Novel 1-Amidino-4-Phenylpiperazines as Potent Agonists at Human TAAR1 Receptor: Rational Design, Synthesis, Biological Evaluation and Molecular Docking Studies

 ,
, 
 ,
, 

Abstract
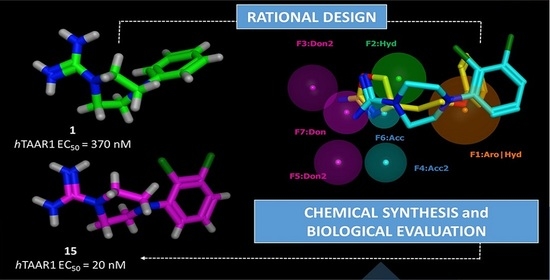
:
1. Introduction
2. Results and Discussion
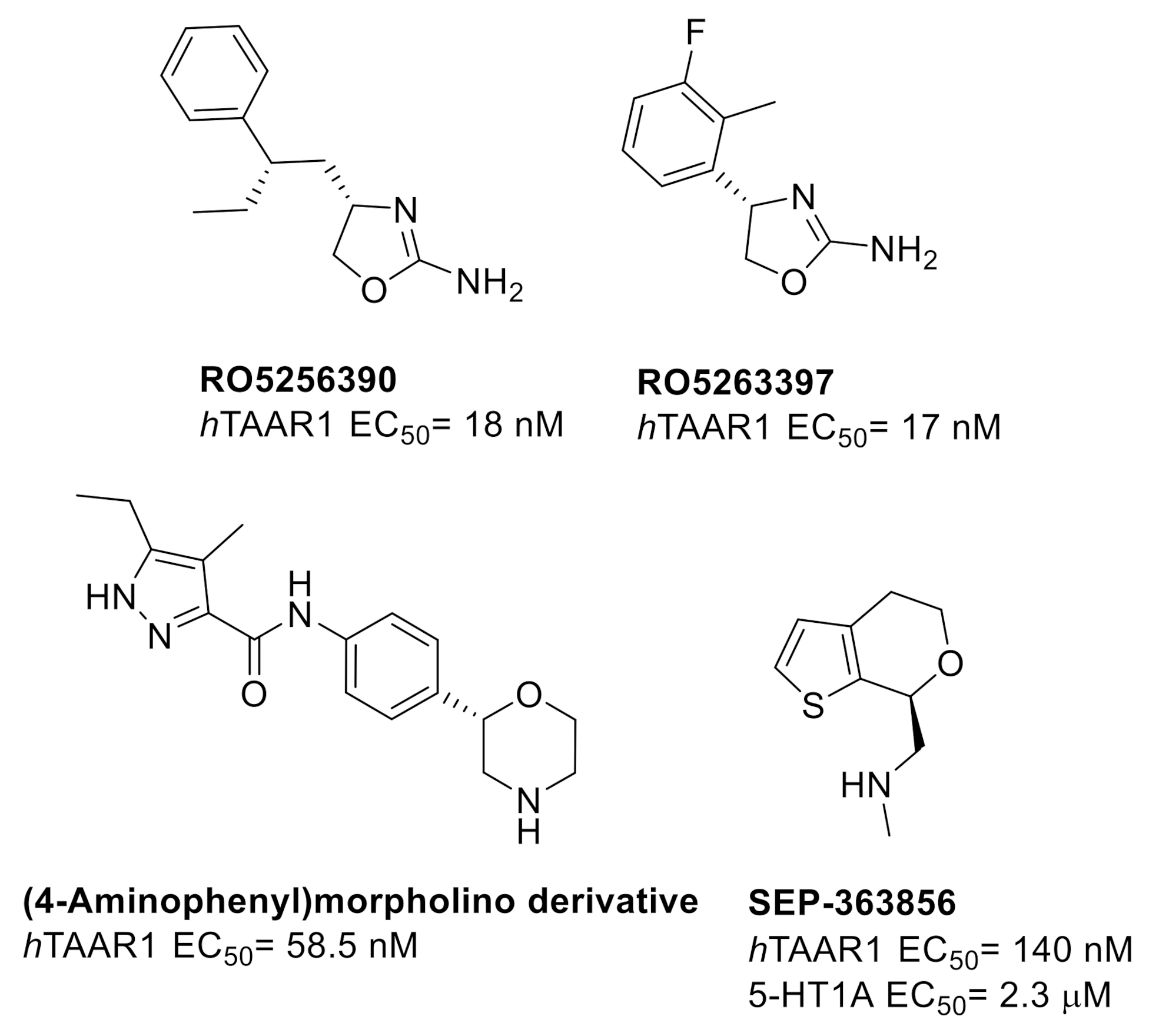
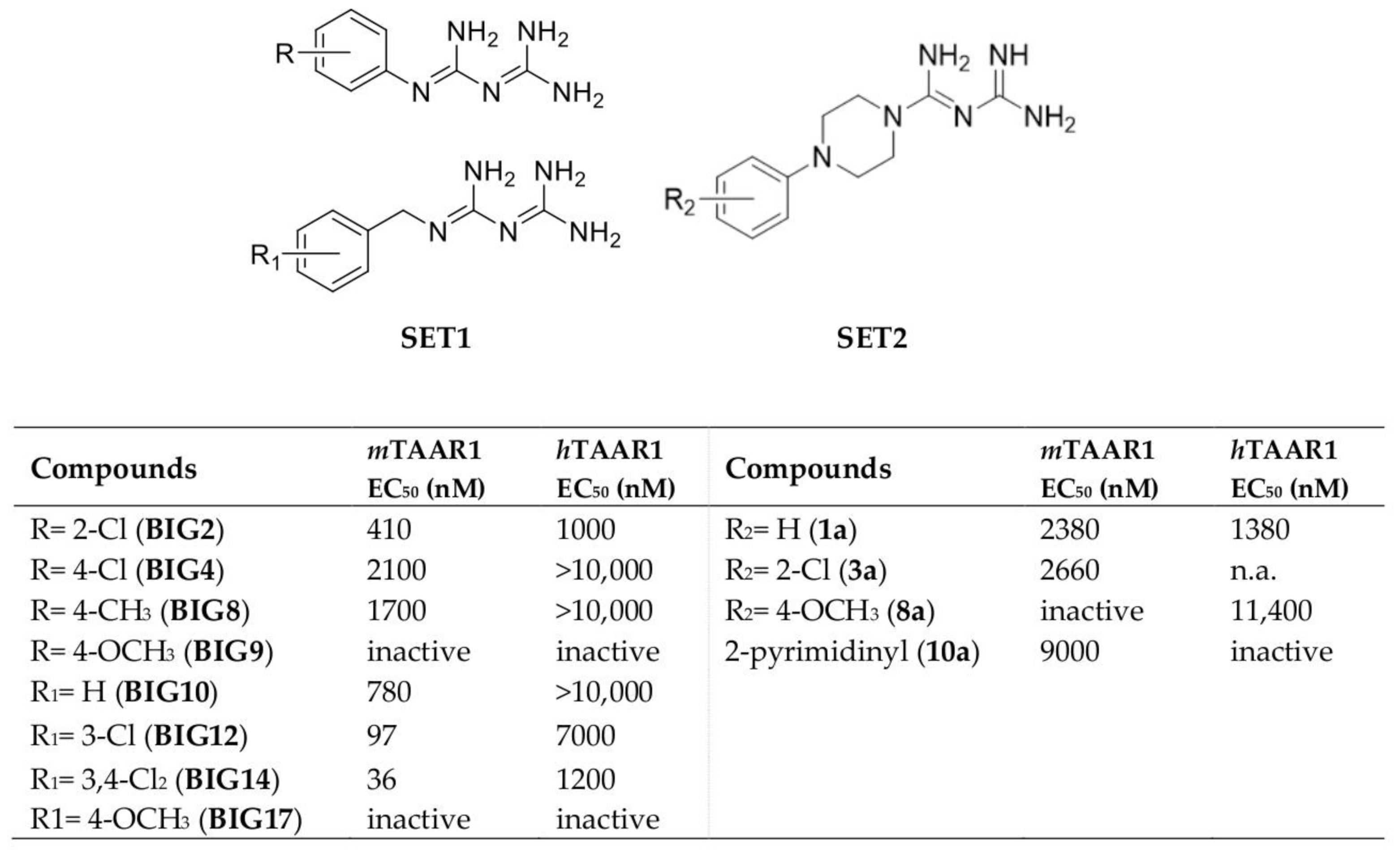
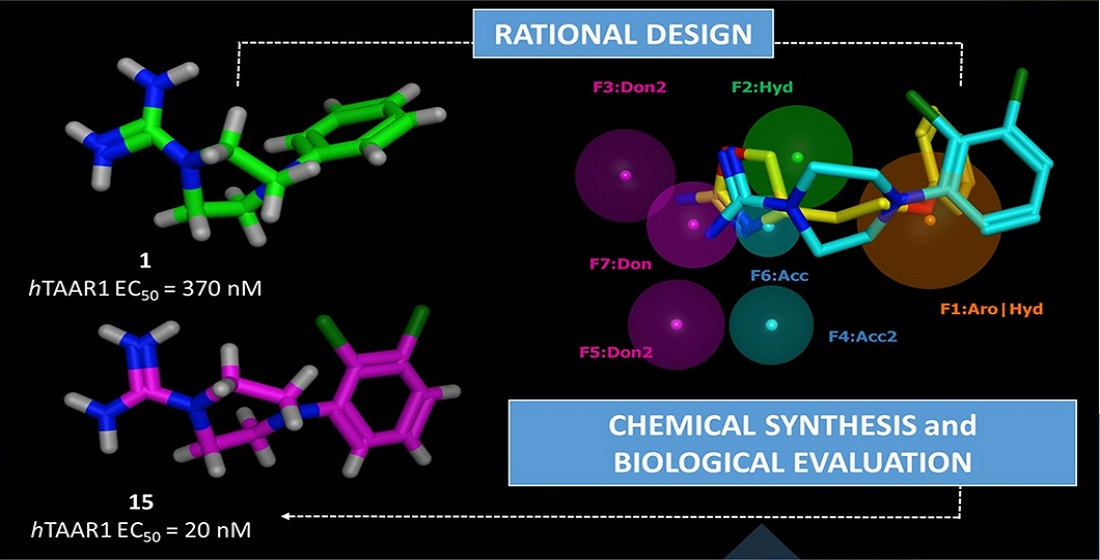
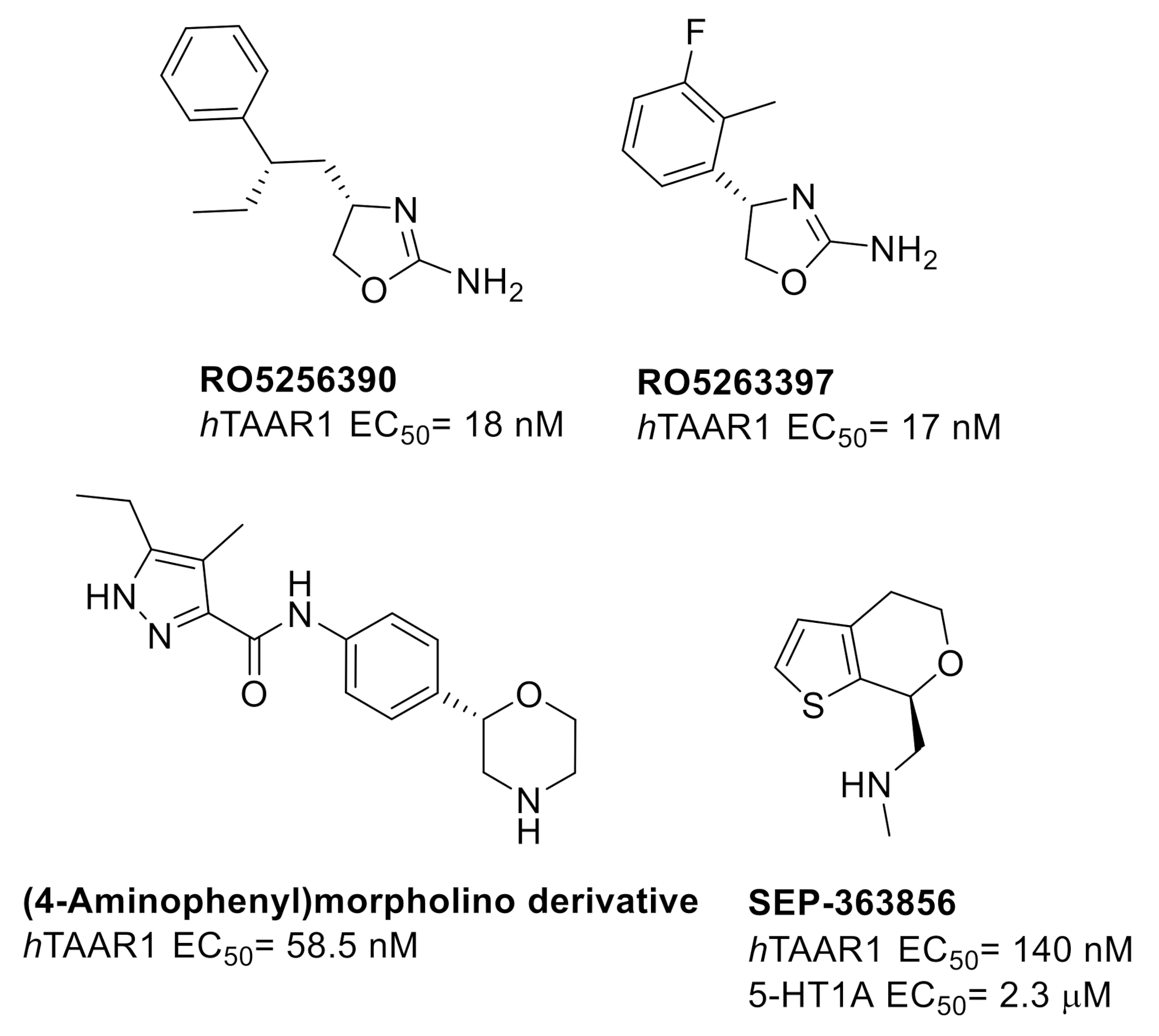
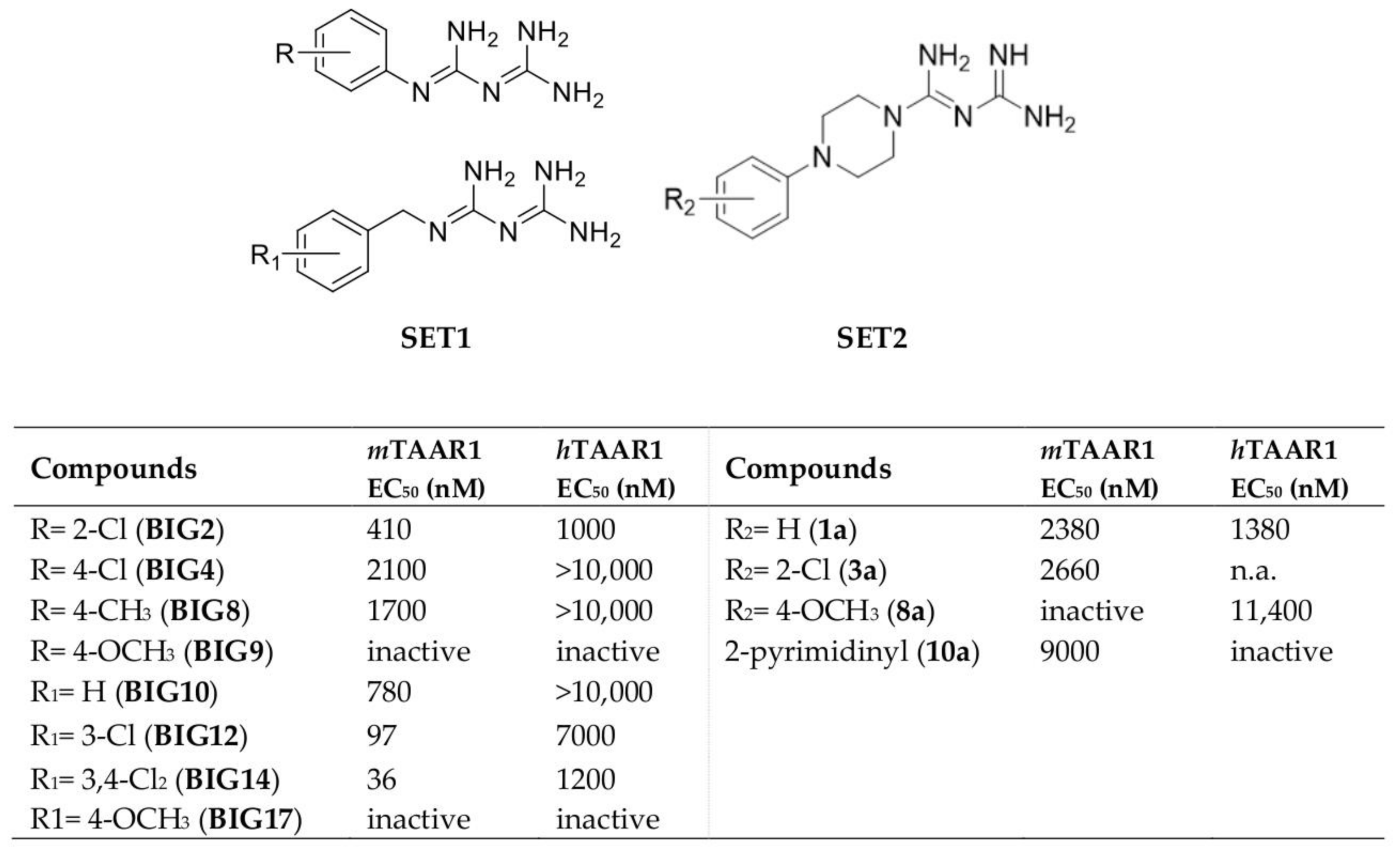
2.1. Design of hTAAR1 Agonists
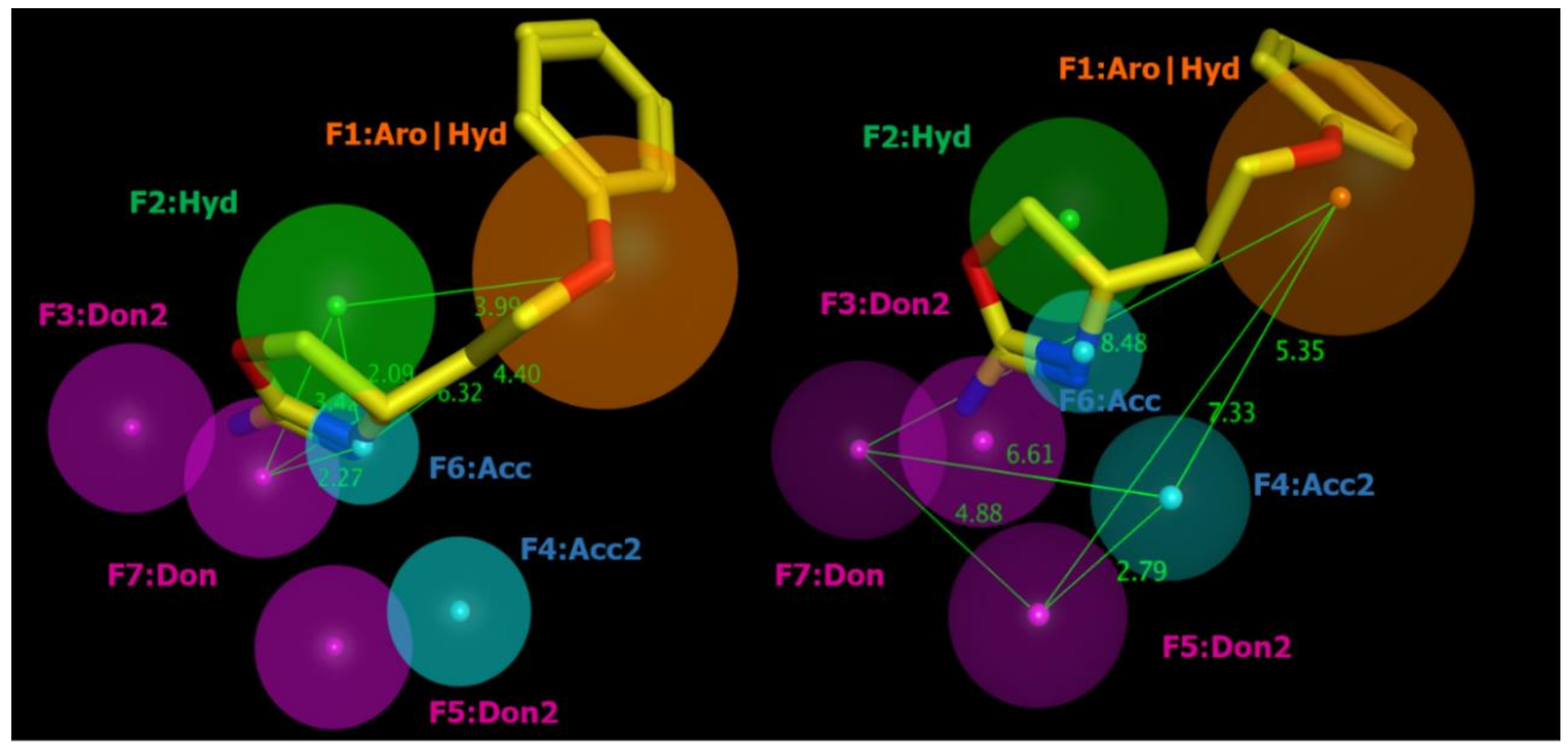
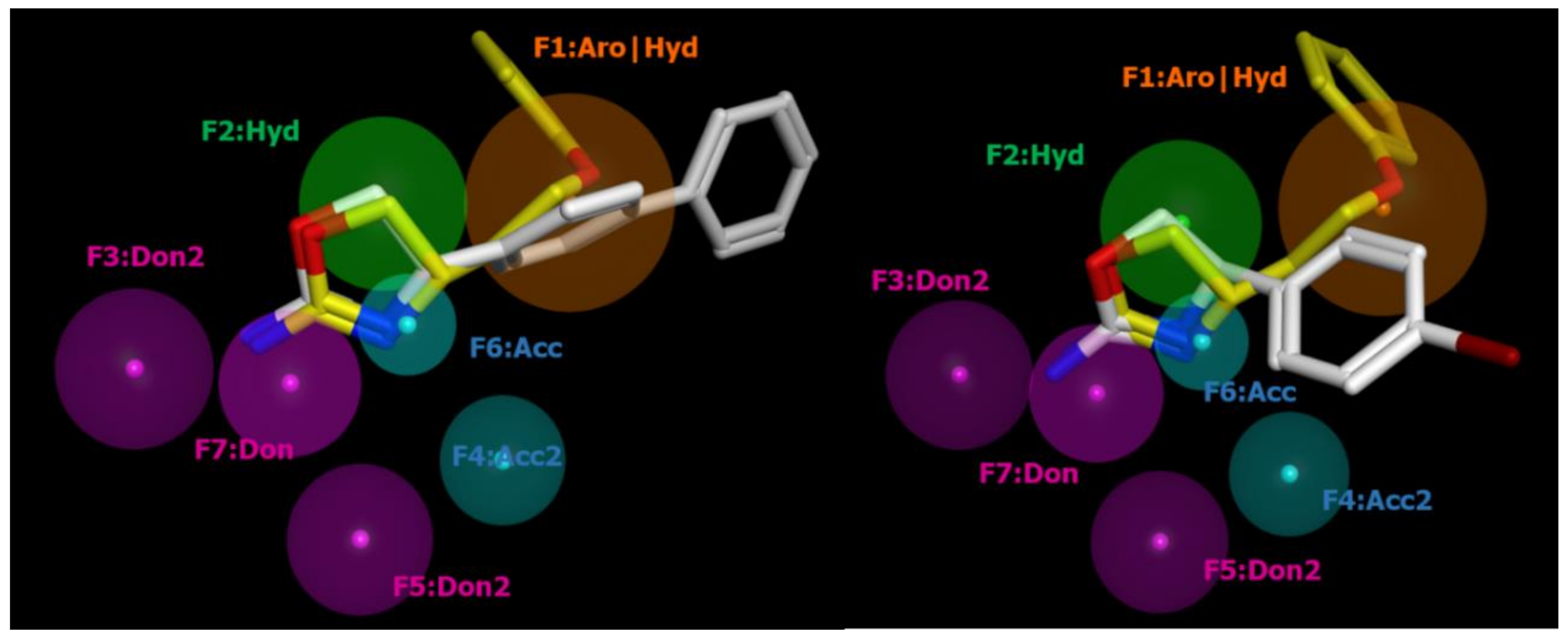
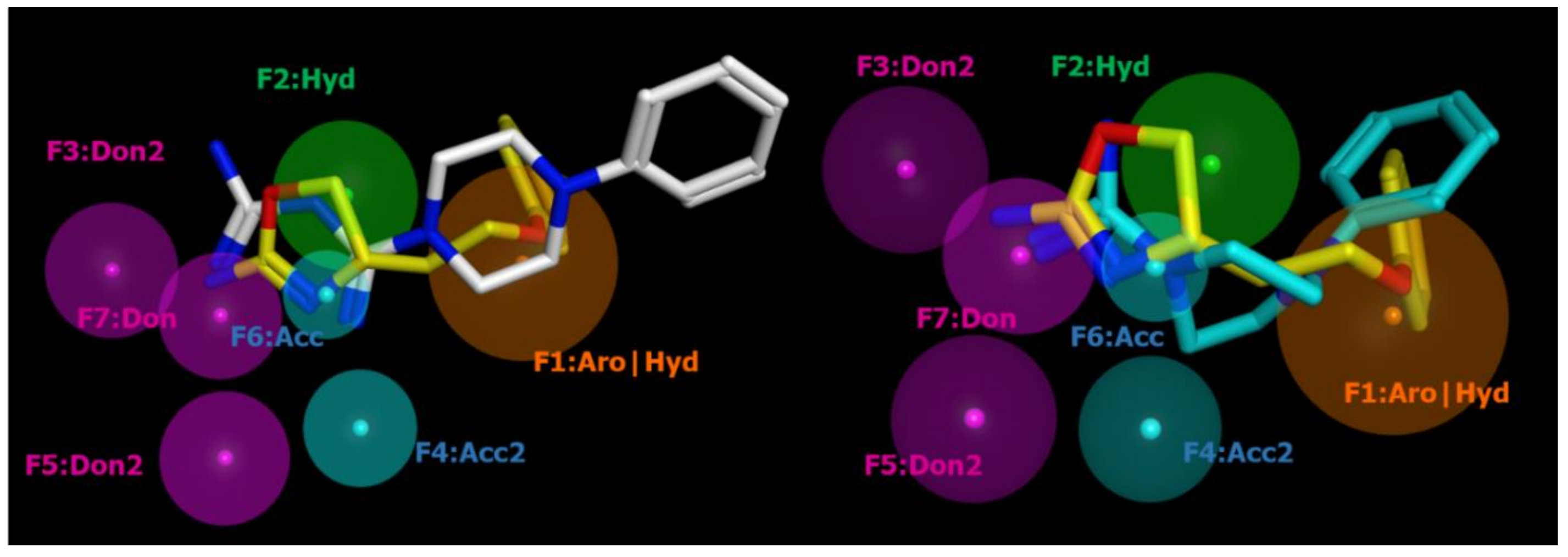
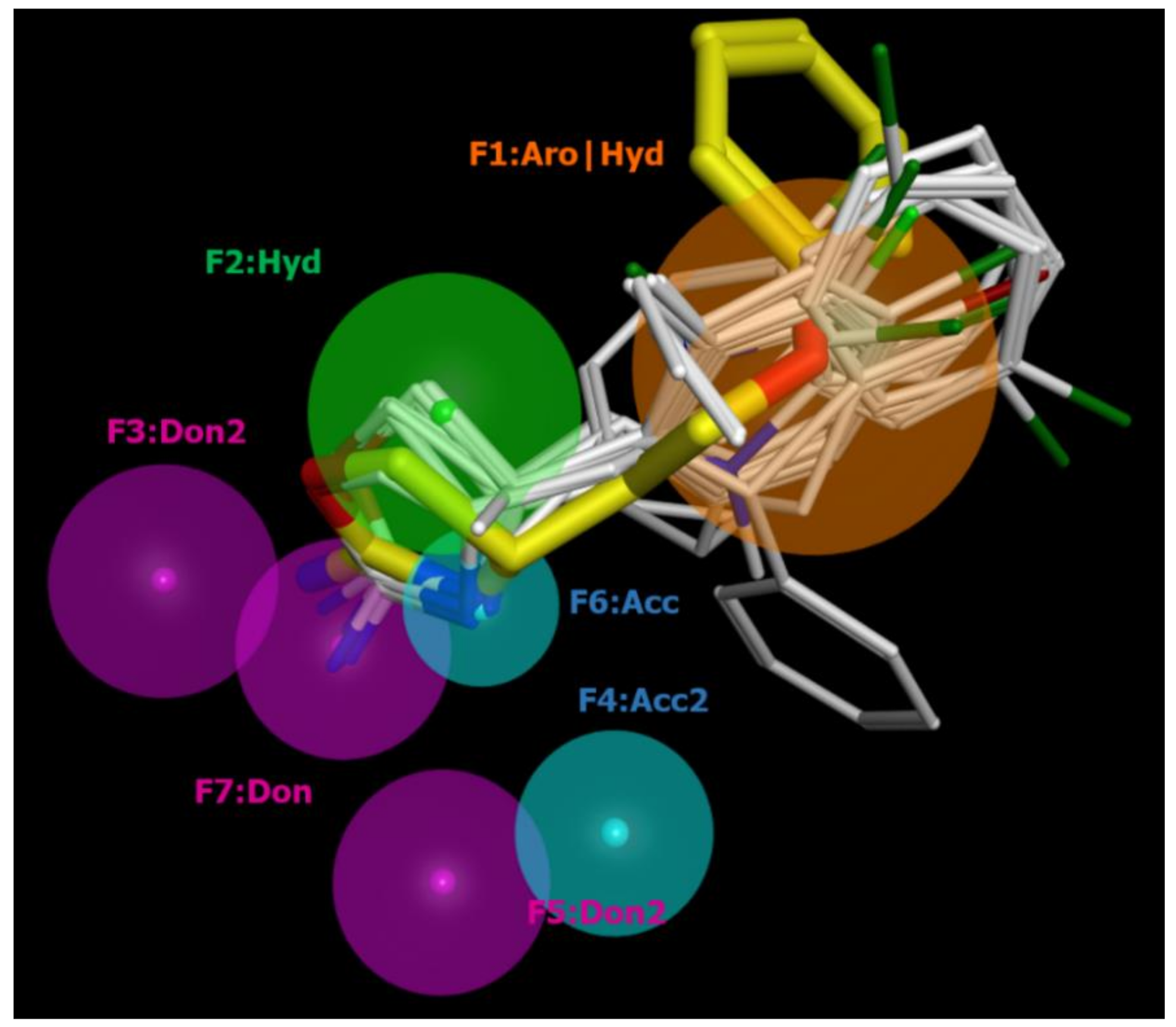
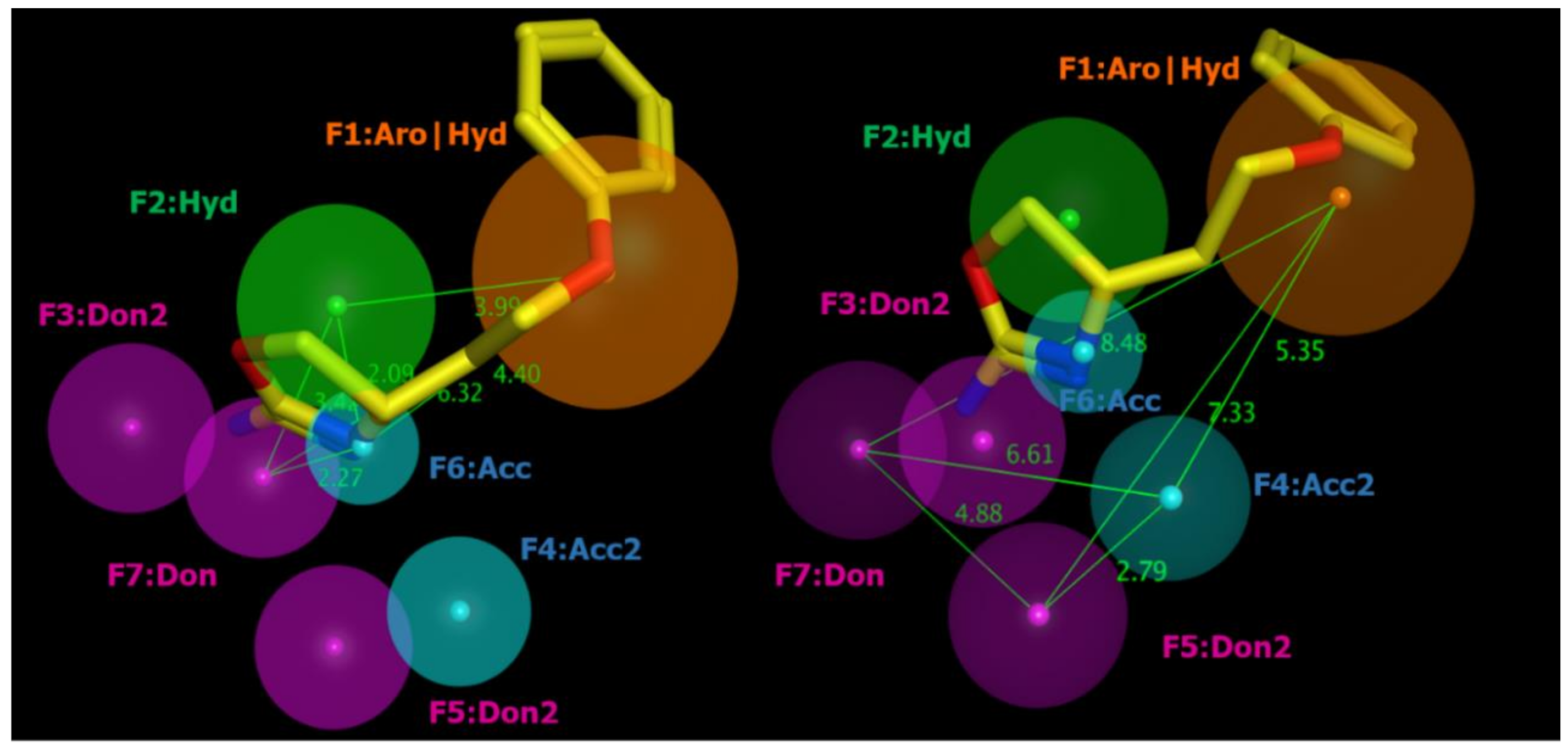
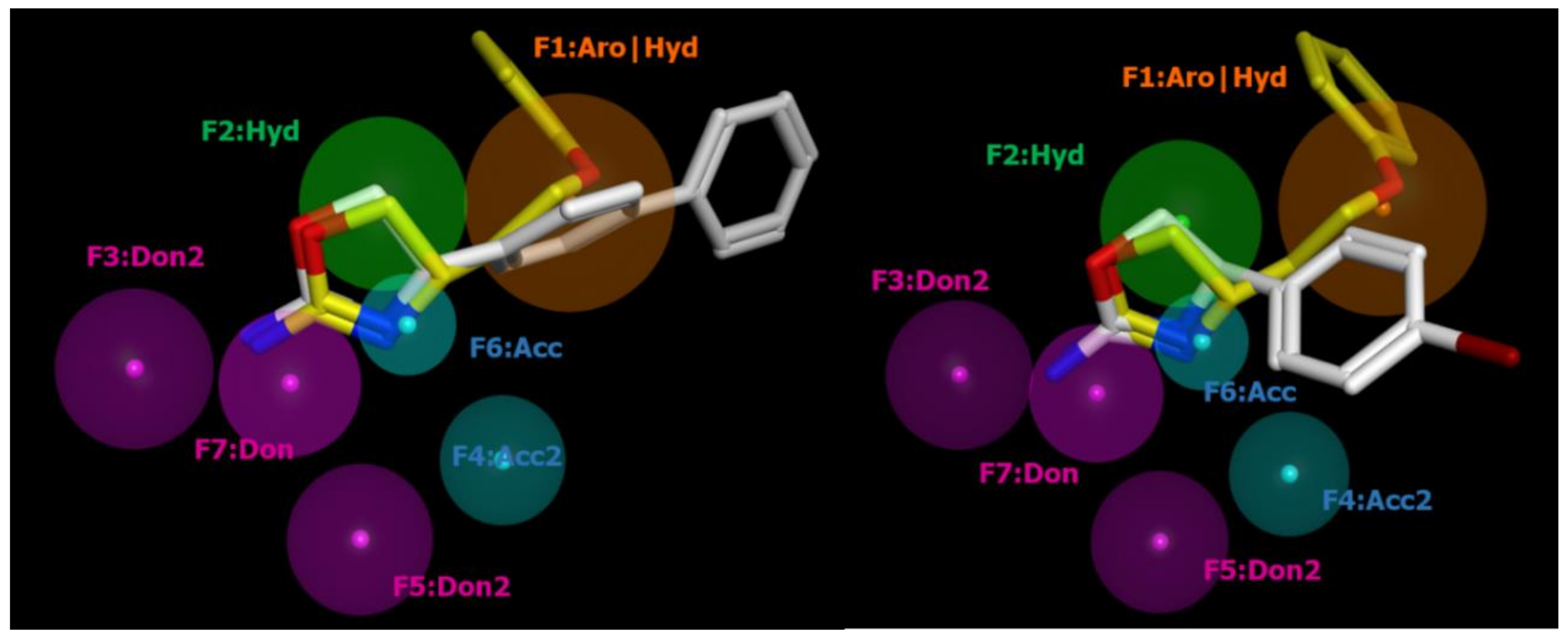
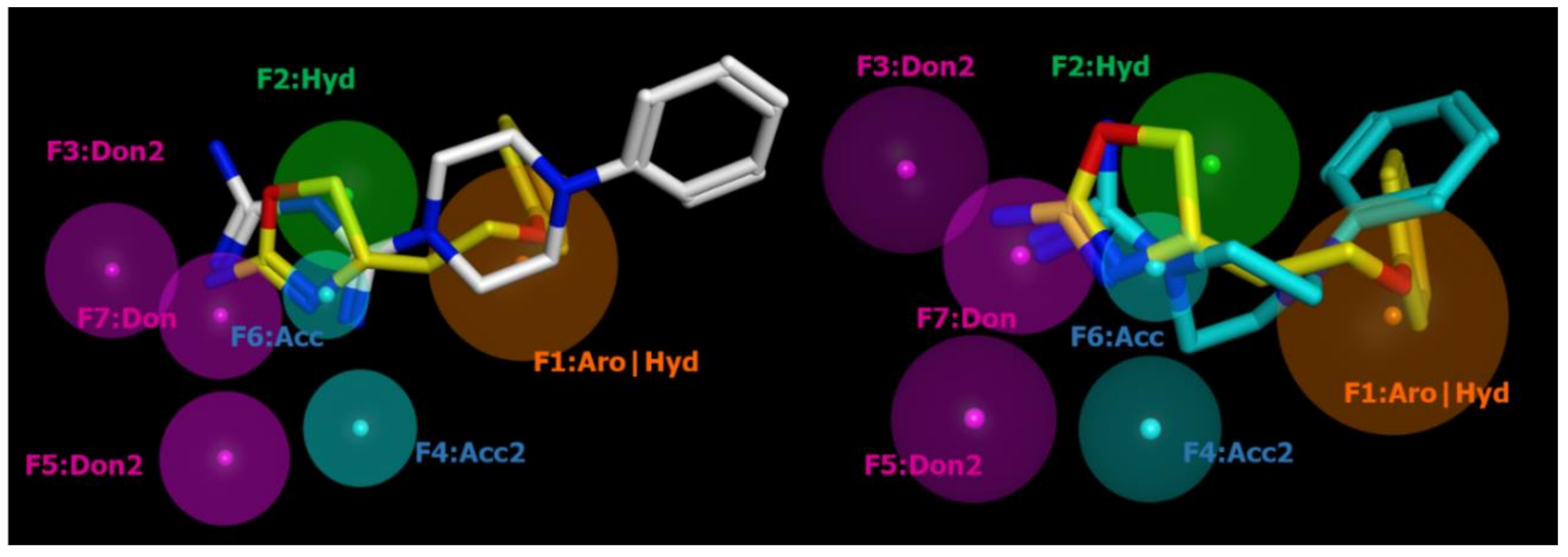
2.2. Pharmacophore Modeling
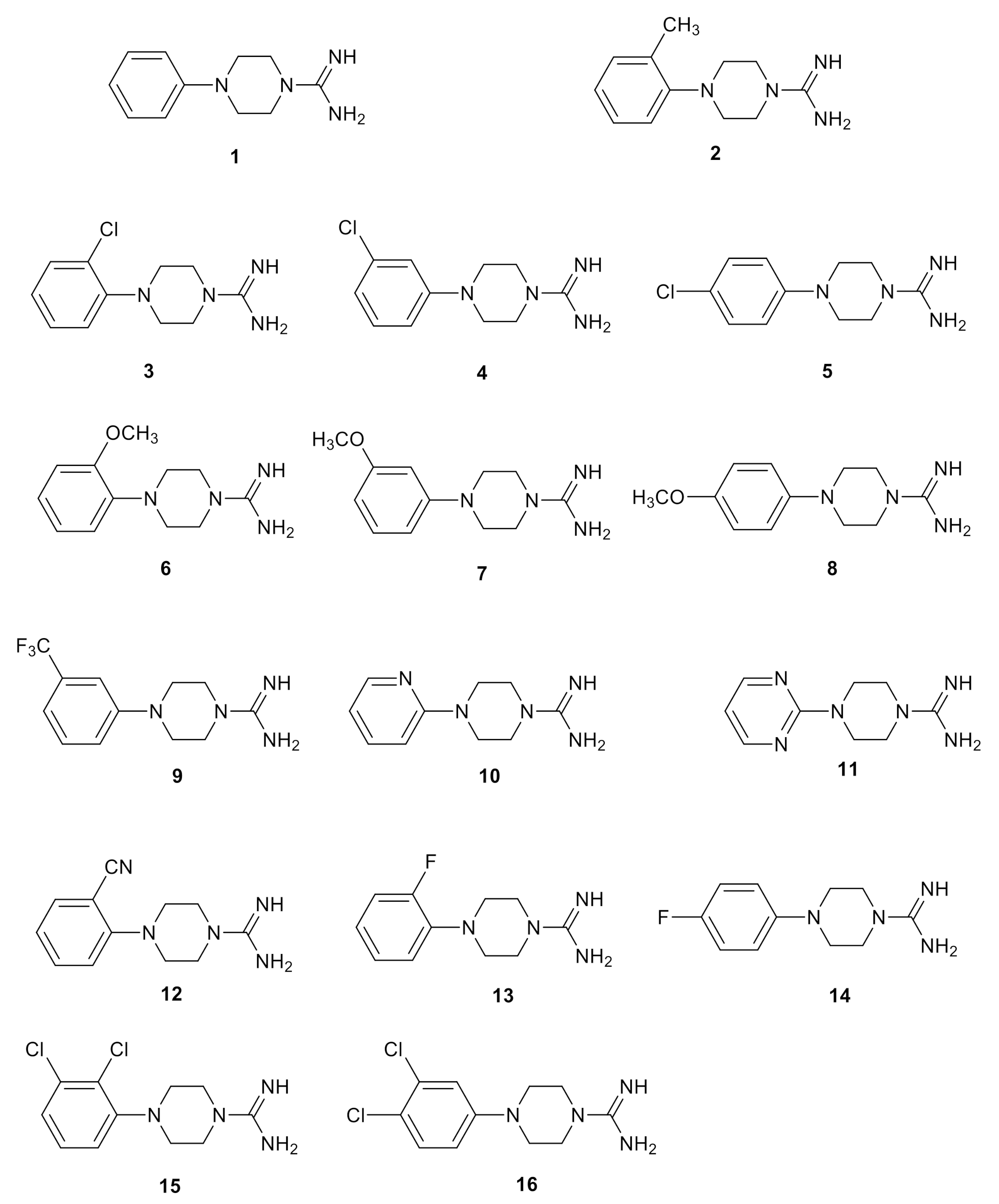
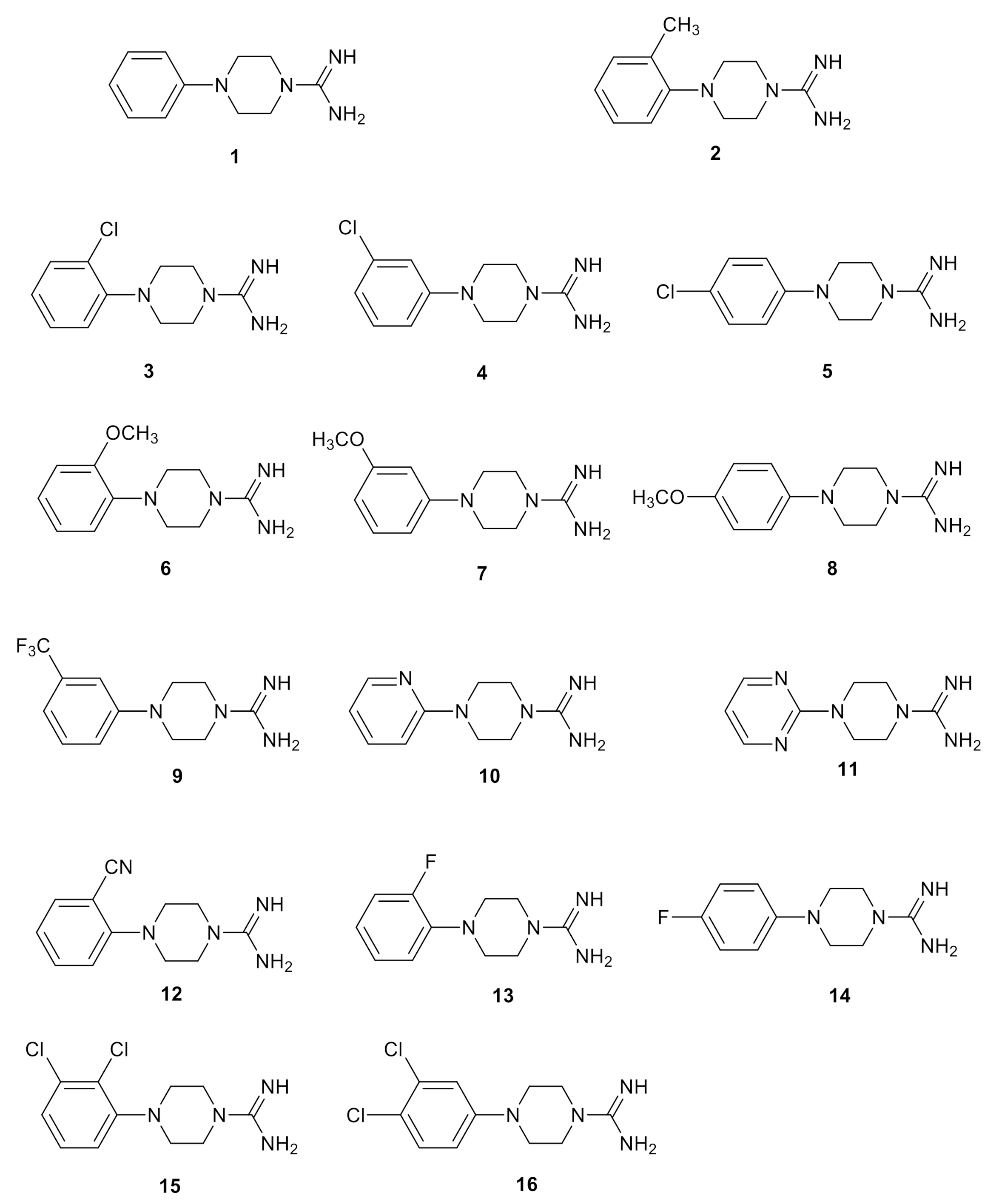
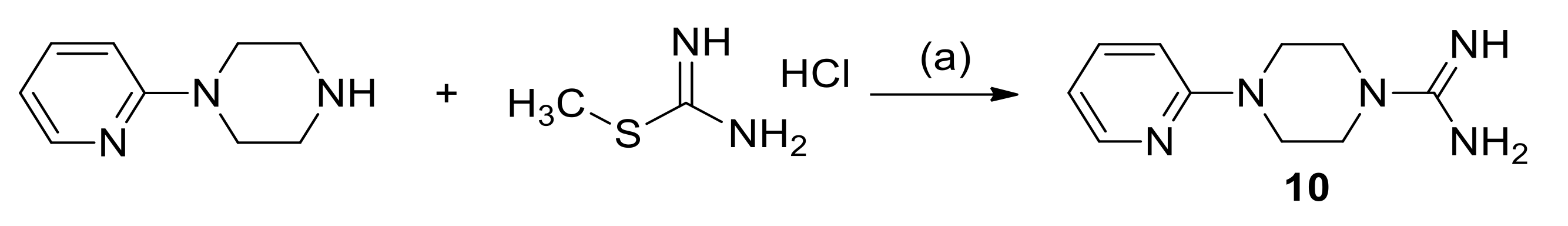
2.3. Chemistry
2.4. Biological Studies and SAR
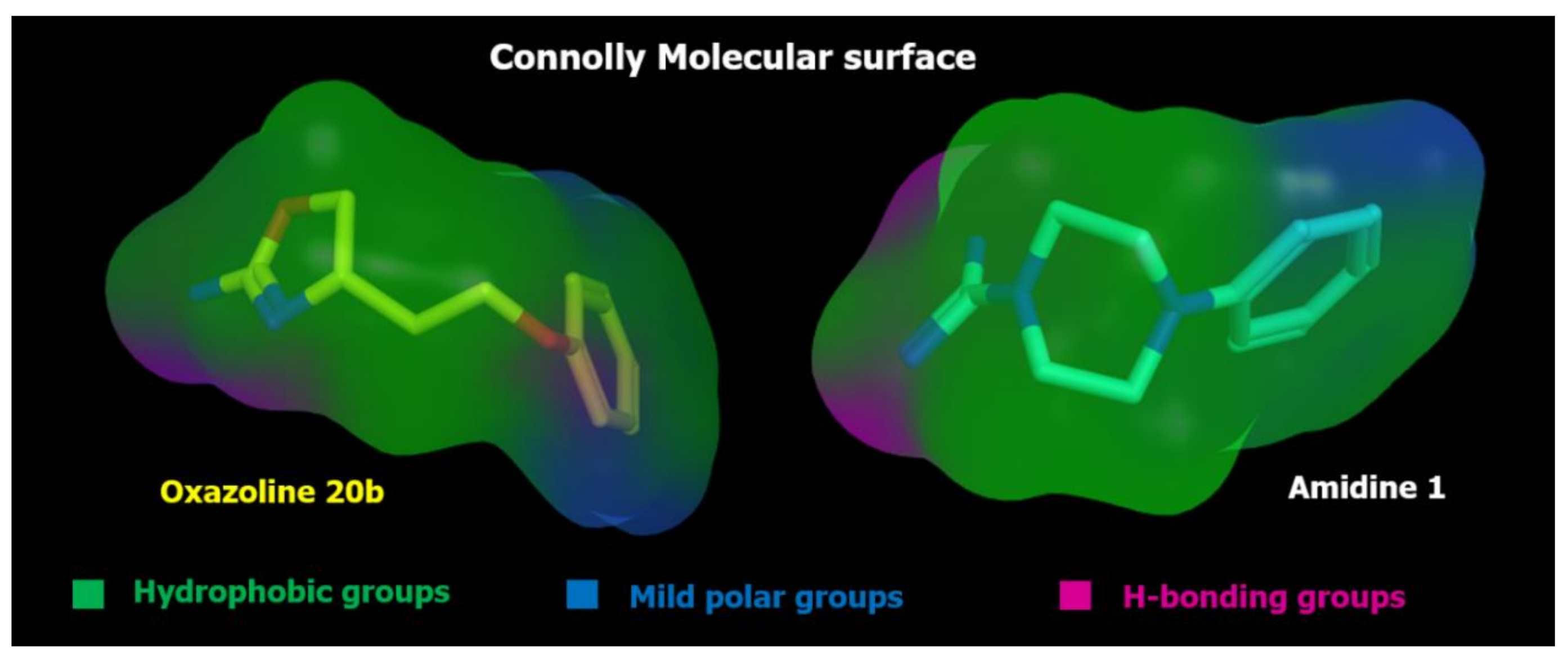
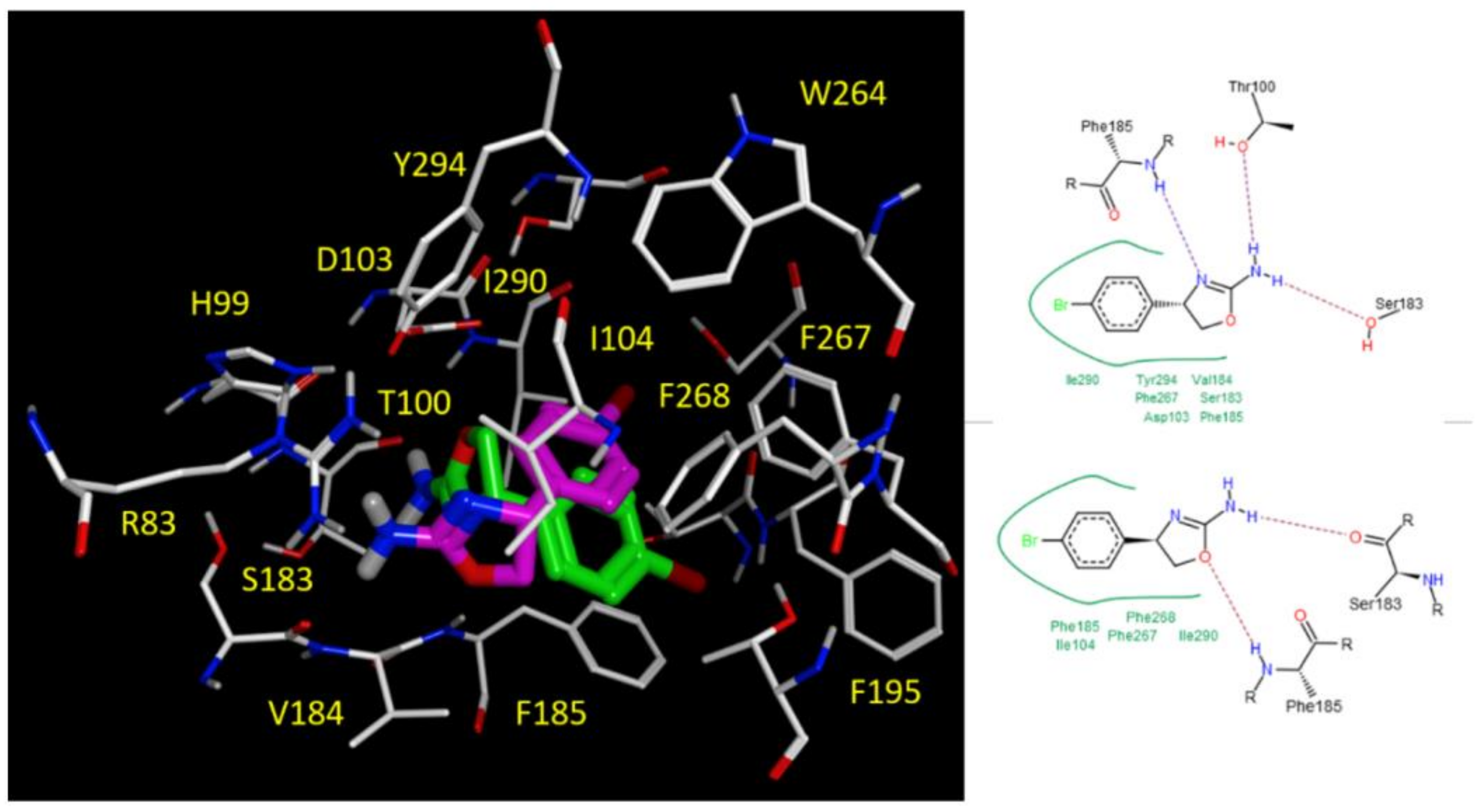
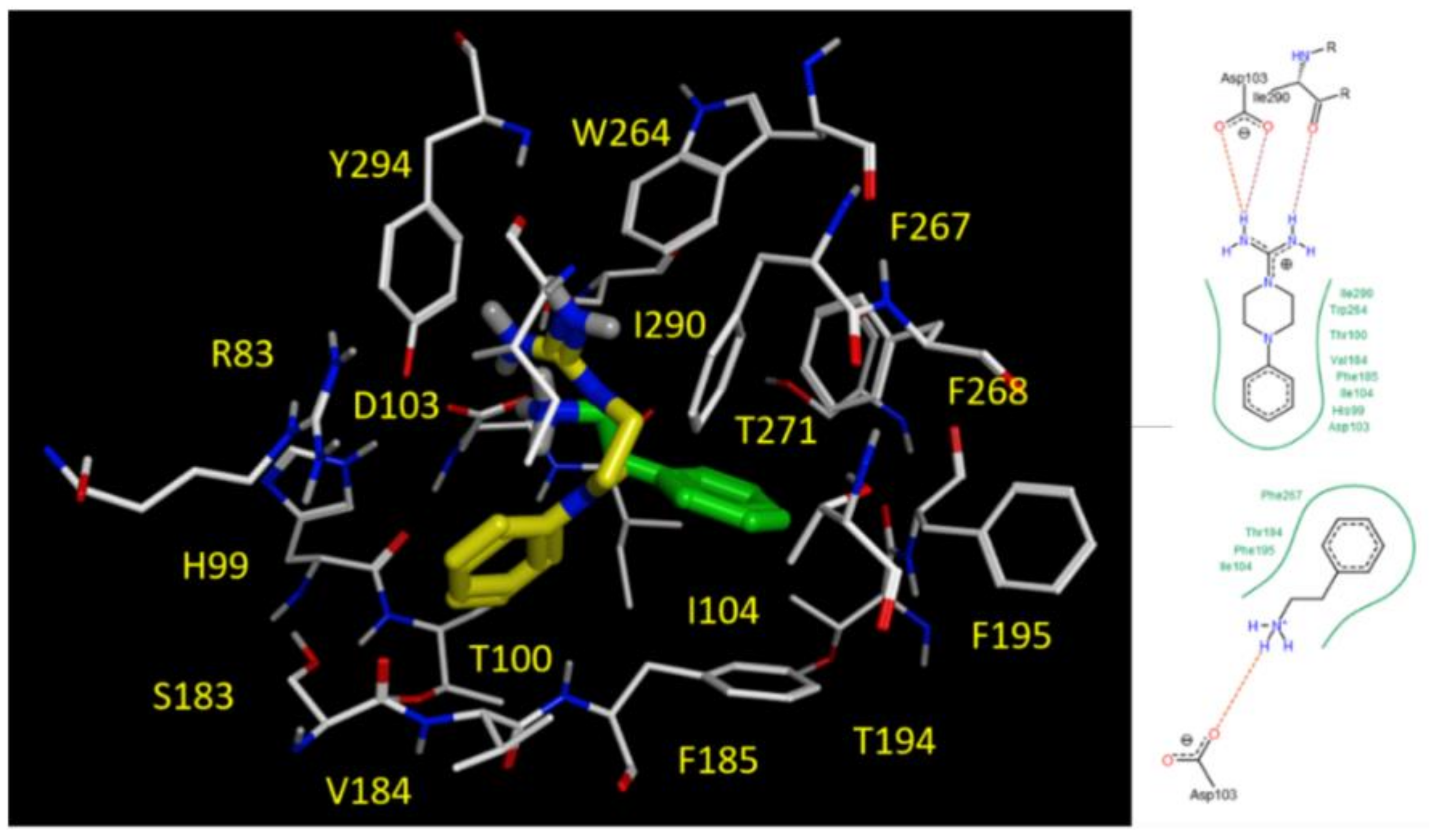
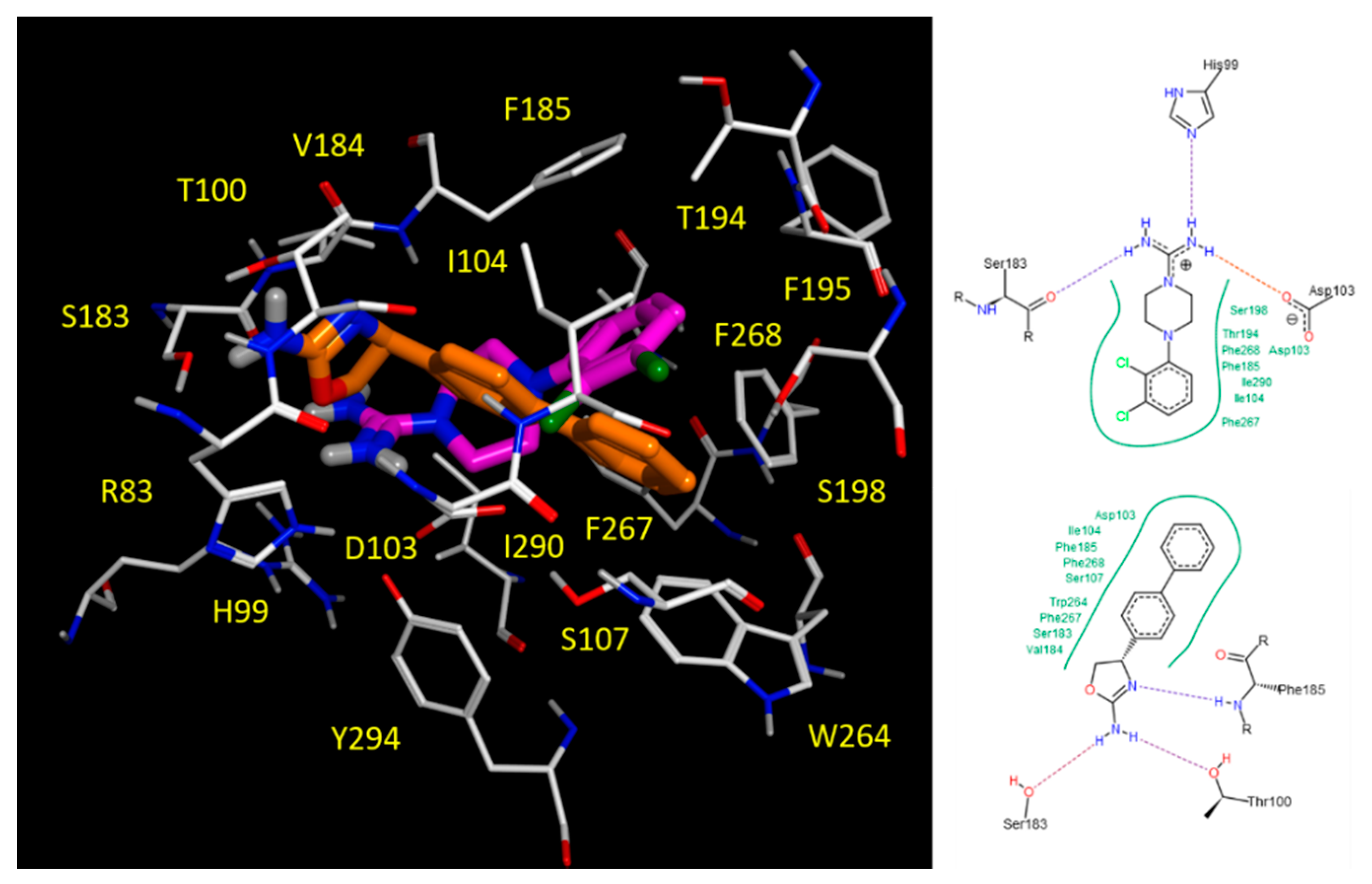
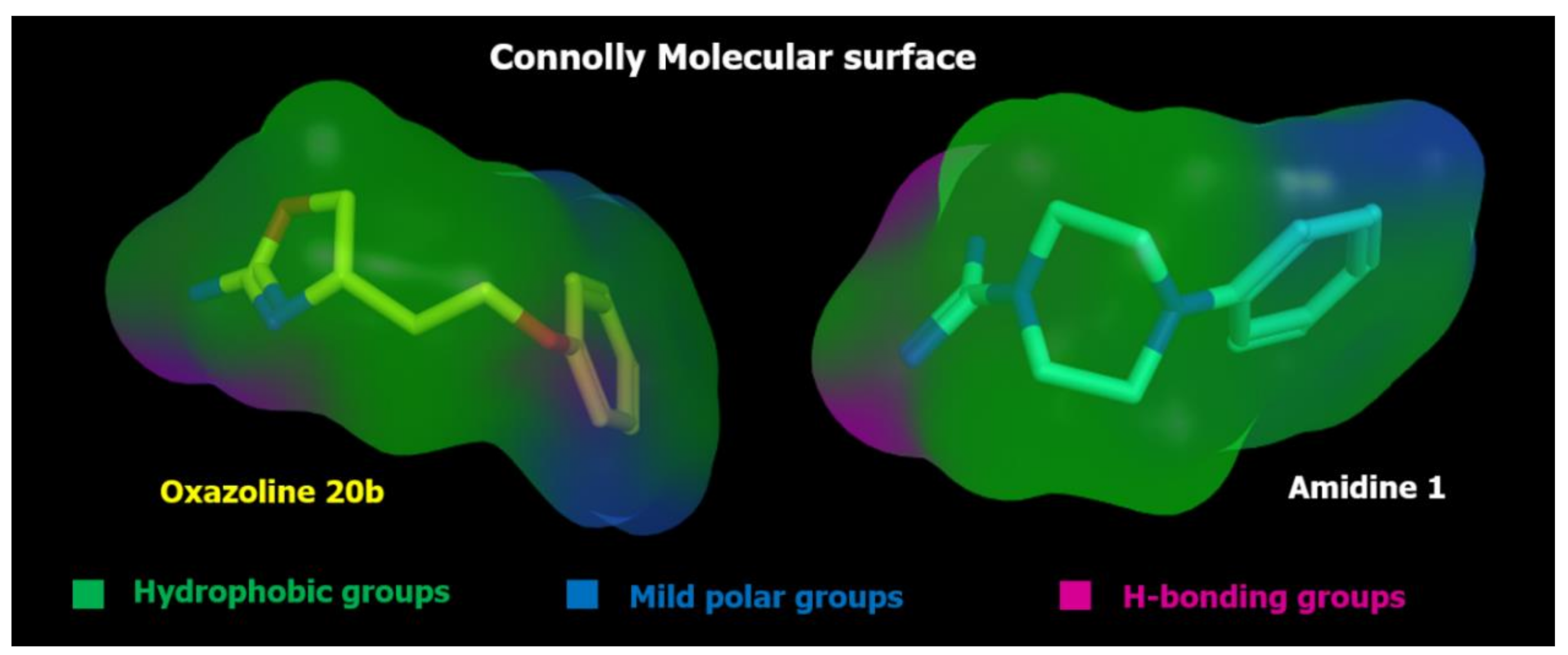
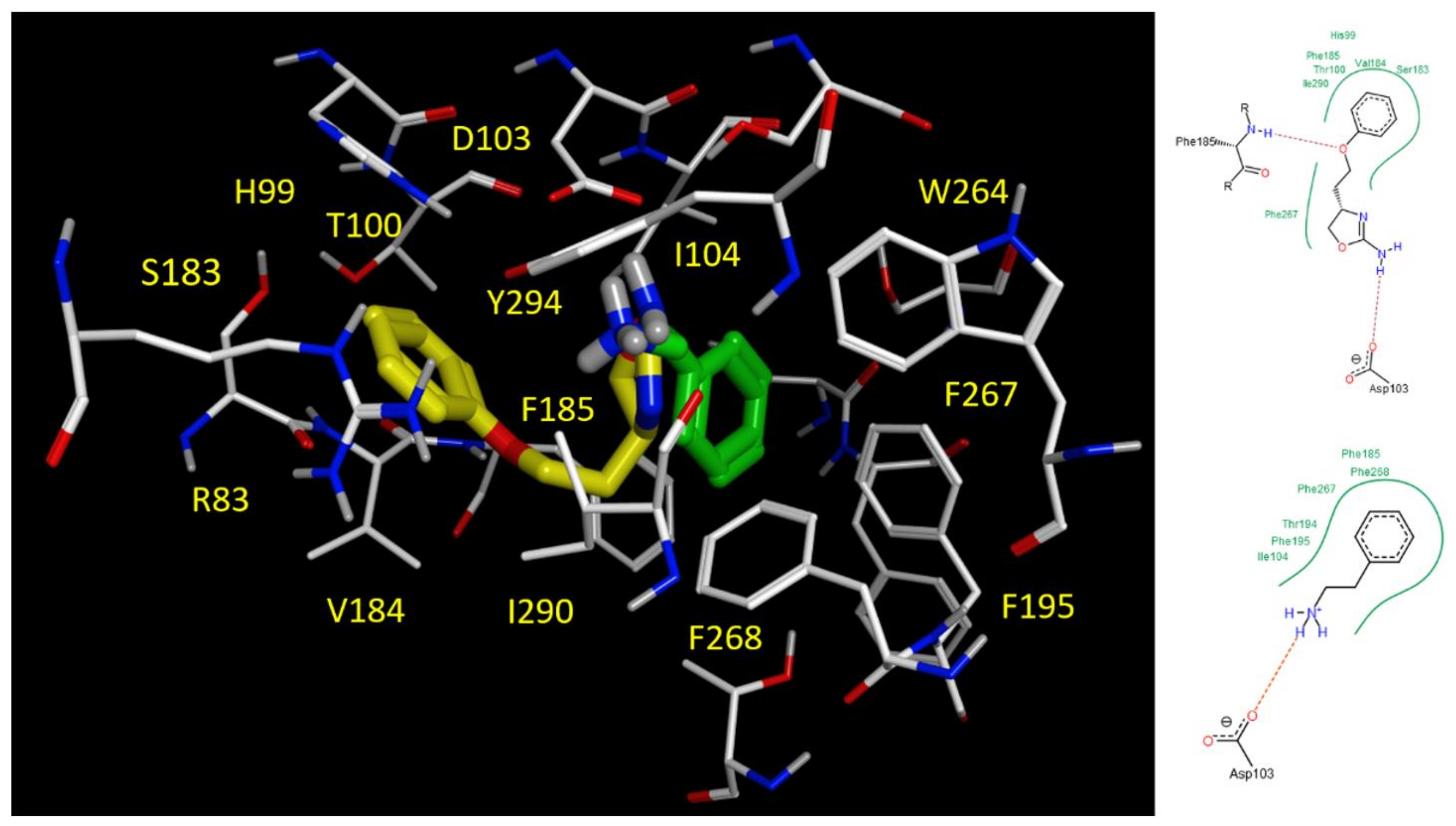
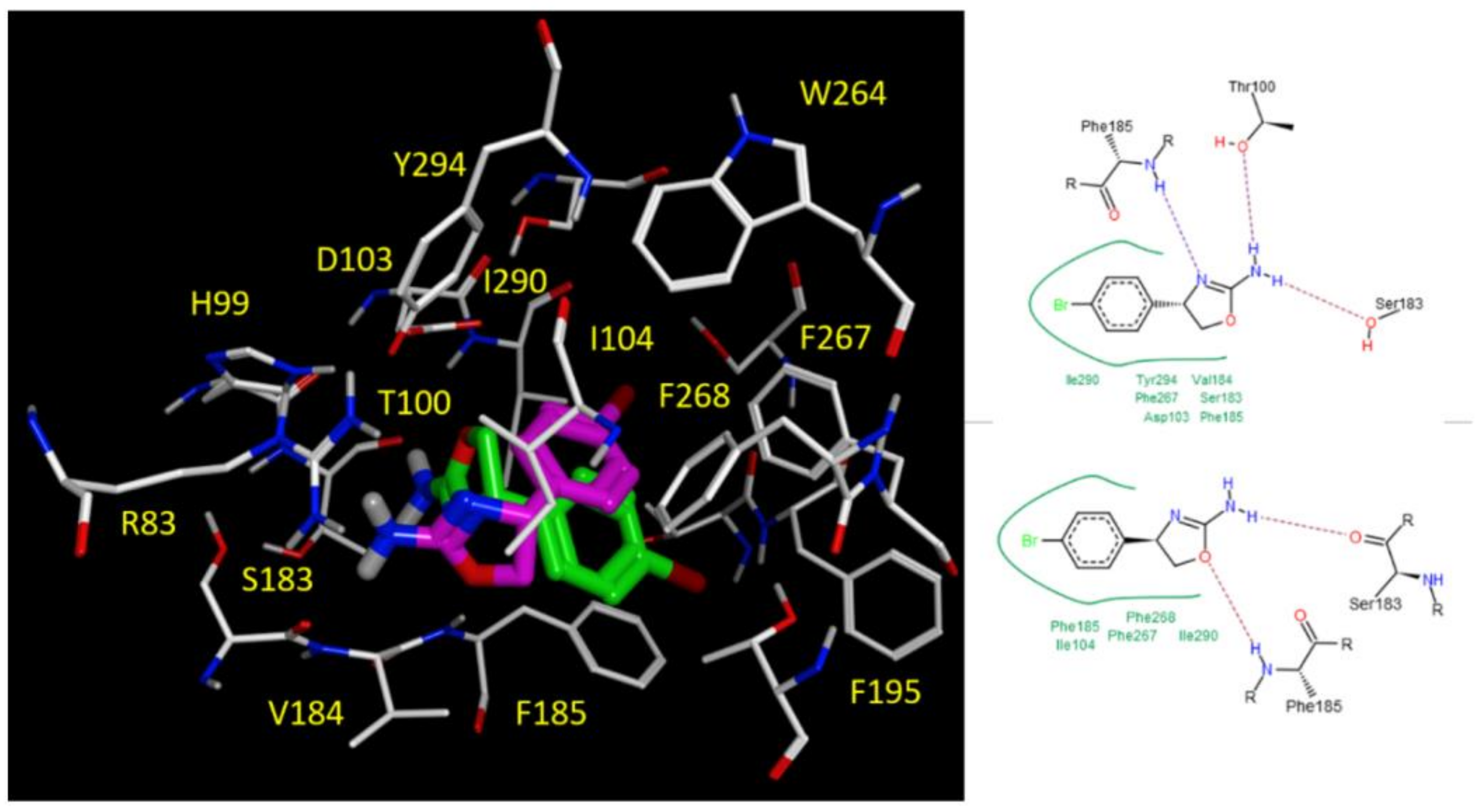
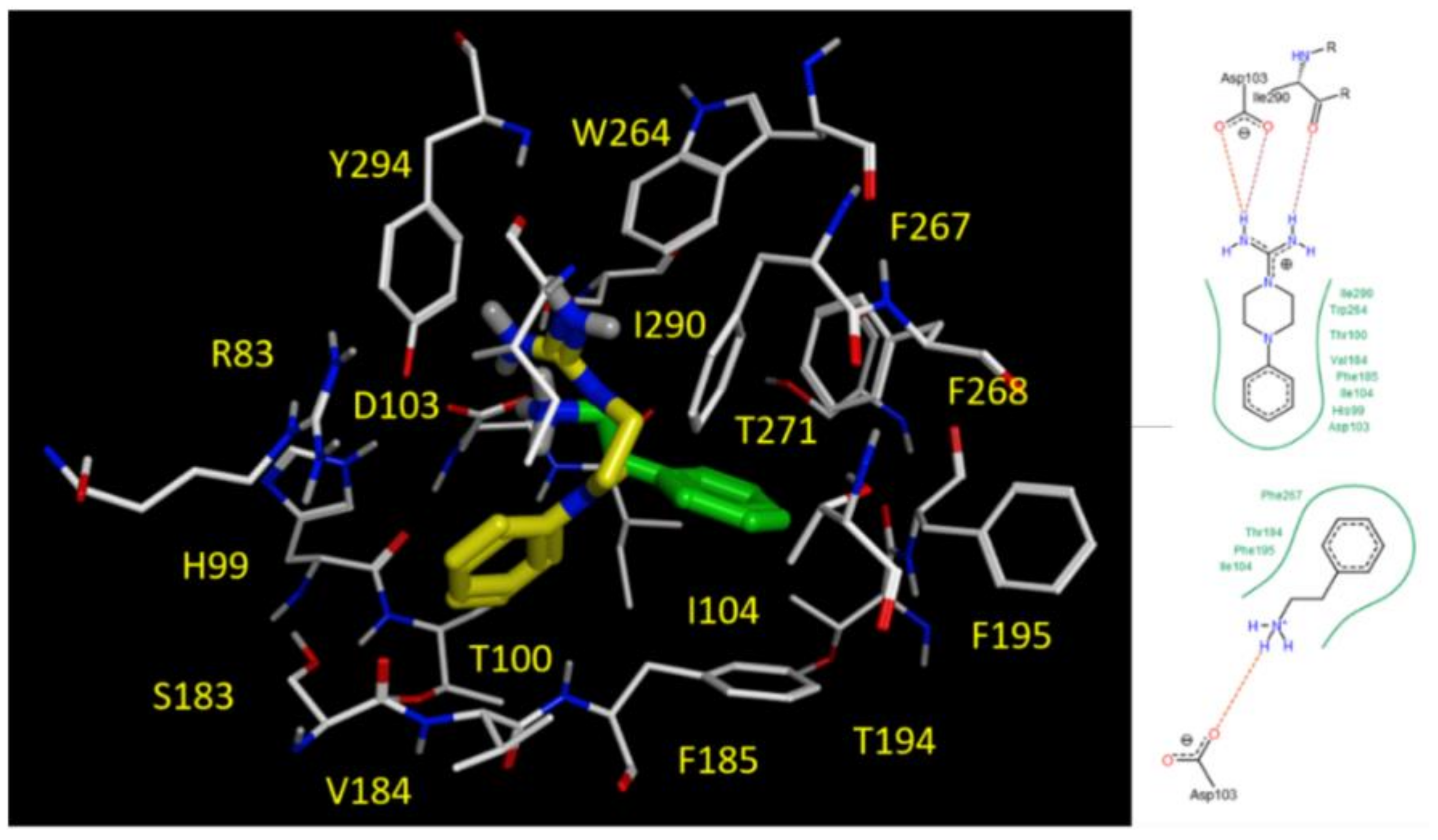
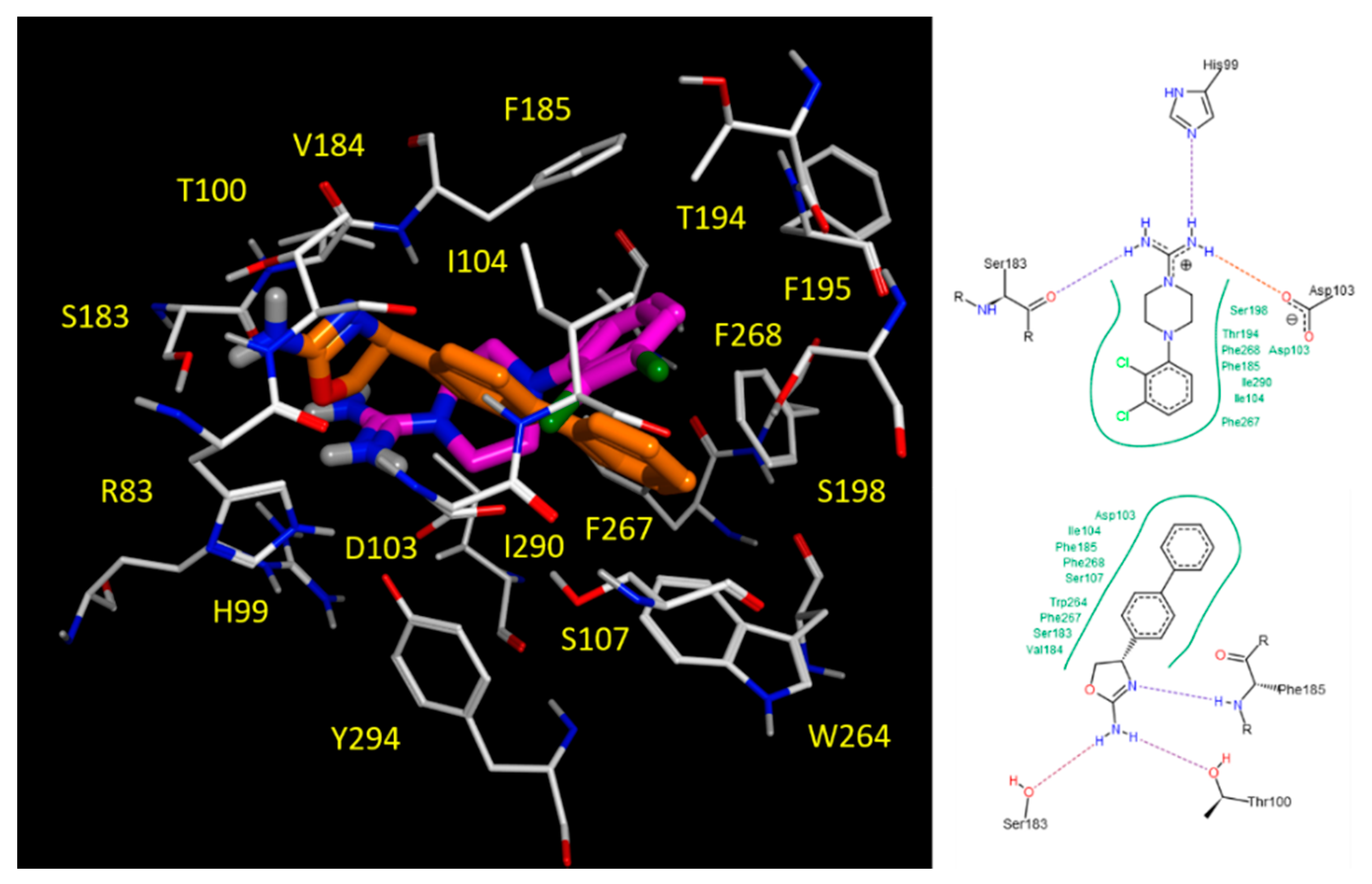
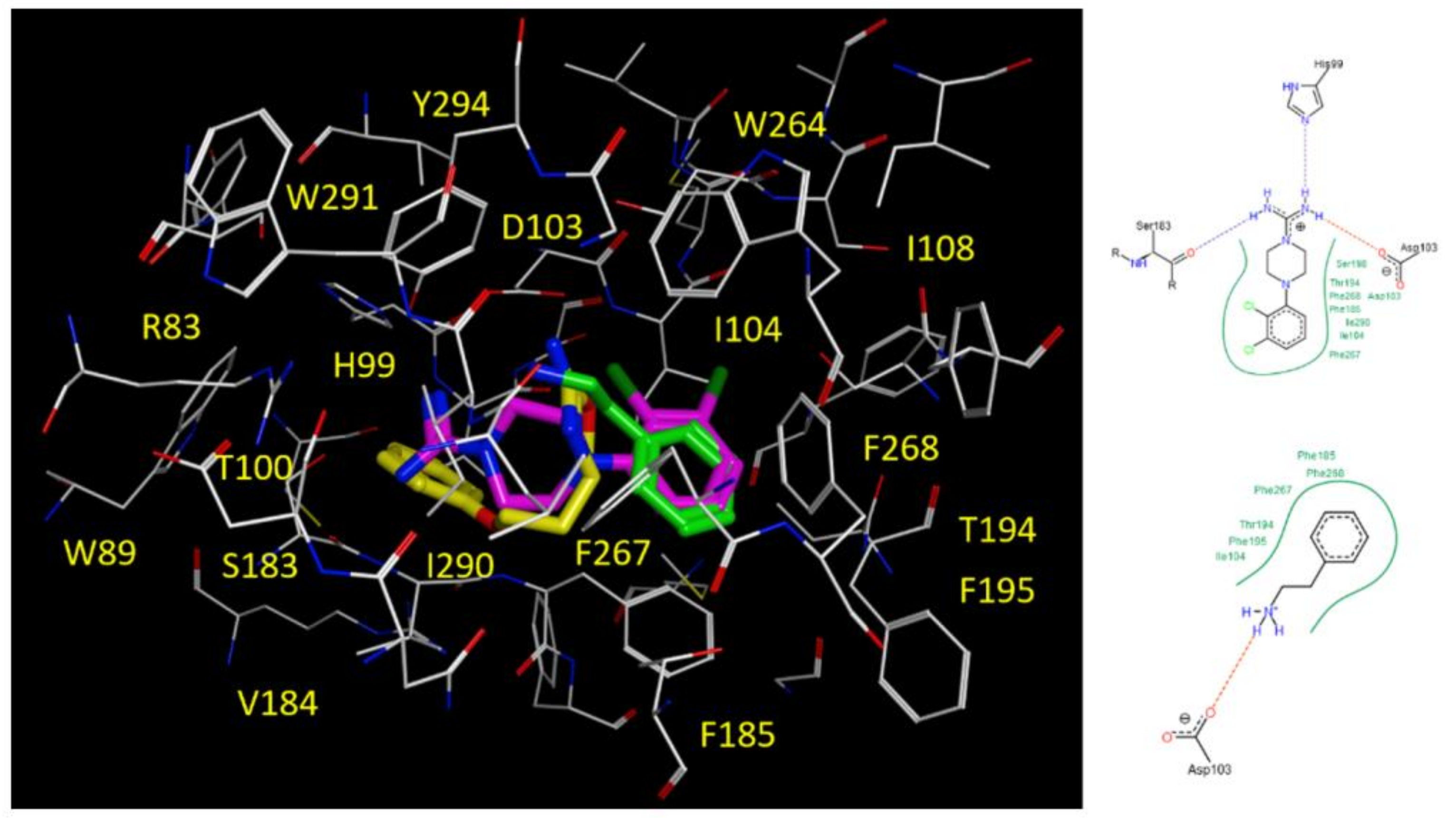
2.5. Docking Studies
2.6. Prediction of ADMET Properties
3. Materials and Methods
3.1. Chemistry
3.1.1. General Information
3.1.2. General Method for the Synthesis of 1-Amidino-4-Arylpiperazine Derivatives
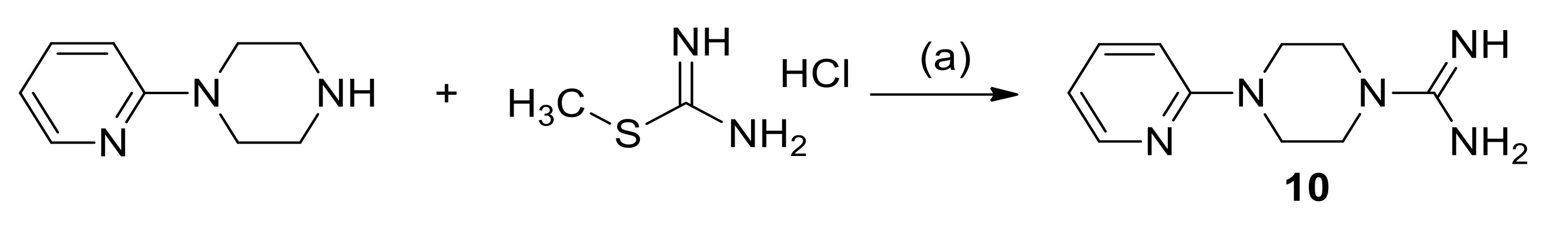
3.1.3. Synthesis of 4-(Pyridin-2-yl)Piperazine-1-Carboximidamide Hydrochloride (10)
3.2. In Vitro Biological Tests
3.2.1. Screening of hTAAR1 Agonists by Means of Bioluminescence Resonance Energy Transfer (BRET) Technology
3.2.2. Cell Cytotoxicity Assay
3.3. Molecular Modelling Studies
3.3.1. Ligand Preparation and Pharmacophore Analysis
