Predictive Power of In Silico Approach to Evaluate Chemicals against M. tuberculosis: A Systematic Review

 ,
, 

Abstract
1. Introduction
2. Results and Discussion
2.1. Mycobacterium tuberculosis Enzyme Targets
2.2. PDB, Organisms, and Expression System
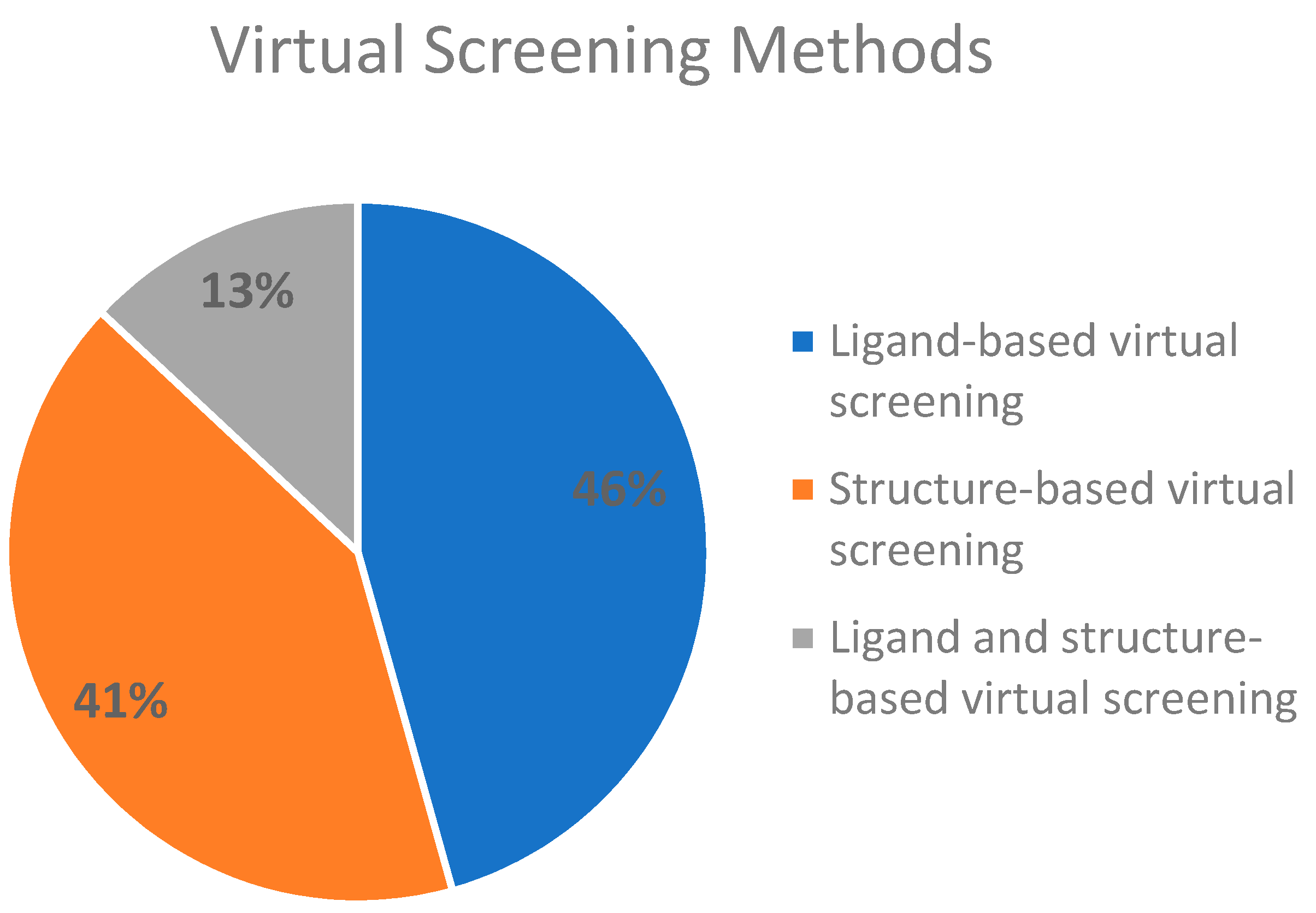
2.3. Virtual Screening Methods Applied
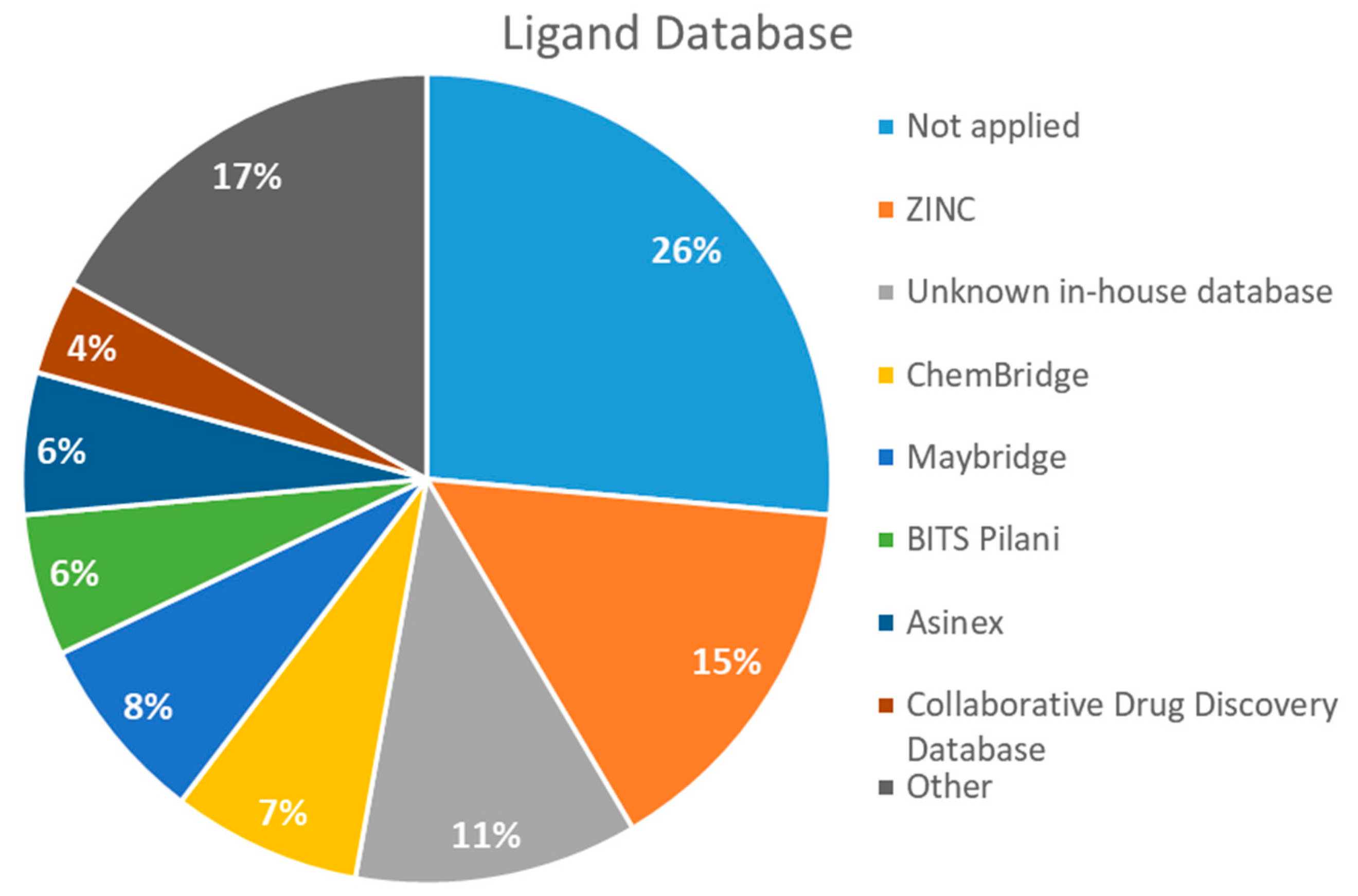
2.4. Databases Screened
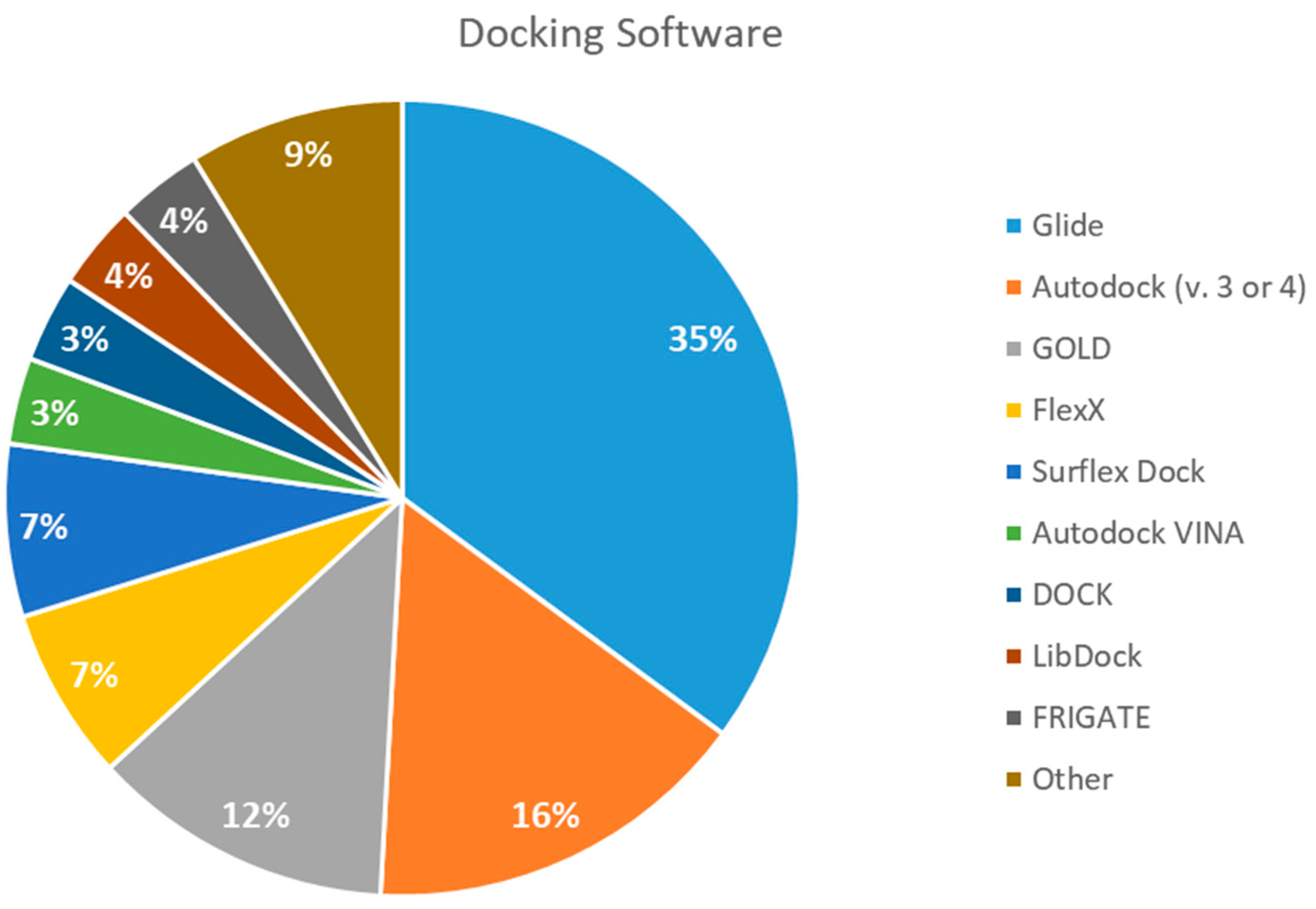
2.5. Docking Software Employed
2.6. In Vitro or In Vivo Testing
2.7. Validation Procedures
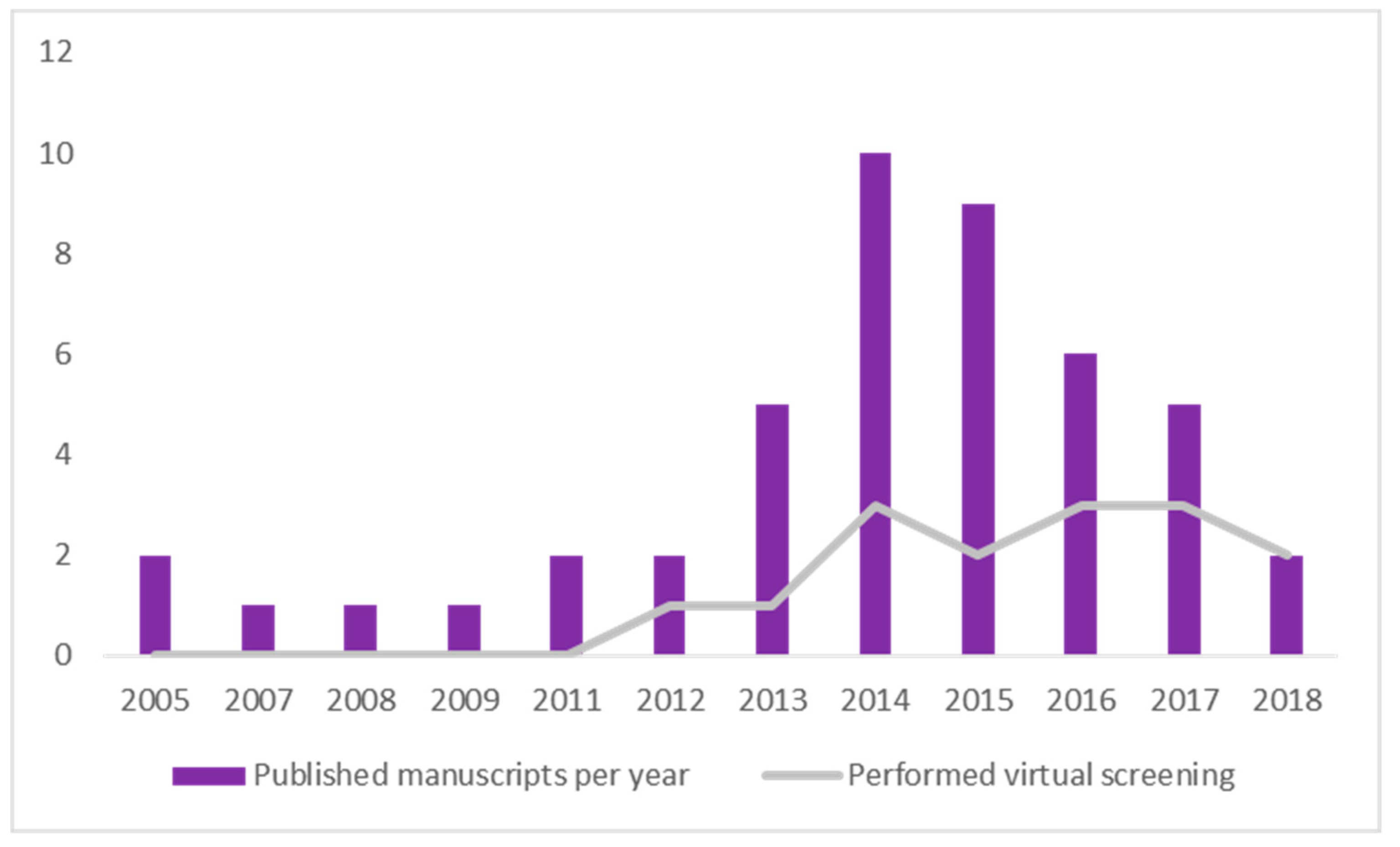
2.8. Timeline Analysis of Retrieved Manuscripts
3. Materials and Methods
3.1. Background Definitions
3.2. Data Sources and Searches
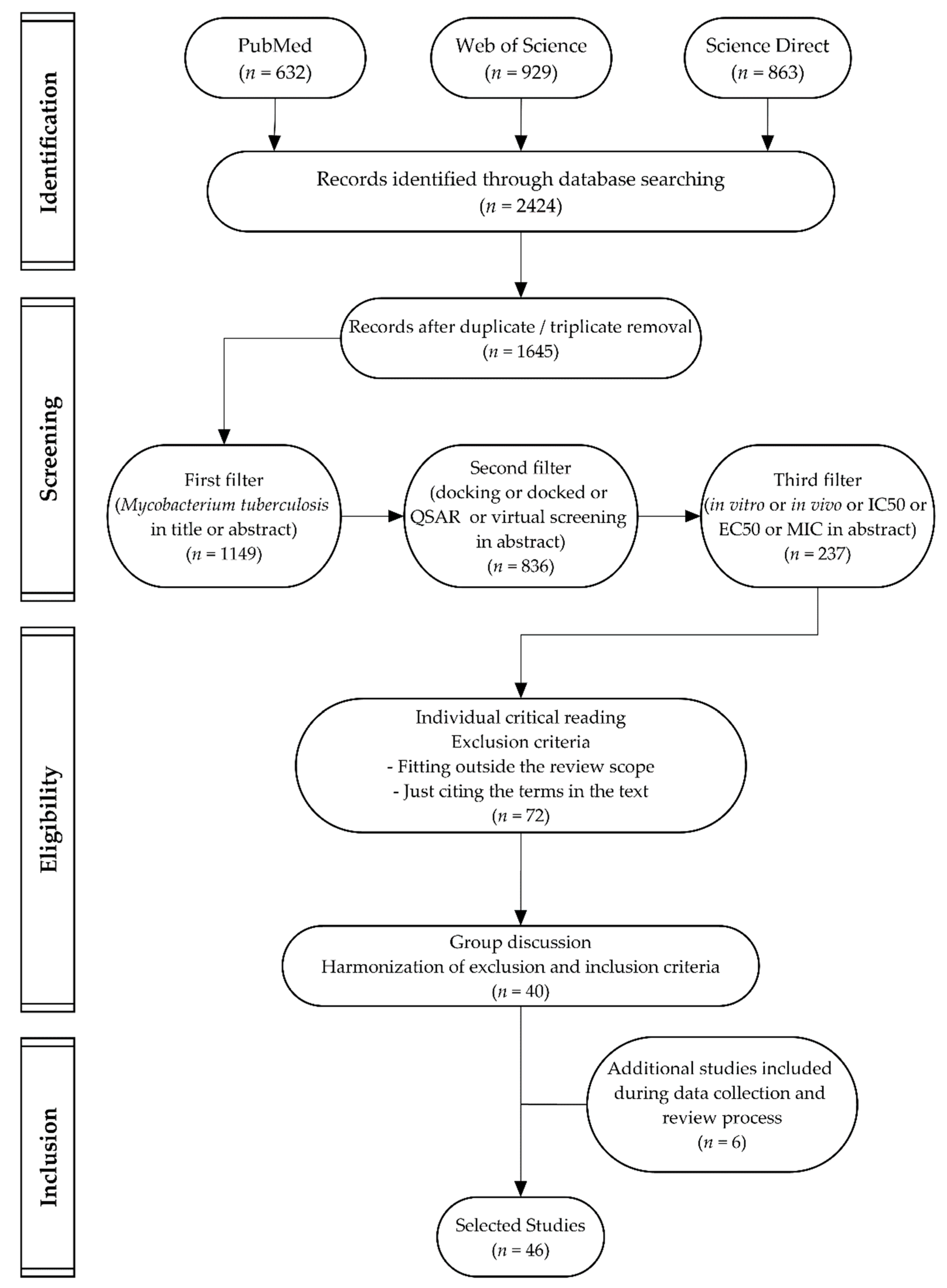
3.3. Study Selection
3.4. Data Extraction Process
- (a)
- Mycobacterium tuberculosis enzymes target, EC code, and accepted nomenclature [85]
- (b)
- PDB, organism and expression system [86]
- (c)
- Virtual screening methods applied (if applied)
- (d)
- Databases screened (if applied)
- (e)
- Docking software (if applied)
- (f)
- In vitro or in vivo assay
- (g)
- Validation procedure (if applied)
- (h)
- Years in which manuscripts were published
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A

References
- WHO. Global Tuberculosis Report; WHO: Geneva, Switzerland, 2018; p. 277. [Google Scholar]
- Gandhi, N.R.; Nunn, P.; Dheda, K.; Schaaf, H.S.; Zignol, M.; van Soolingen, D.; Jensen, P.; Bayona, J. Multidrug-resistant and extensively drug-resistant tuberculosis: a threat to global control of tuberculosis. Lancet 2010, 375, 1830–1843. [Google Scholar] [CrossRef]
- Langer, T.; Wolber, G. Pharmacophore definition and 3D searches. Drug Discov. Today Technol. 2004, 1, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Leelananda, S.P.; Lindert, S. Computational methods in drug discovery. Beilstein J. Org. Chem. 2016, 12, 2694–2718. [Google Scholar] [CrossRef] [PubMed]
- Philip, P.; Anshuman, D.; Anil, K.S. Computer-Aided Drug Design: Integration of Structure-Based and Ligand-Based Approaches in Drug Design. Curr. Comput. -Aided Drug Des. 2007, 3, 133–148. [Google Scholar] [CrossRef]
- Mehra, R.; Chib, R.; Munagala, G.; Yempalla, K.R.; Khan, I.A.; Singh, P.P.; Khan, F.G.; Nargotra, A. Discovery of new Mycobacterium tuberculosis proteasome inhibitors using a knowledge-based computational screening approach. Mol. Divers. 2015, 19, 1003–1019. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.; Mussa, H.Y.; Glen, R.C.; Reiling, S. Similarity searching of chemical databases using atom environment descriptors (MOLPRINT 2D): evaluation of performance. J. Chem. Inf. Comput. Sci. 2004, 44, 1708–1718. [Google Scholar] [CrossRef] [PubMed]
- Lengauer, T.; Rarey, M. Computational methods for biomolecular docking. Curr. Opin. Struct. Biol. 1996, 6, 402–406. [Google Scholar] [CrossRef]
- Srivastava, S.K.; Tripathi, R.P.; Ramachandran, R. NAD(+)-dependent DNA ligase (Rv3014c) from Mycobacterium tuberculosis. J. Biol. Chem. 2005, 280, 30273–30281. [Google Scholar] [CrossRef] [PubMed]
- Kinjo, T.; Koseki, Y.; Kobayashi, M.; Yamada, A.; Morita, K.; Yamaguchi, K.; Tsurusawa, R.; Gulten, G.; Komatsu, H.; Sakamoto, H.; et al. Identification of compounds with potential antibacterial activity against Mycobacterium through structure-based drug screening. J. Chem. Inf. Modeling 2013, 53, 1200–1212. [Google Scholar] [CrossRef] [PubMed]
