoietin and erythropoietin receptors in human cns neurons, astrocytes, microglia, and oligodendrocytes grown in culture. J. Neuropathol. Exp. Neurol. 2001, 60, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.T.; Collins, J.S.; Wiener, H.; Acton, R.; Go, R.C. The role of tnf and its receptors in alzheimer’s disease. Neurobiol. Aging 2001, 22, 873–883. [Google Scholar] [CrossRef]
- Montgomery, S.L.; Bowers, W.J. Tumor necrosis factor-alpha and the roles it plays in homeostatic and degenerative processes within the central nervous system. J. Neuroimmune. Pharmacol. 2012, 7, 42–59. [Google Scholar] [CrossRef]
- Clark, I.; Atwood, C.; Bowen, R.; Paz-Filho, G.; Vissel, B. Tumor necrosis factor-induced cerebral insulin resistance in alzheimer’s disease links numerous treatment rationales. Pharmacol. Rev. 2012, 64, 1004–1026. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kiyota, T.; Horiba, M.; Buescher, J.L.; Walsh, S.M.; Gendelman, H.E.; Ikezu, T. Interferon-gamma and tumor necrosis factor-alpha regulate amyloid-beta plaque deposition and beta-secretase expression in swedish mutant app transgenic mice. Am. J. Pathol. 2007, 170, 680–692. [Google Scholar] [CrossRef]
- Janelsins, M.C.; Mastrangelo, M.A.; Park, K.M.; Sudol, K.L.; Narrow, W.C.; Oddo, S.; LaFerla, F.M.; Callahan, L.M.; Federoff, H.J.; Bowers, W.J. Chronic neuron-specific tumor necrosis factor-alpha expression enhances the local inflammatory environment ultimately leading to neuronal death in 3xtg-ad mice. Am. J. Pathol. 2008, 173, 1768–1782. [Google Scholar] [CrossRef]
- Shen, J.; Wu, Y.; Xu, J.Y.; Zhang, J.; Sinclair, S.H.; Yanoff, M.; Xu, G.; Li, W.; Xu, G.T. Erk- and akt-dependent neuroprotection by erythropoietin (epo) against glyoxal-ages via modulation of bcl-xl, bax, and bad. Investig. Ophthalmol. Vis. Sci. 2010, 51, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Horiuchi, T.; Mitoma, H.; Harashima, S.; Tsukamoto, H.; Shimoda, T. Transmembrane tnf-alpha: Structure, function and interaction with anti-tnf agents. Rheumatology 2010, 49, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, S.L.; Mastrangelo, M.A.; Habib, D.; Narrow, W.C.; Knowlden, S.A.; Wright, T.W.; Bowers, W.J. Ablation of tnf-ri/rii expression in alzheimer’s disease mice leads to an unexpected enhancement of pathology: Implications for chronic pan-tnf-alpha suppressive therapeutic strategies in the brain. Am. J. Pathol. 2011, 179, 2053–2070. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Su, F.; Bai, F.; Zhang, Z. Inflammatory cytokines and alzheimer’s disease: A review from the perspective of genetic polymorphisms. Neurosci. Bull 2016, 32, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Noh, M.Y.; Cho, K.A.; Kim, H.; Kim, S.M.; Kim, S.H. Erythropoietin modulates the immune-inflammatory response of a sod1(g93a) transgenic mouse model of amyotrophic lateral sclerosis (als). Neurosci. Lett. 2014, 574, 53–58. [Google Scholar] [CrossRef]
- He, F.Q.; Qiu, B.Y.; Li, T.K.; Xie, Q.; Cui, D.J.; Huang, X.L.; Gan, H.T. Tetrandrine suppresses amyloid-beta-induced inflammatory cytokines by inhibiting nf-kappab pathway in murine bv2 microglial cells. Int. Immunopharmacol. 2011, 11, 1220–1225. [Google Scholar] [CrossRef]