3.3.2. Molecular Docking Studies
| Subscript | Description |
| hb | Interactions between hydrogen bond donor–acceptor pairs. An optimistic view is taken; for example, two hydroxyl groups are assumed to interact in the most favorable way |
| ion | Ionic interactions. A Coulomb-like term is used to evaluate the interactions between charged groups. This can contribute to or detract from binding affinity |
| mlig | Metal ligation. Interactions between nitrogens/sulfurs and transition metals are assumed to be metal ligation interactions |
| hh | Hydrophobic interactions, for example, between alkane carbons. These interactions are generally favorable |
| hp | Interactions between hydrophobic and polar atoms. These interactions are generally unfavorable |
| aa | An interaction between any two atoms. This interaction is weak and generally favorable |
| S | The final score, which is the score of the last stage of refinement. |
| E_conf | The energy of the conformer. If there is a refinement stage, this is the energy calculated at the end of the refinement |
| E_place | Score from the placement stage |
| E_score1 E_score2 | Score from rescoring stages 1 and 2 |
| E_refine | Score from the refinement stage, calculated to be the sum of the van der Waals electrostatics and solvation energies, under the Generalized Born solvation model (GB/VI) |
3.3.3. In Silico Evaluation of Pharmacokinetic and Toxicity Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tonelli, M.; Cichero, E. Trace amine associated receptor 1 (TAAR1) modulators: A patent review (2010–present). Expert Opin. Ther. Patents 2020, 30, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.D.; Canales, J.J.; Zucchi, R.; Espinoza, S.; Sukhanov, I.; Gainetdinov, R.R. Trace amine-associated receptor 1: A multimodal therapeutic target for neuropsychiatric diseases. Expert Opin. Ther. Targets 2018, 22, 513–526. [Google Scholar] [CrossRef] [PubMed]
