Abstract
Tumor cells are particularly adept at exploiting the immunosuppressive potential of neutrophils as a strategy to achieve uncontrolled proliferation and spread. Recruitment of neutrophils, particularly those of an immature phenotype, known as granulocytic myeloid-derived suppressor cells, is achieved via the production of tumor-derived granulocyte colony-stimulating factor (G-CSF) and neutrophil-selective chemokines. This is not the only mechanism by which G-CSF contributes to tumor-mediated immunosuppression. In this context, the G-CSF receptor is expressed on various cells of the adaptive and innate immune systems and is associated with induction of T cell polarization towards the Th2 and regulatory T cell (Treg) phenotypes. In contrast to the potentially adverse effects of sustained, endogenous production of G-CSF by tumor cells, stringently controlled prophylactic administration of recombinant (r) G-CSF is now a widely practiced strategy in medical oncology to prevent, and in some cases treat, chemotherapy-induced severe neutropenia. Following an overview of the synthesis, structure and function of G-CSF and its receptor, the remainder of this review is focused on: (i) effects of G-CSF on the cells of the adaptive and innate immune systems; (ii) mechanisms by which this cytokine promotes tumor progression and invasion; and (iii) current clinical applications and potential risks of the use of rG-CSF in medical oncology.
1. Introduction
Colony-stimulating factors (CSFs) are hematopoietic growth factors that are released in response to infection or inflammation to stimulate hematopoietic stem cells to proliferate and generate colonies of differentiated progeny such as neutrophils or macrophages. There are four distinct types of CSFs, each with specific types of receptors, with the occurrence of cross-talk between these receptors, if any, being largely unknown. The CSFs are named after the major types of colonies that they initiate, specifically, granulocyte colony-stimulating factor (G-CSF), granulocyte-macrophage colony-stimulating factor (GM-CSF), macrophage colony-stimulating factor (M-CSF) and multipotential colony-stimulating factor (most commonly termed interleukin-3) [1].
G-CSF, the focus of the current review, stimulates the proliferation and differentiation of neutrophil precursors, to maintain the number of circulating mature and functional neutrophils.
In the therapeutic setting, major applications of administration of recombinant (r) G-CSF include: (i) attenuation of the magnitude and duration of chemotherapy-induced neutropenia in cancer patients [2], described in greater detail below; (ii) treatment of cyclic and chronic neutropenias [3,4]; and (iii) mobilization of hematopoietic progenitor cells into peripheral blood to be harvested for stem cell transplantation. In the latter setting, the incidence and severity of experimental acute graft-versus-host disease (aGVHD) are also reduced by administration of rG-CSF [5,6], seemingly due to the immunoregulatory effects of rG-CSF on cells of both the innate and adaptive immune systems, especially T cells, as reviewed earlier by Yang et al. [7], as well as immature neutrophils [7,8]. The current review builds on these earlier studies and also addresses the origins and structure of the growth factor and its receptor. Other topics covered include the involvement of tumor-derived G-CSF in tumorigenesis, as well as the therapeutic applications of rG-CSF in medical oncology, in addition to the associated potential risks.
2. Synthesis and Function of G-CSF
Neutrophils are continuously produced from bone marrow-derived pluripotent hematopoietic stem cells via the process of granulopoiesis in which G-CSF plays a key role not only by regulating the production of neutrophils, but also the activity of these cells [9,10]. G-CSF stimulates the proliferation of hematopoietic progenitors by accelerating their cell cycle rate, thus reducing the transit time through granulopoiesis, via enhancement of the transition of immature metamyelocytes into mature neutrophils and acceleration of their release into the circulation [10]. Apart from mediating the survival of neutrophils and their precursors, G-CSF also promotes the antimicrobial functions of mature neutrophils, such as phagocytosis, superoxide production and pathogen killing [11]. The generation of neutrophils (and other types of granulocytes) from hematopoietic progenitor cells under the influence of various growth factors and cytokines, including G-CSF, is depicted in Figure 1.
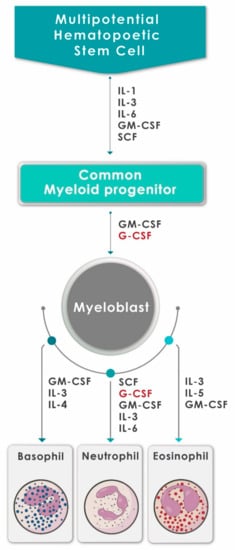
Figure 1.
A schematic diagram showing the generation of neutrophils (and other granulocytes) from hematopoietic progenitor cells under the influence of various growth factors and cytokines, including G-CSF (adapted and reproduced from Mehta et al., J Immunol 2015;195:1341–1349; Copyright 2018. The American Association of Immunologists, Inc., Rockville, MD, USA).
Endogenous production of G-CSF is largely triggered by infection and tissue damage in response to production of several pro-inflammatory stimuli such as the cytokines interleukin-1 beta (IL-1β) and tumor necrosis factor-alpha (TNFα), as well as bacterial lipopolysaccharides (LPS) [12,13]. G-CSF is found in a wide variety of tissue types, but is mainly produced by resting or stimulated stromal cells of the hematopoietic microenvironment (fibroblasts and endothelial cells) and by cells of the innate immune system such as monocytes and macrophages [14].
The importance of G-CSF in the regulation of granulopoiesis became clear following the findings of two early studies based on mice deficient in expression of either G-CSF or the G-CSF receptor (G-CSFR). These mice displayed chronic neutropenia with a corresponding decrease in granulocytic precursors in the bone marrow [15,16]. Despite these abnormalities, mature neutrophils remained detectable, albeit in much lower numbers, in the blood and bone marrow, indicating the existence of both G-CSF-dependent and -independent mechanisms of granulopoiesis [15,16], probably involving GM-CSF, IL-3 and IL-6.
According to Toghraie et al. (2019), four different mRNA isoforms resulting from alternative splicing have been reported for G-CSF (transcript variants 1, 2, 3 and 4) [17]. Transcript variants 1 and 2 encode the mature 177-amino acid G-CSF isoform A and the 174-amino acid G-CSF isoform B, respectively [17]. Isoform B is the major isoform produced in prokaryotic and eukaryotic expression systems and has more potent biological activity and greater stability than isoform A [17,18]. The other recently described G-CSF isoforms are the 141-amino acid isoform C and the 138-amino acid isoform D that are encoded by transcript variants 3 and 4, respectively [17], the activities of which remain to be established.
3. The G-CSF Receptor
The biological activities of G-CSF are mediated via the G-CSFR, which belongs to the class I cytokine receptor superfamily [13]. This receptor is expressed on a range of hematopoietic cells. These include mature neutrophilic granulocytes, myeloid progenitors, hematopoietic stem cells, monocytes and lymphocytes, as well as certain non-hematopoietic tissues [19].
The G-CSFR is a single-chain membrane protein composed of extracellular, transmembrane and intracellular regions [10,19]. The large glycosylated extracellular region contains three distinct functional zones: an N-terminal immunoglobulin (Ig)-like domain, a cytokine receptor homology (CRH) domain and three fibronectin (FBN) type III domains [13]. The CRH domain has four highly conserved cysteines and a Trp-Ser-X-Trp-Ser (WSXWS) motif that are required for ligand recognition and receptor activation [13,20].
The intracellular region of the G-CSFR comprises three distinct box regions. Boxes 1 and 2 are required for activation of proliferation signals, while box 3 promotes the differentiation of myeloid progenitor cells [20]. In addition, phosphorylation of the four tyrosine residues at positions 704, 729, 744 and 764 is also a key event in intracellular signaling, resulting from phosphorylation of multiple SH2 (Src Homology 2)-containing signaling moieties [10,20].
G-CSF binds to the extracellular domains of the G-CSFR, triggering receptor homodimerization, leading to the activation of intracellular signaling cascades involving the Janus kinase-signal transduction and activator of transcription (JAK/STAT), phosphatidylinositol 3-kinase/protein kinase B (PI3K/AKT) and mitogen-activated protein kinase/extracellular signal-regulated kinase 1/2 (MAPK/ERK); these signaling pathways initiate the transcriptional changes that promote proliferation differentiation, migration and survival [19,20,21].
According to Mehta et al. (2014), seven mRNA isoforms encoding the G-CSFR have been isolated with only class 1 (the canonical type) and class IV (differentiation-defective) detectable in hematopoietic cells [22].
4. Effects of rG-CSF on Cells of the Adaptive Immune System
Recombinant G-CSF appears to affect T cell homeostasis by multiple mechanisms. Most importantly, these include induction of T cell polarization toward the T helper 2 (Th2) phenotype, as well as inhibition of T cell proliferation, and enhancement of T cell apoptosis. This cytokine may affect T cells either directly or indirectly, by regulating the activities of various cell types such as dendritic cells, monocytes, myeloid-derived suppressor cells (MDSCs) and others [7].
4.1. T Cell Proliferation
The proliferative responses of T lymphocytes to both allogeneic and mitogenic stimulation are profoundly reduced in patients, as well as healthy stem cell donors, following treatment with rG-CSF [23,24,25]. These responses normalized 14 days after administration of the last dose of rG-CSF [24]. The inhibitory effects of rG-CSF on T cell proliferation were achieved, at least in part, by an indirect mechanism. This contention is based on observations that soluble factors present in serum from healthy donors who had received rG-CSF also inhibited T cell proliferation to mitotic challenge, possibly due to perturbation of lymphocyte mitochondrial function and inhibition of cell cycle progression [26,27]. Other studies have suggested that administration of rG-CSF suppresses T cell proliferative responses indirectly by modulating monocyte function, as depletion of monocytes in the cell co-culture partially reversed the abnormal proliferative capacity of T cells [23,28].
4.2. T Helper 2 Cell Polarization
As alluded to above, rG-CSF may polarize the transition of T cells from the Th1 to Th2 phenotype by switching the cellular cytokine secretion profile. In this context, several studies reported that both murine and human T cells pre-treated with rG-CSF followed by stimulation with mitogens or alloantigens in vitro demonstrated significant decreases in the production of the Th1 cytokines interferon-gamma (IFNγ) and interleukin (IL)-2, while expression of the Th2 cytokines IL-4 and IL-10 was increased [5,29,30,31]. Another study observed that polarization of murine T lymphocytes toward a type 2 cytokine profile was associated with a decrease in severity of experimental GVHD [5]. With respect to a potential molecular mechanism underpinning rG-CSF-mediated Th2 phenotypic transition, interaction of the cytokine with G-CSFR-expressing T cells induced an increase in mRNA expression and protein levels of the transcription factor GATA-3 (GATA3-binding protein) and a decrease in the level of expression of mRNA encoding the γ-subunit of the transcriptional regulator complex ISGF3 (interferon stimulated gene factor 3) [7,25,32]. GATA-3 is a transcription factor that plays critical roles in Th2 differentiation, Th2 cytokine production and selective growth of Th2 cells [33]. In addition, GATA-3 inhibits Th1 differentiation by blocking the IL-12 signaling pathway, neutralizing the function of RUNX3 (Runt-related transcription factor 3) and directly silencing the ifng gene [34]. Moreover, rG-CSF may also limit IFNγ signaling in T cells by suppressing expression of the gene encoding the ISGF3 subunit/p48 in cluster of differentiation (CD)4+ donor T cells [32].