- Livne-Bar, I.; Lam, S.; Chan, D.; Guo, X.; Askar, I.; Nahirnyj, A.; Flanagan, J.G.; Sivak, J.M. Pharmacologic inhibition of reactive gliosis blocks tnf-alpha-mediated neuronal apoptosis. Cell Death Dis. 2016, 7, e2386. [Google Scholar] [CrossRef]
- Neniskyte, U.; Vilalta, A.; Brown, G.C. Tumour necrosis factor alpha-induced neuronal loss is mediated by microglial phagocytosis. FEBS Lett. 2014, 588, 2952–2956. [Google Scholar] [CrossRef]
- Shingo, T.; Sorokan, S.T.; Shimazaki, T.; Weiss, S. Erythropoietin regulates the in vitro and in vivo production of neuronal progenitors by mammalian forebrain neural stem cells. J. Neurosci. 2001, 21, 9733–9743. [Google Scholar] [CrossRef] [PubMed]
- Chin, K.; Yu, X.; Beleslin-Cokic, B.; Liu, C.; Shen, K.; Mohrenweiser, H.W.; Noguchi, C.T. Production and processing of erythropoietin receptor transcripts in brain. Brain Res. Mol. Brain Res. 2000, 81, 29–42. [Google Scholar] [CrossRef]
- Schipper, H.M. Brain iron deposition and the free radical-mitochondrial theory of ageing. Ageing Res. Rev. 2004, 3, 265–301. [Google Scholar] [CrossRef] [PubMed]
- Vinothkumar, G.; Krishnakumar, S.; Venkataraman, P. Correlation between abnormal gsk3beta, beta amyloid, total tau, p-tau 181 levels and neuropsychological assessment total scores in ckd patients with cognitive dysfunction: Impact of rhuepo therapy. J. Clin. Neurosci. 2019, 69, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, D.G. Mitochondrial calcium function and dysfunction in the central nervous system. Biochim. Biophys. Acta 2009, 1787, 1416–1424. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A. Beta-amyloid-associated free radical oxidative stress and neurotoxicity: Implications for alzheimer’s disease. Chem. Res. Toxicol. 1997, 10, 495–506. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G.; Wu, D.; Lambert, M.P.; Fernandez, S.J.; Velasco, P.T.; Lacor, P.N.; Bigio, E.H.; Jerecic, J.; Acton, P.J.; Shughrue, P.J.; et al. Alzheimer’s disease-type neuronal tau hyperphosphorylation induced by a beta oligomers. Neurobiol. Aging 2008, 29, 1334–1347. [Google Scholar] [CrossRef]
- De Felice, F.G.; Velasco, P.T.; Lambert, M.P.; Viola, K.; Fernandez, S.J.; Ferreira, S.T.; Klein, W.L. Abeta oligomers induce neuronal oxidative stress through an n-methyl-d-aspartate receptor-dependent mechanism that is blocked by the alzheimer drug memantine. J. Biol. Chem. 2007, 282, 11590–11601. [Google Scholar] [CrossRef]
- Adamcio, B.; Sargin, D.; Stradomska, A.; Medrihan, L.; Gertler, C.; Theis, F.; Zhang, M.; Muller, M.; Hassouna, I.; Hannke, K.; et al. Erythropoietin enhances hippocampal long-term potentiation and memory. BMC Biol. 2008, 6, 37. [Google Scholar] [CrossRef]
- Almaguer-Melian, W.; Merceron-Martinez, D.; Delgado-Ocana, S.; Pavon-Fuentes, N.; Ledon, N.; Bergado, J.A. Epo induces changes in synaptic transmission and plasticity in the dentate gyrus of rats. Synapse 2016, 70, 240–252. [Google Scholar] [CrossRef]
