Mitogen-Activated Protein Kinase Inhibitors and T-Cell-Dependent Immunotherapy in Cancer



 ,
,
Abstract
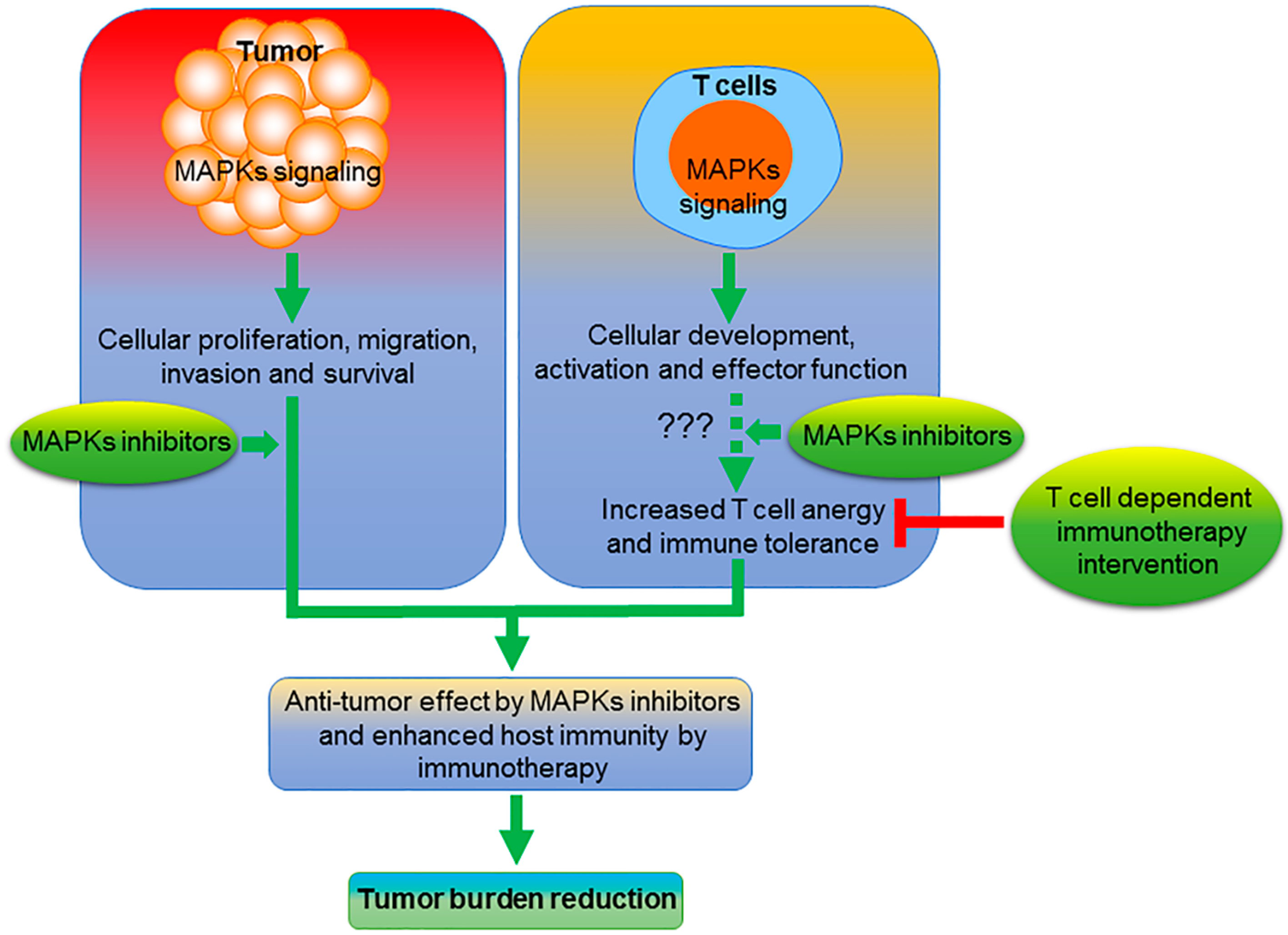
1. Introduction
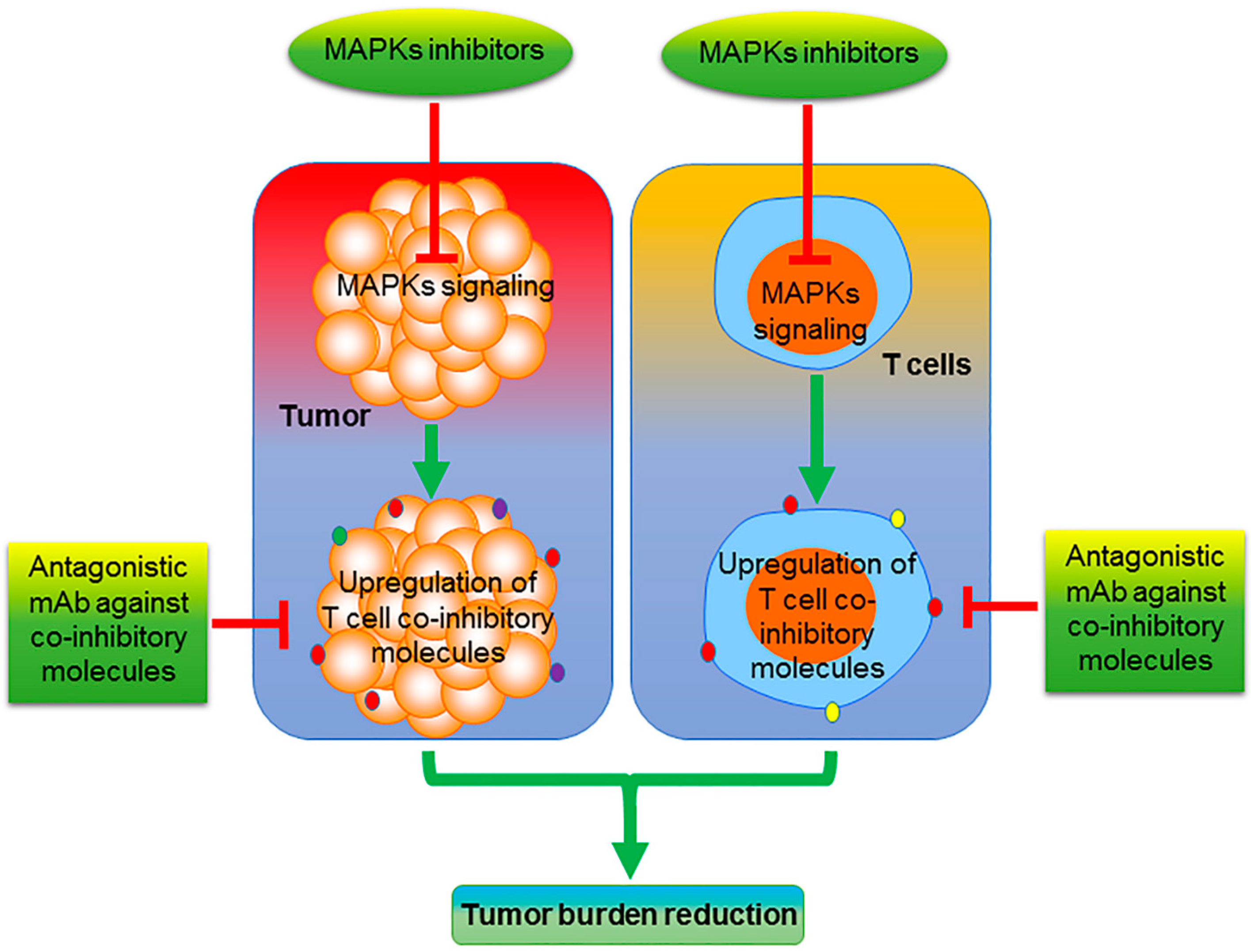
2. MEK/ERK Inhibition
3. JNK Inhibition
4. p38 MAPK Inhibition
5. Other MAPK Family Members
6. Future Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schaeffer, H.J.; Weber, M.J. Mitogen-activated protein kinases: Specific messages from ubiquitous messengers. Mol. Cell. Biol. 1999, 19, 2435–2444. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Jacobson, K.; Schaller, M.D. MAP kinases and cell migration. J. Cell Sci. 2004, 117, 4619–4628. [Google Scholar] [CrossRef] [PubMed]
- Sohn, S.J.; Sarvis, B.K.; Cado, D.; Winoto, A. ERK5 MAPK regulates embryonic angiogenesis and acts as a hypoxia-sensitive repressor of vascular endothelial growth factor expression. J. Biol. Chem. 2002, 277, 43344–43351. [Google Scholar] [CrossRef] [PubMed]
- Krueger, J.S.; Keshamouni, V.G.; Atanaskova, N.; Reddy, K.B. Temporal and quantitative regulation of mitogen-activated protein kinase (MAPK) modulates cell motility and invasion. Oncogene 2001, 20, 4209–4218. [Google Scholar] [CrossRef]
- Hickson, J.A.; Huo, D.Z.; Vander Griend, D.J.; Lin, A.N.; Rinker-Schaeffer, C.W.; Yamada, S.D. The p38 kinases MKK4 and MKK6 suppress metastatic colonization in human ovarian carcinoma. Cancer Res. 2006, 66, 2264–2270. [Google Scholar] [CrossRef]
- Wada, T.; Penninger, J.M. Mitogen-activated protein kinases in apoptosis regulation. Oncogene 2004, 23, 2838–2849. [Google Scholar] [CrossRef]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef]
- Amaral, T.; Sinnberg, T.; Meier, F.; Krepler, C.; Levesque, M.; Niessner, H.; Garbe, C. The mitogen-activated protein kinase pathway in melanoma part I—Activation and primary resistance mechanisms to BRAF inhibition. Eur. J. Cancer 2017, 73, 85–92. [Google Scholar] [CrossRef]
- Ellerhorst, J.A.; Ekmekcioglu, S.; Johnson, M.K.; Cooke, C.P.; Johnson, M.M.; Grimm, E.A. Regulation of iNOS by the p44/42 mitogen-activated protein kinase pathway in human melanoma. Oncogene 2006, 25, 3956–3962. [Google Scholar] [CrossRef]
- Nan, X.; Tamguney, T.M.; Collisson, E.A.; Lin, L.J.; Pitt, C.; Galeas, J.; Lewis, S.; Gray, J.W.; McCormick, F.; Chu, S. Ras-GTP dimers activate the Mitogen-Activated Protein Kinase (MAPK) pathway. Proc. Natl. Acad. Sci. USA 2015, 112, 7996–8001. [Google Scholar] [CrossRef] [PubMed]
- Schonwasser, D.C.; Marais, R.M.; Marshall, C.J.; Parker, P.J. Activation of the mitogen-activated protein kinase/extracellular signal-regulated kinase pathway by conventional, novel, and atypical protein kinase C isotypes. Mol. Cell. Biol. 1998, 18, 790–798. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, A.; Zehorai, E.; Procaccia, S.; Seger, R. The MAPK cascades: Signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim. Biophys. Acta 2011, 1813, 1619–1633. [Google Scholar] [CrossRef] [PubMed]
- Cicenas, J.; Zalyte, E.; Rimkus, A.; Dapkus, D.; Noreika, R.; Urbonavicius, S. JNK, p38, ERK, and SGK1 inhibitors in cancer. Cancers 2017, 10, 1. [Google Scholar] [CrossRef]