4.3. T Helper 17 Cells
A gene profiling study of purified T cells isolated from the blood of rG-CSF-treated peripheral stem cell donors largely confirmed the aforementioned findings by revealing that genes related to Th2 cell-mediated immunity were upregulated, while those associated with Th1 cell function, including cytotoxicity, antigen presentation and GVHD, were downregulated [35]. In addition, overexpression of genes related to T regulatory cell (Treg) differentiation, as well as those that suppress Th17 phenotypic transition, were detected [35]. Although controversial [7], it is noteworthy that immunophenotyping procedures revealed that rG-CSF-exposed stem cell donors had reduced levels of T cells with a Th17 phenotype (CD4+IL-17+CCR6+IL-23R+) but increased expression levels of CD4+CD25highCD45RO+ T cells typical of Tregs [35]. Moreover, findings originating from the same study revealed that levels of mRNA encoding the Th17-specific transcription factor RORγt were significantly decreased in T cells isolated from G-CSF-mobilized peripheral blood stem cell harvests [35]. Further, also from a mechanistic perspective, another study demonstrated that rG-CSF directly modulated CD4+ T cell responses via upregulation of expression of the protein suppressor of cytokine signaling-3 (SOCS3), resulting in attenuation of Th17-mediated aGVHD [36]. In contrast, however, others have reported that utilization of rG-CSF to mobilize stem cells is associated with increased numbers of Th17 cells in the circulation that may exacerbate GVHD [37,38]. Clearly additional research is required to resolve these discrepancies [7,39].
4.4. T Regulatory Cells
As alluded to above, the mechanisms of rG-CSF-induced immune tolerance also include maturation of bone marrow-derived CD4+CD25+Foxp3+ Tregs that produce the immunosuppressive cytokines IL-10 and transforming growth factor-beta (TGFβ) [35]. High donor Treg content is associated with a low risk for the development of GVHD following allogeneic stem cell transplantation [40]. In this context, an early report by Rutella et al. (2002) [41] demonstrated that highly purified CD4+ T cells from healthy donors receiving rG-CSF acquired the functional properties of Treg type 1 (Tr1) cells that secreted high amounts of IL-10 and moderate amounts of TGFβ following activation with alloantigens in the absence of significant release of IL-2 and IL-4. These cells had a low proliferative capacity and mediated active suppression of antigen-driven proliferation of bystander T cells, seemingly by an IL-10/TGFβ-dependent mechanism [41,42]. These effects were consistent with observations derived from a murine model that revealed protection against development of GVHD following pre-treatment of the donors with pegylated rG-CSF that was dependent on the enhanced generation of IL-10-producing Tregs [43]. In addition, animal studies showed that administration of rG-CSF promoted the systemic expansion of natural (n) CD4+ CD25+ Tregs, depletion of which significantly exacerbated GVHD [44]. Importantly, however, depletion of Tregs did not completely negate the immunosuppressive effects of rG-CSF, consistent with the regulatory effects of rG-CSF on other types of immune cells [44]. In this context, it is noteworthy that rG-CSF upregulated several genes or gene families implicated in nTreg survival, including Pias3, an inhibitor of activated STAT3 (an established suppressor of nTreg stability), thereby enhancing nTreg stability and survival [7,44]. Furthermore, rG-CSF has been shown to reduce the expression of the chemokine CXCL12 in bone marrow, thereby inducing the homeostatic trafficking of CXCR4-expressing Tregs from human bone marrow to the periphery [45]. Apart from contributing to a protective effect in aGVHD, rG-CSF-induced Tregs have also been reported to protect against atherosclerosis, lupus nephritis and diabetes in murine studies [46,47,48].
4.5. CD8+ T Cells
Recombinant G-CSF exerts direct, suppressive effects on cytotoxic effector CD8+ T cells, resulting in partial attenuation of T cell function [49]. The effects of rG-CSF on CD8+ T cell function were investigated following antigen-dependent and -independent stimulation in vitro. In both cases, exposure to rG-CSF resulted in: (i) decreased secretion of IFNγ and granzyme B; (ii) reduced surface expression of the activation markers CD25, CD38, CD69 and CD137, as well as human leucocyte antigen D-related (HLA-DR); (iii) a decrease in the expression of microRNA-155, which is a key regulator of effector CD8+ T cells; and (iv) a reduction in the activation of T cell receptor (TCR)-dependent and -independent signaling pathways, as reflected by the phosphorylation of ERK1/2, Lck (lymphocyte-specific protein tyrosine kinase) and the CD3-ζ chain (CD247) [49]. All of these observations demonstrate that rG-GSF also directly suppresses cytotoxic CD8+ T lymphocyte function.
5. Effects of rG-CSF on Cells of the Innate Immune System
5.1. Dendritic Cells
Dendritic cells (DCs) are professional antigen-presenting cells that activate naïve T helper cells. T helper 1- and Th2-promoting DC lineages, known as DC1 and DC2, respectively, have been described [7]. In this context, administration of rG-CSF to healthy human donors resulted in systemic mobilization of large numbers of type 2 DCs, as well as CD4+ T cells expressing the Th2 phenotype [50,51]. Another study reported that administration of rG-CSF to mice resulted in induction of DCs that produced low levels of IL-12 [52]. Although not distinguishing between DC1 and DC2 cells, the authors speculated that their findings were consistent with reduced numbers of the former cell type that are critical for the differentiation of Th1 cells [52].
In addition, isolated human monocytes cultured with autologous serum derived from rG-CSF- treated healthy donors, which contained high levels of IL-10 and IFNα, differentiated into tolerogenic DCs in vitro that exhibited diminished IL-12p70 release and poor allostimulatory capacity, as well as induction of Tregs [53]. It has also been shown that human monocytes differentiated with rG-CSF and IL-4 acquire a DC-like morphology that is associated with spontaneous release of IL-10, leading to induction of anergy in naïve T cells in vitro [54].
The aforementioned findings appear to support the contention that transplantation of rG-CSF-augmented peripheral blood stem cells does not result in overwhelming aGVHD because the stem cell infusions contain predominantly Th2-inducing DCs [50].
5.2. Monocytes
As mentioned above, several earlier studies have indicated that the inhibitory effects of rG-CSF on T cell proliferation and cytokine responses were mediated indirectly by modulation of monocyte function [28,55]. In this context, rG-CSF pre-treatment of both a monocytic cell line (NOMO-1) and isolated human blood monocytes resulted in significant attenuation of LPS-induced secretion of the pro-inflammatory cytokines TNFα and IL-12 while augmenting production of the anti-inflammatory cytokine IL-10 [56]. In addition, the antigen-presenting function of rG-CSF-treated human monocytes was also found to be impaired [57]. A more recent publication reported that administration of rG-CSF to both humans and mice resulted in mobilization of a subset of immunosuppressive CD34+ cells that exhibited the features of mature monocytes [58]. In response to IFNγ released by activated T cells, these CD34+ cells induced allogeneic T cell death, resulting in expansion of Tregs and immunosuppression [58]. Moreover, and somewhat predictably, the presence of CD34+ monocytes in peripheral blood correlated inversely with the incidence of aGVHD in humans [58].
5.3. Myeloid-Derived Suppressor Cells
Myeloid-derived suppressor cells (MDSCs) are a heterogeneous group of myeloid cells of monocytic and granulocytic origin that suppress both innate and adaptive immune responses and undergo expansion during cancer, infection and inflammatory diseases [59]. It has been reported that rG-CSF-induced immune tolerance may be mediated by various MDSC subsets and that these are important regulators of the development of aGVHD in allogeneic hematopoietic stem cell transplantation [60]. In this context, expansion of myeloid cells in the peripheral blood of rG-CSF-treated human donors resulted in increased numbers of MDSC subtypes of both monocytic and granulocytic origin that significantly regulated alloreactive T cell responses in vitro [61]. In addition, a recent study revealed the existence of an unidentified, immature cell population of monocytic origin defined by the cell surface expression of HLA-DR−/lowCD33+CD16− in healthy donors that appeared following administration of rG-CSF [62]. This early MDSC-like population suppressed T cell proliferation in a TGFβ-dependent manner, skewed the T helper cell balance from a Th1 to Th2 predominance and promoted generation of Tregs [62]. Importantly, adoptive transfer of these cells prevented development of aGVHD in a humanized mouse model [62]. These findings are in agreement with those of earlier studies that identified the presence of immunosuppressive monocytic MDSCs that predicted the risk of aGVHD following infusion of rG-CSF-expanded peripheral blood stem cells [63,64].
5.4. Natural Killer Cells
Natural killer (NK) cells isolated from the blood of human rG-CSF-expanded stem cell donors, as well as following exposure to the cytokine in vitro, were found to be dysfunctional with respect to downregulation of expression of various activating receptors [NKp44, NKp46, NKGD2 (natural killer receptor group 2, member D)], as well as the inhibitory killer immunoglobulin-like receptors (KIRs) KIR2DL1 and 2 [65]. Additional abnormalities included a reduction in granzyme B levels that was associated with decreased cytotoxic/lytic capacity of NK cells both in vitro and in vivo [65,66,67], as well as decreased production of pro-inflammatory cytokines (IFNγ, TNFα, GM-CSF, IL-6, IL-8) and proliferative capacity [65]. More recently, Yu et al., who explored the effects of rG-CSF on NK cell subpopulations, found that administration of the growth factor not only decreased the percentage of circulating and bone marrow NK cells, but also altered the balance of NK subpopulations, resulting in a high ratio of CD56bri to CD56dim subsets in the setting of low levels of NK1 cells (IFNγ-secreting NK cells) [68].
5.5. Platelets
Administration of rG-CSF to humans prior to autologous stem cell transplantation is associated with decreased platelet counts [69]. In this context, experimental administration of rG-CSF to mice was found to induce thrombocytopenia by interfering with the differentiation of hematopoietic precursors into megakaryocytes [70]. Although this activity of rG-CSF may have implications for impairment of anti-viral and antimicrobial immunity mediated by both megakaryocytes [71] and platelets [72], the relevance, if any, to the clinical applications of rG-CSF remains to be established.