- Nirmal, C.R.; Rao, R.; Hopper, W. Inhibition of 3-deoxy-D-arabino-heptulosonate 7-phosphate synthase from Mycobacterium tuberculosis: in silico screening and in vitro validation. Eur. J. Med. Chem. 2015, 105, 182–193. [Google Scholar] [CrossRef]
- Singh, N.; Tiwari, S.; Srivastava, K.K.; Siddiqi, M.I. Identification of Novel Inhibitors of Mycobacterium tuberculosis PknG Using Pharmacophore Based Virtual Screening, Docking, Molecular Dynamics Simulation, and Their Biological Evaluation. J. Chem. Inf. Modeling 2015, 55, 1120–1129. [Google Scholar] [CrossRef] [PubMed]
- Mehra, R.; Rajput, V.S.; Gupta, M.; Chib, R.; Kumar, A.; Wazir, P.; Khan, I.A.; Nargotra, A. Benzothiazole Derivative as a Novel Mycobacterium tuberculosis Shikimate Kinase Inhibitor: Identification and Elucidation of Its Allosteric Mode of Inhibition. J. Chem. Inf. Modeling 2016, 56, 930–940. [Google Scholar] [CrossRef] [PubMed]
- Saxena, S.; Devi, P.B.; Soni, V.; Yogeeswari, P.; Sriram, D. Identification of novel inhibitors against Mycobacterium tuberculosis L-alanine dehydrogenase (MTB-AlaDH) through structure-based virtual screening. J. Mol. Graph. Model. 2014, 47, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Cinu, T.A.; Sidhartha, S.K.; Indira, B.; Varadaraj, B.G.; Vishnu, P.S.; Shenoy, G.G. Design, synthesis and evaluation of antitubercular activity of Triclosan analogues. Arab. J. Chem. 2015. [Google Scholar] [CrossRef]
- Samala, G.; Nallangi, R.; Devi, P.B.; Saxena, S.; Yadav, R.; Sridevi, J.P.; Yogeeswari, P.; Sriram, D. Identification and development of 2-methylimidazo[1,2-a]pyridine-3-carboxamides as Mycobacterium tuberculosis pantothenate synthetase inhibitors. Bioorganic Med. Chem. 2014, 22, 4223–4232. [Google Scholar] [CrossRef]
- Menchon, G., Maveyraud; Czaplicki, G. Molecular Dynamics as a Tool for Virtual Ligand Screening. In Computational Drug Discovery and Design. Methods in Molecular Biology; Humana Press: New York, NY, USA, 2018; Volume 1762, pp. 145–178. [Google Scholar]
- Feher, M. Consensus scoring for protein–ligand interactions. Drug Discov. Today 2006, 11, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Seifert, M.H.J. Targeted scoring functions for virtual screening. Drug Discov. Today 2009, 14, 562–569. [Google Scholar] [CrossRef]
- Li, Y.; Su, M.; Liu, Z.; Li, J.; Liu, J.; Han, L.; Wang, R. Assessing protein–ligand interaction scoring functions with the CASF-2013 benchmark. Nat. Protoc. 2018, 13, 666. [Google Scholar] [CrossRef]
- Rohilla, A.; Khare, G.; Tyagi, A.K. Virtual Screening, pharmacophore development and structure based similarity search to identify inhibitors against IdeR, a transcription factor of Mycobacterium tuberculosis. Sci. Rep. 2017, 7, 4653. [Google Scholar] [CrossRef]
- Salimizand, H.; Jamehdar, S.A.; Nik, L.B.; Sadeghian, H. Design of peptides interfering with iron-dependent regulator (IdeR) and evaluation of Mycobacterium tuberculosis growth inhibition. Iran. J. Basic Med. Sci. 2017, 20, 722–728. [Google Scholar] [CrossRef]
- Barot, K.P.; Jain, S.V.; Gupta, N.; Kremer, L.; Singh, S.; Takale, V.B.; Joshi, K.; Ghate, M.D. Design, synthesis and docking studies of some novel (R)-2-(4’-chlorophenyl)-3-(4’-nitrophenyl)-1,2,3,5-tetrahydrobenzo[4,5] imidazo [1,2-c]pyrimidin-4-ol derivatives as antitubercular agents. Eur. J. Med. Chem. 2014, 83, 245–255. [Google Scholar] [CrossRef]
