The Antitumor Didox Acts as an Iron Chelator in Hepatocellular Carcinoma Cells






 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
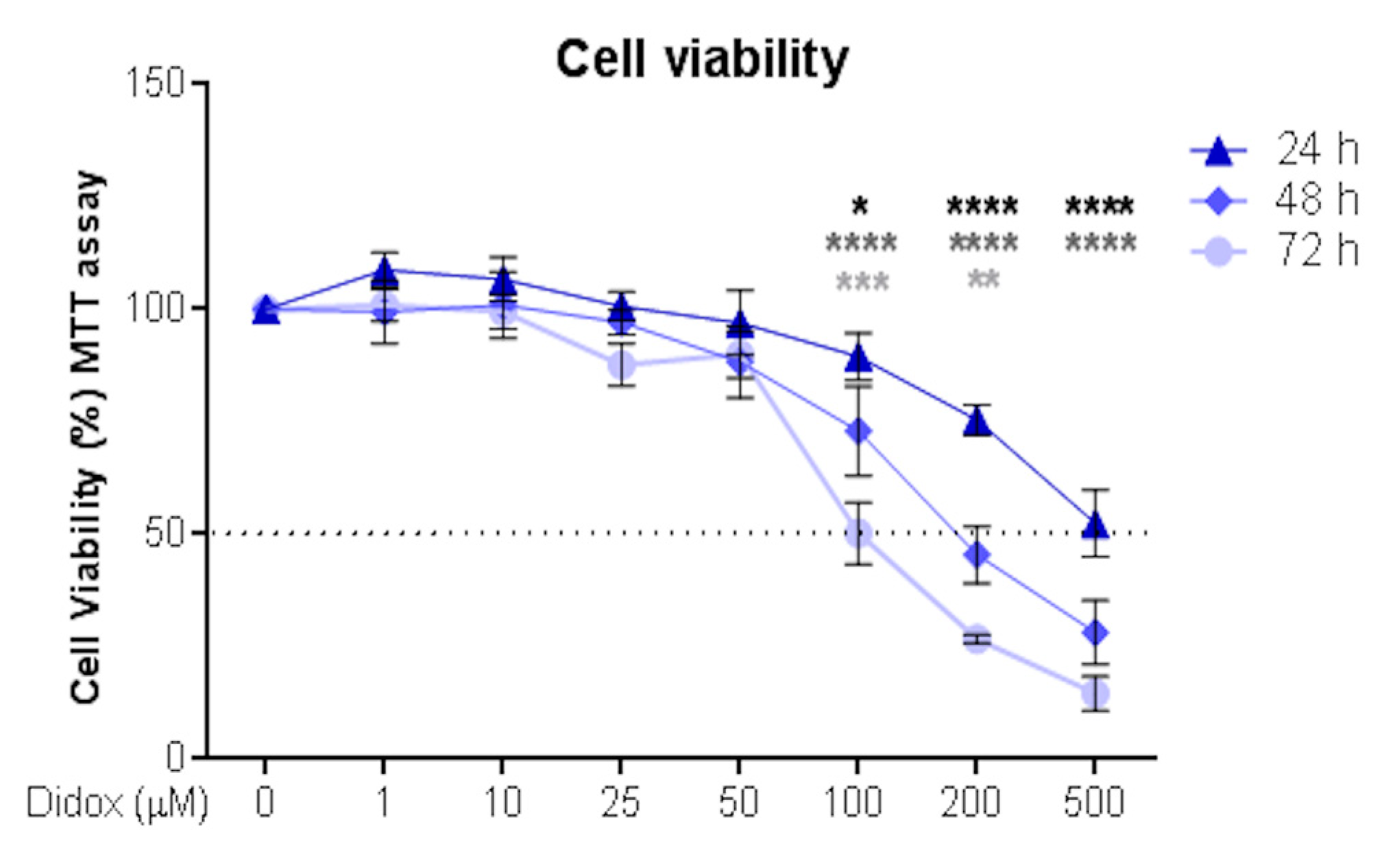
2.1. Didox Suppresses the Viability of Hepatocellular Carcinoma HA22T/VGH Cell Line
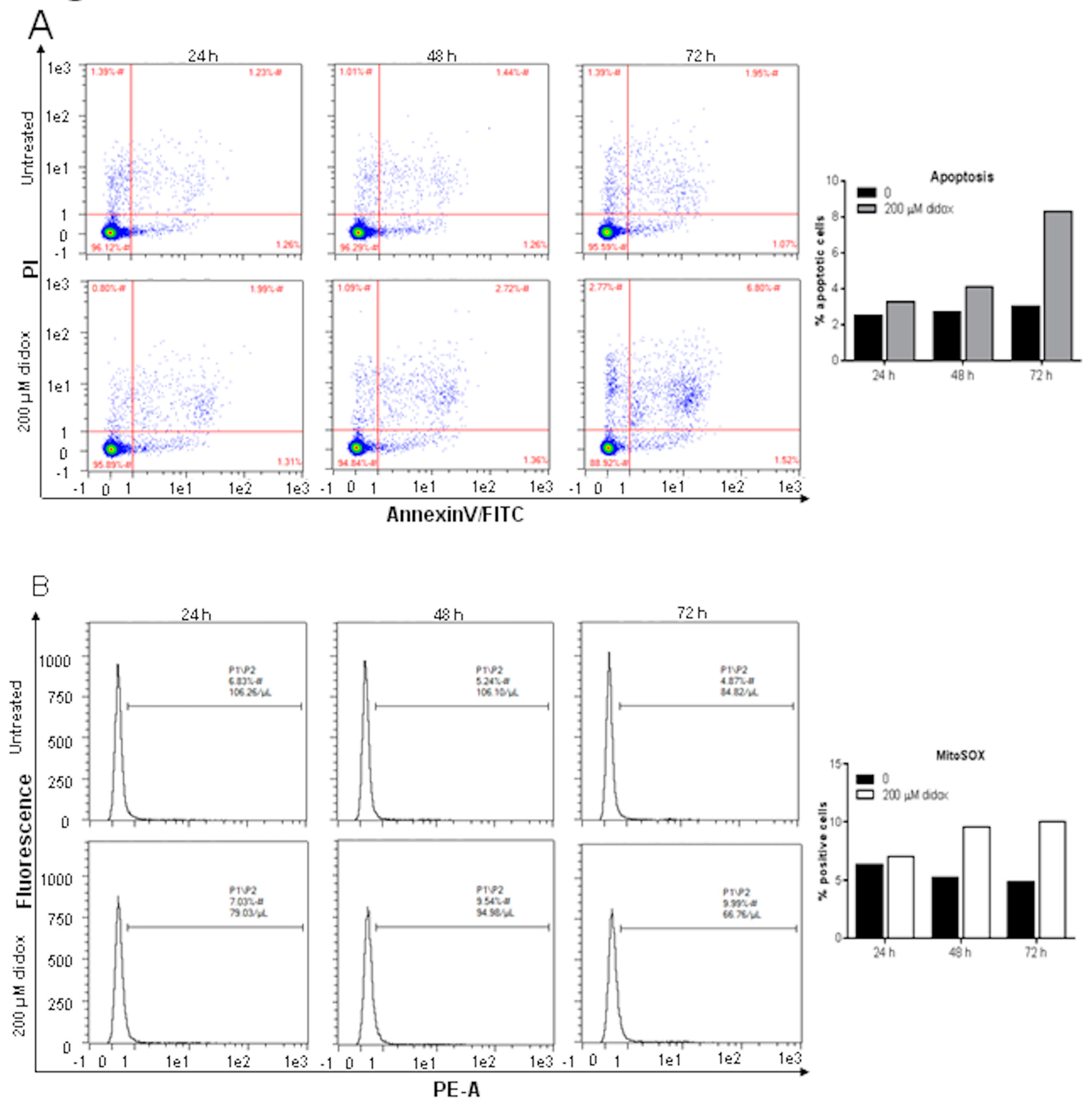
2.2. Didox Induces Apoptosis and Increases Mitochondrial ROS
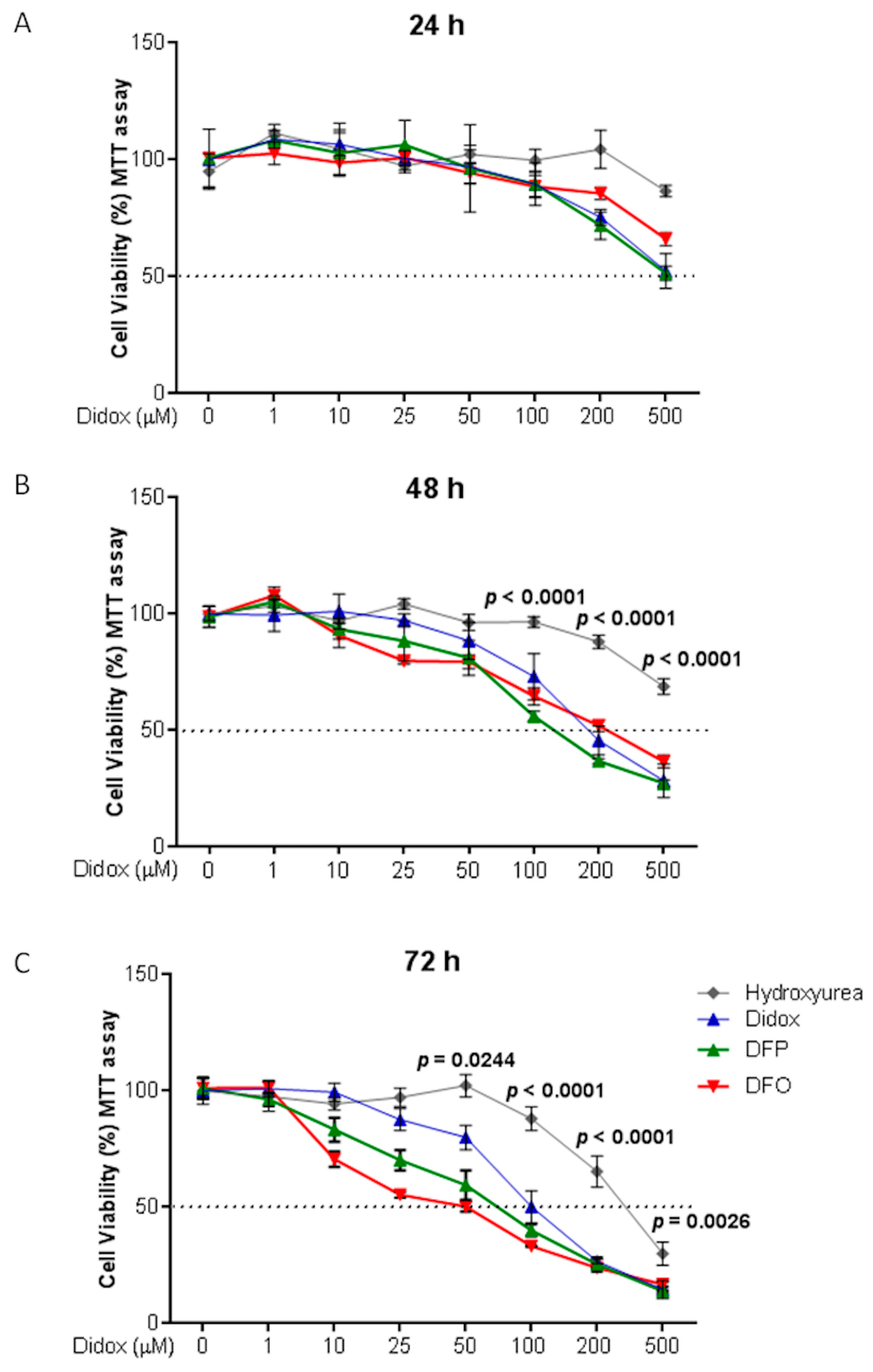
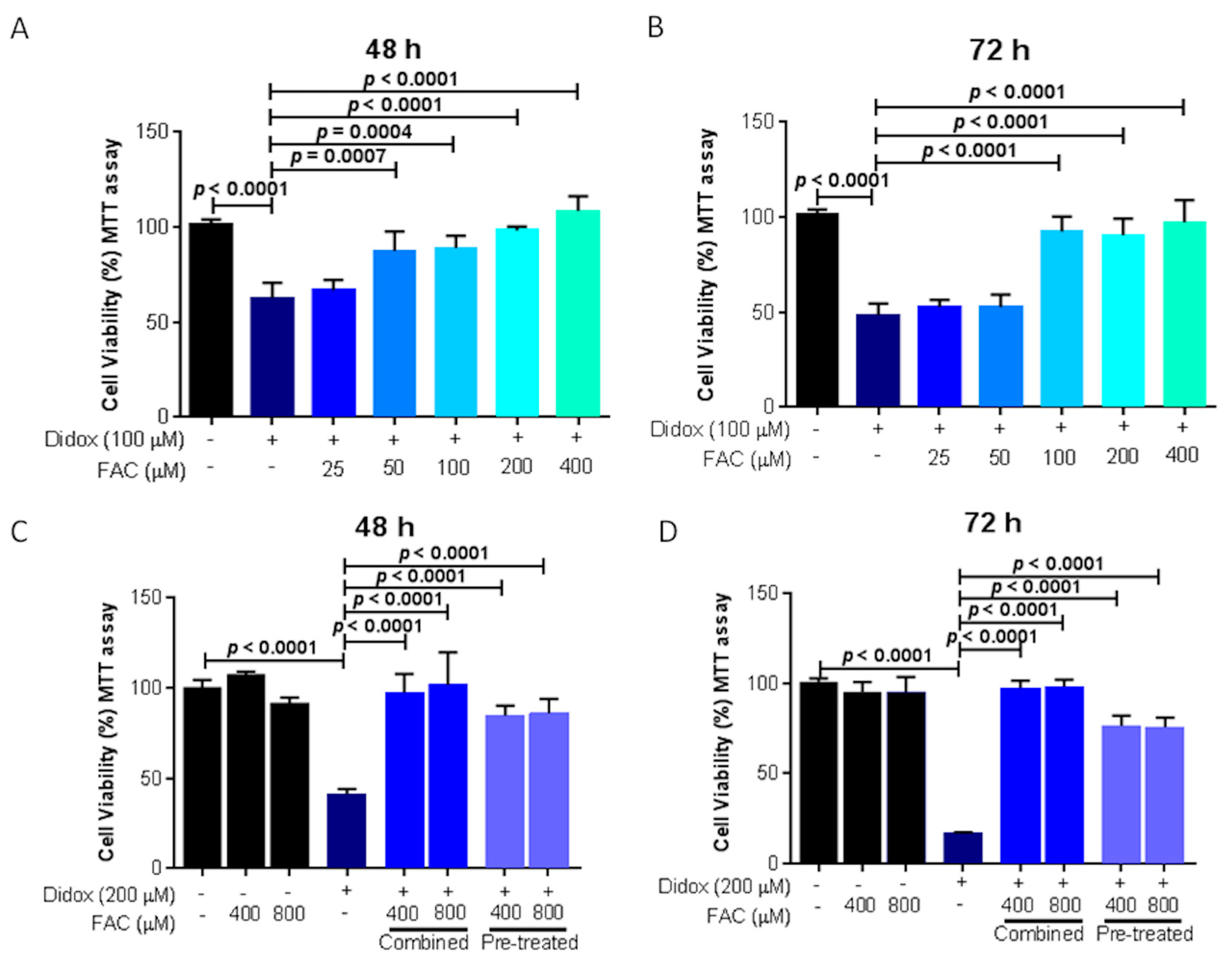
2.3. Didox Inhibits Cell Viability Similarly to DFO and DFP
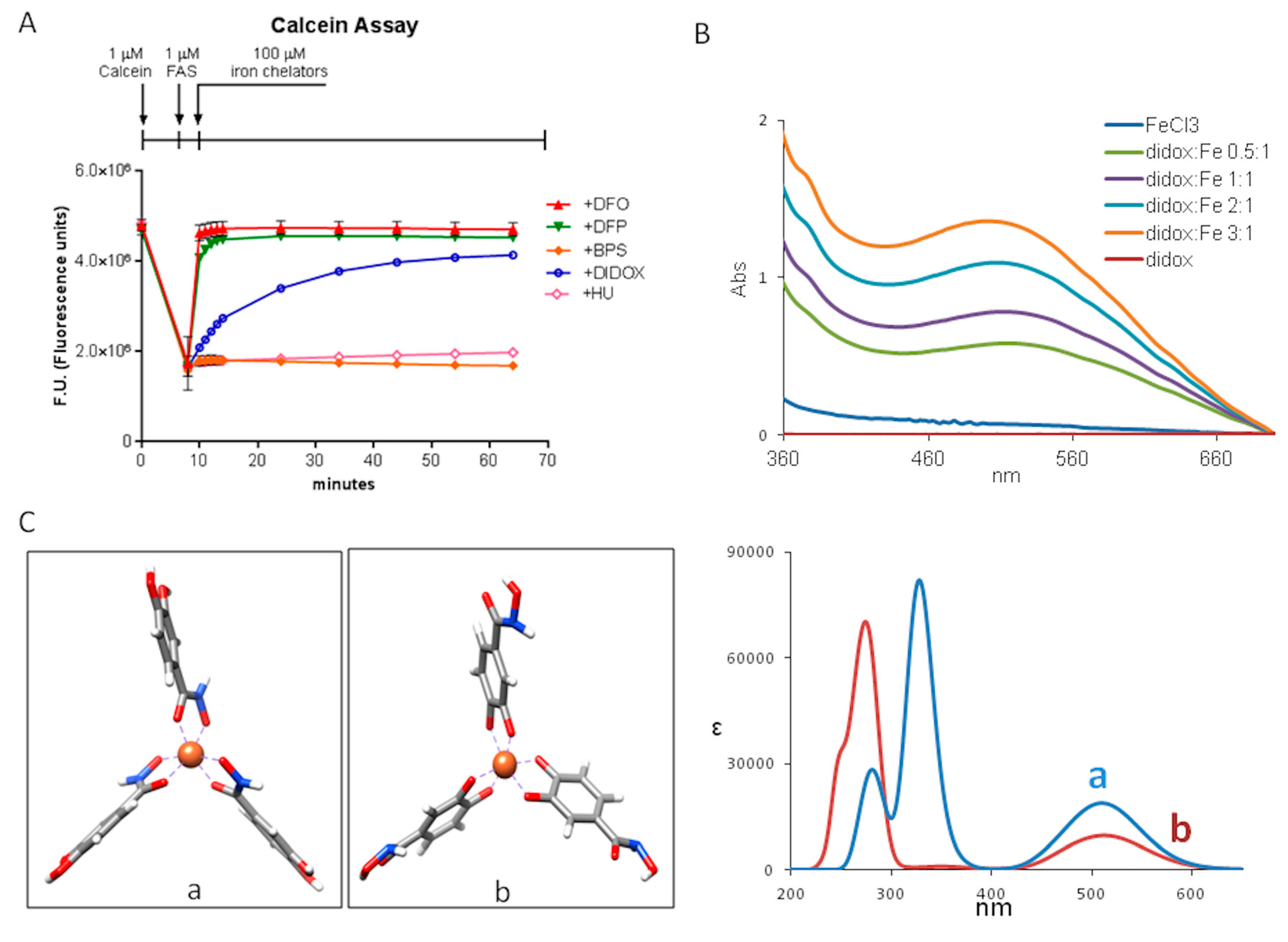
2.4. Didox Binds Fe3+ in a Time-Dependent Manner
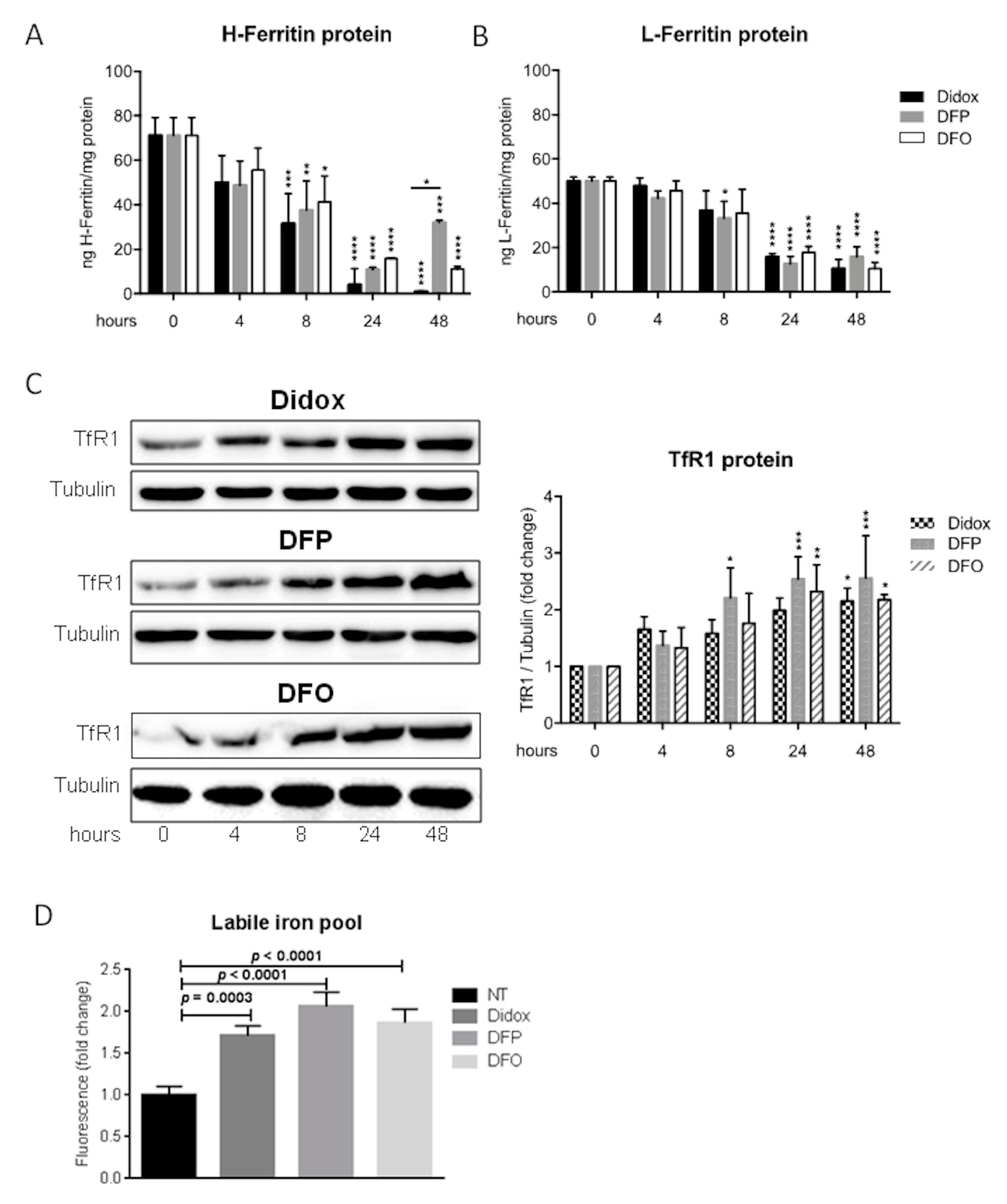
