Quality-by-Design Is a Tool for Quality Assurance in the Assessment of Enantioseparation of a Model Active Pharmaceutical Ingredient




Abstract
1. Introduction
2. Results
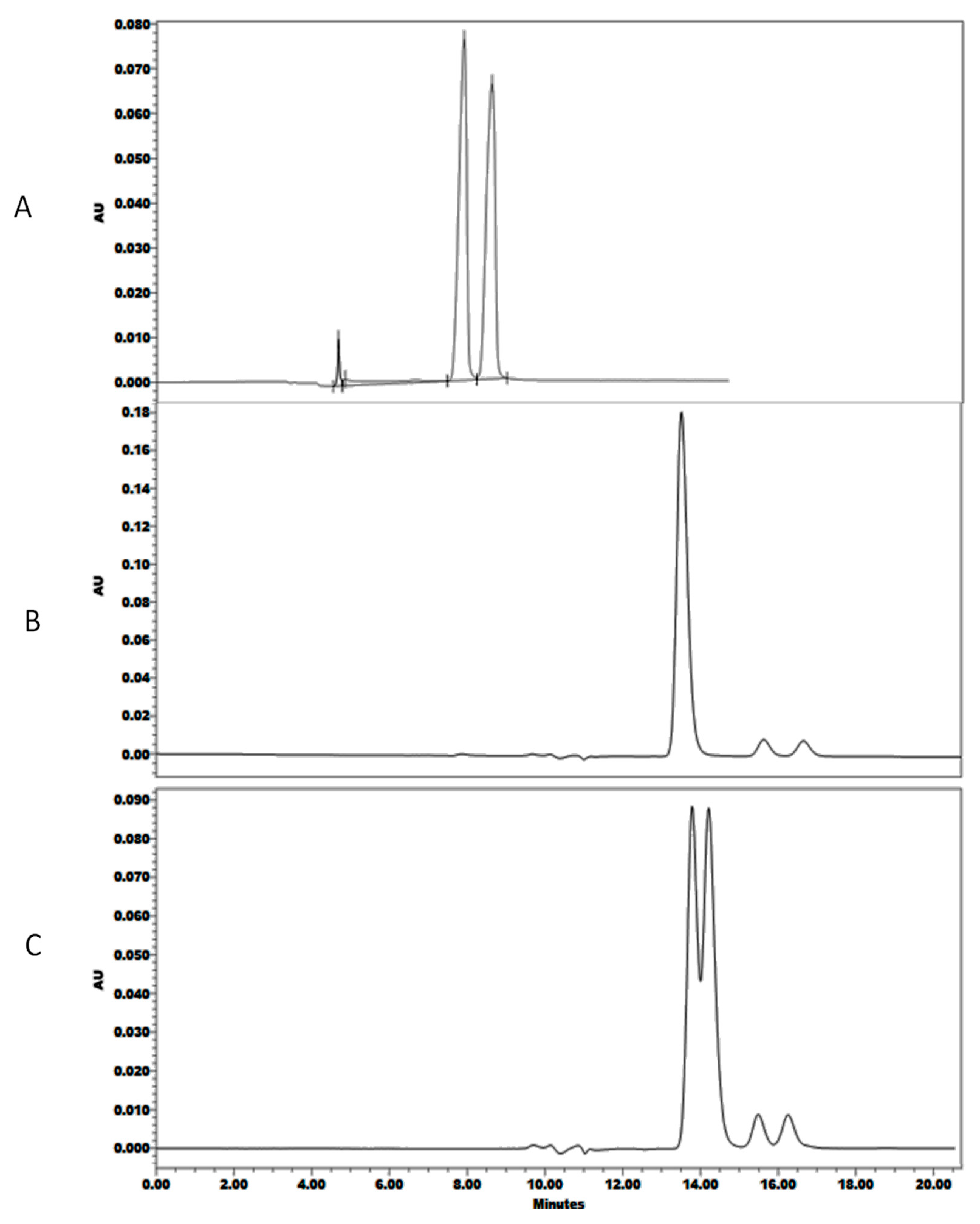
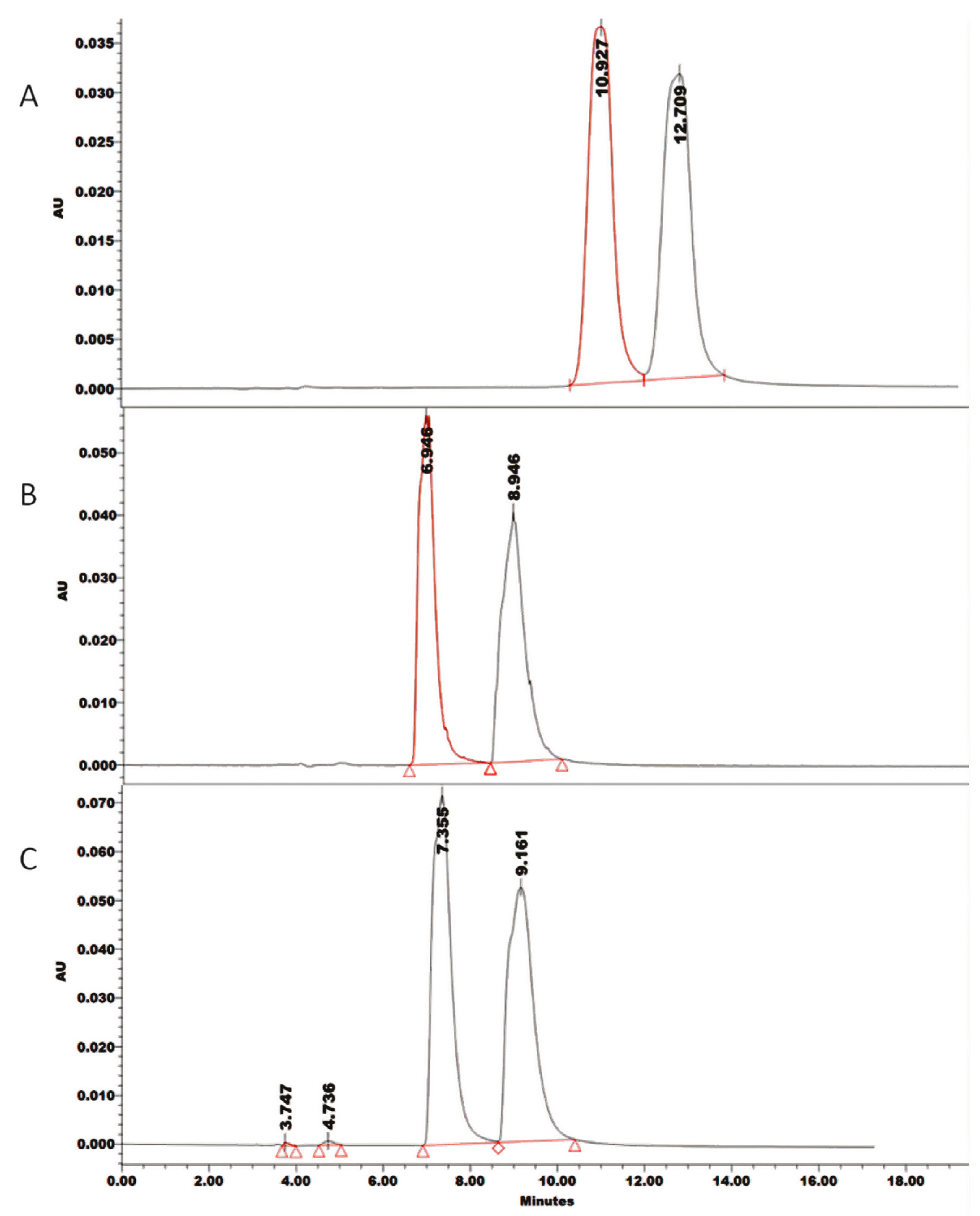
2.1. Screening Design
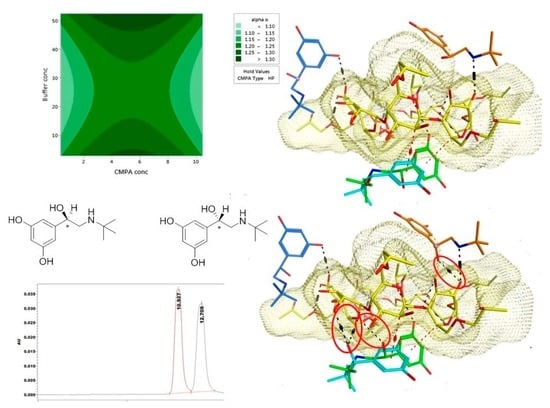
2.2. Optimization Design
2.3. Multiple Linear Regression Models
3. Discussion
4. Materials and Methods
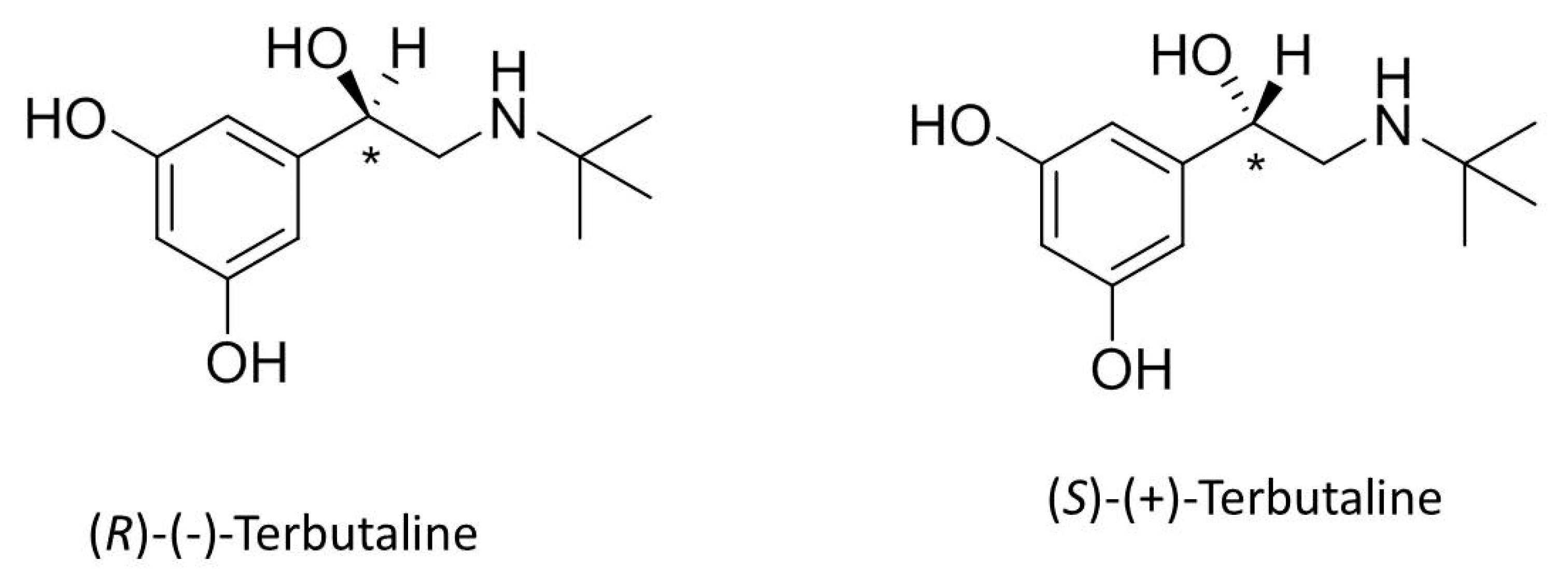
4.1. Standards and Reagents
4.2. Instrumentation
4.3. Software
4.4. Standard Preparation
4.5. Analytical Target Profile
4.6. Plackett-Burman Screening Design for UHPLC Analysis
4.7. Multiple Linear Regression Analysis
4.8. 3D Molecular Modeling
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Peraman, R.; Bhadraya, K.; Padmanabha Reddy, Y. Analytical quality by design: A tool for regulatory flexibility and robust analytics. Int. J. Anal. Chem. 2015, 2015, 868727. [Google Scholar] [CrossRef] [PubMed]
- Torbeck, L.D. OOS, OOT, OOC, and OOSC. Pharm. Technol. 2011, 35, 46–47. [Google Scholar]
- Parr, M.K.; Schmidt, A.H. Life cycle management of analytical methods. J. Pharm. Biomed. Anal. 2018, 147, 506–517. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, P.S.; Beg, S.; Katare, O.P.; Singh, B. QbD-Driven Development and Validation of a HPLC Method for Estimation of Tamoxifen Citrate with Improved Performance. J. Chromatogr. Sci. 2016, 54, 1373–1384. [Google Scholar] [CrossRef] [PubMed]
- Hibbert, D.B. Experimental design in chromatography: A tutorial review. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2012, 910, 2–13. [Google Scholar] [CrossRef]