- Lapinski, P.E.; King, P.D. Regulation of Ras signal transduction during T cell development and activation. Am. J. Clin. Exp. Immunol. 2012, 1, 147–153. [Google Scholar]
- Schafer, P.H.; Wang, L.; Wadsworth, S.A.; Davis, J.E.; Siekierka, J.J. T cell activation signals up-regulate p38 mitogen-activated protein kinase activity and induce TNF-alpha production in a manner distinct from LPS activation of monocytes. J. Immunol. 1999, 162, 659–668. [Google Scholar]
- D’Souza, W.N.; Chang, C.F.; Fischer, A.M.; Li, M.; Hedrick, S.M. The Erk2 MAPK regulates CD8 T cell proliferation and survival. J. Immunol. 2008, 181, 7617–7629. [Google Scholar] [CrossRef]
- Atsaves, V.; Leventaki, V.; Rassidakis, G.Z.; Claret, F.X. AP-1 transcription factors as regulators of immune responses in cancer. Cancers 2019, 11, 1037. [Google Scholar] [CrossRef]
- Dushyanthen, S.; Teo, Z.L.; Caramia, F.; Savas, P.; Mintoff, C.P.; Virassamy, B.; Henderson, M.A.; Luen, S.J.; Mansour, M.; Kershaw, M.H.; et al. Agonist immunotherapy restores T cell function following MEK inhibition improving efficacy in breast cancer. Nat. Commun. 2017, 8, 606. [Google Scholar] [CrossRef]
- Boulton, T.G.; Nye, S.H.; Robbins, D.J.; Ip, N.Y.; Radziejewska, E.; Morgenbesser, S.D.; Depinho, R.A.; Panayotatos, N.; Cobb, M.H.; Yancopoulos, G.D. Erks—A family of protein-serine threonine kinases that are activated and tyrosine phosphorylated in response to insulin and Ngf. Cell 1991, 65, 663–675. [Google Scholar] [CrossRef]
- Li, L.; Zhao, G.D.; Shi, Z.; Qi, L.L.; Zhou, L.Y.; Fu, Z.X. The Ras/Raf/MEK/ERK signaling pathway and its role in the occurrence and development of HCC. Oncol. Lett. 2016, 12, 3045–3050. [Google Scholar] [CrossRef] [PubMed]
- Yujiri, T.; Sather, S.; Fanger, G.R.; Johnson, G.L. Role of MEKK1 in cell survival and activation of JNK and ERK pathways defined by targeted gene disruption. Science 1998, 282, 1911–1914. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.M.; Katayama, C.D.; Pages, G.; Pouyssegur, J.; Hedrick, S.M. The role of erk1 and erk2 in multiple stages of T cell development. Immunity 2005, 23, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Lafont, V.; Ottones, F.; Liautard, J.; Favero, J. Evidence for a p21 (ras)/Raf-1/MEK-1/ERK-2-independent pathway in stimulation of IL-2 gene transcription in human primary T lymphocytes. J. Biol. Chem. 1999, 274, 25743–25748. [Google Scholar] [CrossRef] [PubMed]
- Nekrasova, T.; Shive, C.; Gao, Y.H.; Kawamura, K.; Guardia, R.; Landreth, G.; Forsthuber, T.G. ERK1-deficient mice show normal T cell effector function and are highly susceptible to experimental autoimmune encephalomyelitis. J. Immunol. 2005, 175, 2374–2380. [Google Scholar] [CrossRef] [PubMed]
- Dillon, T.J.; Carey, K.D.; Wetzel, S.A.; Parker, D.C.; Stork, P.J. Regulation of the small GTPase Rap1 and extracellular signal-regulated kinases by the costimulatory molecule CTLA-4. Mol. Cell. Biol. 2005, 25, 4117–4128. [Google Scholar] [CrossRef]