2.5. Didox Alters the Iron Status of HA22T/VGH Cells Similarly to DFO and DFP
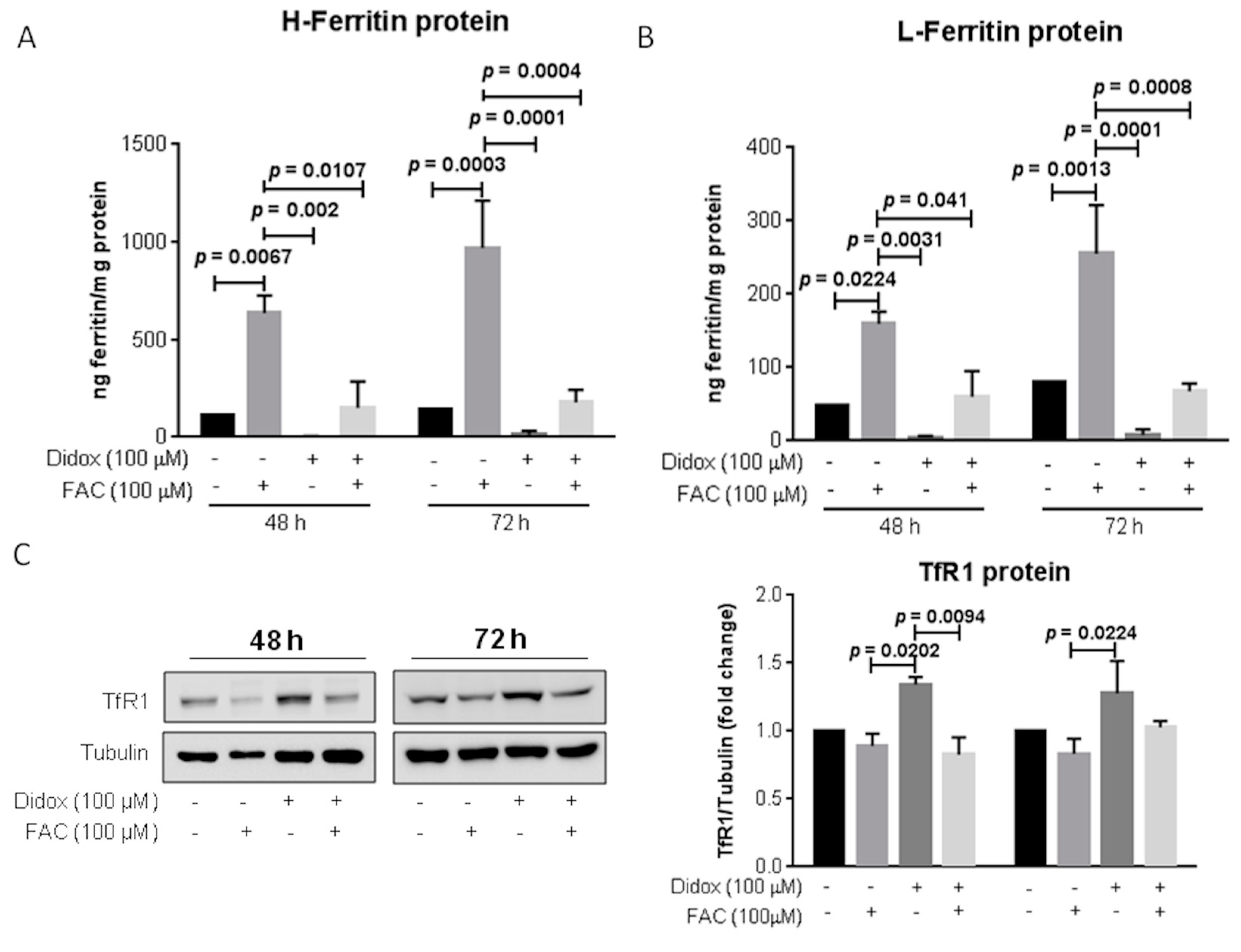
2.6. Iron Supplementation Suppresses the Cell Toxicity of Didox in HA22T/VGH
3. Discussion
4. Materials and Methods
4.1. Antibodies and Chemicals
4.2. Cell Culture
4.3. Cell Treatments and Cell Viability Analysis
4.4. Protein Extraction
4.5. Western Blot Analysis
4.6. ELISA Assay
4.7. Labile Iron Pool Assay and Calcein-AM Assay
4.8. MitoSOX™ Red Mitochondrial Superoxide Indicator Assay
4.9. AnnexinV/Propidium Iodide Assay
4.10. Dequencing of Calcein Fluorescence In Vitro
4.11. UV-Vis Spectroscopy
4.12. Computational Method