- Kumar, R.; Jaggi, A.S.; Singh, N. Effects of erythropoietin on memory deficits and brain oxidative stress in the mouse models of dementia. Korean J. Physiol. Pharmacol. 2010, 14, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Merelli, A.; Czornyj, L.; Lazarowski, A. Erythropoietin: A neuroprotective agent in cerebral hypoxia, neurodegeneration, and epilepsy. Curr. Pharm. Des. 2013, 19, 6791–6801. [Google Scholar] [CrossRef] [PubMed]
- Yazihan, N.; Karakurt, O.; Ataoglu, H. Erythropoietin reduces lipopolysaccharide-induced cell damage and midkine secretion in u937 human histiocytic lymphoma cells. Adv. Ther. 2008, 25, 502–514. [Google Scholar] [CrossRef] [PubMed]
- Barichello, T.; Simoes, L.R.; Generoso, J.S.; Sangiogo, G.; Danielski, L.G.; Florentino, D.; Dominguini, D.; Comim, C.M.; Petronilho, F.; Quevedo, J. Erythropoietin prevents cognitive impairment and oxidative parameters in wistar rats subjected to pneumococcal meningitis. Transl. Res. 2014, 163, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Kumral, A.; Gonenc, S.; Acikgoz, O.; Sonmez, A.; Genc, K.; Yilmaz, O.; Gokmen, N.; Duman, N.; Ozkan, H. Erythropoietin increases glutathione peroxidase enzyme activity and decreases lipid peroxidation levels in hypoxic-ischemic brain injury in neonatal rats. Biol. Neonate 2005, 87, 15–18. [Google Scholar] [CrossRef]
- Lv, H.Y.; Wu, S.J.; Wang, Q.L.; Yang, L.H.; Ren, P.S.; Qiao, B.J.; Wang, Z.Y.; Li, J.H.; Gu, X.L.; Li, L.X. Effect of erythropoietin combined with hypothermia on serum tau protein levels and neurodevelopmental outcome in neonates with hypoxic-ischemic encephalopathy. Neural. Regen. Res. 2017, 12, 1655–1663. [Google Scholar] [PubMed]
- Bailey, D.M.; Lundby, C.; Berg, R.M.; Taudorf, S.; Rahmouni, H.; Gutowski, M.; Mulholland, C.W.; Sullivan, J.L.; Swenson, E.R.; McEneny, J.; et al. On the antioxidant properties of erythropoietin and its association with the oxidative-nitrosative stress response to hypoxia in humans. Acta. Physiol. 2014, 212, 175–187. [Google Scholar] [CrossRef]
- Malhotra, S.; Nijhawan, S.; Rosenbaum, D.M. Erythropoietin (epoetin) as a protective factor for the brain. Curr. Atheroscler. Rep. 2004, 6, 301–306. [Google Scholar] [CrossRef]
- Doble, A. The role of excitotoxicity in neurodegenerative disease: Implications for therapy. Pharmacol. Ther. 1999, 81, 163–221. [Google Scholar] [CrossRef]
- Prentice, H.; Modi, J.P.; Wu, J.Y. Mechanisms of neuronal protection against excitotoxicity, endoplasmic reticulum stress, and mitochondrial dysfunction in stroke and neurodegenerative diseases. Oxid. Med. Cell. Longev. 2015, 2015, 964518. [Google Scholar] [CrossRef]
- Lipton, S.A.; Rosenberg, P.A. Excitatory amino acids as a final common pathway for neurologic disorders. N. Engl. J. Med. 1994, 330, 613–622. [Google Scholar] [PubMed]
- Dong, X.X.; Wang, Y.; Qin, Z.H. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.W. Excitotoxic cell death. J. Neurobiol. 1992, 23, 1261–1276. [Google Scholar] [CrossRef] [PubMed]
- Stanika, R.I.; Pivovarova, N.B.; Brantner, C.A.; Watts, C.A.; Winters, C.A.; Andrews, S.B. Coupling diverse routes of calcium entry to mitochondrial dysfunction and glutamate excitotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 9854–9859. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Mattson, M.P. Free radicals, calcium, and the synaptic plasticity-cell death continuum: Emerging roles of the transcription factor nf kappa b. Int. Rev. Neurobiol. 1998, 42, 103–168. [Google Scholar]