- Martins, F.; Santos, S.; Ventura, C.; Elvas-Leitão, R.; Santos, L.; Vitorino, S.; Reis, M.; Miranda, V.; Correia, H.F.; Aires-de-Sousa, J.; et al. Design, synthesis and biological evaluation of novel isoniazid derivatives with potent antitubercular activity. Eur. J. Med. Chem. 2014, 81, 119–138. [Google Scholar] [CrossRef] [PubMed]
- Minor, W.; Dauter, Z.; Jaskolski, M. The young person’s guide to the PDB. Postepy Biochem. 2016, 62, 242–249. [Google Scholar] [PubMed]
- Koch, O.; Jager, T.; Heller, K.; Khandavalli, P.C.; Pretzel, J.; Becker, K.; Flohe, L.; Selzer, P.M. Identification of M. tuberculosis thioredoxin reductase inhibitors based on high-throughput docking using constraints. J. Med. Chem. 2013, 56, 4849–4859. [Google Scholar] [CrossRef] [PubMed]
- Massengo-Tiasse, R.P.; Cronan, J.E. Diversity in enoyl-acyl carrier protein reductases. Cell. Mol. Life Sci. 2009, 66, 1507–1517. [Google Scholar] [CrossRef]
- Quemard, A.; Sacchettini, J.C.; Dessen, A.; Vilcheze, C.; Bittman, R.; Jacobs, W.R., Jr.; Blanchard, J.S. Enzymatic characterization of the target for isoniazid in Mycobacterium tuberculosis. Biochemistry 1995, 34, 8235–8241. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.; Wang, L.; David, H.L. Effect of isoniazid on the in vivo mycolic acid synthesis, cell growth, and viability of Mycobacterium tuberculosis. Antimicrob. Agents Chemotherapy. 1972, 2, 29–35. [Google Scholar] [CrossRef]
- Wang, F.; Langley, R.; Gulten, G.; Dover, L.G.; Besra, G.S.; Jacobs, W.R., Jr.; Sacchettini, J.C. Mechanism of thioamide drug action against tuberculosis and leprosy. J. Exp. Med. 2007, 204, 73–78. [Google Scholar] [CrossRef]
- Seifert, M.; Catanzaro, D.; Catanzaro, A.; Rodwell, T.C. Genetic mutations associated with isoniazid resistance in Mycobacterium tuberculosis: a systematic review. PLoS ONE 2015, 10, e0119628. [Google Scholar] [CrossRef]
- Morlock, G.P.; Metchock, B.; Sikes, D.; Crawford, J.T.; Cooksey, R.C. ethA, inhA, and katG loci of ethionamide-resistant clinical Mycobacterium tuberculosis isolates. Antimicrob. Agents Chemother. 2003, 47, 3799–3805. [Google Scholar] [CrossRef]
- Inturi, B.; Pujar, G.V.; Purohit, M.N.; Iyer, V.B.; Sowmya, G.S.; Kulkarni, M. Design, synthesis and evaluation of diphenyl ether analogues as antitubercular agents. RSC Adv. 2016, 6, 110571–110582. [Google Scholar] [CrossRef]
- Izumizono, Y.; Arevalo, S.; Koseki, Y.; Kuroki, M.; Aoki, S. Identification of novel potential antibiotics for tuberculosis by in silico structure-based drug screening. Eur. J. Med. Chem. 2011, 46, 1849–1856. [Google Scholar] [CrossRef] [PubMed]
- Mathew, B.; Mathew, G.E.; Sonia, G.; Kumar, A.; Charles, N.P.; Kumar, P. Design of 1-(furan-2-yl)-N-(5-substituted phenyl-1, 3, 4-thiadiazol-2-yl) methanimine derivatives as Enoyl-ACP reductase inhibitors: Synthesis, molecular docking studies and anti-tubercular activity. Bangladesh J. Pharmacol. 2013, 8, 242–248. [Google Scholar] [CrossRef]
- Mohan, S.B.; Ravi Kumar, B.V.; Dinda, S.C.; Naik, D.; Prabu Seenivasan, S.; Kumar, V.; Rana, D.N.; Brahmkshatriya, P.S. Microwave-assisted synthesis, molecular docking and antitubercular activity of 1,2,3,4-tetrahydropyrimidine-5-carbonitrile derivatives. Bioorganic Med. Chem. Lett. 2012, 22, 7539–7542. [Google Scholar] [CrossRef] [PubMed]
- Mohire, P.P.; Chandam, D.R.; Patil, R.B.; Kumbhar, D.R.; Jadhav, S.J.; Patravale, A.A.; Godase, V.P.; Ghosh, J.S.; Deshmukh, M.B. Protic Ionic Liquid Promoted One Pot Synthesis of 2-amino-4-(phenyl)-7-methyl-5-oxo-4H, 5H-pyrano 4,3-b pyran-3-carbonitrile Derivatives in Water and Their Antimycobacterial Activity. J. Heterocycl. Chem. 2018, 55, 1010–1023. [Google Scholar] [CrossRef]