- Ferreira, S.L.; Bruns, R.E.; Ferreira, H.S.; Matos, G.D.; David, J.M.; Brandao, G.C.; da Silva, E.G.; Portugal, L.A.; dos Reis, P.S.; Souza, A.S.; et al. Box-Behnken design: An alternative for the optimization of analytical methods. Anal. Chim. Acta 2007, 597, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Szekely, G.; Henriques, B.; Gil, M.; Alvarez, C. Experimental design for the optimization and robustness testing of a liquid chromatography tandem mass spectrometry method for the trace analysis of the potentially genotoxic 1,3-diisopropylurea. Drug Test. Anal. 2014, 6, 898–908. [Google Scholar] [CrossRef] [PubMed]
- Eliasson, M.; Rannar, S.; Madsen, R.; Donten, M.A.; Marsden-Edwards, E.; Moritz, T.; Shockcor, J.P.; Johansson, E.; Trygg, J. Strategy for optimizing LC-MS data processing in metabolomics: A design of experiments approach. Anal. Chem. 2012, 84, 6869–6876. [Google Scholar] [CrossRef] [PubMed]
- George, N.; Herz, M.; Aboul-Enein, H.Y.; Shihata, L.; Hanafi, R. Surface Design of Enantiomeric HPLC Separation on Vancomycin and Teicoplanin-Based Stationary Phases, a Tool for Chiral Recognition of Model beta-Blockers. J. Chromatogr. Sci. 2019, 57, 485–494. [Google Scholar] [CrossRef]
- Brandmaier, S.; Tetko, I.V. Robustness in experimental design: A study on the reliability of selection approaches. Comput. Struct. Biotechnol. J. 2013, 7, e201305002. [Google Scholar] [CrossRef][Green Version]