The immunomodulatory effects of rG-CSF, which presumably mimic those of endogenously generated G-CSF, are summarized in Table 1.

Table 1.
The immunomodulatory effects of recombinant granulocyte colony-stimulating factor (rG-CSF).
6. The Role of Endogenous G-CSF in the Pathogenesis of Cancer Progression and Invasion
Various types of advanced, solid malignancies produce G-CSF and also express its receptor, enabling autocrine proliferation of tumor cells, as well as conferring a strategy to intensify the immunosuppressive milieu of the tumor microenvironment (TME) via recruitment of immature and mature neutrophils that function as MDSCs [73]. Approximately 10% of patients with advanced, often metastatic, solid malignancies exhibit tumor-related leukocytosis, including, but not limited to, those of the lung, gastrointestinal tract, pancreas, breast and bladder [74]. In these settings, tumor-derived G-CSF induces accelerated myelopoiesis that results in a moderate and, in some instances, a profound leukocytosis that is associated with increased numbers of immature myeloid cells. These cells are found in the bone marrow, blood and spleen and their presence is predictive of a poor prognosis [10,73,74,75,76,77,78,79].
Particularly high levels of expression (~90%) of the G-CSFR have been reported in human gastric and colon cancers, a setting in which G-CSF originates not only from tumor cells per se, but also from stromal myofibroblasts/fibroblasts in the tumor microenvironment (TME) [80]. In the case of breast cancer, systemic levels of G-CSF are significantly increased in patients with advanced but not early disease, being highest in those with aggressive, invasive N3 tumors [81], with expression of the G-CSFR detected in 71% of patients with invasive ductal adenocarcinomas (stage T1-2 NOMO) [82]. In these and other types of advanced breast cancers such as triple negative breast cancer [83], as well as the high-risk luminal A dominant breast cancer subtype (C3) [84], high-level expression of G-CSF in biopsy specimens is associated with poor overall survival (OS) and invasive potential.
7. Mechanisms by Which G-CSF Promotes Tumor Progression and Invasion
Enhancement of tumor cell proliferation and immunosuppression represent the predominant mechanisms of G-CSF-driven tumor progression, while G-CSF-mediated pro-metastatic activity is achieved via several mechanisms that include augmentation of release of pro-angiogenic factors by various cell types in the TME, as well as via the involvement of neutrophil extracellular traps (NETs).
7.1. Tumor Cell Proliferation
The mechanisms by which G-CSF drives tumor progression vary according to the type of malignancy. For example, in the case of aggressive tumors with high-level expression of G-CSF/G-CSFR, such as breast cancer, gastric and colon cancers, squamous cell cancers of the head and neck and neuroblastoma, data derived from pre-clinical in vitro studies and animal models of tumorigenesis, as well as from histological/genotypic analysis of biopsy specimens from patients, have revealed the existence of autocrine mechanisms of tumor proliferation [10,80,85,86,87]. Intracellular signaling pathways involved in G-CSF/G-CSFR-activated tumor cell proliferation were, however, found to vary somewhat between tumor types. These mechanisms included the JAK/STAT pathway in the case of neuroblastoma cell lines [86,87] and the ERK1/2 and p90 ribosomal S6 kinase 1 pathways in gastric and colorectal cancer cell lines [80].
Alternative mechanisms, independent of G-CSFR-driven autocrine tumor cell proliferation, appear to promote G-CSF-mediated tumor progression in other types of malignancies such as experimental metastatic bone tumors [88] and Ewing sarcoma (ES) [89]. In the case of the former, a murine bone metastasis model was used that involved intra-tibial injection of a murine melanoma or a murine breast cancer cell line. Following subcutaneous administration of rG-CSF or vehicle (control) for eight days, tumor growth was assessed on the ninth day. The authors of the study observed that administration of rG-CSF resulted in significantly increased tumor growth that was dependent on the secretion of the pro-osteoclastogenic factor osteoprotegerin by osteoclasts, indicating that G-CSF-mediated osteoclastogenesis may enhance tumor growth in bone [88]. The relevance, if any, of these observations to the potential risk of short-term administration of rG-CSF in the clinical setting of chemotherapy-induced neutropenia in cancer patients has not, however, been established.
With respect to ES, analysis of biopsy material from 83 patients with this vascular malignancy revealed expression of G-CSF and G-CSFR protein and mRNA in all biopsy specimens, as well as in four ES cell lines [89]. Treatment of ES cell lines with rG-CSF in vitro failed to trigger proliferation of the cells. However, in a murine model of experimental tumorigenesis, administration of rG-CSF to nude mice for five days prior to injection of an ES cell line (TC/7-1) and continued administration of the growth factor for a further 14 day period resulted in significantly increased tumor growth relative to that of rG-CSF-untreated, control mice [89]. Although somewhat contentious [90], the authors speculate that the pro-tumorigenic effects of rG-CSF in this setting are linked to the release of progenitor cells from the bone marrow that promote tumor vascular expansion and growth [85]. Again, however, the clinical relevance of this proposed mechanism of endogenous G-CSF-associated tumorigenesis, if any, remains to be established.
7.2. Induction of Myeloid-Derived Suppressor Cells (MDSCs)
Notwithstanding the myelopoiesis-independent mechanisms of G-CSF-mediated immunosuppression mentioned in an earlier section of this review, proliferation and mobilization of MDSCs, specifically those of granulocytic leukocyte origin, represent the most probable mechanisms by which tumor-derived G-CSF promotes tumor progression [91]. In this setting, G-CSF operates in partnership with tumor-derived neutrophil chemoattractants, most prominently the chemokines CXCL5 (also known as epithelial neutrophil-activating peptide-78; ENA-78) and CXCL8 (IL-8) [92,93]. The resultant accumulation of MDSCs in the TME is conducive to the establishment of a highly immunosuppressive milieu [91]. In this setting, MDSCs are exposed to cytokines that promote differentiation of these cells and augmentation of their suppressive effects on the anti-tumor activities of tumor-infiltrating lymphocytes (TILs). In addition to G-CSF, cytokines encountered by MDSCs in the TME include most prominently TGFβ1 derived from various cell types including tumor cells per se, tumor-associated macrophages (TAMs) of the M2 phenotype, epithelial cells and fibroblasts [94,95,96,97]. In this setting, TGFβ1 is proteolytically cleaved to yield the bioactive protein by matrix metalloproteinase-9 (MMP-9) released by MDSCs [98].
Mechanisms utilized by MDSCs to suppress the functions of TILs have recently been described in detail elsewhere [99] and are summarized as follows:
- High-level production of anti-proliferative, cytotoxic reactive oxygen species (ROS) [99];
- Depletion of arginine and tryptophan via extracellular release of the enzymes arginase and indoleamine-2,3-dioxygenase [100,101];
- Expansion of Tregs via production of IL-10 [102];
- Interference with the recognition of tumor antigens via nitrosative modification of the T cell receptor for antigen [103];
- Physically impede the access of TILs to tumor cells in the TME [104];
- Via potentiation of neutrophil extracellular trap (NET) formation within the TME, which also obstructs the access of TILs to tumor cells [105].
In addition to promoting immunosuppression-associated tumor progression, the G-CSF/MDSC axis also contributes to tumor invasion and metastasis by driving NETosis within the TME.
8. Tumor Invasion/Metastasis
In addition to NETosis, other pro-invasive mechanisms involving the G-CSF/MDSC axis include MMP-9-driven epithelial-to-mesenchymal transition [106] and production of pro-angiogenic factors such as vascular endothelial growth factor (VEGF) and Bv8, as well as MMP-9 [106,107,108,109,110]. Furthermore, G-CSF released by tumor cells creates a pre-metastatic environment via mobilization of immature neutrophils from the bone marrow [111]. In this setting, these cells migrate to the metastatic site where they create an inflammatory, pro-metastatic milieu conducive to tumor cell invasion and proliferation, remodeling of the extracellular matrix and suppression of the anti-tumor immune response. In addition, the overexpression of G-CSF by various types of tumor cells has been shown to act as a “priming factor” that induces neutrophils to undergo NETosis [112,113].
Role of Neutrophil Extracellular Traps in Tumor Invasion
NETosis is a highly regulated sequence of events that leads to cell death in response to various microbial and inflammatory stimuli. As a result of NETosis, neutrophils extrude their chromatin material leading to the formation of NETs [114]. Production of ROS by activated neutrophils leads to the disassembly of the nuclear envelope. This is followed by decondensation of the chromatin which is driven via activation of myeloperoxidase (MPO) [115], neutrophil elastase (NE) [116] and protein arginine deiminase type 4 (PAD4) [117]. The released chromatin, in turn, is “decorated” with various granular components including MPO, α-defensins, elastase, cathepsin G and lactoferrin, as well as histones [118,119]. NETs, therefore, aid in the immobilization and killing of invading microorganisms. However, the involvement of NETs in non-infectious inflammatory pathologies, including cancer, is now well recognized. NETs have been identified in patients with sarcoma [120], pancreatic cancer [121], breast cancer [122] and ovarian cancer [123]. In the context of cancer, NETs aid cancer cells in evading detection by cytotoxic TILs, or by forming a physical barrier between them [124]. They also play a role in metastasis through the trapping of tumor cells in vessels, thereby facilitating extravasation and tumor progression [125]. The progression of tumors by NETs is reported to be achieved through the promotion of thrombosis [113] and angiogenesis [126]. Due to the involvement of G-CSF, albeit indirect, in promoting NETosis in the TME, G-CSF has been proposed as a potential marker for individuals with cancer who are at risk of developing metastasis due to the formation of NETs [122].
9. Therapeutic Usage of Recombinant G-CSF
Chemotherapy-induced febrile neutropenia (FN) is a life-threatening after-effect of cancer treatment that poses a significant risk for development of infection-related morbidity and mortality, as well as substantial dose-limiting toxicity. The risk of infection increases with the severity and duration of neutropenia.