- Pauli, I.; dos Santos, R.N.; Rostirolla, D.C.; Martinelli, L.K.; Ducati, R.G.; Timmers, L.F.; Basso, L.A.; Santos, D.S.; Guido, R.V.; Andricopulo, A.D.; et al. Discovery of new inhibitors of Mycobacterium tuberculosis InhA enzyme using virtual screening and a 3D-pharmacophore-based approach. J. Chem. Inf. Modeling 2013, 53, 2390–2401. [Google Scholar] [CrossRef] [PubMed]
- Saharan, V.D.; Mahajan, S.S. Development of gallic acid formazans as novel enoyl acyl carrier protein reductase inhibitors for the treatment of tuberculosis. Bioorganic Med. Chem. Lett. 2017, 27, 808–815. [Google Scholar] [CrossRef] [PubMed]
- Champoux, J.J. DNA Topoisomerases: Structure, Function, and Mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. [Google Scholar] [CrossRef] [PubMed]
- Corbett, K.D.; Berger, J.M. Structure, Molecular Mechanisms, and Evolutionary Relationships in DNA Topoisomerases. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 95–118. [Google Scholar] [CrossRef] [PubMed]
- Forterre, P.; Gribaldo, S.; Gadelle, D.; Serre, M.-C. Origin and evolution of DNA topoisomerases. Biochimie 2007, 89, 427–446. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA Topoisomerases and Their Poisoning by Anticancer and Antibacterial Drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Jeankumar, V.U.; Renuka, J.; Kotagiri, S.; Saxena, S.; Kakan, S.S.; Sridevi, J.P.; Yellanki, S.; Kulkarni, P.; Yogeeswari, P.; Sriram, D. Gyrase ATPase domain as an antitubercular drug discovery platform: structure-based design and lead optimization of nitrothiazolyl carboxamide analogues. ChemMedChem 2014, 9, 1850–1859. [Google Scholar] [CrossRef] [PubMed]
- Saxena, S.; Renuka, J.; Jeankumar, V.U.; Yogeeswari, P.; Sriram, D. Mycobacterial DNA gyrB inhibitors: Ligand based pharmacophore modelling and in vitro enzyme inhibition studies. Curr. Top. Med. Chem. 2014, 14, 1990–2005. [Google Scholar] [CrossRef] [PubMed]
- Saxena, S.; Renuka, J.; Yogeeswari, P.; Sriram, D. Discovery of Novel Mycobacterial DNA Gyrase B Inhibitors: In Silico and In Vitro Biological Evaluation. Mol. Inform. 2014, 33, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Saxena, S.; Samala, G.; Renuka, J.; Sridevi, J.P.; Yogeeswari, P.; Sriram, D. Development of 2-amino-5-phenylthiophene-3-carboxamide derivatives as novel inhibitors of Mycobacterium tuberculosis DNA GyrB domain. Bioorganic Med. Chem. 2015, 23, 1402–1412. [Google Scholar] [CrossRef] [PubMed]
- Sridevi, J.P.; Suryadevara, P.; Janupally, R.; Sridhar, J.; Soni, V.; Anantaraju, H.S.; Yogeeswari, P.; Sriram, D. Identification of potential Mycobacterium tuberculosis topoisomerase I inhibitors: a study against active, dormant and resistant tuberculosis. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2015, 72, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Godbole, A.A.; Ahmed, W.; Bhat, R.S.; Bradley, E.K.; Ekins, S.; Nagaraja, V. Inhibition of Mycobacterium tuberculosis topoisomerase I by m-AMSA, a eukaryotic type II topoisomerase poison. Biochem. Biophys. Res. Commun. 2014, 446, 916–920. [Google Scholar] [CrossRef]
- Godbole, A.A.; Ahmed, W.; Bhat, R.S.; Bradley, E.K.; Ekins, S.; Nagaraja, V. Targeting Mycobacterium tuberculosis Topoisomerase I by Small-Molecule Inhibitors. Antimicrob. Agents Chemother. 2015, 59, 1549. [Google Scholar] [CrossRef] [PubMed]