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chen, Y.R.; Tsou, B.; Hu, S.; Ma, H.; Liu, X.; Yen, Y.; Ann, D.K. Autophagy induction causes a synthetic lethal sensitization to ribonucleotide reductase inhibition in breast cancer cells. Oncotarget 2016, 7, 1984–1999. [Google Scholar] [CrossRef]
- Elford, H.L.; Freese, M.; Passamani, E.; Morris, H.P. Ribonucleotide reductase and cell proliferation. I. Variations of ribonucleotide reductase activity with tumor growth rate in a series of rat hepatomas. J. Biol. Chem. 1970, 245, 5228–5233. [Google Scholar]
- Elford, H.L.; Wampler, G.L.; Riet, B.V. New ribonucleotide reductase inhibitors with antineoplastic activity. Cancer Res. 1979, 39, 844–851. [Google Scholar] [PubMed]
- Hsu, N.Y.; Wu, J.Y.; Liu, X.; Yen, Y.; Chen, C.Y.; Chou, M.C.; Lin, C.H.; Lee, H.; Cheng, Y.W. Expression status of ribonucleotide reductase small subunits hRRM2/p53R2 as prognostic biomarkers in stage I and II non-small cell lung cancer. Anticancer Res. 2011, 31, 3475–3481. [Google Scholar] [PubMed]
- Takeda, E.; Weber, G. Role of ribonucleotide reductase in expression in the neoplastic program. Life Sci. 1981, 28, 1007–1014. [Google Scholar] [CrossRef]
- Eklund, H.; Uhlin, U.; Färnegårdh, M.; Logan, D.T.; Nordlund, P. Structure and function of the radical enzyme ribonucleotide reductase. Prog. Biophys. Mol. Biol. 2001, 77, 177–268. [Google Scholar] [CrossRef]