- Ohnishi, H.; Takeda, K.; Domenico, J.; Lucas, J.J.; Miyahara, N.; Swasey, C.H.; Dakhama, A.; Gelfand, E.W. Mitogen-activated protein kinase/extracellular signal-regulated kinase 1/2-dependent pathways are essential for CD8(+) T cell-mediated airway hyperresponsiveness and inflammation. J. Allergy Clin. Immun. 2009, 123, 249–257. [Google Scholar] [CrossRef]
- Ohori, M.; Kinoshita, T.; Okubo, M.; Sato, K.; Yamazaki, A.; Arakawa, H.; Nishimura, S.; Inamura, N.; Nakajima, H.; Neya, M.; et al. Identification of a selective ERK inhibitor and structural determination of the inhibitor-ERK2 complex. Biochem. Biophys. Res. Commun. 2005, 336, 357–363. [Google Scholar] [CrossRef]
- Sullivan, R.J.; Infante, J.R.; Janku, F.; Wong, D.J.L.; Sosman, J.A.; Keedy, V.; Patel, M.R.; Shapiro, G.I.; Mier, J.W.; Tolcher, A.W.; et al. First-in-class ERK1/2 inhibitor ulixertinib (BVD-523) in patients with MAPK mutant advanced solid tumors: Results of a phase I dose-escalation and expansion study. Cancer Discov. 2018, 8, 184–195. [Google Scholar] [CrossRef]
- Aaron, C.P.; Tandri, H.; Barr, R.G.; Johnson, W.C.; Bagiella, E.; Chahal, H.; Jain, A.; Kizer, J.R.; Bertoni, A.G.; Lima, J.A.; et al. Physical activity and right ventricular structure and function: The MESA-right ventricle study. Am. J. Respir. Crit. Care Med. 2011, 183, 396–404. [Google Scholar] [CrossRef]
- Kirouac, D.; Schaefer, G.; Chan, J.; Merchant, M.; Orr, C.; Liu, L.; Huang, A.; Moffat, J.; Gadkar, K.; Ramanujan, S. Clinical responses to ERK inhibitor (GDC-0994) treatment combinations predicted using a Quantitative Systems Pharmacology model of MAPK signaling in BRAF(V600E)-mutant colorectal cancer. Eur. J. Cancer 2016, 69, S20. [Google Scholar] [CrossRef]
- Moschos, S.J.; Sullivan, R.J.; Hwu, W.J.; Ramanathan, R.K.; Adjei, A.A.; Fong, P.C.; Shapira-Frommer, R.; Tawbi, H.A.; Rubino, J.; Rush, T.S.; et al. Development of MK-8353, an orally administered ERK1/2 inhibitor, in patients with advanced solid tumors. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Bavaria, M.N.; Jin, S.; Ray, R.M.; Johnson, L.R. The mechanism by which MEK/ERK regulates JNK and p38 activity in polyamine depleted IEC-6 cells during apoptosis. Apoptosis 2014, 19, 467–479. [Google Scholar] [CrossRef] [PubMed]
- Ritt, D.A.; Abreu-Blanco, M.T.; Bindu, L.; Durrant, D.E.; Zhou, M.; Specht, S.I.; Stephen, A.G.; Holderfield, M.; Morrison, D.K. Inhibition of Ras/Raf/MEK/ERK pathway signaling by a stress-induced phospho-regulatory circuit. Mol. Cell 2016, 64, 875–887. [Google Scholar] [CrossRef] [PubMed]
- Abe, H.; Kikuchi, S.; Hayakawa, K.; Iida, T.; Nagahashi, N.; Maeda, K.; Sakamoto, J.; Matsumoto, N.; Miura, T.; Matsumura, K.; et al. Discovery of a highly potent and selective MEK inhibitor: GSK1120212 (JTP-74057 DMSO solvate). ACS Med. Chem. Lett. 2011, 2, 320–324. [Google Scholar] [CrossRef]
- Shapiro, G.I.; LoRusso, P.; Kwak, E.; Pandya, S.; Rudin, C.M.; Kurkjian, C.; Cleary, J.M.; Pilat, M.J.; Jones, S.; de Crespigny, A.; et al. Phase Ib study of the MEK inhibitor cobimetinib (GDC-0973) in combination with the PI3K inhibitor pictilisib (GDC-0941) in patients with advanced solid tumors. Investig. New Drugs 2019. [Google Scholar] [CrossRef]