- Rutigliano, G.; Accorroni, A.; Zucchi, R. The case for TAAR1 as a modulator of central nervous system function. Front. Pharmacol. 2018, 8, 987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scanlan, T.S.; Suchland, K.L.; Hart, M.E.; Chiellini, G.; Huang, Y.; Kruzich, P.J.; Frascarelli, S.; Crossley, D.A.; Bunzow, J.R.; Ronca-Testoni, S.; et al. 3-Iodothyronamine is an endogenous and rapid-acting derivative of thyroid hormone. Nat. Med. 2004, 10, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Gainetdinov, R.R.; Hoener, M.C.; Berry, M.D. Trace amines and their receptors. Pharmacol. Rev. 2018, 70, 549–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, M.D.; Gainetdinov, R.R.; Hoener, M.C.; Shahid, M. Pharmacology of human trace amine-associated receptors: Therapeutic opportunities and challenges. Pharmacol. Ther. 2017, 180, 161–180. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Asif-Malik, A.; Canales, J.J. Trace amines and the trace amine-associated Receptor 1: Pharmacology, neurochemistry, and clinical implications. Front. Neurosci. 2016, 10, 148. [Google Scholar] [CrossRef] [Green Version]
- Leo, D.; Mus, L.; Espinoza, S.; Hoener, M.C.; Sotnikova, T.D.; Gainetdinov, R.R. TAAR1-mediated modulation of presynaptic dopaminergic neurotransmission: Role of D2 dopamine autoreceptors. Neuropharmacology 2014, 81, 283–291. [Google Scholar] [CrossRef]
- Espinoza, S.; Ghisi, V.; Emanuele, M.; Leo, D.; Sukhanov, I.; Sotnikova, T.D.; Chieregatti, E.; Gainetdinov, R.R. Postsynaptic D2 dopamine receptor supersensitivity in the striatum of mice lacking TAAR1. Neuropharmacology 2015, 93, 308–313. [Google Scholar] [CrossRef]
- Bräunig, J.; Dinter, J.; Höfig, C.; Paisdzior, S.; Szczepek, M.; Scheerer, P.; Rosowski, M.; Mittag, J.; Kleinau, G.; Biebermann, H. The trace amine-associated receptor 1 agonist 3-iodothyronamine induces biased signaling at the serotonin 1b receptor. Front. Pharmacol. 2018, 9, 222. [Google Scholar] [CrossRef] [Green Version]