- Letzel, T.E. Design of Experiment Strategy for an Electrospray Ionization Optimization in Supercritical Fluid Chromatography–Mass Spectrometry Coupling. LCGC Eur. 2019, 32, 526–535. [Google Scholar]
- Flaherty, P.; Davis, R.W. Robust Optimization of Biological Protocols. Technometrics 2015, 57, 234–244. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Torres-Lapasio, J.R.; Pous-Torres, S.; Baeza-Baeza, J.J.; Garcia-Alvarez-Coque, M.C. Optimal experimental designs in RPLC at variable solvent content and pH based on prediction error surfaces. Anal. Bioanal. Chem. 2011, 400, 1217–1230. [Google Scholar] [CrossRef]
- Hanafi, R.S.; Lammerhofer, M. Response surface methodology for the determination of the design space of enantiomeric separations on cinchona-based zwitterionic chiral stationary phases by high performance liquid chromatography. J. Chromatogr. A 2018, 1534, 55–63. [Google Scholar] [CrossRef] [PubMed]
- De Klerck, K.; Vander Heyden, Y.; Mangelings, D. Pharmaceutical-enantiomers resolution using immobilized polysaccharide-based chiral stationary phases in supercritical fluid chromatography. J. Chromatogr. A 2014, 1328, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Novakova, L.; Dousa, M. General screening and optimization strategy for fast chiral separations in modern supercritical fluid chromatography. Anal. Chim. Acta 2017, 950, 199–210. [Google Scholar] [CrossRef]
- Zhang, T.; Nguyen, D.; Franco, P. Enantiomer resolution screening strategy using multiple immobilised polysaccharide-based chiral stationary phases. J. Chromatogr. A 2008, 1191, 214–222. [Google Scholar] [CrossRef]