- Elford, H.L.; Riet, B.V. Inhibition of nucleoside diphosphate reductase by hydroxybenzohydroxamic acid derivatives. Pharmacol. Ther. 1985, 29, 239–254. [Google Scholar] [CrossRef]
- Reichard, P. Interactions Between Deoxyribonucleotide and DNA Synthesis. Annu. Rev. Biochem. 1988, 57, 349–374. [Google Scholar] [CrossRef] [PubMed]
- Bepler, G.; Zheng, Z.; Gautam, A.; Sharma, S.; Cantor, A.; Sharma, A.; Cress, W.D.; Kim, Y.C.; Rosell, R.; McBride, C.; et al. Ribonucleotide reductase M1 gene promoter activity, polymorphisms, population frequencies, and clinical relevance. Lung Cancer 2005, 47, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Liu, X.; Mo, X.; Xue, L.; Darwish, D.; Qiu, W.; Shih, J.; Hwu, E.B.; Luh, F.; Yen, Y. The human ribonucleotide reductase subunit hRRM2 complements p53R2 in response to UV-induced DNA repair in cells with mutant p53. Cancer Res. 2003, 63, 6583–6594. [Google Scholar] [PubMed]
- Szekeres, T.; Fritzer-Szekeres, M.; Elford, H.L.; Jayaram, H.M. The Enzyme Ribonucleotide Reductase: Target for Antitumor and Anti-HIV Therapy. Crit. Rev. Clin. Lab. Sci. 1997, 34, 503–528. [Google Scholar] [CrossRef] [PubMed]
- Riet, B.V.; Wampler, G.L.; Elford, H.L. Synthesis of hydroxy- and amino-substituted benzohydroxamic acids: Inhibition of ribonucleotide reductase and antitumor activity. J. Med. Chem. 1979, 22, 589–592. [Google Scholar] [CrossRef] [PubMed]