Patients developing grades 3 and 4 neutropenia (severe and life threatening, respectively) or FN during chemotherapy commonly require dose reductions and/or delayed administration of their chemotherapy, resulting in decreased relative dose intensity (RDI). Treatment outcomes may be compromised, mostly when the goal of treatment intent is either curative or to prolong survival. Accordingly, prophylactic use of rG-CSF is a therapeutic strategy for maintaining the RDI during treatment by decreasing the incidence of severe neutropenia and FN, thereby improving treatment outcomes and quality of life. Most commonly, rG-CSF is utilized to increase circulating neutrophil counts in patients facing severe and prolonged neutropenic episodes following chemotherapy. Additionally, as mentioned earlier, rG-CSF is used in patients and donors to mobilize peripheral blood stem cells for harvesting in the settings of autologous and allogeneic stem cell transplantation, respectively, as well as to support post-transplantation neutrophil recovery. The American Society of Clinical Oncology (ASCO), the National Comprehensive Cancer Network (NCCN) and the European Society for Medical Oncology (ESMO) have developed evidence-based guidelines defining the appropriate use of rG-CSF in cancer patients [127,128,129]. The main indications for the usage of rG-CSF are summarized in the following sections.
9.1. Indications for the Usage of rG-CSF
9.1.1. Primary Prophylaxis
Primary prophylaxis is defined as the usage of rG-CSF in the first and subsequent cycles of chemotherapy. Most rG-CSF guidelines recommend primary prophylaxis for patients receiving chemotherapy regimens with an overall risk of FN that is greater than 20%. Patients receiving myelotoxic chemotherapy with curative or radical intent (including adjuvant/neoadjuvant settings) are included in this category. Additionally, primary prophylaxis should be considered for patients receiving myelotoxic chemotherapy with a documented FN incidence rate of 10–20% and who have one or more of the risk factors listed in Table 2 [129].
Table 2.
Risk factors associated with development of febrile neutropenia associated with an overall risk of 10–20%.
Primary prophylaxis is supported by the fact that approximately half of the neutropenic episodes occur during the first cycle of chemotherapy [130].
9.1.2. Secondary Prophylaxis
Secondary prophylaxis is defined as the usage of rG-SCF in subsequent cycles after initial episodes of severe neutropenia and/or FN. This strategy may be considered if dose reduction or treatment delay may result in a decrease in disease-free or overall survival. Examples of types of malignancies in this category include adjuvant breast cancer, non-Hodgkin’s lymphoma, Hodgkin’s disease, testicular cancer and other germ cell tumors, as well as patients undergoing neoadjuvant chemotherapy with curative intention. For patients undergoing chemotherapy with palliative intent, dose modifications are considered a reasonable alternative.
9.1.3. Supportive Therapy for Neutropenic Sepsis
Most guidelines do not recommend the use of rG-CSF for the treatment of patients with low-risk or uncomplicated FN, or afebrile neutropenia. Recombinant G-CSF is recommended for patients with severe or complicated FN as summarized in Table 3.
Table 3.
Complicated febrile neutropenia.
9.1.4. Short-Acting vs. Long-Acting rG-CSF
Short-acting and long-acting rG-CSF are approved for the prophylaxis of FN [131]. Although observational, non-randomized studies have shown a greater efficacy of long-acting rG-CSF, most randomized studies have shown that there are no differences with respect to the duration of action between these formulations. The differences shown in observational studies may be a result of under-dosing patients with short-acting rG-CSFs as may occur in the real-world setting of general practice usage [132]. Importantly, biosimilar formulations of both forms of rG-CSF are available [133,134]. These represent a significant saving to healthcare systems [135].
9.1.5. Other Clinical Recommendations for rG-CSFs
Additional clinical indications for the therapeutic usage of rG-CSF include idiosyncratic drug-induced neutropenia and agranulocytosis, congenital neutropenic syndromes and neutropenia-associated primary immunodeficiency disorders [136,137,138]. However, the clinical utility of rG-CSF in patients with myelodysplastic syndromes (MDS) remains unclear and controversial due to the effect of this agent on malignant clones. Some studies reported preferential stimulation of malignant clones, while others did not. There is a lack of significant evidence for the prophylactic use of rG-CSF in MDS, and most guidelines do not recommend its usage on a routine basis [139].
Colony-stimulating factors should be avoided in patients receiving concurrent chemotherapy and radiation therapy, particularly involving the mediastinum. In the absence of chemotherapy treatment, therapeutic use of CSFs may be considered in patients receiving radiation therapy alone if prolonged delays secondary to neutropenia are expected [121].
Hematopoietic syndrome is a clinical diagnosis given to people who present with equal or more than one new-onset cytopenia in the background of acute radiation exposure [140]. Recombinant G-CSF has been successfully used in this clinical setting. Additionally, a broad spectrum of agents has been investigated in the management of lethal acute radiation syndrome. These agents include immunomodulators, prostaglandins, inhibitors of prostaglandin synthesis, agonists of adenosine cell receptors, herbal extracts, flavonoids and vitamins [141].
Finally and reassuringly, long-term follow-up of healthy donors who had received rG-CSF in the setting of allogeneic stem cell transplantation failed to reveal a causal link with future development of hematological malignancies [142], underscoring the distinction between the clinical applications of rG-CSF and prolonged production of endogenous G-CSF by advanced malignancies. Nevertheless, the increasing prophylactic use of rG-CSF in the setting of medical oncology and hematology, together with the availability of more cost-effective biosimilars, clearly necessitates ongoing vigilance with respect to pro-tumorigenic potential [99,143,144,145].
10. Conclusions
Like many other cytokines, G-CSF is a rather enigmatic protein. On the one hand, controlled and appropriate production of G-CSF plays a critical role in anti-infective host defense, while on the other hand, inappropriate production by tumor cells appears to contribute to tumor growth and invasion. In the latter scenario, of which many clinicians may be unaware, the pro-tumorigenic effects are achieved via a dual mechanism of immunosuppression, seemingly by directing T cell polarization towards the Th2 and Treg phenotypes, and, most prominently, via induction of production of MDSCs. Given its key role in host defense, the most effective strategies to neutralize the pro-tumorigenic effects of endogenous G-CSF in the setting of adjunctive anti-cancer therapy are likely to involve pharmacologic targeting of the CXCR1 and CXCR2 chemokine receptors. The involvement of endogenously produced, tumor-derived G-CSF in tumorigenesis contrasts with the beneficial administration of rG-CSF in the prevention of chemotherapy-induced severe and life-threatening infection. Recent innovations in this setting include prophylactic use of rG-CSF, rather than delayed therapeutic administration following the onset of FN, thereby alleviating development of infection and treatment disruption. Although this strategy does not appear to pose the risk of development of hematological and other types of malignancies, ongoing vigilance is necessary, particularly in the context of increasing prophylactic utilization of rG-CSF and the availability of more cost-effective biosimilars. Future studies should be undertaken to monitor the emergence of MDSCs and/or alterations in the levels of their soluble, systemic mediators of immunosuppression in patients with cancer who receive prolonged/frequent administration of rG-CSF.
Author Contributions
All authors were involved in conceptualizing the manuscript; the introductory section and overview of the effects of G-CSF on cells of the adaptive and innate immune systems were compiled by A.J.T.; the involvement of G-CSF in tumor progression and invasion by R.A. and H.C.S., and the therapeutic applications of G-CSF were by B.L.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
B.L.R. is supported by the Cancer Association of South Africa.
Conflicts of Interest
B.L. Rapoport reports grants, personal fees, non-financial support, advisory boards and speaker engagement from Sandoz Novartis AG, as well as speaker engagement from Amgen South Africa and Mylan South Africa. None of the other authors have a conflict of interest to declare.
References
- Metcalf, D. The colony-stimulating factors and cancer. Cancer Immunol. Res. 2013, 1, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Mhaskar, R.; Clark, O.A.; Lyman, G.; Engel Ayer Botrel, T.; Morganti Paladini, L.; Djulbegovic, B. Colony-stimulating factors for chemotherapy-induced febrile neutropenia. Cochrane Database Syst. Rev. 2014, 2014, CD003039. [Google Scholar] [CrossRef] [PubMed]