- Aneja, R.; Luthra, P.M.; Ahuja, S. High-performance liquid chromatography separation of enantiomers of mandelic acid and its analogs on a chiral stationary phase. Chirality 2010, 22, 479–485. [Google Scholar] [CrossRef]
- Peng, Y.; He, Q.S.; Cai, J. Enantioseparation of Citalopram by RP-HPLC, Using Sulfobutyl Ether-beta-Cyclodextrin as a Chiral Mobile Phase Additive. Int. J. Anal. Chem. 2016, 2016, 1231386. [Google Scholar] [CrossRef]
- Ye, J.; Yu, W.; Chen, G.; Shen, Z.; Zeng, S. Enantiomeric separation of 2-arylpropionic acid nonsteroidal anti-inflammatory drugs by HPLC with hydroxypropyl-beta-cyclodextrin as chiral mobile phase additive. Biomed. Chromatogr. 2010, 24, 799–807. [Google Scholar] [CrossRef]
- Tong, S.; Zhang, H.; Yan, J. Analytical Enantioseparation of β-Substituted-2-Phenylpropionic Acids by High-Performance Liquid Chromatography with Hydroxypropyl-β-Cyclodextrin as Chiral Mobile Phase Additive. J. Chromatogr. Sci. 2016, 54, 593–597. [Google Scholar] [CrossRef]
- Jandera, P.; Buncekova, S.; Planeta, J. Separation of isomeric naphthalenesulphonic acids by micro high-performance liquid chromatography with mobile phases containing cyclodextrin. J. Chromatogr. A 2000, 871, 139–152. [Google Scholar] [CrossRef]
- Shimada, K.; Masue, T.; Toyoda, K.; Takani, M.; Nambara, T. The Utility of Cyclodextrin in Mobile Phase for High-Performance Liquid Chromatographic Separation of Isomeric Estrogens. J. Liq. Chromatogr. 1988, 11, 1475–1484. [Google Scholar] [CrossRef]