- Bardia, A.; Gounder, M.; Rodon, J.; Janku, F.; Lolkema, M.P.; Stephenson, J.J.; Bedard, P.L.; Schuler, M.; Sessa, C.; LoRusso, P.; et al. Phase Ib Study of Combination Therapy with MEK Inhibitor Binimetinib and Phosphatidylinositol 3-Kinase Inhibitor Buparlisib in Patients with advanced solid tumors with RAS/RAF Alterations. Oncologist 2019. [Google Scholar] [CrossRef]
- Banerjee, A.; Jakacki, R.I.; Onar-Thomas, A.; Wu, S.; Nicolaides, T.; Young Poussaint, T.; Fangusaro, J.; Phillips, J.; Perry, A.; Turner, D.; et al. A phase I trial of the MEK inhibitor selumetinib (AZD6244) in pediatric patients with recurrent or refractory low-grade glioma: A Pediatric Brain Tumor Consortium (PBTC) study. Neuro-Oncology 2017, 19, 1135–1144. [Google Scholar] [CrossRef]
- Stutvoet, T.S.; Kol, A.; de Vries, E.G.E.; de Bruyn, M.; Fehrmann, R.S.N.; van Scheltinga, A.G.T.T.; de Jong, S. MAPK pathway activity plays a key role in PD-L1 expression of lung adenocarcinoma cells. J. Pathol. 2019, 249, 52–64. [Google Scholar] [CrossRef]
- Ebert, P.J.R.; Cheung, J.; Yang, Y.; McNamara, E.; Hong, R.; Moskalenko, M.; Gould, S.E.; Maecker, H.; Irving, B.A.; Kim, J.M.; et al. MAP kinase inhibition promotes T cell and anti-tumor activity in combination with PD-L1 checkpoint blockade. Immunity 2016, 44, 609–621. [Google Scholar] [CrossRef]
- Davis, R.J. Signal transduction by the JNK group of MAP kinases. Cell 2000, 103, 239–252. [Google Scholar] [CrossRef]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [PubMed]
- O’Hayre, M.; Degese, M.S.; Gutkind, J.S. Novel insights into G protein and G protein-coupled receptor signaling in cancer. Curr. Opin. Cell Biol. 2014, 27, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; Brenner, D.A.; Karin, M. A liver full of JNK: Signaling in regulation of cell function and disease pathogenesis, and clinical approaches. Gastroenterology 2012, 143, 307–320. [Google Scholar] [CrossRef]
- Dong, C.; Yang, D.D.; Tournier, C.; Whitmarsh, A.J.; Xu, J.; Davis, R.J.; Flavell, R.A. JNK is required for effector T-cell function but not for T-cell activation. Nature 2000, 405, 91–94. [Google Scholar] [CrossRef]
- Behrens, A.; Sabapathy, K.; Graef, I.; Cleary, M.; Crabtree, G.R.; Wagner, E.F. Jun N-terminal kinase 2 modulates thymocyte apoptosis and T cell activation through c-Jun and nuclear factor of activated T cell (NF-AT). Proc. Natl. Acad. Sci. USA 2001, 98, 1769–1774. [Google Scholar] [CrossRef]
- Su, B.; Cheng, J.K.; Yang, J.H.; Guo, Z.J. MEKK2 is required for T-cell receptor signals in JNK activation and interleukin-2 gene expression. J. Biol. Chem. 2001, 276, 14784–14790. [Google Scholar] [CrossRef]
- Conze, D.; Krahl, T.; Kennedy, N.; Weiss, L.; Lumsden, J.; Hess, P.; Flavell, R.A.; Le Gros, G.; Davis, R.J.; Rincon, M. C-jun NH2-terminal kinase (JNK)1 and JNK2 have distinct roles in CD8(+) T cell activation. J. Exp. Med. 2002, 195, 811–823. [Google Scholar] [CrossRef]