- Sinor, A.D.; Greenberg, D.A. Erythropoietin protects cultured cortical neurons, but not astroglia, from hypoxia and ampa toxicity. Neurosci. Lett. 2000, 290, 213–215. [Google Scholar] [CrossRef]
- Sinor, A.D.; Irvin, S.M.; Cobbs, C.S.; Chen, J.; Graham, S.H.; Greenberg, D.A. Hypoxic induction of vascular endothelial growth factor (vegf) protein in astroglial cultures. Brain Res. 1998, 812, 289–291. [Google Scholar] [CrossRef]
- Masliah, E.; Alford, M.; DeTeresa, R.; Mallory, M.; Hansen, L. Deficient glutamate transport is associated with neurodegeneration in alzheimer’s disease. Ann. Neurol. 1996, 40, 759–766. [Google Scholar] [CrossRef]
- Li, S.; Mallory, M.; Alford, M.; Tanaka, S.; Masliah, E. Glutamate transporter alterations in alzheimer disease are possibly associated with abnormal app expression. J. Neuropathol. Exp. Neurol. 1997, 56, 901–911. [Google Scholar] [CrossRef]
- Adam-Vizi, V.; Starkov, A.A. Calcium and mitochondrial reactive oxygen species generation: How to read the facts. J. Alzheimers Dis. 2010, 20 (Suppl. 2), S413–S426. [Google Scholar] [CrossRef]
- Li, M.X.; Dewson, G. Mitochondria and apoptosis: Emerging concepts. F1000Prime Rep. 2015, 7, 42. [Google Scholar] [CrossRef] [PubMed]
- Ehrenreich, H.; Degner, D.; Meller, J.; Brines, M.; Behe, M.; Hasselblatt, M.; Woldt, H.; Falkai, P.; Knerlich, F.; Jacob, S.; et al. Erythropoietin: A candidate compound for neuroprotection in schizophrenia. Mol. Psychiatry 2004, 9, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Haljan, G.; Maitland, A.; Buchan, A.; Arora, R.C.; King, M.; Haigh, J.; Culleton, B.; Faris, P.; Zygun, D. The erythropoietin neuroprotective effect: Assessment in cabg surgery (tenpeaks): A randomized, double-blind, placebo controlled, proof-of-concept clinical trial. Stroke 2009, 40, 2769–2775. [Google Scholar] [CrossRef] [PubMed]
- Wiese, L.; Hempel, C.; Penkowa, M.; Kirkby, N.; Kurtzhals, J.A. Recombinant human erythropoietin increases survival and reduces neuronal apoptosis in a murine model of cerebral malaria. Malar J. 2008, 7, 3. [Google Scholar] [CrossRef]
- Miskowiak, K.W.; Ehrenreich, H.; Christensen, E.M.; Kessing, L.V.; Vinberg, M. Recombinant human erythropoietin to target cognitive dysfunction in bipolar disorder: A double-blind, randomized, placebo-controlled phase 2 trial. J. Clin. Psychiatry 2014, 75, 1347–1355. [Google Scholar] [CrossRef]
- Sargin, D.; Friedrichs, H.; El-Kordi, A.; Ehrenreich, H. Erythropoietin as neuroprotective and neuroregenerative treatment strategy: Comprehensive overview of 12 years of preclinical and clinical research. Best Pract. Res. Clin. Anaesthesiol. 2010, 24, 573–594. [Google Scholar] [CrossRef]
- Ehrenreich, H.; Hinze-Selch, D.; Stawicki, S.; Aust, C.; Knolle-Veentjer, S.; Wilms, S.; Heinz, G.; Erdag, S.; Jahn, H.; Degner, D.; et al. Improvement of cognitive functions in chronic schizophrenic patients by recombinant human erythropoietin. Mol. Psychiatry 2007, 12, 206–220. [Google Scholar] [CrossRef]