- Olaru, I.D.; von Groote-Bidlingmaier, F.; Heyckendorf, J.; Yew, W.W.; Lange, C.; Chang, K.C. Novel drugs against tuberculosis: a clinician’s perspective. Eur. Respir. J. 2015, 45, 1119–1131. [Google Scholar] [CrossRef]
- Hoagland, D.T.; Liu, J.; Lee, R.B.; Lee, R.E. New agents for the treatment of drug-resistant Mycobacterium tuberculosis. Adv. Drug Deliv. Rev. 2016, 102, 55–72. [Google Scholar] [CrossRef]
- Srivastava, S.K.; Dube, D.; Tewari, N.; Dwivedi, N.; Tripathi, R.P.; Ramachandran, R. Mycobacterium tuberculosis NAD+-dependent DNA ligase is selectively inhibited by glycosylamines compared with human DNA ligase I. Nucleic Acids Res. 2005, 33, 7090–7101. [Google Scholar] [CrossRef] [PubMed]
- Krishnasamy, S.K.; Namasivayam, V.; Mathew, S.; Eakambaram, R.S.; Ibrahim, I.A.; Natarajan, A.; Palaniappan, S. Design, Synthesis, and Characterization of Some Hybridized Pyrazolone Pharmacophore Analogs against Mycobacterium tuberculosis. Arch. Der Pharm. 2016, 349, 383–397. [Google Scholar] [CrossRef] [PubMed]
- Scheich, C.; Szabadka, Z.; Vertessy, B.; Putter, V.; Grolmusz, V.; Schade, M. Discovery of novel MDR-Mycobacterium tuberculosis inhibitor by new FRIGATE computational screen. PLoS ONE 2011, 6, e28428. [Google Scholar] [CrossRef] [PubMed]
- Petersen, G.O.; Saxena, S.; Renuka, J.; Soni, V.; Yogeeswari, P.; Santos, D.S.; Bizarro, C.V.; Sriram, D. Structure-based virtual screening as a tool for the identification of novel inhibitors against Mycobacterium tuberculosis 3-dehydroquinate dehydratase. J. Mol. Graph. Model. 2015, 60, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Gudzera, O.I.; Golub, A.G.; Bdzhola, V.G.; Volynets, G.P.; Lukashov, S.S.; Kovalenko, O.P.; Kriklivyi, I.A.; Yaremchuk, A.D.; Starosyla, S.A.; Yarmoluk, S.M.; et al. Discovery of potent anti-tuberculosis agents targeting leucyl-tRNA synthetase. Bioorganic Med. Chem. 2016, 24, 1023–1031. [Google Scholar] [CrossRef]
- Harer, S.L.; Bhatia, M.S. In-silico docking based design and synthesis of [1H,3H] imidazo[4,5-b] pyridines as lumazine synthase inhibitors for their effective antimicrobial activity. J. Pharm. Bioallied Sci. 2014, 6, 285–296. [Google Scholar] [CrossRef]
- Swain, S.S.; Paidesetty, S.K.; Padhy, R.N. Development of antibacterial conjugates using sulfamethoxazole with monocyclic terpenes: A systematic medicinal chemistry based computational approach. Comput. Methods Programs Biomed. 2017, 140, 185–194. [Google Scholar] [CrossRef]
- Agrawal, H.; Kumar, A.; Bal, N.C.; Siddiqi, M.I.; Arora, A. Ligand based virtual screening and biological evaluation of inhibitors of chorismate mutase (Rv1885c) from Mycobacterium tuberculosis H37Rv. Bioorganic Med. Chem. Lett. 2007, 17, 3053–3058. [Google Scholar] [CrossRef]
- Billones, J.B.; Carrillo, M.C.; Organo, V.G.; Macalino, S.J.; Sy, J.B.; Emnacen, I.A.; Clavio, N.A.; Concepcion, G.P. Toward antituberculosis drugs: in silico screening of synthetic compounds against Mycobacterium tuberculosisl,d-transpeptidase 2. Drug Des. Dev. Ther. 2016, 10, 1147–1157. [Google Scholar] [CrossRef]
- Puranik, N.V.; Srivastava, P.; Swami, S.; Choudhari, A.; Sarkar, D. Molecular modeling studies and in vitro screening of dihydrorugosaflavonoid and its derivatives against Mycobacterium tuberculosis. Rsc Adv. 2018, 8, 10634–10643. [Google Scholar] [CrossRef]
- Horvati, K.; Bacsa, B.; Szabo, N.; David, S.; Mezo, G.; Grolmusz, V.; Vertessy, B.; Hudecz, F.; Bosze, S. Enhanced cellular uptake of a new, in silico identified antitubercular candidate by peptide conjugation. Bioconjugate Chem. 2012, 23, 900–907. [Google Scholar] [CrossRef] [PubMed]