- Bennett, B.L.; Sasaki, D.T.; Murray, B.W.; O’Leary, E.C.; Sakata, S.T.; Xu, W.; Leisten, J.C.; Motiwala, A.; Pierce, S.; Satoh, Y.; et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. USA 2001, 98, 13681–13686. [Google Scholar] [CrossRef]
- Zhang, T.; Inesta-Vaquera, F.; Niepel, M.; Zhang, J.; Ficarro, S.B.; Machleidt, T.; Xie, T.; Marto, J.A.; Kim, N.; Sim, T.; et al. Discovery of potent and selective covalent inhibitors of JNK. Chem. Biol. 2012, 19, 140–154. [Google Scholar] [CrossRef]
- Ma, F.Y.; Flanc, R.S.; Tesch, G.H.; Han, Y.; Atkins, R.C.; Bennett, B.L.; Friedman, G.C.; Fan, J.H.; Nikolic-Paterson, D.J. A pathogenic role for c-Jun amino-terminal kinase signaling in renal fibrosis and tubular cell apoptosis. J. Am. Soc. Nephrol. 2007, 18, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Kuramoto, K.; Takeda, H.; Watarai, H.; Sakaki, H.; Seino, S.; Seino, M.; Suzuki, S.; Kitanaka, C. The novel JNK inhibitor AS602801 inhibits cancer stem cells in vitro and in vivo. Oncotarget 2016, 7, 27021–27032. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Spigolon, G.; Bonny, C.; Culman, J.; Vercelli, A.; Herdegen, T. The JNK inhibitor D-JNKI-1 blocks apoptotic JNK signaling in brain mitochondria. Mol. Cell. Neurosci. 2012, 49, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Stebbins, J.L.; De, S.K.; Machleidt, T.; Becattini, B.; Vazquez, J.; Kuntzen, C.; Chen, L.H.; Cellitti, J.F.; Riel-Mehan, M.; Emdadi, A.; et al. Identification of a new JNK inhibitor targeting the JNK-JIP interaction site. Proc. Natl. Acad. Sci. USA 2008, 105, 16809–16813. [Google Scholar] [CrossRef]
- Bubici, C.; Papa, S. JNK signalling in cancer: In need of new, smarter therapeutic targets. Br. J. Pharmacol. 2014, 171, 24–37. [Google Scholar] [CrossRef]
- Mehrotra, S.; Chhabra, A.; Chattopadhyay, S.; Dorsky, D.I.; Chakraborty, N.G.; Mukherji, B. Rescuing melanoma epitope-specific cytolytic T lymphocytes from activation-induced cell death, by SP600125, an inhibitor of JNK: Implications in cancer immunotherapy. J. Immunol. 2004, 173, 6017–6024. [Google Scholar] [CrossRef]
- Hu, M.C.; Wang, Y.P.; Mikhail, A.; Qiu, W.R.; Tan, T.H. Murine p38-delta mitogen-activated protein kinase, a developmentally regulated protein kinase that is activated by stress and proinflammatory cytokines. J. Biol. Chem. 1999, 274, 7095–7102. [Google Scholar] [CrossRef]
- Cuenda, A.; Rousseau, S. P38 MAP-kinases pathway regulation, function and role in human diseases. Biochim. Biophys. Acta 2007, 1773, 1358–1375. [Google Scholar] [CrossRef]
- Donnelly, S.M.; Paplomata, E.; Peake, B.M.; Sanabria, E.; Chen, Z.; Nahta, R. P38 MAPK contributes to resistance and invasiveness of HER2—Overexpressing breast cancer. Curr. Med. Chem. 2014, 21, 501–510. [Google Scholar] [CrossRef]
- Farhat, F.; Daulay, E.R.; Chrestella, J.; Asnir, R.A.; Yudhistira, A.; Susilo, R.R. Correlation of P38 mitogen-activated protein kinase expression to clinical stage in nasopharyngeal carcinoma. Open Access Maced. J. Med. Sci. 2018, 6, 1982–1985. [Google Scholar] [CrossRef]