- Dale, D.C.; Bolyard, A.; Marrero, T.; Makaryan, V.; Bonilla, M.; Link, D.C.; Newburger, P.; Shimamura, A.; Boxer, L.A.; Spiekerman, C. Long-term effects of G-CSF therapy in cyclic neutropenia. N. Engl. J. Med. 2017, 377, 2290–2292. [Google Scholar] [CrossRef] [PubMed]
- Christensen, R.D. Recombinant G-CSF treatment of severe chronic neutropenia in neonates and infants. In Neonatology; Buonocore, G., Bracci, R., Weindling, M., Eds.; Springer: Cham, Switzerland, 2018. [Google Scholar] [CrossRef]
- Pan, L.; Delmonte, J., Jr.; Jalonen, C.K.; Ferrara, J.L. Pretreatment of donor mice with granulocyte colony-stimulating factor polarizes donor T lymphocytes toward type-2 cytokine production and reduces severity of experimental graft-versus-host disease. Blood 1995, 86, 4422–4429. [Google Scholar] [CrossRef] [PubMed]
- Zeng, D.; Dejbakhsh-Jones, S.; Strober, S. Granulocyte colony-stimulating factor reduces the capacity of blood mononuclear cells to induce graft-versus-host disease: Impact on blood progenitor cell transplantation. Blood 1997, 90, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.Z.; Zhang, J.Q.; Sun, L.X. Mechanisms for T cell tolerance induced with granulocyte colony-stimulating factor. Mol. Immunol. 2016, 70, 56–62. [Google Scholar] [CrossRef]
- Mackey, J.B.G.; Coffelt, S.B.; Carlin, L.M. Neutrophil maturity in cancer. Front. Immunol. 2019, 10, 1912. [Google Scholar] [CrossRef]
- Fuchs, O. Development of neutrophils and their role in hematopoetic microenvironment regulation. In Cells of the Immune System; IntechOpen: London, UK, 2020. [Google Scholar] [CrossRef]
- Liu, L.; Liu, Y.; Yan, X.; Zhou, C.; Xiong, X. The role of granulocyte colony-stimulating factor in breast cancer development: A review. Mol. Med. Rep. 2020, 21, 2019–2029. [Google Scholar] [CrossRef]
- Carulli, G. Effects of recombinant human granulocyte colony-stimulating factor administration on neutrophil phenotype and functions. Haematologica 1997, 82, 606–616. [Google Scholar]
- Panopoulos, A.D.; Watowich, S.S. Granulocyte colony-stimulating factor: Molecular mechanisms of action during steady state and ‘emergency’ hematopoiesis. Cytokine 2008, 42, 277–288. [Google Scholar] [CrossRef]
- Wright, C.R.; Ward, A.C.; Russell, A.P. Granulocyte colony-stimulating factor and its potential application for skeletal muscle repair and regeneration. Mediat. Inflamm. 2017, 2017, 7517350. [Google Scholar] [CrossRef] [PubMed]
- Cetean, S.; Căinap, C.; Constantin, A.M.; Căinap, S.; Gherman, A.; Oprean, L.; Hangan, A.; Oprean, R. The importance of the granulocyte-colony stimulating factor in oncology. Clujul Med. 2015, 88, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Lieschke, G.J.; Grail, D.; Hodgson, G.; Metcalf, D.; Stanley, E.; Cheers, C.; Fowler, K.J.; Basu, S.; Zhan, Y.F.; Dunn, A.R. Mice lacking granulocyte colony-stimulating factor have chronic neutropenia, granulocyte and macrophage progenitor cell deficiency, and impaired neutrophil mobilization. Blood 1994, 84, 1737–1746. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Wu, H.Y.; Wesselschmidt, R.; Kornaga, T.; Link, D.C. Impaired production and increased apoptosis of neutrophils in granulocyte colony-stimulating factor receptor-deficient mice. Immunity 1996, 5, 491–501. [Google Scholar] [CrossRef]
- Toghraie, F.S.; Yazdanpanah-Samani, M.; Mahmoudi Maymand, E.; Hosseini, A.; Asgari, A.; Ramezani, A.; Ghaderi, A. Molecular cloning, expression and purification of G-CSF isoform D, an alternative splice variant of human G-CSF. Iran. J. Allergy Asthma Immunol. 2019, 18, 419–426. [Google Scholar] [CrossRef]
- Gianoncelli, A.; Bertuzzi, M.; Guarienti, M.; Vezzoli, S.; Bonini, S.A.; Mastinu, A.; Sigala, S.; Memo, M. Parallelism of chemicostructural properties between filgrastim originator and three of its biosimilar drugs. J. Chem. 2019, 2019, 2751461. [Google Scholar] [CrossRef]
- Liongue, C.; Wright, C.; Russell, A.P.; Ward, A.C. Granulocyte colony-stimulating factor receptor: Stimulating granulopoiesis and much more. Int. J. Biochem. Cell Biol. 2009, 41, 2372–2375. [Google Scholar] [CrossRef]
- Dwivedi, P.; Greis, K.D. Granulocyte colony-stimulating factor receptor signaling in severe congenital neutropenia, chronic neutrophilic leukemia, and related malignancies. Exp. Hematol. 2017, 46, 9–20. [Google Scholar] [CrossRef]
- Geissler, K.; Gunzer, M.; Ostermann, H. How safe is the administration of long-acting granulocyte colony-stimulating factor in cancer patients? Oncol. Res. Treat. 2018, 41, 316–326. [Google Scholar] [CrossRef]
- Mehta, H.M.; Futami, M.; Glaubach, T.; Lee, D.W.; Andolina, J.R.; Yang, Q.; Whichard, Z.; Quinn, M.; Lu, H.F.; Kao, W.M.; et al. Alternatively spliced, truncated GCSF receptor promotes leukemogenic properties and sensitivity to JAK inhibition. Leukemia 2014, 28, 1041–1051. [Google Scholar] [CrossRef]
- Mielcarek, M.; Martin, P.J.; Torok-Storb, B. Suppression of alloantigen-induced T-cell proliferation by CD14+ cells derived from granulocyte colony-stimulating factor-mobilized peripheral blood mononuclear cells. Blood 1997, 89, 1629–1634. [Google Scholar] [CrossRef] [PubMed]
- Reyes, E.; García-Castro, I.; Esquivel, F.; Hornedo, J.; Cortes-Funes, H.; Solovera, J.; Alvarez-Mon, M. Granulocyte colony-stimulating factor (G-CSF) transiently suppresses mitogen-stimulated T-cell proliferative response. Br. J. Cancer 1999, 80, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Franzke, A. The role of G-CSF in adaptive immunity. Cytokine Growth Factor Rev. 2006, 17, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Rutella, S.; Rumi, C.; Lucia, M.B.; Sica, S.; Cauda, R.; Leone, G. Serum of healthy donors receiving granulocyte colony-stimulating factor induces T cell unresponsiveness. Exp. Hematol. 1998, 26, 1024–1033. [Google Scholar] [PubMed]
- Rutella, S.; Rumi, C.; Pierelli, L.; Morosetti, R.; Sica, S.; Bonanno, G.; Scambia, G.; Leone, G. Granulocyte colony-stimulating factor perturbs lymphocyte mitochondrial function and inhibits cell cycle progression. Exp. Hematol. 2000, 28, 612–625. [Google Scholar] [CrossRef]
- Nawa, Y.; Teshima, T.; Sunami, K.; Hiramatsu, Y.; Maeda, Y.; Yano, T.; Shinagawa, K.; Ishimaru, F.; Omoto, E.; Harada, M. G-CSF reduces IFN-γ and IL-4 production by T cells after allogeneic stimulation by indirectly modulating monocyte function. Bone Marrow Transplant. 2000, 25, 1035–1040. [Google Scholar] [CrossRef][Green Version]
- Sloand, E.M.; Kim, S.; Maciejewski, J.P.; Van Rhee, F.; Chaudhuri, A.; Barrett, J.; Young, N.S. Pharmacologic doses of granulocyte colony-stimulating factor affect cytokine production by lymphocytes in vitro and in vivo. Blood 2000, 95, 2269–2274. [Google Scholar] [CrossRef]
- Mifsud, N.; Ismail, A.; de Valle, E.; Spencer, A.; Patil, S.S.; Grigoriadis, G.; Gugasyan, R. The Effect of granulocyte-colony-stimulating factor (G-CSF) on T cell polarization in vitro: A direct comparison between Nivestim® and Neupogen®. Blood 2014, 124, 5803. [Google Scholar] [CrossRef]
- Malashchenko, V.V.; Meniailo, M.E.; Shmarov, V.A.; Gazatova, N.D.; Melashchenko, O.B.; Goncharov, A.G.; Seledtsova, G.V.; Seledtsov, V.I. Direct anti-inflammatory effects of granulocyte colony-stimulating factor (G-CSF) on activation and functional properties of human T cell subpopulations in vitro. Cell. Immunol. 2018, 325, 23–32. [Google Scholar] [CrossRef]
- Franzke, A.; Piao, W.; Lauber, J.; Gatzlaff, P.; Könecke, C.; Hansen, W.; Schmitt-Thomsen, A.; Hertenstein, B.; Buer, J.; Ganser, A. G-CSF as immune regulator in T cells expressing the G-CSF receptor: Implications for transplantation and autoimmune diseases. Blood 2003, 102, 734–739. [Google Scholar] [CrossRef]
- Zhu, J.; Yamane, H.; Cote-Sierra, J.; Guo, L.; Paul, W.E. GATA-3 promotes Th2 responses through three different mechanisms: Induction of Th2 cytokine production, selective growth of Th2 cells and inhibition of Th1 cell-specific factors. Cell Res. 2006, 16, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Yagi, R.; Zhu, J.; Paul, W.E. An updated view on transcription factor GATA3-mediated regulation of Th1 and Th2 cell differentiation. Int. Immunol. 2011, 23, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Toh, H.C.; Sun, L.; Soe, Y.; Wu, Y.; Phoon, Y.P.; Chia, W.K.; Wu, J.; Wong, K.Y.; Tan, P. G-CSF induces a potentially tolerant gene and immunophenotype profile in T cells in vivo. Clin. Immunol. 2009, 132, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Joo, Y.D.; Lee, W.S.; Won, H.J.; Lee, S.M.; Kim, H.R.; Park, J.K.; Park, S.G.; Choi, I.W.; Choi, I.; Seo, S.K. G-CSF-treated donor CD4+ T cells attenuate acute GVHD through a reduction in Th17 cell differentiation. Cytokine 2012, 60, 277–283. [Google Scholar] [CrossRef]
- Sun, L.X.; Ren, H.Y.; Shi, Y.J.; Wang, L.H.; Qiu, Z.X. Recombinant human granulocyte colony-stimulating factor significantly decreases the expression of CXCR3 and CCR6 on T cells and preferentially induces T helper cells to a T helper 17 phenotype in peripheral blood harvests. Biol. Blood Marrow Transplant. 2009, 15, 835–843. [Google Scholar] [CrossRef][Green Version]