- Espinoza, S.; Lignani, G.; Caffino, L.; Maggi, S.; Sukhanov, I.; Leo, D.; Mus, L.; Emanuele, M.; Ronzitti, G.; Harmeier, A.; et al. TAAR1 modulates cortical glutamate NMDA receptor function. Neuropsychopharmacology 2015, 40, 2217–2227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Li, Q. TAAR1 agonists. Cell Mol. Neurobiol. 2020, 40, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Cichero, E.; Tonelli, M. Targeting species-specific trace amine-associated receptor 1 ligands: To date perspective of the rational drug design process. Future Med. Chem. 2017, 9, 1507–1527. [Google Scholar] [CrossRef] [PubMed]
- Chiellini, G.; Nesi, G.; Digiacomo, M.; Malvasi, R.; Espinoza, S.; Sabatini, M.; Frascarelli, S.; Laurino, A.; Cichero, E.; Macchia, M.; et al. Design, Synthesis, and evaluation of thyronamine analogues as novel potent mouse trace amine associated receptor 1 (mTAAR1) agonists. J. Med. Chem. 2015, 58, 5096–5107. [Google Scholar] [CrossRef] [PubMed]
- Chiellini, G.; Nesi, G.; Sestito, S.; Chiarugi, S.; Runfola, M.; Espinoza, S.; Sabatini, M.; Bellusci, L.; Laurino, A.; Cichero, E.; et al. Hit-to-lead optimization of mouse trace amine associated receptor 1 (mTAAR1) agonists with a diphenylmethane-scaffold: Design, synthesis, and biological study. J. Med. Chem. 2016, 59, 9825–9836. [Google Scholar] [CrossRef] [Green Version]
- Regard, J.B.; Kataoka, H.; Cano, D.A.; Camerer, E.; Yin, L.; Zheng, Y.W.; Scanlan, T.S.; Hebrok, M.; Coughlin, S.R. Probing cell type-specific functions of Gi in vivo identifies GPCR regulators of insulin secretion. J. Clin. Investig. 2007, 117, 4034–4043. [Google Scholar] [CrossRef] [Green Version]
- Hoefig, C.S.; Zucchi, R.; Köhrle, J. Thyronamines and derivatives: Physiological relevance, pharmacological actions and future research directions. Thyroid 2016, 26, 1656–1673. [Google Scholar] [CrossRef]
- Galley, G.; Beurier, A.; Décoret, G.; Goergler, A.; Hutter, R.; Mohr, S.; Pähler, A.; Schmid, P.; Türck, D.; Unger, R.; et al. Discovery and characterization of 2-aminooxazolines as highly potent, selective, and orally active TAAR1 agonists. ACS Med. Chem. Lett. 2015, 7, 192–197. [Google Scholar] [CrossRef] [Green Version]