- Ehrenreich, H.; Weissenborn, K.; Prange, H.; Schneider, D.; Weimar, C.; Wartenberg, K.; Schellinger, P.D.; Bohn, M.; Becker, H.; Wegrzyn, M.; et al. Recombinant human erythropoietin in the treatment of acute ischemic stroke. Stroke 2009, 40, e647–e656. [Google Scholar] [CrossRef]
- Miskowiak, K.W.; Favaron, E.; Hafizi, S.; Inkster, B.; Goodwin, G.M.; Cowen, P.J.; Harmer, C.J. Effects of erythropoietin on emotional processing biases in patients with major depression: An exploratory fmri study. Psychopharmacology 2009, 207, 133–142. [Google Scholar] [CrossRef]
- Velly, L.; Pellegrini, L.; Guillet, B.; Bruder, N.; Pisano, P. Erythropoietin 2nd cerebral protection after acute injuries: A double-edged sword? Pharmacol. Ther. 2010, 128, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Rozga, M.; Bittner, T.; Hoglund, K.; Blennow, K. Accuracy of cerebrospinal fluid abeta1-42 measurements: Evaluation of pre-analytical factors using a novel elecsys immunosassay. Clin. Chem. Lab. Med. 2017, 55, 1545–1554. [Google Scholar] [CrossRef] [PubMed]
- Zetterberg, H. Applying fluid biomarkers to alzheimer’s disease. Am. J. Physiol. Cell Physiol. 2017, 313, C3–C10. [Google Scholar] [CrossRef] [PubMed]
- Shaw, L.M.; Vanderstichele, H.; Knapik-Czajka, M.; Clark, C.M.; Aisen, P.S.; Petersen, R.C.; Blennow, K.; Soares, H.; Simon, A.; Lewczuk, P.; et al. Cerebrospinal fluid biomarker signature in alzheimer’s disease neuroimaging initiative subjects. Ann. Neurol. 2009, 65, 403–413. [Google Scholar] [CrossRef]
- Trombetta, B.A.; Carlyle, B.C.; Koenig, A.M.; Shaw, L.M.; Trojanowski, J.Q.; Wolk, D.A.; Locascio, J.J.; Arnold, S.E. The technical reliability and biotemporal stability of cerebrospinal fluid biomarkers for profiling multiple pathophysiologies in alzheimer’s disease. PLoS ONE 2018, 13, e0193707. [Google Scholar] [CrossRef]
- Lund, A.; Lundby, C.; Olsen, N.V. High-dose erythropoietin for tissue protection. Eur. J. Clin. Investig. 2014, 44, 1230–1238. [Google Scholar] [CrossRef]
- Shi, J.Q.; Shen, W.; Chen, J.; Wang, B.R.; Zhong, L.L.; Zhu, Y.W.; Zhu, H.Q.; Zhang, Q.Q.; Zhang, Y.D.; Xu, J. Anti-tnf-alpha reduces amyloid plaques and tau phosphorylation and induces cd11c-positive dendritic-like cell in the app/ps1 transgenic mouse brains. Brain Res. 2011, 1368, 239–247. [Google Scholar] [CrossRef]
- Sun, Y.; Calvert, J.W.; Zhang, J.H. Neonatal hypoxia/ischemia is associated with decreased inflammatory mediators after erythropoietin administration. Stroke 2005, 36, 1672–1678. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Epo Molecule | Molecular Modification | Hematocrit Effects | Protective Effects | References |
|---|---|---|---|---|
| rhEPO | None | High | Yes | [22,13] |
| Asialo-EPO | Deglycosylated form of EPO | Low | Yes | [51,52] |
| CEPO | Conformational modification (replaces lysines with homocitrulines) | Low | Yes | [54] |
| EPOL | Different glycosylation pattern (low sialylated bi-antennary structures) | None | Yes | [55,56] |
| Neuro-EPO | Low sialic acid content | None | Yes | [40,57] |
| cTfRMAb-EPO | Fusion of chimeric antibody with EPO | Low | Yes | [59] |
| In Vitro Model | Noxious Stimulus | EPO Molecule | Timing and Doses of Administration | Effects | References |