- Hill, G.R.; Olver, S.D.; Kuns, R.D.; Varelias, A.; Raffelt, N.C.; Don, A.L.; Markey, K.A.; Wilson, Y.A.; Smyth, M.J.; Iwakura, Y.; et al. Stem cell mobilization with G-CSF induces type 17 differentiation and promotes scleroderma. Blood 2010, 116, 819–828. [Google Scholar] [CrossRef]
- Martins, A.; Han, J.; Kim, S.O. The multifaceted effects of granulocyte colony-stimulating factor in immunomodulation and potential roles in intestinal immune homeostasis. IUBMB Life 2010, 62, 611–617. [Google Scholar] [CrossRef]
- Rezvani, K.; Mielke, S.; Ahmadzadeh, M.; Kilical, Y.; Savani, B.N.; Zeilah, J.; Keyvanfar, K.; Montero, A.; Hensel, N.; Kurlander, R.; et al. High donor FOXP3-positive regulatory T-cell (Treg) content is associated with a low risk of GVHD following HLA-matched allogeneic SCT. Blood 2006, 108, 1291–1297. [Google Scholar] [CrossRef]
- Rutella, S.; Pierelli, L.; Bonanno, G.; Sica, S.; Ameglio, F.; Capoluongo, E.; Mariotti, A.; Scambia, G.; d’Onofrio, G.; Leone, G. Role for granulocyte colony-stimulating factor in the generation of human T regulatory type 1 cells. Blood 2002, 100, 2562–2571. [Google Scholar] [CrossRef]
- Rutella, S.; Zavala, F.; Danese, S.; Kared, H.; Leone, G. Granulocyte colony-stimulating factor: A novel mediator of T cell tolerance. J. Immunol. 2005, 175, 7085–7091. [Google Scholar] [CrossRef]
- Morris, E.S.; MacDonald, K.P.; Rowe, V.; Johnson, D.H.; Banovic, T.; Clouston, A.D.; Hill, G.R. Donor treatment with pegylated G-CSF augments the generation of IL-10-producing regulatory T cells and promotes transplantation tolerance. Blood 2004, 103, 3573–3581. [Google Scholar] [CrossRef]
- MacDonald, K.P.; Le Texier, L.; Zhang, P.; Morris, H.; Kuns, R.D.; Lineburg, K.E.; Leveque, L.; Don, A.L.; Markey, K.A.; Vuckovic, S.; et al. Modification of T cell responses by stem cell mobilization requires direct signaling of the T cell by G-CSF and IL-10. J. Immunol. 2014, 192, 3180–3189. [Google Scholar] [CrossRef]
- Zou, L.; Barnett, B.; Safah, H.; Larussa, V.F.; Evdemon-Hogan, M.; Mottram, P.; Wei, S.; David, O.; Curiel, T.J.; Zou, W. Bone marrow is a reservoir for CD4+CD25+ regulatory T cells that traffic through CXCL12/CXCR4 signals. Cancer Res. 2004, 64, 8451–8455. [Google Scholar] [CrossRef]
- Kared, H.; Masson, A.; Adle-Biassette, H.; Bach, J.-F.; Chatenoud, L.; Zavala, F. Treatment with granulocyte colony-stimulating factor prevents diabetes in NOD mice by recruiting plasmacytoid dendritic cells and functional CD4+CD25+ regulatory T-cells. Diabetes 2005, 54, 78–84. [Google Scholar] [CrossRef]
- Uchiyama, R.; Hasegawa, H.; Kameda, Y.; Ueda, K.; Kobayashi, Y.; Komuro, I.; Takano, H. Role of regulatory T cells in atheroprotective effects of granulocyte colony stimulating factor. J. Mol. Cell. Cardiol. 2012, 52, 1038–1047. [Google Scholar] [CrossRef]
- Yan, J.-J.; Jambaldorj, E.; Lee, J.-G.; Jang, J.Y.; Shim, J.M.; Han, M.; Koo, T.Y.; Ahn, C.; Yang, J. Granulocyte colony-stimulating factor treatment ameliorates lupus nephritis through the expansion of regulatory T cells. BMC Nephrol. 2016, 17, 175. [Google Scholar] [CrossRef]
- Bunse, C.E.; Tischer, S.; Lahrberg, J.; Oelke, M.; Figueiredo, C.; Blasczyk, R.; Eiz-Vesper, B. Granulocyte colony-stimulating factor impairs CD8+ T cell functionality by interfering with central activation elements. Clin. Exp. Immunol. 2016, 185, 107–118. [Google Scholar] [CrossRef]
- Arpinati, M.; Green, C.L.; Heimfeld, S.; Heuser, J.E.; Anasetti, C. Granulocyte-colony stimulating factor mobilizes T helper 2-inducing dendritic cells. Blood 2000, 95, 2484–2490. [Google Scholar] [CrossRef]
- Klangsinsirikul, P.; Russell, N.H. Peripheral blood stem cell harvests from G-CSF-stimulated donors contain a skewed Th2 CD4 phenotype and a predominance of type 2 dendritic cells. Exp. Hematol. 2002, 30, 495–501. [Google Scholar] [CrossRef]
- Reddy, V.; Hill, G.R.; Gerbitz, A.; Teshima, T.; Brinson, Y.; Ferrara, J.L. G-CSF modulates cytokine profile of dendritic cells and decreases acute graft-versus-host disease through effects on donor rather than the recipient. Transplantation 2000, 69, 691–693. [Google Scholar] [CrossRef]
- Rutella, S.; Bonanno, G.; Pierelli, L.; Mariotti, A.; Capoluongo, E.; Contemi, A.M.; Ameglio, F.; Curti, A.; De Ritis, D.G.; Voso, M.T.; et al. Granulocyte colony-stimulating factor promotes the generation of regulatory DC through induction of IL-10 and IFN-α. Eur. J. Immunol. 2004, 34, 1291–1302. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, M.; Gregori, S.; Roncarolo, M.G. Granulocyte-colony stimulating factor drives the in vitro differentiation of human dendritic cells that induce anergy in naïve T cells. Eur. J. Immunol. 2010, 40, 3097–3106. [Google Scholar] [CrossRef] [PubMed]
- Mielcarek, M.; Graf, L.; Johnson, G.; Torok-Storb, B. Production of interleukin-10 by granulocyte colony-stimulating factor-mobilized blood products: A mechanism for monocyte-mediated suppression of T-cell proliferation. Blood 1998, 92, 215–222. [Google Scholar] [CrossRef]
- Saito, M.; Kiyokawa, N.; Taguchi, T.; Suzuki, K.; Sekino, T.; Mimori, K.; Suzuki, T.; Nakajima, H.; Katagiri, Y.U.; Fujimura, J.; et al. Granulocyte colony-stimulating factor directly affects human monocytes and modulates cytokine secretion. Exp. Hematol. 2002, 30, 1115–1123. [Google Scholar] [CrossRef]
- Sunami, K.; Teshima, T.; Nawa, Y.; Hiramatsu, Y.; Maeda, Y.; Takenaka, K.; Shinagawa, K.; Ishimaru, F.; Ikeda, K.; Niiya, K.; et al. Administration of granulocyte colony-stimulating factor induces hyporesponsiveness to lipopolysaccharide and impairs antigen-presenting function of peripheral blood monocytes. Exp. Hematol. 2001, 29, 1117–1124. [Google Scholar] [CrossRef]
- D’Aveni, M.; Rossignol, J.; Coman, T.; Sivakumaran, S.; Henderson, S.; Manzo, T.; Santos e Sousa, P.; Bruneau, J.; Fouquet, G.; Zavala, F.; et al. G-CSF mobilizes CD34+ regulatory monocytes that inhibit graft-versus-host disease. Sci. Transl. Med. 2015, 7, 281ra42. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Lv, M.; Zhao, X.S.; Hu, Y.; Chang, Y.J.; Zhao, X.Y.; Kong, Y.; Zhang, X.H.; Xu, L.P.; Liu, K.Y.; Huang, X.J. Monocytic and promyelocytic myeloid-derived suppressor cells may contribute to G-CSF-induced immune tolerance in haplo-identical allogeneic hematopoietic stem cell transplantation. Am. J. Hematol. 2015, 90, E9–E16. [Google Scholar] [CrossRef]
- Luyckx, A.; Schouppe, E.; Rutgeerts, O.; Lenaerts, C.; Fevery, S.; Devos, T.; Dierickx, D.; Waer, M.; Van Ginderachter, J.A.; Billiau, A.D. G-CSF stem cell mobilization in human donors induces polymorphonuclear and mononuclear myeloid-derived suppressor cells. Clin. Immunol. 2012, 143, 83–87. [Google Scholar] [CrossRef]
- Wang, K.; Lv, M.; Chang, Y.J.; Zhao, X.Y.; Zhao, X.S.; Zhang, Y.Y.; Sun, Y.Q.; Wang, Z.D.; Suo, P.; Zhou, Y.; et al. Early myeloid-derived suppressor cells (HLA-DR-/lowCD33+CD16−) expanded by granulocyte colony-stimulating factor prevent acute graft-versus-host disease (GVHD) in humanized mouse and might contribute to lower GVHD in patients post allo-HSCT. J. Hematol. Oncol. 2019, 12, 31. [Google Scholar] [CrossRef]
- Mougiakakos, D.; Jitschin, R.; von Bahr, L.; Poschke, I.; Gary, R.; Sundberg, B.; Gerbitz, A.; Ljungman, P.; Le Blanc, K. Immunosuppressive CD14+HLA-DRlow/neg IDO+ myeloid cells in patients following allogeneic hematopoietic stem cell transplantation. Leukemia 2013, 27, 377–388. [Google Scholar] [CrossRef]
- Vendramin, A.; Gimondi, S.; Bermema, A.; Longoni, P.; Rizzitano, S.; Corradini, P.; Carniti, C. Graft monocytic myeloid-derived suppressor cell content predicts the risk of acute graft-versus-host disease after allogeneic transplantation of granulocyte colony-stimulating factor-mobilized peripheral blood stem cells. Biol. Blood Marrow Transplant. 2014, 20, 2049–2055. [Google Scholar] [CrossRef]
- Schlahsa, L.; Jaimes, Y.; Blasczyk, R.; Figueiredo, C. Granulocyte-colony-stimulatory factor: A strong inhibitor of natural killer cell function. Transfusion 2011, 51, 293–305. [Google Scholar] [CrossRef]
- Su, Y.C.; Li, S.C.; Hsu, C.K.; Yu, C.C.; Lin, T.J.; Lee, C.Y.; Liao, H.F. G-CSF downregulates natural killer cell-mediated cytotoxicity in donors for hematopoietic SCT. Bone Marrow Transplant. 2012, 47, 73–81. [Google Scholar] [CrossRef]
- Xiong, Y.; Mouginot, M.; Reppel, L.; Qian, C.; Stoltz, J.F.; Bensoussan, D.; Decot, V. Modification of NK cell subset repartition and functions in granulocyte colony-stimulating factor-mobilized leukapheresis after expansion with IL-15. Immunol. Res. 2017, 65, 1130–1138. [Google Scholar] [CrossRef]
- Yu, X.X.; Han, T.T.; Xu, L.L.; Chang, Y.J.; Huang, X.J.; Zhao, X.Y. Effect of the in vivo application of granulocyte colony-stimulating factor on NK cells in bone marrow and peripheral blood. J. Cell. Mol. Med. 2018, 22, 3025–3034. [Google Scholar] [CrossRef]
- Bakeer, M.S.; Zubair, A.; Roy, V. G-CSF causes decrease in peripheral blood platelet counts unrelated to leukapheresis during autologous stem cell mobilization. Cytotherapy 2019, 21 (Suppl. S59). [Google Scholar] [CrossRef]