- Revel, F.G.; Moreau, J.L.; Gainetdinov, R.R.; Bradaia, A.; Sotnikova, T.D.; Mory, R.; Durkin, S.; Zbinden, K.G.; Norcross, R.; Meyer, C.A.; et al. TAAR1 activation modulates monoaminergic neurotransmission, preventing hyperdopaminergic and hypoglutamatergic activity. Proc. Natl. Acad. Sci. USA 2011, 108, 8485–8490. [Google Scholar] [CrossRef] [Green Version]
- Revel, F.G.; Moreau, J.L.; Gainetdinov, R.R.; Ferragud, A.; Velázquez-Sánchez, C.; Sotnikova, T.D.; Morairty, S.R.; Harmeier, A.; Groebke Zbinden, K.; Norcross, R.D.; et al. Trace amine-associated receptor 1 partial agonism reveals novel paradigm for neuropsychiatric therapeutics. Biol. Psychiatry 2012, 72, 934–942. [Google Scholar] [CrossRef]
- Revel, F.G.; Moreau, J.L.; Pouzet, B.; Mory, R.; Bradaia, A.; Buchy, D.; Metzler, V.; Chaboz, S.; Groebke Zbinden, K.; Galley, G.; et al. A new perspective for schizophrenia: TAAR1 agonists reveal antipsychotic- and antidepressant-like activity, improve cognition and control body weight. Mol. Psychiatry 2013, 18, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann-La Roche, A.G.F. Combinations Comprising Atypical Antipsychotics and TAAR1 Agonists. WO2012/016879 A1, 9 February 2012. [Google Scholar]
- Hoffmann-La Roche, A.G.F. 5-Ethyl-4-Methyl-Pyrazole-3-Carboxamide Derivative Having Activity as Agonist of TAAR. WO2017/157873 A1, 21 September 2017. [Google Scholar]
- Michael, E.S.; Covic, L.; Kuliopulos, A. Trace amine-associated receptor 1 (TAAR1) promotes anti-diabetic signaling in insulin-secreting cells. J. Biol. Chem. 2019, 294, 4401–4411. [Google Scholar] [CrossRef] [PubMed]