|---|---|---|---|---|---|
| PC12 | Aβ40 Oligomers Aβ25-35 peptide | rhEPO or EPOL rhEPO | Pre-treatment for 1 h or co-incubation for 24 h; doses used 10, 50 or 100 ng/mL Pre-treatment for 1 h; doses used 0.5-10 IU/mL |
| [56,63,79] |
| SH-SY5Y | Aβ25-35 Tau-p | rhEPO | Pre-treatment for 1 h; used doses 5, 10 or 20 IU/g |
| [84,88] |
| Primary culture of hippocampal neurons | Aβ40 oligomers Tau-p | rhEPO or EPOL | Co-incubation for 24 h; dose used 100 ng/mL |
| [55,56,94] |
| Organotypic rat hippocampus culture | Aβ25-35 | rhEPO or EPOL | Co-incubation for 4 days; dose used 100 ng/mL |
| [56] |
| In Vivo Model | Noxious Stimulus | EPO Molecule | Doses and Route of Administration | Effects | References |
|---|---|---|---|---|---|
| ICV-STZ (rat) | Injection of STZ | rhEPO | Dose used 5000 IU/kg, every other day for 2 weeks; i.p |
| [41,97] |
| ICV-Aβ25-35 (mouse) | Injection of Aβ25-35 | rhEPO or Neuro-EPO | Dose used 1250, 2500 and 5000 UI/kg once a day or 62, 125 and 250 IU/kg three times a day; i.p and i.n (respectively) |
| [40,101] |
| Tg2576* (mouse) | Overexpression of human APP (Swedish double mutation) | rhEPO or Neuro-EPO | Dose used 5000 IU/kg once a day for five days or 125 and 250 IU/kg, three times a week for 2 months; i.p and i.n (respectively) |
| [72,101] |
| AβPP/PS1* (mouse) | Expression of human APP and mutant human presinilin 1 (PS1-dE9) | rhEPO or CEPO cTfRMAb-EPO | Dose used 2500 IU/kg or 2500 and 5000 IU/kg administered three days a week for four weeks respectively; i.p Dose used 3 mg/kg administered three days a week for eight weeks; i.p |
| [104,59] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jarero-Basulto, J.J.; Rivera-Cervantes, M.C.; Gasca-Martínez, D.; García-Sierra, F.; Gasca-Martínez, Y.; Beas-Zárate, C. Current Evidence on the Protective Effects of Recombinant Human Erythropoietin and Its Molecular Variants against Pathological Hallmarks of Alzheimer’s Disease. Pharmaceuticals 2020, 13, 424. https://doi.org/10.3390/ph13120424
Jarero-Basulto JJ, Rivera-Cervantes MC, Gasca-Martínez D, García-Sierra F, Gasca-Martínez Y, Beas-Zárate C. Current Evidence on the Protective Effects of Recombinant Human Erythropoietin and Its Molecular Variants against Pathological Hallmarks of Alzheimer’s Disease. Pharmaceuticals. 2020; 13(12):424. https://doi.org/10.3390/ph13120424
Chicago/Turabian StyleJarero-Basulto, José J., Martha C. Rivera-Cervantes, Deisy Gasca-Martínez, Francisco García-Sierra, Yadira Gasca-Martínez, and Carlos Beas-Zárate. 2020. "Current Evidence on the Protective Effects of Recombinant Human Erythropoietin and Its Molecular Variants against Pathological Hallmarks of Alzheimer’s Disease" Pharmaceuticals 13, no. 12: 424. https://doi.org/10.3390/ph13120424
APA StyleJarero-Basulto, J. J., Rivera-Cervantes, M. C., Gasca-Martínez, D., García-Sierra, F., Gasca-Martínez, Y., & Beas-Zárate, C. (2020). Current Evidence on the Protective Effects of Recombinant Human Erythropoietin and Its Molecular Variants against Pathological Hallmarks of Alzheimer’s Disease. Pharmaceuticals, 13(12), 424. https://doi.org/10.3390/ph13120424