- Guo, X.L.; Ma, N.N.; Wang, J.; Song, J.R.; Bu, X.X.; Cheng, Y.; Sun, K.; Xiong, H.Y.; Jiang, G.C.; Zhang, B.H.; et al. Increased p38-MAPK is responsible for chemotherapy resistance in human gastric cancer cells. BMC Cancer 2008, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Naito, Y.; Cope, L.; Naranjo-Suarez, S.; Saunders, T.; Hong, S.M.; Goggins, M.G.; Herman, J.M.; Wolfgang, C.L.; Iacobuzio-Donahue, C.A. Functional p38 MAPK identified by biomarker profiling of pancreatic cancer restrains growth through JNK inhibition and correlates with improved survival. Clin. Cancer Res. 2014, 20, 6200–6211. [Google Scholar] [CrossRef] [PubMed]
- Dodeller, F.; Schulze-Koops, H. The p38 mitogen-activated protein kinase signaling cascade in CD4 T cells. Arthritis Res. Ther. 2006, 8, 205. [Google Scholar] [CrossRef] [PubMed]
- Noubade, R.; Krementsov, D.N.; Del Rio, R.; Thornton, T.; Nagaleekar, V.; Saligrama, N.; Spitzack, A.; Spach, K.; Sabio, G.; Davis, R.J.; et al. Activation of p38 MAPK in CD4 T cells controls IL-17 production and autoimmune encephalomyelitis. Blood 2011, 118, 3290–3300. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Hsu, S.C.; Shih, H.M.; Lai, M.Z. Nuclear factor of activated T cells c is a target of p38 mitogen-activated protein kinase in T cells. Mol. Cell. Biol. 2003, 23, 6442–6454. [Google Scholar] [CrossRef]
- Klein-Hessling, S.; Muhammad, K.; Klein, M.; Pusch, T.; Rudolf, R.; Floter, J.; Qureischi, M.; Beilhack, A.; Vaeth, M.; Kummerow, C.; et al. NFATc1 controls the cytotoxicity of CD8+ T cells. Nat. Commun. 2017, 8, 511. [Google Scholar] [CrossRef]
- Ohkusu-Tsukada, K.; Toda, M.; Udono, H.; Kawakami, Y.; Takahashi, K. Targeted inhibition of IL-10-secreting CD25- Treg via p38 MAPK suppression in cancer immunotherapy. Eur. J. Immunol. 2010, 40, 1011–1021. [Google Scholar] [CrossRef]
- Laufer, S.; Lehmann, F. Investigations of SCIO-469-like compounds for the inhibition of p38 MAP kinase. Bioorg. Med. Chem. Lett. 2009, 19, 1461–1464. [Google Scholar] [CrossRef]
- Kuma, Y.; Sabio, G.; Bain, J.; Shpiro, N.; Marquez, R.; Cuenda, A. BIRB796 inhibits all p38 MAPK isoforms in vitro and in vivo. J. Biol. Chem. 2005, 280, 19472–19479. [Google Scholar] [CrossRef]
- Campbell, R.M.; Anderson, B.D.; Brooks, N.A.; Brooks, H.B.; Chan, E.M.; De Dios, A.; Gilmour, R.; Graff, J.R.; Jambrina, E.; Mader, M.; et al. Characterization of LY2228820 dimesylate, a potent and selective inhibitor of p38 MAPK with antitumor activity. Mol. Cancer Ther. 2014, 13, 364–374. [Google Scholar] [CrossRef]
- Duffy, J.P.; Harrington, E.M.; Salituro, F.G.; Cochran, J.E.; Green, J.; Gao, H.A.; Bemis, G.W.; Evindar, G.; Galullo, V.P.; Ford, P.J.; et al. The discovery of VX-745: A Novel and selective p38 alpha kinase inhibitor. ACS Med. Chem. Lett. 2011, 2, 758–763. [Google Scholar] [CrossRef] [PubMed]
- Barancik, M.; Bohacova, V.; Kvackajova, J.; Hudecova, S.; Krizanova, O.; Breier, A. SB203580, a specific inhibitor of p38-MAPK pathway, is a new reversal agent of P-glycoprotein-mediated multidrug resistance. Eur. J. Pharm. Sci. 2001, 14, 29–36. [Google Scholar] [CrossRef]