- Li, Y.; Gou, R.; Wang, L.; Li, S.; Zhu, Z.; Tu, P. G-CSF administration results in thrombocytopenia by inhibiting the differentiation of hematopoetic progenitors into megakaryocytes. Biochem. Pharmacol. 2019, 169, 113624. [Google Scholar] [CrossRef]
- Campbell, R.A.; Schwertz, H.; Hottz, E.D.; Rowley, J.W.; Manne, B.K.; Washington, A.V.; Hunter-Mellado, R.; Tolley, N.D.; Christensen, M.; Eustes, A.S.; et al. Human megakaryocytes possess intrinsic antiviral immunity through regulated induction of IFITM3. Blood 2019, 133, 2013–2026. [Google Scholar] [CrossRef]
- Page, M.J.; Pretorius, E. A champion of host defense: A generic large-scale cause for platelet dysfunction and depletion in infection. Semin. Thromb. Hemost. 2020, 46, 302–319. [Google Scholar] [CrossRef]
- Almand, B.; Clark, J.I.; Nikitina, E.; van Beynen, J.; English, N.R.; Knight, S.C.; Carbone, D.P.; Gabrilovich, D.I. Increased production of immature myeloid cells in cancer patients: A mechanism of immunosuppression in cancer. J. Immunol. 2001, 166, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Granger, J.M.; Kontoyiannis, D.P. Etiology and outcome of extreme leukocytosis in 758 nonhematologic cancer patients: A retrospective, single-institution study. Cancer 2009, 115, 3919–3923. [Google Scholar] [CrossRef] [PubMed]
- Thomson, S.P.; Kessler, J.F.; Miller, T.P. Leukocyte concentrations in discrimination of benign from malignant lung lesions. Am. J. Med. 1986, 80, 1035–1040. [Google Scholar] [CrossRef]
- Kasuga, I.; Makino, S.; Kiyokawa, H.; Katoh, H.; Ebihara, Y.; Ohyashiki, K. Tumor-related leukocytosis is linked with poor prognosis in patients with lung carcinoma. Cancer 2001, 92, 2399–2405. [Google Scholar] [CrossRef]
- Connolly, G.C.; Khorana, A.A.; Kuderer, N.M.; Culakova, E.; Francis, C.W.; Lyman, G.H. Leukocytosis, thrombosis and early mortality in cancer patients initiating chemotherapy. Thromb. Res. 2010, 126, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Bahar, B.; Acedil Ayc Iota, B.; Çoşkun, U.; Büyükberber, S.; Benekli, M.; Yildiz, R. Granulocyte colony stimulating factor (G-CSF) and macrophage colony stimulating factor (M-CSF) as potential tumor markers in non small cell lung cancer diagnosis. Asian Pac. J. Cancer Prev. 2010, 11, 709–712. [Google Scholar]
- Wu, C.; Ning, H.; Liu, M.; Lin, J.; Luo, S.; Zhu, W.; Xu, J.; Wu, W.C.; Liang, J.; Shao, C.K.; et al. Spleen mediates a distinct hematopoietic progenitor response supporting tumor-promoting myelopoiesis. J. Clin. Investig. 2018, 128, 3425–3438. [Google Scholar] [CrossRef]
- Morris, K.T.; Khan, H.; Ahmad, A.; Weston, L.L.; Nofchissey, R.A.; Pinchuk, I.V.; Beswick, E.J. G-CSF and G-CSFR are highly expressed in human gastric and colon cancers and promote carcinoma cell proliferation and migration. Br. J. Cancer 2014, 110, 1211–1220. [Google Scholar] [CrossRef]
- Bordbar, E.; Malekzadeh, M.; Ardekani, M.T.; Doroudchi, M.; Ghaderi, A. Serum levels of G-CSF and IL-7 in Iranian breast cancer patients. Asian Pac. J. Cancer Prev. 2012, 13, 5307–5312. [Google Scholar] [CrossRef]
- Wojtukiewicz, M.Z.; Sierko, E.; Skalij, P.; Kamińska, M.; Zimnoch, L.; Brekken, R.A.; Thorpe, P.E. Granulocyte-colony stimulating factor receptor, tissue factor, and VEGF-R bound VEGF in human breast cancer in loco. Adv. Clin. Exp. Med. 2016, 25, 505–511. [Google Scholar] [CrossRef]
- Hollmén, M.; Karaman, S.; Schwager, S.; Lisibach, A.; Christiansen, A.J.; Maksimow, M.; Varga, Z.; Jalkanen, S.; Detmar, M. G-CSF regulates macrophage phenotype and associates with poor overall survival in human triple-negative breast cancer. Oncoimmunology 2015, 5, e1115177. [Google Scholar] [CrossRef]
- Guo, L.; Chen, G.; Zhang, W.; Zhou, L.; Xiao, T.; Di, X.; Wang, Y.; Feng, L.; Zhang, K. A high-risk luminal a dominant breast cancer subtype with increased mobility. Breast Cancer Res. Treat. 2019, 175, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Gutschalk, C.M.; Herold-Mende, C.C.; Fusenig, N.E.; Mueller, M.M. Granulocyte colony-stimulating factor and granulocyte-macrophage colony-stimulating factor promote malignant growth of cells from head and neck squamous cell carcinomas in vivo. Cancer Res. 2006, 66, 8026–8036. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Lakoma, A.; Chen, Z.; Hicks, J.; Metelitsa, L.S.; Kim, E.S.; Shohet, J.M. G-CSF promotes neuroblastoma tumorigenicity and metastasis via STAT3-dependent cancer stem cell activation. Cancer Res. 2015, 75, 2566–2579. [Google Scholar] [CrossRef] [PubMed]
- Paul, M.R.; Huo, Y.; Liu, A.; Lesperance, J.; Garancher, A.; Wechsler-Reya, R.J.; Zage, P.E. Characterization of G-CSF receptor expression in medulloblastoma. Neurooncol. Adv. 2020, 2, vdaa062. [Google Scholar] [CrossRef] [PubMed]
- Hirbe, A.C.; Uluçkan, O.; Morgan, E.A.; Eagleton, M.C.; Prior, J.L.; Piwnica-Worms, D.; Trinkaus, K.; Apicelli, A.; Weilbaecher, K. Granulocyte colony-stimulating factor enhances bone tumor growth in mice in an osteoclast-dependent manner. Blood 2007, 109, 3424–3431. [Google Scholar] [CrossRef]
- Morales-Arias, J.; Meyers, P.A.; Bolontrade, M.F.; Rodriguez, N.; Zhou, Z.; Reddy, K.; Chou, A.J.; Koshkina, N.V.; Kleinerman, E.S. Expression of granulocyte-colony-stimulating factor and its receptor in human Ewing sarcoma cells and patient tumor specimens: Potential consequences of granulocyte-colony-stimulating factor administration. Cancer 2007, 110, 1568–1577. [Google Scholar] [CrossRef]
- Fujisawa, T.; Tura-Ceide, O.; Hunter, A.; Mitchell, A.; Vesey, A.; Medine, C.; Gallogly, S.; Hadoke, P.W.F.; Keith, C.; Sproul, A.; et al. Endothelial progenitor cells do not originate from the bone marrow. Circulation 2019, 140, 1524–1526. [Google Scholar] [CrossRef]
- Tavakkoli, M.; Wilkins, C.R.; Mones, J.V.; Mauro, M.J. A novel paradigm between leukocytosis, G-CSF secretion, neutrophil-to-lymphocyte ratio, myeloid-derived suppressor cells, and prognosis in non-small cell lung cancer. Front. Oncol. 2019, 9, 295. [Google Scholar] [CrossRef]
- Najjar, Y.G.; Rayman, P.; Jia, X.; Pavicic, P.G., Jr.; Rini, B.I.; Tannenbaum, C.; Ko, J.; Haywood, S.; Cohen, P.; Hamilton, T.; et al. Myeloid-derived suppressor cell subset accumulation in renal cell carcinoma parenchyma is associated with intratumoral expression of IL1β, IL8, CXCL5, and Mip-1α. Clin. Cancer Res. 2017, 23, 2346–2355. [Google Scholar] [CrossRef]
- David, J.M.; Dominguez, C.; Hamilton, D.H.; Palena, C. The IL-8/IL-8R axis: A double agent in tumor immune resistance. Vaccines 2016, 4, 22. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.R.; Lee, W.; Cho, S.K.; Park, S.G. Characterization of multiple cytokine combinations and TGF-β on differentiation and functions of myeloid-derived suppressor cells. Int. J. Mol. Sci. 2018, 19, 869. [Google Scholar] [CrossRef] [PubMed]
- Pickup, M.; Novitskiy, S.; Moses, H.L. The roles of TGFβ in the tumour microenvironment. Nat. Rev. Cancer 2013, 13, 788–799. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Qiu, X.; Li, J.; Zheng, S.; Li, L.; Zhao, H. TGF-β secreted by tumor-associated macrophages promotes proliferation and invasion of colorectal cancer via miR-34a-VEGF axis. Cell Cycle 2018, 17, 2766–2778. [Google Scholar] [CrossRef]
- Calon, A.; Tauriello, D.V.; Batlle, E. TGF-beta in CAF-mediated tumor growth and metastasis. Semin. Cancer Biol. 2014, 25, 15–22. [Google Scholar] [CrossRef]
- Germann, M.; Zangger, N.; Sauvain, M.O.; Sempoux, C.; Bowler, A.D.; Wirapati, P.; Kandalaft, L.E.; Delorenzi, M.; Tejpar, S.; Coukos, G.; et al. Neutrophils suppress tumor-infiltrating T cells in colon cancer via matrix metalloproteinase-mediated activation of TGFβ. EMBO Mol. Med. 2020, 12, e10681. [Google Scholar] [CrossRef]
- Rapoport, B.L.; Steel, H.C.; Theron, A.J.; Smit, T.; Anderson, R. Role of the neutrophil in the pathogenesis of advanced cancer and impaired responsiveness to therapy. Molecules 2020, 25, 1618. [Google Scholar] [CrossRef]
- Rodriguez, P.; Ochoa, A.C.; Al-Khami, A.A. Arginine metabolism in myeloid cells shapes innate and adaptive immunity. Front. Immunol. 2017, 8, 93. [Google Scholar] [CrossRef]
- Yu, J.; Du, W.; Yan, F.; Wang, Y.; Li, H.; Cao, S.; Yu, W.; Shen, C.; Liu, J.; Ren, X. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J. Immunol. 2013, 190, 3783–3797. [Google Scholar] [CrossRef]
- Huang, B.; Pan, P.Y.; Li, Q.; Sato, A.I.; Levy, D.E.; Bromberg, J.; Divino, C.M.; Chen, S.H. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006, 66, 1123–1131. [Google Scholar] [CrossRef]
- Nagaraj, S.; Gupta, K.; Pisarev, V.; Kinarsky, L.; Sherman, S.; Kang, L.; Herber, D.L.; Schneck, J.; Gabrilovich, D.I. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat. Med. 2007, 13, 828–835. [Google Scholar] [CrossRef] [PubMed]
- Kargl, J.; Zhu, X.; Zhang, H.; Yang, G.H.Y.; Friesen, T.J.; Shipley, M.; Maeda, D.Y.; Zebala, J.A.; McKay-Fleisch, J.; Meredith, G.; et al. Neutrophil content predicts lymphocyte depletion and anti-PD1 treatment failure in NSCLC. JCI Insight 2019, 4, e130850. [Google Scholar] [CrossRef] [PubMed]
- Cedervall, J.; Hamidi, A.; Olsson, A.K. Platelets, NETs and cancer. Thromb. Res. 2018, 164, S148–S152. [Google Scholar] [CrossRef] [PubMed]