- Dube, D.; Tripathi, S.M.; Ramachandran, R. Identification of in vitro inhibitors of Mycobacterium tuberculosis Lysine epsilon-aminotransferase by pharmacophore mapping and three-dimensional flexible searches. Med. Chem. Res. 2008, 17, 182–188. [Google Scholar] [CrossRef]
- Kumar, A.; Chaturvedi, V.; Bhatnagar, S.; Sinha, S.; Siddiqi, M.I. Knowledge based identification of potent antitubercular compounds using structure based virtual screening and structure interaction fingerprints. J. Chem. Inf. Modeling 2009, 49, 35–42. [Google Scholar] [CrossRef]
- Hamza, A.; Wagner, J.M.; Evans, T.J.; Frasinyuk, M.S.; Kwiatkowski, S.; Zhan, C.G.; Watt, D.S.; Korotkov, K.V. Novel Mycosin Protease MycP(1) Inhibitors Identified by Virtual Screening and 4D Fingerprints. J. Chem. Inf. Modeling 2014, 54, 1166–1173. [Google Scholar] [CrossRef] [PubMed]
- Dkhar, H.K.; Gopalsamy, A.; Loharch, S.; Kaur, A.; Bhutani, I.; Saminathan, K.; Bhagyaraj, E.; Chandra, V.; Swaminathan, K.; Agrawal, P.; et al. Discovery of Mycobacterium tuberculosis alpha-1,4-glucan branching enzyme (GlgB) inhibitors by structure- and ligand-based virtual screening. J. Biol. Chem. 2015, 290, 76–89. [Google Scholar] [CrossRef]
- Wang, X.; Ahn, Y.M.; Lentscher, A.G.; Lister, J.S.; Brothers, R.C.; Kneen, M.M.; Gerratana, B.; Boshoff, H.I.; Dowd, C.S. Design, synthesis, and evaluation of substituted nicotinamide adenine dinucleotide (NAD(+)) synthetase inhibitors as potential antitubercular agents. Bioorganic Med. Chem. Lett. 2017, 27, 4426–4430. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.E.; Matarlo, J.S.; Tonge, P.J.; Tan, D.S. Stereoselective Synthesis, Docking, and Biological Evaluation of Difluoroindanediol-Based MenE Inhibitors as Antibiotics. Org. Lett. 2016, 18, 6384–6387. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhu, X.; Cui, C.; Dong, M.; Jiang, H.; Li, Z.; Liu, Z.; Zhu, W.; Wang, J.G. Discovery of novel acetohydroxyacid synthase inhibitors as active agents against Mycobacterium tuberculosis by virtual screening and bioassay. J. Chem. Inf. Modeling 2013, 53, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Shirude, P.S.; Madhavapeddi, P.; Tucker, J.A.; Murugan, K.; Patil, V.; Basavarajappa, H.; Raichurkar, A.V.; Humnabadkar, V.; Hussein, S.; Sharma, S.; et al. Aminopyrazinamides: novel and specific GyrB inhibitors that kill replicating and nonreplicating Mycobacterium tuberculosis. Acs Chem. Biol. 2013, 8, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Humnabadkar, V.; Madhavapeddi, P.; Basavarajappa, H.; Sheikh, M.G.; Rane, R.; Basu, R.; Verma, P.; Sundaram, A.; Mukherjee, K.; de Sousa, S.M. Assays, surrogates, and alternative technologies for a TB lead identification program targeting DNA gyrase ATPase. J. Biomol. Screen. 2015, 20, 265–274. [Google Scholar] [CrossRef] [PubMed][Green Version]
- He, X.; Alian, A.; Stroud, R.; Ortiz de Montellano, P.R. Pyrrolidine carboxamides as a novel class of inhibitors of enoyl acyl carrier protein reductase from Mycobacterium tuberculosis. J. Med. Chem. 2006, 49, 6308–6323. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.A.; Maggiora, G.M. Concepts and Applications of Molecular Similarity; Wiley: New York, NY, USA, 1990. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J. ZINC 15. Available online: http://zinc.docking.org/ (accessed on 14 June 2019).