- Selness, S.R.; Devraj, R.V.; Devadas, B.; Walker, J.K.; Boehm, T.L.; Durley, R.C.; Shieh, H.; Xing, L.; Rucker, P.V.; Jerome, K.D.; et al. Discovery of PH-797804, a highly selective and potent inhibitor of p38 MAP kinase. Bioorg. Med. Chem. Lett. 2011, 21, 4066–4071. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Mo, Q.; Zhang, Y.; Gao, Y.; Wu, Y.; Li, J.; Hao, X.; Ma, D.; Gao, Q.; Chen, P. The p38 MAPK inhibitor BIRB796 enhances the antitumor effects of VX680 in cervical cancer. Cancer Biol. Ther. 2016, 17, 566–576. [Google Scholar] [CrossRef]
- Malm, S.W.; Hanke, N.T.; Gill, A.; Carbajal, L.; Baker, A.F. The anti-tumor efficacy of 2-deoxyglucose and D-allose are enhanced with p38 inhibition in pancreatic and ovarian cell lines. J. Exp. Clin. Cancer Res. 2015, 34, 31. [Google Scholar] [CrossRef]
- Kuhnol, C.; Herbarth, M.; Foll, J.; Staege, M.S.; Kramm, C. CD137 stimulation and p38 MAPK inhibition improve reactivity in an in vitro model of glioblastoma immunotherapy. Cancer Immunol. Immunother. 2013, 62, 1797–1809. [Google Scholar] [CrossRef]
- Lu, Y.; Zhang, M.; Wang, S.; Hong, B.; Wang, Z.; Li, H.; Zheng, Y.; Yang, J.; Davis, R.E.; Qian, J.; et al. P38 MAPK-inhibited dendritic cells induce superior antitumour immune responses and overcome regulatory T-cell-mediated immunosuppression. Nat. Commun. 2014, 5, 4229. [Google Scholar] [CrossRef]
- Hu, M.C.; Wang, Y.; Qiu, W.R.; Mikhail, A.; Meyer, C.F.; Tan, T.H. Hematopoietic progenitor kinase-1 (HPK1) stress response signaling pathway activates IkappaB kinases (IKK-alpha/beta) and IKK-beta is a developmentally regulated protein kinase. Oncogene 1999, 18, 5514–5524. [Google Scholar] [CrossRef]
- Alzabin, S.; Bhardwaj, N.; Kiefer, F.; Sawasdikosol, S.; Burakoff, S. Hematopoietic progenitor kinase 1 is a negative regulator of dendritic cell activation. J. Immunol. 2009, 182, 6187–6194. [Google Scholar] [CrossRef]
- Wang, H.; Song, X.; Logsdon, C.; Zhou, G.; Evans, D.B.; Abbruzzese, J.L.; Hamilton, S.R.; Tan, T.H.; Wang, H. Proteasome-mediated degradation and functions of hematopoietic progenitor kinase 1 in pancreatic cancer. Cancer Res. 2009, 69, 1063–1070. [Google Scholar] [CrossRef]
- Wang, J.J.; Song, L.J.; Yang, S.; Zhang, W.J.; Lu, P.W.; Li, S.L.; Li, H.X.; Wang, L.X. HPK1 positive expression associated with longer overall survival in patients with estrogen receptor-positive invasive ductal carcinoma-not otherwise specified. Mol. Med. Rep. 2017, 16, 4634–4642. [Google Scholar] [CrossRef] [PubMed]
- Shui, J.W.; Boomer, J.S.; Han, J.; Xu, J.; Dement, G.A.; Zhou, G.; Tan, T.H. Hematopoietic progenitor kinase 1 negatively regulates T cell receptor signaling and T cell-mediated immune responses. Nat. Immunol. 2007, 8, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Curtin, J.; You, D.; Hillerman, S.; Li-Wang, B.; Eraslan, R.; Xie, J.; Swanson, J.; Ho, C.P.; Oppenheimer, S.; et al. Critical role of kinase activity of hematopoietic progenitor kinase 1 in anti-tumor immune surveillance. PLoS ONE 2019, 14, e0212670. [Google Scholar] [CrossRef] [PubMed]