- Shojaei, F.; Wu, X.; Qu, X.; Kowanetz, M.; Yu, L.; Tan, M.; Meng, Y.G.; Ferrara, N. G-CSF-initiated myeloid cell mobilization and angiogenesis mediate tumor refractoriness to anti-VEGF therapy in mouse models. Proc. Natl. Acad. Sci. USA 2009, 106, 6742–6747. [Google Scholar] [CrossRef] [PubMed]
- Shojaei, F.; Singh, M.; Thompson, J.D.; Ferrara, N. Role of Bv8 in neutrophil-dependent angiogenesis in a transgenic model of cancer progression. Proc. Natl. Acad. Sci. USA 2008, 105, 2640–2645. [Google Scholar] [CrossRef] [PubMed]
- Condamine, T.; Ramachandran, I.; Youn, J.I.; Gabrilovich, D.I. Regulation of tumor metastasis by myeloid-derived suppressor cells. Annu. Rev. Med. 2015, 66, 97–110. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Fenselau, C. Myeloid-derived suppressor cells: Immune-suppressive cells that impair antitumor immunity and are sculpted by their environment. J. Immunol. 2018, 200, 422–431. [Google Scholar] [CrossRef]
- Mizuno, R.; Kawada, K.; Itatani, Y.; Ogawa, R.; Kiyasu, Y.; Sakai, Y. The role of tumor-associated neutrophils in colorectal cancer. Int. J. Mol. Sci. 2019, 20, 529. [Google Scholar] [CrossRef]
- Mouchmore, K.A.; Anderson, R.L.; Hamilton, J.A. Neutrophils, G-CSF and their contribution to breast cancer metastasis. FEBS J. 2018, 285, 665–679. [Google Scholar] [CrossRef]
- Park, S.; Kim, E.S.; Noh, D.Y.; Hwang, K.T.; Moon, A. H-Ras-specific upregulation of granulocyte colony-stimulating factor promotes human breast cell invasion via matrix metalloproteinase-2. Cytokine 2011, 55, 126–133. [Google Scholar] [CrossRef]
- Demers, M.; Wagner, D.D. Neutrophil extracellular traps: A new link to cancer-associated thrombosis and potential implications for tumor progression. Oncoimmunology 2013, 2, e22946. [Google Scholar] [CrossRef] [PubMed]
- Metzler, K.D.; Goosmann, C.; Lubojemska, A.; Zychlinsky, A.; Papayannopoulos, V. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Rep. 2014, 8, 883–896. [Google Scholar] [CrossRef] [PubMed]
- Metzler, K.D.; Fuchs, T.A.; Nauseef, W.M.; Reumaux, D.; Roesler, J.; Schulze, I.; Wahn, V.; Papayannopoulos, V.; Zychlinsky, A. Myeloperoxidase is required for neutrophil extracellular trap formation: Implications for innate immunity. Blood 2011, 117, 953–959. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef]
- Rohrbach, A.S.; Slade, D.J.; Thompson, P.R.; Mowen, K.A. Activation of PAD4 in NET formation. Front. Immunol. 2012, 3, 360. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Kaplan, M.J.; Radic, M. Neutrophil extracellular traps: Double-edged swords of innate immunity. J. Immunol. 2012, 189, 2689–2695. [Google Scholar] [CrossRef]
- Berger-Achituv, S.; Brinkmann, V.; Abed, U.A.; Kühn, L.I.; Ben-Ezra, J.; Elhasid, R.; Zychlinsky, A. A proposed role for neutrophil extracellular traps in cancer immunoediting. Front. Immunol. 2013, 4, 48. [Google Scholar] [CrossRef]
- Boone, B.A.; Orlichenko, L.; Schapiro, N.E.; Loughran, P.; Gianfrate, G.C.; Ellis, J.T.; Singhi, A.D.; Kang, R.; Tang, D.; Lotze, M.T.; et al. The receptor for advanced glycation end products (RAGE) enhances autophagy and neutrophil extracellular traps in pancreatic cancer. Cancer Gene Ther. 2015, 22, 326–334. [Google Scholar] [CrossRef]
- Park, J.; Wysocki, R.W.; Amoozgar, Z.; Maiorino, L.; Fein, M.R.; Jorns, J.; Schott, A.F.; Kinugasa-Katayama, Y.; Lee, Y.; Won, N.H.; et al. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci. Transl. Med. 2016, 8, 361ra138. [Google Scholar] [CrossRef]
- Lee, W.; Ko, S.Y.; Mohamed, M.S.; Kenny, H.A.; Lengyel, E.; Naora, H. Neutrophils facilitate ovarian cancer premetastatic niche formation in the omentum. J. Exp. Med. 2019, 216, 176–194. [Google Scholar] [CrossRef] [PubMed]
- Garley, M.; Jabłońska, E.; Dąbrowska, D. NETs in cancer. Tumour Biol. 2016, 37, 14355–14361. [Google Scholar] [CrossRef] [PubMed]
- Cools-Lartigue, J.; Spicer, J.; McDonald, B.; Gowing, S.; Chow, S.; Giannias, B.; Bourdeau, F.; Kubes, P.; Ferri, L. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J. Clin. Investig. 2013, 123, 3446–3458. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.S.; Gu, J.; Kim, J.E.; Nam, Y.; Song, J.W.; Kim, H.K. Cancer cell-induced neutrophil extracellular traps promote both hypercoagulability and cancer progression. PLoS ONE 2019, 14, e0216055. [Google Scholar] [CrossRef]
- Smith, T.J.; Bohlke, K.; Armitage, J.O. Recommendations for the use of white blood cell growth factors: American Society of Clinical Oncology Clinical Practice Guideline Update. J. Oncol. Pract. 2015, 11, 511–513. [Google Scholar] [CrossRef]
- Klastersky, J.; de Naurois, J.; Rolston, K.; Rapoport, B.; Maschmeyer, G.; Aapro, M.; Herrstedt, J. ESMO Guidelines Committee. Management of febrile neutropaenia: ESMO Clinical Practice Guidelines. Ann. Oncol. 2016, 27, v111–v118. [Google Scholar] [CrossRef]
- NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) Hematopoietic Growth Factors Version 2.2020—January 27 2020. Available online: https://www.nccn.org/professionals/physician_gls/pdf/growthfactors.pdf (accessed on 6 September 2020).
- Crawford, J.; Dale, D.C.; Kuderer, N.M.; Culakova, E.; Poniewierski, M.S.; Wolff, D.; Lyman, G.H. Risk and timing of neutropenic events in adult cancer patients receiving chemotherapy: The results of a prospective nationwide study of oncology practice. J. Natl. Compr. Cancer Netw. 2008, 6, 109–118. [Google Scholar] [CrossRef]
- Lambertini, M.; Ferreira, A.R.; Del Mastro, L.; Danesi, R.; Pronzato, P. Pegfilgrastim for the prevention of chemotherapy-induced febrile neutropenia in patients with solid tumors. Expert Opin. Biol. Ther. 2015, 15, 1799–1817. [Google Scholar] [CrossRef]
- Cornes, P.; Gascon, P.; Chan, S.; Hameed, K.; Mitchell, C.R.; Field, P.; Latymer, M.; Arantes, L.H., Jr. Systematic review and meta-analysis of short-versus long-acting granulocyte colony-stimulating factors for reduction of chemotherapy-induced febrile neutropenia. Adv. Ther. 2018, 35, 1816–1829. [Google Scholar] [CrossRef]
- Schulz, M.; Bonig, H. Update on biosimilars of granulocyte colony-stimulating factor-when no news is good news. Curr. Opin. Hematol. 2016, 23, 61–66. [Google Scholar] [CrossRef]
- Cornes, P.; Gascon, P.; Vulto, A.G.; Aapro, M. Biosimilar pegfilgrastim: Improving access and optimising practice to supportive care that enables cure. BioDrugs 2020, 34, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Mansell, K.; Bhimji, H.; Eurich, D.; Mansell, H. Potential cost-savings from the use of the biosimilars filgrastim, infliximab and insulin glargine in Canada: A retrospective analysis. BMC Health Serv. Res. 2019, 19, 827. [Google Scholar] [CrossRef]
- Andrès, E.; Villalba, N.L.; Zulfiqar, A.A.; Serraj, K.; Mourot-Cottet, R.; Gottenberg, A.J. State of art of idiosyncratic drug-induced neutropenia or agranulocytosis, with a focus on biotherapies. J. Clin. Med. 2019, 8, 1351. [Google Scholar] [CrossRef] [PubMed]
- Sokolic, R. Neutropenia in primary immunodeficiency. Curr. Opin. Hematol. 2013, 20, 55–65. [Google Scholar] [CrossRef]
- Skokowa, J.; Dale, D.C.; Touw, I.P.; Zeidler, C.; Welte, K. Severe congenital neutropenias. Nat. Rev. Dis. Primers 2017, 3, 17032. [Google Scholar] [CrossRef]
- Radsak, M.; Platzbecker, U.; Schmidt, C.S.; Hofmann, W.K.; Nolte, F. Infectious complications in patients with myelodysplastic syndromes: A review of the literature with emphasis on patients treated with 5-azacitidine. Eur. J. Haematol. 2017, 99, 112–118. [Google Scholar] [CrossRef]
- Dainiak, N.; Gent, R.N.; Carr, Z.; Schneider, R.; Bader, J.; Buglova, E.; Chao, N.; Coleman, C.N.; Ganser, A.; Gorin, C.; et al. First global consensus for evidence-based management of the hematopoietic syndrome resulting from exposure to ionizing radiation. Disaster Med. Public Health Prep. 2011, 5, 202–212. [Google Scholar] [CrossRef]
- Hofer, M.; Hoferová, Z.; Falk, M. Pharmacological modulation of radiation damage. Does it exist a chance for other substances than hematopoietic growth factors and cytokines? Int. J. Mol. Sci. 2017, 18, 1385. [Google Scholar] [CrossRef]
- Shaw, B.E.; Confer, D.L.; Hwang, W.; Pulsipher, M.A. A review of the genetic and long-term effects of G-CSF injections in healthy donors: A reassuring lack of evidence for the development of Haematological malignancies. Bone Marrow Transplant. 2015, 50, 334–340. [Google Scholar] [CrossRef]
- Yeo, B.; Redfern, A.D.; Mouchmore, K.A.; Hamilton, J.A.; Anderson, R.L. The dark side of granulocyte-colony stimulating factor: A supportive therapy with potential to promote tumour progression. Clin. Exp. Metastasis 2018, 35, 255–267. [Google Scholar] [CrossRef]
- Pilatova, K.; Bencsikova, B.; Demlova, R.; Valik, D.; Zdrazilova-Dubska, L. Myeloid-derived suppressor cells (MDSCs) in patients with solid tumors: Considerations for granulocyte colony-stimulating factor treatment. Cancer Immunol. Immunother. 2018, 67, 1919–1929. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Ma, M.; Tan, Z.; Zheng, H.; Liu, X. Neutrophil: A new player in metastatic cancers. Front. Immunol. 2020, 11, 565165. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).