- ChemBridge. The Gold Standard in Small Molecule Screening Libraries and Building Blocks. Available online: https://www.chembridge.com/screening_libraries/ (accessed on 14 June 2019).
- MAYBRIDGE. Part of Thermo Fisher Scientific. Available online: https://www.maybridge.com/portal/alias__Rainbow/lang__en/tabID__177/DesktopDefault.aspx (accessed on 14 June 2019).
- Tiwari, R.; Mahasenan, K.; Pavlovicz, R.; Li, C.; Tjarks, W. Carborane clusters in computational drug design: a comparative docking evaluation using AutoDock, FlexX, Glide, and Surflex. J. Chem. Inf. Modeling 2009, 49, 1581–1589. [Google Scholar] [CrossRef] [PubMed]
- Perola, E.; Walters, W.P.; Charifson, P.S. A detailed comparison of current docking and scoring methods on systems of pharmaceutical relevance. Proteins 2004, 56, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Kontoyianni, M.; McClellan, L.M.; Sokol, G.S. Evaluation of docking performance: comparative data on docking algorithms. J. Med. Chem. 2004, 47, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.C.; Murray, C.W.; Nissink, J.W.; Taylor, R.D.; Taylor, R. Comparing protein-ligand docking programs is difficult. Proteins 2005, 60, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Sander, T.; Freyss, J.; von Korff, M.; Reich, J.R.; Rufener, C. OSIRIS, an entirely in-house developed drug discovery informatics system. J. Chem. Inf. Modeling 2009, 49, 232–246. [Google Scholar] [CrossRef]
- Hospital, A.; Goni, J.R.; Orozco, M.; Gelpi, J.L. Molecular dynamics simulations: Advances and applications. Adv. Appl. Bioinform. Chem. Aabc 2015, 8, 37–47. [Google Scholar] [CrossRef]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. 2009, 6, e1000097. [Google Scholar] [CrossRef]
- Enzyme ExPASy. Bioinformatics Resource Portal. Available online: https://enzyme.expasy.org/ (accessed on 7 November 2018).
- RCSB PDB. Protein Data Bank. Available online: http://www.rcsb.org/ (accessed on 7 November 2018).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme Targeted | PDB | Resolution (Å) | References | EC Code | Quantity |
|---|---|---|---|---|---|
| Enoyl-[acyl-carrier-protein] reductase (NADH) | 4U0J | 1.62 Å | [10,34,36] | EC 1.3.1.9 | 9 |
| 4TZK | 1.62 Å | [35] | |||
| 2NSD | 1.90 Å | [39] | |||
| 2AQ8 | 1.92 Å | [37] | |||
| 3FNG | 1.97 Å | [33] | |||
| 1P45 | 2.60 Å | [15] | |||
| 1P44 | 2.70 Å | [38] | |||
| DNA topoisomerase (ATP-hydrolyzing) | 4B6C | 2.20 Å | [44,45,46,47] | EC 5.6.2.3 | 4 |
| DNA topoisomerase I | 1MW9 | 1.67 Å | [49,50] | EC 5.6.2.2 | 3 |
| 1ECL | 1.90 Å | [49,50] | |||
| 1MW8 | 1.90 Å | [49,50] | |||
| 3PX7 | 2.30 Å | [48] | |||
| DNA ligase (NAD (+)) | 1TAE | 2.70 Å | [53] | EC 6.5.1.2 | 2 |
| 1ZAU | 3.15 Å | [9] | |||
| Shikimate kinase | 2IYQ | 1.80 Å | [13,54] | EC 2.7.1.71 | 2 |
| 1WE2 | 2.30 Å | [13,54] | |||
| 2IYZ | 2.30 Å | [13,54] | |||
| Diacylglycerol O-acyltransferase | 5KWI | 1.30 Å | [55] | EC 2.3.1.20 | 1 |
| 3-dehydroquinate dehydratase | 2Y71 | 1.50 Å | [56] | EC 4.2.1.10 | 1 |
| Leucine-tRNA ligase | 2VOC | 1.50 Å | [57] | EC 6.1.1.4 | 1 |
| 6,7-dimethyl-8-ribityllumazine synthase | 2C92 | 1.60 Å | [58] | EC 2.5.1.78 | 1 |
| Dihydropteroate synthase | 1EYE | 1.70 Å | [59] | EC 2.5.1.15 | 1 |
| Chorismate mutase | 2F6L | 1.70 Å | [60] | EC 5.4.99.5 | 1 |
| D-glutamyltransferase | 3TUR | 1.72 Å | [61] | EC 2.3.2.- | 1 |
| Pantoate-beta-alanine ligase (AMP-forming) | 3IVX | 1.73 Å | [16] | EC 6.3.2.1 | 1 |
| 3-oxoacyl-[acyl-carrier-protein] reductase | 1UZN | 1.91 Å | [62] | EC 1.1.1.100 | 1 |
| dUTP diphosphatase | 1MQ7 | 1.95 Å | [63] | EC 3.6.1.23 | 1 |
| L-lysine 6-transaminase | 2CIN | 1.98 Å | [64] | EC 2.6.1.36 | 1 |
| dTMP kinase | 1W2H | 2.00 Å | [65] | EC 2.7.4.9 | 1 |
| Alanine dehydrogenase | 2VHW | 2.00 Å | [14] | EC 1.4.1.1 | 1 |
| Thermitase | 4HVL | 2.00 Å | [66] | EC 3.4.21.66 | 1 |
| 3-deoxy-7-phosphoheptulonate synthase | 2B7O | 2.30 Å | [11] | EC 2.5.1.54 | 1 |
| 1,4-alpha-glucan branching enzyme | 3K1D | 2.33 Å | [67] | EC 2.4.1.18 | 1 |
| NAD (+) synthase (glutamine-hydrolyzing) | 3DLA | 2.35 Å | [68] | EC 6.3.5.1 | 1 |
| Pantothenate kinase | 3AF3 | 2.35 Å | [62] | EC 2.7.1.33 | 1 |
| o-succinylbenzoate-CoA ligase | 5C5H | 2.40 Å | [69] | EC 6.2.1.26 | 1 |
| Nonspecific serine/threonine protein kinase | 2PZI | 2.40 Å | [12] | EC 2.7.11.1 | 1 |
| Acetolactate synthase | 1N0H | 2.80 Å | [70] | EC 2.2.1.6 | 1 |
| Thioredoxin-disulfide reductase | 2A87 | 3.00 Å | [26] | EC 1.8.1.9 | 1 |
| Proteasome endopeptidase complex | 2FHG | 3.23 Å | [6] | EC 3.4.25.1 | 1 |