- Raab, S.; Wang, H.; Uhles, S.; Cole, N.; Alvarez-Sanchez, R.; Künnecke, B.; Ullmer, C.; Matile, H.; Bedoucha, M.; Norcross, R.D.; et al. Incretin-like effects of small molecule trace amine-associated receptor 1 agonists. Mol. Metab. 2016, 5, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Ferragud, A.; Howell, A.D.; Moore, C.F.; Ta, T.L.; Hoener, M.C.; Sabino, V.; Cottone, P. The trace amine-associated receptor 1 agonist RO5256390 blocks compulsive, binge-like eating in rats. Neuropsychopharmacology 2017, 42, 1458–1470. [Google Scholar] [CrossRef] [PubMed]
- Dedic, N.; Jones, P.G.; Hopkins, S.C.; Lew, R.; Shao, L.; Campbell, J.E.; Spear, K.L.; Large, T.H.; Campbell, U.C.; Hanania, T.; et al. SEP-363856, a novel psychotropic agent with a unique, non-D2 receptor mechanism of action. J. Pharmacol. Exp. Ther. 2019, 371, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koblan, K.S.; Kent, J.; Hopkins, S.C.; Krystal, J.H.; Cheng, H.; Goldman, R.; Loebel, A. A non-D2-receptor-binding drug for the treatment of schizophrenia. N. Engl. J. Med. 2020, 382, 1497–1506. [Google Scholar] [CrossRef]
- Tonelli, M.; Espinoza, S.; Gainetdinov, R.R.; Cichero, E. Novel biguanide-based derivatives scouted as TAAR1 agonists: Synthesis, biological evaluation, ADME prediction and molecular docking studies. Eur. J. Med. Chem. 2017, 127, 781–792. [Google Scholar] [CrossRef]
- Cichero, E.; Espinoza, S.; Tonelli, M.; Franchini, S.; Gerasimov, A.S.; Sorbi, C.; Gainetdinov, R.R.; Brasili, L.; Fossa, P. Homology modelling-driven study leading to the discovery of the first mouse Trace Amine-Associated Receptor 5 (TAAR5) antagonists. Med. Chem. Comm. 2016, 7, 353–364. [Google Scholar] [CrossRef]
- Guariento, S.; Tonelli, M.; Espinoza, S.; Gerasimov, A.S.; Gainetdinov, R.R.; Cichero, E. Rational design, chemical synthesis and biological evaluation of novel biguanides exploring species-specificity responsiveness of TAAR1 agonists. Eur. J. Med. Chem. 2018, 146, 171–184. [Google Scholar] [CrossRef]
- Guisado, O.; Martınez, S.; Pastor, J. A novel, facile methodology for the synthesis of N,N′-bis(tert-butoxycarbonyl)-protected guanidines using polymer-supported carbodiimide. Tetrahedron Lett. 2002, 43, 7105–7109. [Google Scholar] [CrossRef]