- de Boer, T.; de Zeeuw, R.A.; de Jong, G.J.; Ensing, K. Recent innovations in the use of charged cyclodextrins in capillary electrophoresis for chiral separations in pharmaceutical analysis. ELECTROPHORESIS 2000, 21, 3220–3239. [Google Scholar] [CrossRef]
- Gratz, S.R.; Stalcup, A.M. Enantiomeric separations of terbutaline by CE with a sulfated beta-cyclodextrin chiral selector: A quantitative binding study. Anal. Chem. 1998, 70, 5166–5171. [Google Scholar] [CrossRef]
- Abdel-Megied, A.M.; Hanafi, R.S.; Aboul-Enein, H.Y. A chiral enantioseparation generic strategy for anti-Alzheimer and antifungal drugs by short end injection capillary electrophoresis using an experimental design approach. Chirality 2018, 30, 165–176. [Google Scholar] [CrossRef]
- Zhou, J.; Ai, F.; Zhou, B.; Tang, J.; Ng, S.C.; Tang, W. Hydroxyethylammonium monosubstituted cyclodextrin as chiral selector for capillary electrophoresis. Anal. Chim. Acta 2013, 800, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Borgstrom, L.; Liu, C.X.; Walhagen, A. Pharmacokinetics of the Enantiomers of Terbutaline after Repeated Oral Dosing with Racemic Terbutaline. Chirality 1989, 1, 174–177. [Google Scholar] [CrossRef]
- Beng, H.; Zhang, H.; Jayachandra, R.; Li, J.; Wu, J.; Tan, W. Enantioselective resolution of Rac-terbutaline and evaluation of optically pure R-terbutaline hydrochloride as an efficient anti-asthmatic drug. Chirality 2018, 30, 759–768. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. FDA’s Policy Statement for the Development of New Stereoisomeric Drugs. Available online: https://onlinelibrary.wiley.com/doi/10.1002/chir.530040513 (accessed on 26 April 2019).