| Compound | MIC (µM) | MIC Ratio (Cmp/Ctrl) | IC50 (µM) | Docking Score (Software) | Reference |
|---|---|---|---|---|---|
 G7650246 | - | - | 35.3 | NS (Glide) | [6] |
 2 | 14.7 | 18.8 (Isoniazid 1) | 4.0 | −14.4 kcal/mol (Autodock) | [9] |
 KES4 | - | - | 4.8 | 83.5 (GoldScore) | [10] |
 Alpha-tocopherol | - | - | 21.0 | −7.2 (Glide Score) | [11] |
 NRB04248 | - | - | - | 5.7 (Surflex Score) −20.0 KJ/mol (FlexX) −9.5 Kcal/mol (Autodock) | [12] * |
 5489375 | - | - | 10.7 | NS (Glide) | [13] |
 Lead 1 | - | - | 35.5 | −9.9 (Glide Score) | [14] |
 4h | 80.0 | 219. 5 (Isoniazid) | - | −9.1 (Glide Score) | [15] |
 5b | 4.53 | 6.3 (Isoniazid) | 1.9 | −8.6 (Glide Score) | [16] |
 I-108 | 45.8 | 754.1 (Rifampicin) | 63.6 | −6.2 kcal/mol (Autodock) | [21] |
 GVPG  RPR | 200.0 | 256.4 (Isoniazid 1) | - | −5.28 Kd (GVPG) −5.78 Kd (RPR) (Autodock) | [22] |
 7b | 7.3 | 49.8 (Isoniazid) | - | 3.9 (Surflex Score -logKd) | [23] |
 1 | - | - | 12.5 | Consensus using GoldScore, Chemscore and ASPscore (GOLD) | [26] |
 I1 | - | - | 5.3 | 83.0 (GoldScore) | [34] |
 2g | - | - | - | −5 to −6 (Glide-XP Score) | [36] * |
 C9 | 2.0 | 2.8 (Isoniazid) 0.3 (Ethambutol) 0.9 (Ofloxacin) | 3.4 | −9.5 (Glide Score) | [39] |
 DE3 | 8.5 | 0.4 (Isoniazid) | - | 7.0 (Surflex Score -logKd) | [33] |
 Fb  Fe | 10.7 10.3 | 13.7 13.2 (Isoniazid 1) | - | −9.3 kcal/mol (Fb) −9.2 kcal/mol (Fe) (Argus Dock) | [35] |
 PA | 4.0 | 0.04 (Pyrazinamid) | - | −9.0 kcal/mol (Autodock VINA) | [37] |
 ZINC09137707 | - | - | - | NS GoldScore (GOLD) | [38] * |
 14 | 7.5 | 11.4 (Isoniazid) | 0.5 | −5.9 (Glide Score) | [44] |
 Ex-355 | - | - | - | NP | [45] * |
 Lead 11 | - | - | 1.5 | 62.1 (GoldScore) −10.3 (Glide XP Score) | [46] |
 23 | 4.8 | 7.3 (Isoniazid) 21.0 (Rifampicin) 2.2 (Ofloxacin) 0.6 (Ethambutol) | 0.8 | −10.6 kcal/mol (Glide) | [47] |
 m-AMSA | 125.0 | 160.3 (Isoniazid 1) | - | 94.6 (Libdock Score 21–150) | [49] |
 Norclomipramine | 60.0 | 76.9 (Isoniazid 1) | - | 95.2 (Libdock Score ~46.4–126.3) | [50] |
 3b | 5.9 | 8.2 (Isoniazid) 39.5 (Rifampicin) 0.8 (Ethambutol) 2.74 (Moxifloxacin) | 2.9 | −5.6 (Glide Score) | [48] |
 1 | 12 | 15.4 (Isoniazid 1) | 46.2 | −15.8 (Autodock) | [53] |
 8b | 0.4 | 0.03 (Pyrazinamide) 0.08 (Ciprofloxacin) 0.07 (Streptomycin) | - | NS (Autodock) | [54] |
 1 | - | - | 5.7 | −20.3 (FlexX Score) | [60] |
 MB16695 | 67,8 | 37.2 (Isoniazid) | - | −6.6 kcal/mol (Glide) | [67] |
 (1R,3S)-2 | 26.6 | 57.6 (AMS 2) | 5 | −11.9 (Glide Score) | [69] |
 2j | 38.5 | 0.5 (Ciprofloxacin) | - | −55.3 (Biopredicta Score) | [58] |
 TB8 | 3.7 | 3.1 (Isoniazid) 0.25 (Norfloxacin) | - | NS (FRIGATE) | [63] |
 BTB13566 | 9.7 | 12.8 (Isoniazid 1) | - | −28.9 (FlexX Score) | [65] |
 Lead 1a | 56.75 | 72.8 (Isoniazid 1) | 17.1 | 72.2 (GOLD Score) | [56] |
 1 | 113.5 | 2.5 (Ampicilin) | - | NS (FRIGATE) | [55] |
 Conjugate-5 | 38.9 | 0.34 (Ampicilin) | - | −10.8 (Autodock VINA) | [59] |
 4e | 44.14 | 122.6 (Isoniazid) | 90 | −6.8 kcal/mol (Glide) | [68] |
 Compound 2 | 25.0 | 125.0 (Rifampicin) | - | −164.7 (CDOCKER) | [61] |
 A | - | - | - | 7.2 kcal/mol (Autodock) | [64] * |
 Compound 1 | 25.0 | 32.1 (Isoniazid) | 6 | Less than −40.0 (DOCK) | [57] |
 10 | - | - | 48 | NS (SABRE) | [66] |
 5c | 81.9 | 5617.8 (Isoniazid) | 25.34 | −8.37 (Glide) | [62] |
 15 | 11.5 | 0.42 (Sulfometuron methyl) | 1.85 | Less than −7.0 (Glide Score) | [70] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Timo, G.O.; Reis, R.S.S.V.d.; Melo, A.F.d.; Costa, T.V.L.; Magalhães, P.d.O.; Homem-de-Mello, M. Predictive Power of In Silico Approach to Evaluate Chemicals against M. tuberculosis: A Systematic Review. Pharmaceuticals 2019, 12, 135. https://doi.org/10.3390/ph12030135
Timo GO, Reis RSSVd, Melo AFd, Costa TVL, Magalhães PdO, Homem-de-Mello M. Predictive Power of In Silico Approach to Evaluate Chemicals against M. tuberculosis: A Systematic Review. Pharmaceuticals. 2019; 12(3):135. https://doi.org/10.3390/ph12030135
Chicago/Turabian StyleTimo, Giulia Oliveira, Rodrigo Souza Silva Valle dos Reis, Adriana Françozo de Melo, Thales Viana Labourdette Costa, Pérola de Oliveira Magalhães, and Mauricio Homem-de-Mello. 2019. "Predictive Power of In Silico Approach to Evaluate Chemicals against M. tuberculosis: A Systematic Review" Pharmaceuticals 12, no. 3: 135. https://doi.org/10.3390/ph12030135
APA StyleTimo, G. O., Reis, R. S. S. V. d., Melo, A. F. d., Costa, T. V. L., Magalhães, P. d. O., & Homem-de-Mello, M. (2019). Predictive Power of In Silico Approach to Evaluate Chemicals against M. tuberculosis: A Systematic Review. Pharmaceuticals, 12(3), 135. https://doi.org/10.3390/ph12030135