- Guo, D.X.; Liu, Y.; Wang, N.; Hu, C.; Gong, P. Synthesis and antitumor activities of a new series of 4,5-dihydro-1H-thiochromeno[4,3-d]pyrimidine derivatives. Sci. China Chem. 2012, 55, 347–351. [Google Scholar] [CrossRef]
- Bristol-Myers Squibb Pharma Company. Ligands for Imaging Cardiac Innervation. WO2008/83056, 10 July 2008.
- Protiva, M.; Rajšner, M.; Trčka, V.; Vaněček, M.; Němec, J.; Šedivý, Z. 1-Aryl- and 1-(arylmethyl)-4-guanylpiperazines and other heterocyclic and alicyclic guanidine derivatives. Collect Czechoslov. Chem. Commun. 1975, 40, 3904–3923. [Google Scholar] [CrossRef]
- Ozawa, H.; Iwatsuki, K. Pharmacological properties of heterocyclic amidine derivatives. II. Pharmacological studies of phenylguanylpiperazine derivatives. Chem. Pharm. Bull. 1968, 16, 2482–2487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conroy, E.A.; Denton, J.J. Piperazine. III. 1-Heterocyclic-4-guanyl-, carbamyl-, and thiocarbamyl-piperazines. J. Organic Chem. 1953, 18, 1489–1491. [Google Scholar] [CrossRef]
- Ozawa, H.; Iwatsuki, K. Pharmacological properties of heterocyclic amidine derivatives. IV. The action of 1-phenyl-4-guanylpiperazine sulfate (PGP) on the concentration of norepinephrine in brain and heart of rat. Yakugaku Zasshi 1971, 12, 1381–1383. [Google Scholar] [CrossRef] [Green Version]
- Waterbeemd, H.; Gifford, E. ADMET in silico modelling: Towards prediction paradise? Nat. Rev. Drug Discov. 2003, 2, 192–204. [Google Scholar] [CrossRef]
- Barak, L.S.; Salahpour, A.; Zhang, X.; Masri, B.; Sotnikova, T.D.; Ramsey, A.J.; Violin, J.D.; Lefkowitz, R.J.; Caron, M.G.; Gainetdinov, R.R. Pharmacological characterization of membrane-expressed human trace amine-associated receptor 1 (TAAR1) by a bioluminescence resonance energy transfer cAMP Biosensor. Mol. Pharmacol. 2008, 74, 585–594. [Google Scholar] [CrossRef] [Green Version]
- MOE: Chemical Computing Group Inc. Montreal. H3A 2R7 Canada. Available online: http://www.chemcomp.com/ (accessed on 14 November 2020).