- Diener, K.; Wang, X.S.; Chen, C.; Meyer, C.F.; Keesler, G.; Zukowski, M.; Tan, T.H.; Yao, Z. Activation of the c-Jun N-terminal kinase pathway by a novel protein kinase related to human germinal center kinase. Proc. Natl. Acad. Sci. USA 1997, 94, 9687–9692. [Google Scholar] [CrossRef] [PubMed]
- Chuang, H.C.; Tan, T.H. MAP4K3/GLK in autoimmune disease, cancer and aging. J. Biomed. Sci. 2019, 26, 82. [Google Scholar] [CrossRef]
- Hsu, C.P.; Chuang, H.C.; Lee, M.C.; Tsou, H.H.; Lee, L.W.; Li, J.P.; Tan, T.H. GLK/MAP4K3 overexpression associates with recurrence risk for non-small cell lung cancer. Oncotarget 2016, 7, 41748–41757. [Google Scholar] [CrossRef]
- Chuang, H.C.; Tsai, C.Y.; Hsueh, C.H.; Tan, T.H. GLK-IKKbeta signaling induces dimerization and translocation of the AhR-RORgammat complex in IL-17A induction and autoimmune disease. Sci. Adv. 2018, 4, eaat5401. [Google Scholar] [CrossRef]
- Chuang, H.C.; Chen, Y.M.; Chen, M.H.; Hung, W.T.; Yang, H.Y.; Tseng, Y.H.; Tan, T.H. AhR-ROR-gamma t complex is a therapeutic target for MAP4K3/GLK(high)IL-17A(high) subpopulation of systemic lupus erythematosus. FASEB J. 2019, 33, 11469–11480. [Google Scholar] [CrossRef]
- Xu, W.W.; Dong, J.; Zheng, Y.W.; Zhou, J.; Yuan, Y.; Ta, H.M.; Miller, H.E.; Olson, M.; Rajasekaran, K.; Ernstoff, M.S.; et al. Immune-Checkpoint Protein VISTA Regulates Antitumor Immunity by Controlling Myeloid Cell-Mediated Inflammation and Immunosuppression. Cancer Immunol. Res. 2019, 7, 1497–1510. [Google Scholar] [CrossRef]
- Seimetz, D.; Heller, K.; Richter, J. Approval of first CAR-Ts: Have we solved all hurdles for ATMPs? Cell Med. 2019, 11. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| MEK/ERK Member | Inhibitor | Combination with Immunotherapy | Cancer Type |
|---|---|---|---|
| MEK1/2 | Trametinib | 4-1BB and OX40 agonist antibodies | Breast cancer [19] |
| Selumetinib | Anti-EGFR antibody | Lung adenocarcinoma [39] | |
| G-38963 | Anti-PD-L1 antibody | Colon carcinoma [40] | |
| ERK1/2 | BVD523 | Positive outcomes in patients previously treated with immunotherapy | NRAS-, BRAF V600–, and non–V600 BRAF-mutant solid tumors [29] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, S.; Principe, D.R.; Singh, S.K.; Viswakarma, N.; Sondarva, G.; Rana, B.; Rana, A. Mitogen-Activated Protein Kinase Inhibitors and T-Cell-Dependent Immunotherapy in Cancer. Pharmaceuticals 2020, 13, 9. https://doi.org/10.3390/ph13010009
Kumar S, Principe DR, Singh SK, Viswakarma N, Sondarva G, Rana B, Rana A. Mitogen-Activated Protein Kinase Inhibitors and T-Cell-Dependent Immunotherapy in Cancer. Pharmaceuticals. 2020; 13(1):9. https://doi.org/10.3390/ph13010009
Chicago/Turabian StyleKumar, Sandeep, Daniel R. Principe, Sunil Kumar Singh, Navin Viswakarma, Gautam Sondarva, Basabi Rana, and Ajay Rana. 2020. "Mitogen-Activated Protein Kinase Inhibitors and T-Cell-Dependent Immunotherapy in Cancer" Pharmaceuticals 13, no. 1: 9. https://doi.org/10.3390/ph13010009
APA StyleKumar, S., Principe, D. R., Singh, S. K., Viswakarma, N., Sondarva, G., Rana, B., & Rana, A. (2020). Mitogen-Activated Protein Kinase Inhibitors and T-Cell-Dependent Immunotherapy in Cancer. Pharmaceuticals, 13(1), 9. https://doi.org/10.3390/ph13010009