- Ameyibor, E.; Stewart, J.T. Enantiomeric HPLC Separation of Selected Chiral Drugs Using Native and Derivatized β- Cyclodextrins as Chiral Mobile Phase Additives. J. Liq. Chromatogr. Relat. Technol. 1997, 20, 855–869. [Google Scholar] [CrossRef]
- Walhagen, A.; Edholm, L.E. Chiral separation on achiral stationary phases with different functionalities using β-cyclodextrin in the mobile phase and application to bioanalysis and coupled columns. Chromatographia 1991, 32, 215–223. [Google Scholar] [CrossRef]
- Ngim, K.K.; Zhong, Q.; Mistry, K.; Chetwyn, N. Effect of sulfobutyl ether beta-cyclodextrin modifier on selectivity of reversed phase hplc separations. J. Liq Chromatogr Relat. Technol. 2012, 35, 2845–2859. [Google Scholar] [CrossRef]
- Servais, A.C.; Fillet, M.; Chiap, P.; Dewe, W.; Hubert, P.; Crommen, J. Enantiomeric separation of basic compounds using heptakis(2,3-di-O-methyl-6-O-sulfo)-beta-cyclodextrin in combination with potassium camphorsulfonate in nonaqueous capillary electrophoresis: Optimization by means of an experimental design. Electrophoresis 2004, 25, 2701–2710. [Google Scholar] [CrossRef] [PubMed]
- Salvador, A.; Herbreteau, B.; Dreux, M.; Karlsson, A.; Gyllenhaal, O. Chiral supercritical fluid chromatography on porous graphitic carbon using commercial dimethyl beta-cyclodextrins as mobile phase additive. J. Chromatogr. A 2001, 929, 101–112. [Google Scholar] [CrossRef]
- Williams, K.L.; Sander, L.C.; Wise, S.A. Comparison of liquid and supercritical fluid chromatography for the separation of enantiomers on chiral stationary phases. J. Pharm. Biomed. Anal. 1997, 15, 1789–1799. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. FDA’s Pharmaceutical cGMPs for the 21st Century—A Risk-Based Approach. Final Report; September 2014. Available online: http://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/Manufacturing/QuestionsandAnswersonCurrentGoodManufacturingPracticescGMPforDrugs/UCM176374.pdf (accessed on 26 April 2019).
- Nethercote, P.W.; Ermer, J. Analytical Validation within the Pharmaceutical Lifecycle. In Method Validation in Pharmaceutical Analysis: A Guide to Best Practice, 2nd ed.; Ermer, J., Nethercote, P.W., Eds.; Wiley-VCH: Weinheim, Germany, 2014; pp. 1–10. [Google Scholar]
- Sun, P.; Wang, C.; Breitbach, Z.S.; Zhang, Y.; Armstrong, D.W. Development of new HPLC chiral stationary phases based on native and derivatized cyclofructans. Anal. Chem. 2009, 81, 10215–10226. [Google Scholar] [CrossRef]
- Moskaľová, M.; Kozlov, O.; Gondová, T.; Budovská, M.; Armstrong, D.W. HPLC Enantioseparation of Novel Spirobrassinin Analogs on the Cyclofructan Chiral Stationary Phases. Chromatographia 2017, 80, 53–62. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Run Order | Chiral Mobile Phase Additive CMPA Type | CMPA Conc. (mM) | pH | Buffer Conc. (mM) | Flow Rate (mL/min) | %B Change | tRLast (min) | α |
|---|---|---|---|---|---|---|---|---|
| 1 | SBE-β-CD | 0.6 | 6 | 50 | 0.05 | 0.04 | 152.41 | 1.00 |
| 2 | HP-β-CD | 10 | 6 | 50 | 0.05 | 0.04 | 35.17 | 1.16 |
| 3 | HP β-CD | 0.6 | 2.5 | 5 | 0.05 | 0.02 | 69.82 | 1.27 |
| 4 | HP-β-CD | 10 | 6 | 5 | 0.3 | 0.02 | 6.20 | 1.16 |