- Wolber, G.; Seidel, T.; Bendix, F.; Langer, T. Molecule-pharmacophore superpositioning and pattern matching in computational drug design. Drug Discov. Today 2008, 13, 23–29. [Google Scholar] [CrossRef]
- Khalid, S.; Hanif, R.; Jabeen, I.; Mansoor, Q.; Ismail, M. Pharmacophore modeling for identification of anti-IGF-1R drugs and in-vitro validation of fulvestrant as a potential inhibitor. PLoS ONE 2018, 13, e0196312. [Google Scholar] [CrossRef] [Green Version]
- Haidar, S.; Bouaziz, Z.; Marminon, C.; Laitinen, T.; Poso, A.; Le Borgne, M.; Jose, J. Development of pharmacophore model for indeno[1,2-b]indoles as human protein kinase CK2 inhibitors and database mining. Pharmaceuticals 2017, 10, 8. [Google Scholar] [CrossRef]
- Cichero, E.; Menozzi, G.; Spallarossa, A.; Mosti, L.; Fossa, P. Exploring the binding features of rimonabant analogues and acyclic CB1 antagonists: Docking studies and QSAR analysis. J. Mol. Model. 2008, 14, 1131–1145. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, M.; Naesens, L.; Gazzarrini, S.; Santucci, M.; Cichero, E.; Tasso, B.; Moroni, A.; Costi, M.P.; Loddo, R. Host dihydrofolate reductase (DHFR)-directed cycloguanil analogues endowed with activity against influenza virus and respiratory syncytial virus. Eur. J. Med. Chem. 2017, 135, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Reese, E.A.; Norimatsu, Y.; Grandy, M.S.; Suchland, K.L.; Bunzow, J.R.; Grandy, D.K. Exploring the determinants of trace amine-associated receptor 1’s functional selectivity for the stereoisomers of amphetamine and methamphetamine. J. Med. Chem. 2014, 57, 378–390. [Google Scholar] [CrossRef] [PubMed]

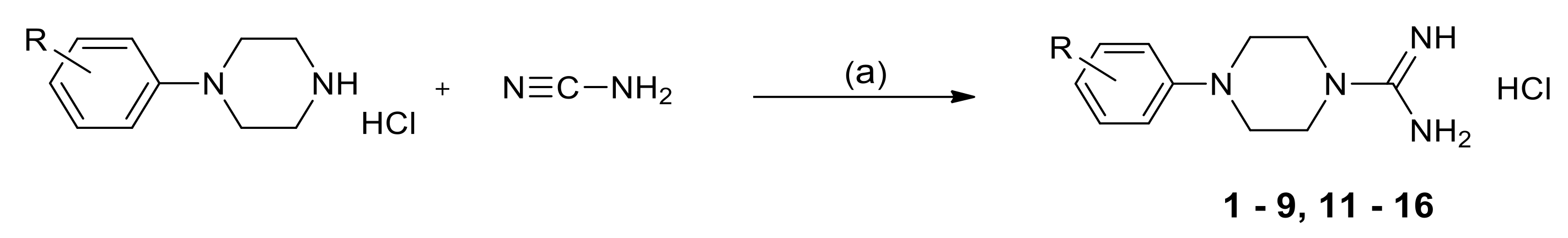



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
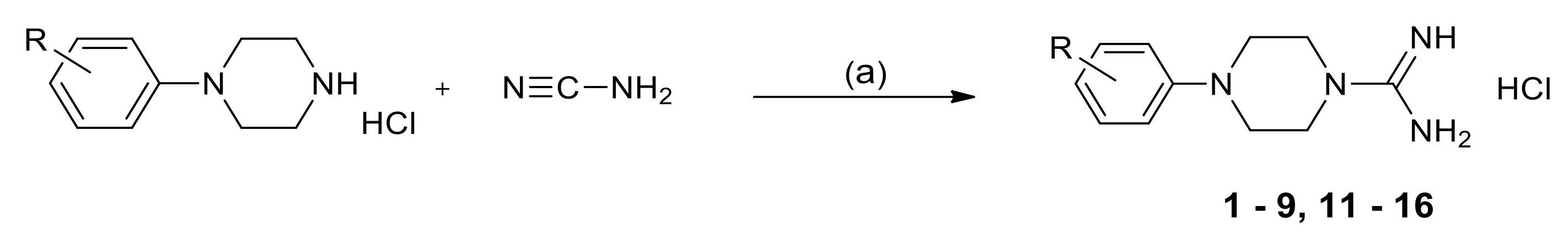{kind=link}
{kind=link}
| ID | SCORE | RADIUS | EXPRESSION |
|---|---|---|---|
| F1 | 89% | 1.96 | Aro|Hyd |
| F2 | 83% | 1.46 | Hyd |
| F3 | 94% | 1.36 | Don2 |
| F4 | 89% | 1.08 | Acc2 |
| F5 | 89% | 1.26 | Don2 |
| F6 | 100% | 0.85 | Acc |
| F7 | 100% | 1.23 | Don |
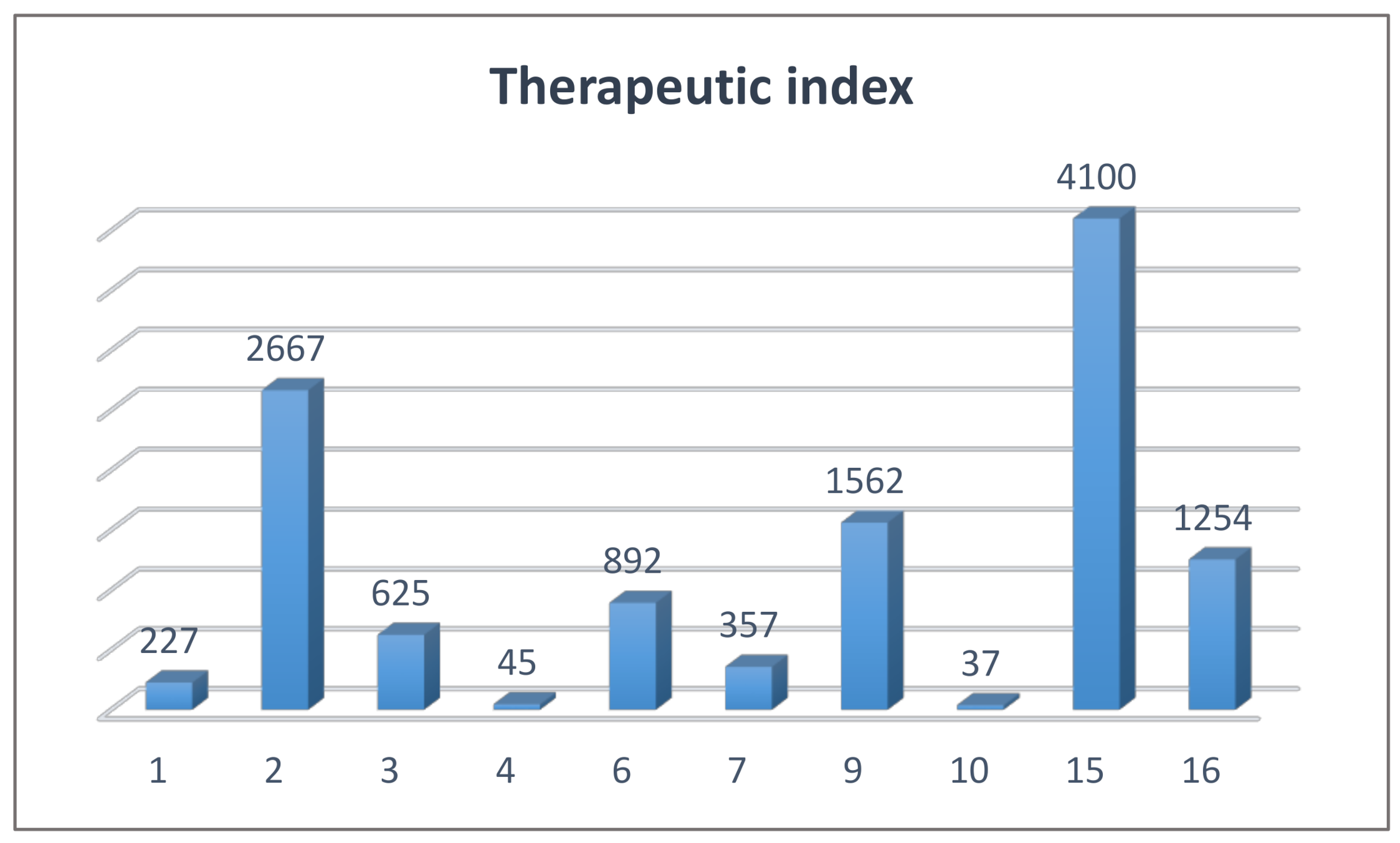
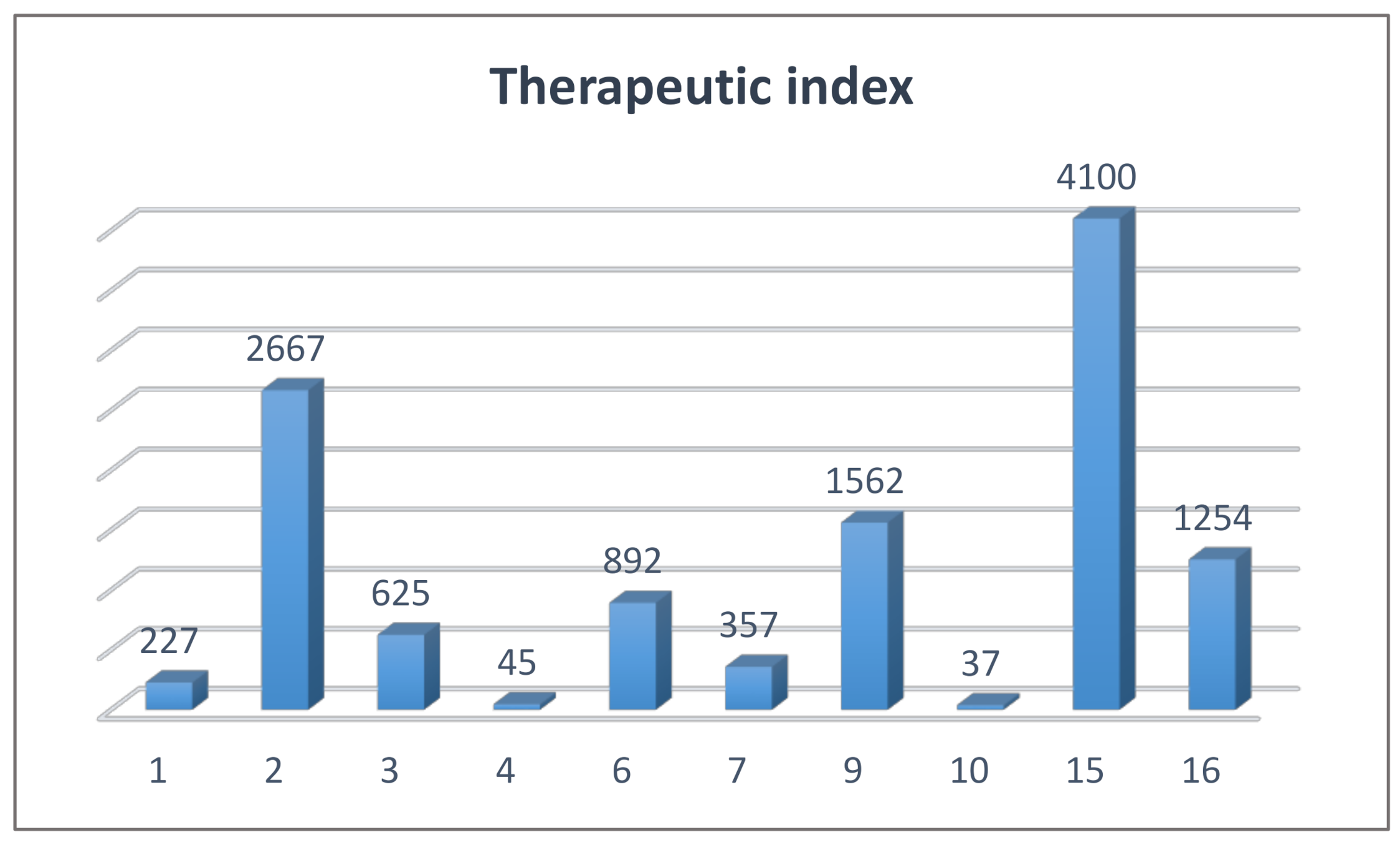
| Compound | hTAAR1 Activity a | hTAAR1 b EC50, nM | Vero-76 Cells c CC50, µM |
|---|---|---|---|
| 1 | 71% | 370 | 84 ± 5 |
| 2 | 100% | 30 | 80 ± 5 |
| 3 | 90% | 160 | > 100 |
| 4 | 35% | >2000 | 90 ± 3 |
| 5 | inactive | - | 95 ± 3 |
| 6 | 64% | 93 | 83 ± 5 |
| 7 | 32% | 244 | 87 ± 4 |
| 8 | inactive | - | 88 ± 2 |
| 9 | 80% | 64 | >100 |
| 10 | 30% | >2000 | 74 ± 5 |
| 11 | inactive | - | 64 ± 2 |
| 12 | inactive | - | 78 ± 2 |
| 13 | inactive | - | 87 ± 5 |
| 14 | inactive | - | 94 ± 6 |
| 15 | 81% | 20 | 82 ± 5 |
| 16 | 49% | 71 | 89 ± 4 |
| TYR | 100% | 66 | - |
| Comp. | cLogP | LogBB a | LogPS b | HIA (%) c | Vd (l/kg) d | %PPB | LogKa HSA | %F (oral) |
|---|---|---|---|---|---|---|---|---|
| β-PEA | 1.56 | 0.19 | −2.4 | 99 | 3.0 | 34.79 | 2.78 | 91.1 |
| 1 | 0.31 | −0.08 | −3.6 | 50 | 1.2 | 33.14 | 2.84 | 28.9 |
| 2 | 0.71 | −0.03 | −3.4 | 58 | 1.4 | 33.91 | 2.85 | 35.7 |
| 3 | 1.15 | −0.04 | −3.2 | 74 | 1.3 | 48.25 | 3.19 | 49.7 |
| 4 | 0.99 | −0.08 | −3.2 | 68 | 1.5 | 49.24 | 3.11 | 44.5 |
| 5 | 0.81 | −0.13 | −3.3 | 61 | 1.4 | 49.93 | 3.15 | 38.5 |
| 6 | 0.33 | −0.08 | −3.7 | 45 | 1.2 | 33.69 | 2.90 | 27.1 |
| 7 | 0.36 | −0.08 | −3.7 | 46 | 1.3 | 33.00 | 2.82 | 27.1 |
| 8 | 0.16 | −0.09 | −3.9 | 41 | 1.1 | 31.42 | 2.86 | 23.2 |
| 9 | 1.26 | −0.05 | −3.1 | 77 | 1.6 | 52.86 | 3.34 | 54.1 |
| 10 | −0.31 | −0.13 | −4.0 | 43 | 1.0 | 32.87 | 2.68 | 25.2 |
| 11 | −0.43 | −0.09 | −4.1 | 42 | 0.7 | 25.03 | 2.49 | 25.2 |
| 12 | 0.04 | −0.04 | −3.8 | 40 | 1.2 | 21.76 | 2.51 | 22.9 |
| 13 | 0.45 | −0.03 | −3.5 | 53 | 1.2 | 29.16 | 2.97 | 32.3 |
| 14 | 0.37 | −0.01 | −3.5 | 51 | 1.1 | 23.28 | 2.51 | 30.6 |
| 15 | 1.72 | 0.13 | −2.9 | 92 | 1.5 | 50.18 | 3.57 | 66.9 |
| 16 | 1.88 | 0.19 | −2.8 | 95 | 1.6 | 50.85 | 3.52 | 76.9 |
| RO5263397 | 1.85 | 0.35 | −2.0 | 100 | 2.4 | 27.57 | 3.12 | 99.0 |
| Comp. | hERG Inhibitor | Endocrine System Disruption a | CYP3A4 and CYP2D6 | |||
|---|---|---|---|---|---|---|
| Inhibitor | Reliability Index (R.I.) | LogRBA > −3 (R.I. ≥ 0.3) | LogRBA > 0 (R.I. ≥ 0.3) | Inhibitor < 10 mM (R.I. > 0.3) | Substrate (R.I. ≥ 0.3) | |
| β-PEA | 0.06 | 0.40 | No binder | No binder | 0.02 | >50% |
| 1 | 0.07 | 0.34 | No binder | No binder | 0.01 | >50% |
| 2 | 0.08 | 0.35 | No binder | No binder | 0.01 | >50% |
| 3 | 0.11 | 0.29 | No binder | No binder | 0.01 | >50% |
| 4 | 0.11 | 0.29 | No binder | No binder | 0.01 | >50% |
| 5 | 0.15 | 0.29 | No binder | No binder | 0.01 | >50% |
| 6 | 0.10 | 0.29 | No binder | No binder | 0.02 | >50% |
| 7 | 0.09 | 0.29 | No binder | No binder | 0.02 | >50% |
| 8 | 0.15 | 0.22 | No binder | No binder | 0.02 | >50% |
| 9 | 0.43 | 0.56 | No binder | No binder | 0.01 | >50% |
| 10 | 0.07 | 0.35 | No binder | No binder | 0.01 | >50% |
| 11 | 0.05 | 0.33 | No binder | No binder | 0.02 | >50% |
| 12 | 0.07 | 0.30 | No binder | No binder | 0.01 | >50% |
| 13 | 0.12 | 0.25 | No binder | No binder | 0.01 | >50% |
| 14 | 0.14 | 0.27 | No binder | No binder | 0.01 | >50% |
| 15 | 0.21 | 0.24 | No binder | No binder | 0.01 | >50% |
| 16 | 0.27 | 0.30 | No binder | No binder | 0.01 | >50% |
| RO5263397 | 0.06 | 0.36 | No binder | No binder | 0.01 | 33% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Francesconi, V.; Cichero, E.; Kanov, E.V.; Laurini, E.; Pricl, S.; Gainetdinov, R.R.; Tonelli, M. Novel 1-Amidino-4-Phenylpiperazines as Potent Agonists at Human TAAR1 Receptor: Rational Design, Synthesis, Biological Evaluation and Molecular Docking Studies. Pharmaceuticals 2020, 13, 391. https://doi.org/10.3390/ph13110391
Francesconi V, Cichero E, Kanov EV, Laurini E, Pricl S, Gainetdinov RR, Tonelli M. Novel 1-Amidino-4-Phenylpiperazines as Potent Agonists at Human TAAR1 Receptor: Rational Design, Synthesis, Biological Evaluation and Molecular Docking Studies. Pharmaceuticals. 2020; 13(11):391. https://doi.org/10.3390/ph13110391
Chicago/Turabian StyleFrancesconi, Valeria, Elena Cichero, Evgeny V. Kanov, Erik Laurini, Sabrina Pricl, Raul R. Gainetdinov, and Michele Tonelli. 2020. "Novel 1-Amidino-4-Phenylpiperazines as Potent Agonists at Human TAAR1 Receptor: Rational Design, Synthesis, Biological Evaluation and Molecular Docking Studies" Pharmaceuticals 13, no. 11: 391. https://doi.org/10.3390/ph13110391
APA StyleFrancesconi, V., Cichero, E., Kanov, E. V., Laurini, E., Pricl, S., Gainetdinov, R. R., & Tonelli, M. (2020). Novel 1-Amidino-4-Phenylpiperazines as Potent Agonists at Human TAAR1 Receptor: Rational Design, Synthesis, Biological Evaluation and Molecular Docking Studies. Pharmaceuticals, 13(11), 391. https://doi.org/10.3390/ph13110391