| 5 | SBE-β-CD | 0.6 | 6 | 5 | 0.05 | 0.02 | >240 | 1.00 |
| 6 | SBE-β-CD | 10 | 6 | 5 | 0.3 | 0.04 | 70.65 | 1.14 |
| 7 | SBE-β-CD | 0.6 | 2.5 | 5 | 0.3 | 0.04 | 6.68 | 1.20 |
| 8 | HP-β-CD | 10 | 2.5 | 5 | 0.05 | 0.04 | 39.70 | 1.18 |
| 9 | HP-β-CD | 0.6 | 6 | 50 | 0.3 | 0.02 | 6.97 | 1.19 |
| 10 | SBE-β-CD | 10 | 2.5 | 50 | 0.05 | 0.02 | 241.06 | 1.11 |
| 11 | SBE-β-CD | 10 | 2.5 | 50 | 0.3 | 0.02 | 100.35 | 1.14 |
| 12 | HP-β-CD | 0.6 | 2.5 | 50 | 0.3 | 0.04 | 66.12 | 1.00 |
| Run Order | CMPA Conc. (mM) | Buffer Conc. (mM) | CMPA Type | tRLast (min) | α |
|---|---|---|---|---|---|
| 1 | 5.40 | 52.25 | HP-β-CD | 11.11 | 1.32 |
| 2 | 10.46 | 27.50 | HP-β-CD | 34.50 | 1.01 |
| 3 | 5.40 | 27.50 | SBE-β-CD | 69.20 | 1.10 |
| 4 | 5.40 | 27.50 | HP-β-CD | 9.62 | 1.23 |
| 5 | 10.00 | 50.00 | HP-β-CD | 7.89 | 1.25 |
| 6 | 0.80 | 50.00 | SBE-β-CD | 18.61 | 1.00 |
| 7 | 5.40 | 27.50 | HP-β-CD | 7.98 | 1.26 |
| 8 | 5.40 | 2.75 | HP-β-CD | 8.58 | 1.35 |
| 9 | 0.34 | 27.50 | SBE-β-CD | 89.16 | 1.00 |
| 10 | 5.40 | 27.50 | SBE-β-CD | 75.71 | 1.23 |
| 11 | 5.40 | 27.50 | HP-β-CD | 9.69 | 1.22 |
| 12 | 10.00 | 50.00 | SBE-β-CD | 9.74 | 1.00 |
| 13 | 0.80 | 5.00 | HP-β-CD | 11.91 | 1.19 |
| 14 | 0.34 | 27.50 | HP-β-CD | 13.45 | 1.16 |
| 15 | 0.80 | 50.00 | HP-β-CD | 10.09 | 1.24 |
| 16 | 5.40 | 27.50 | HP-β-CD | 7.63 | 1.24 |
| 17 | 10.00 | 5.00 | HP-β-CD | 6.43 | 1.17 |
| 18 | 5.40 | 27.50 | SBE-β-CD | 12.19 | 1.12 |
| 19 | 5.40 | 2.75 | SBE-β-CD | 42.30 | 1.23 |
| 20 | 10.46 | 27.50 | SBE-β-CD | 24.01 | 1.22 |
| 21 | 10.00 | 5.00 | SBE-β-CD | 8.14 | 1.00 |
| 22 | 5.40 | 27.50 | SBE-β-CD | 65.12 | 1.22 |
| 23 | 5.40 | 27.50 | HP-β-CD | 9.38 | 1.25 |
| 24 | 0.80 | 5.00 | SBE-β-CD | 6.50 | 1.07 |
| 25 | 5.40 | 27.50 | SBE-β-CD | 45.58 | 1.20 |
| 26 | 5.40 | 52.25 | SBE-β-CD | 56.04 | 1.19 |
| Factor Level | CMPA Type | CMPA Conc. (mM) | pH | Buffer Conc. (mM) | Flow Rate (mL/min) | %B Change/min |
|---|---|---|---|---|---|---|
| Low (−1) | HP-β-CD | 0.6 | 2.5 | 5 | 0.05 | 0.02 |
| High (+1) | SBE-β-CD | 10 | 6 | 50 | 0.3 | 0.04 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aboushady, D.; Parr, M.K.; Hanafi, R.S. Quality-by-Design Is a Tool for Quality Assurance in the Assessment of Enantioseparation of a Model Active Pharmaceutical Ingredient. Pharmaceuticals 2020, 13, 364. https://doi.org/10.3390/ph13110364
Aboushady D, Parr MK, Hanafi RS. Quality-by-Design Is a Tool for Quality Assurance in the Assessment of Enantioseparation of a Model Active Pharmaceutical Ingredient. Pharmaceuticals. 2020; 13(11):364. https://doi.org/10.3390/ph13110364
Chicago/Turabian StyleAboushady, Dina, Maria Kristina Parr, and Rasha S. Hanafi. 2020. "Quality-by-Design Is a Tool for Quality Assurance in the Assessment of Enantioseparation of a Model Active Pharmaceutical Ingredient" Pharmaceuticals 13, no. 11: 364. https://doi.org/10.3390/ph13110364
APA StyleAboushady, D., Parr, M. K., & Hanafi, R. S. (2020). Quality-by-Design Is a Tool for Quality Assurance in the Assessment of Enantioseparation of a Model Active Pharmaceutical Ingredient. Pharmaceuticals, 13(11), 364. https://doi.org/10.3390/ph13110364