- Al-Abd, A.M.; Al-Abbasi, F.A.; Asaad, G.F.; Abdel-Naim, A.B. Didox potentiates the cytotoxic profile of doxorubicin and protects from its cardiotoxicity. Eur. J. Pharmacol. 2013, 718, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Grusch, M. Activation of caspases and induction of apoptosis by novel ribonucleotide reductase inhibitors amidox and didox. Exp. Hematol. 2001, 29, 623–632. [Google Scholar] [CrossRef]
- Raje, N.; Kumar, S.; Hideshima, T.; Ishitsuka, K.; Yasui, H.; Chhetri, S.; Vallet, S.; Vonescu, E.; Shiraishi, N.; Kiziltepe, T.; et al. Didox, a ribonucleotide reductase inhibitor, induces apoptosis and inhibits DNA repair in multiple myeloma cells. Br. J. Haematol. 2006, 135, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Inayat, M.S.; Chendil, D.; Mohiuddin, M.; Elford, H.L.; Gallicchio, V.S.; Ahmed, M.M. Didox (A Novel Ribonucleotide Reductase Inhibitor) Overcomes bcl-2 Mediated Radiation Resistance in Prostate Cancer Cell Line PC-3. Cancer Biol. Ther. 2002, 1, 539–545. [Google Scholar] [CrossRef]
- Shah, K.; Wilson, E.A.; Malla, R.; Elford, H.L.; Faridi, J.S. Targeting ribonucleotide reductase M2 and NF- B activation with Didox to circumvent tamoxifen resistance in breast cancer. Mol. Cancer Ther. 2015, 14, 2411–2421. [Google Scholar] [CrossRef]
- Cook, G.J.; Caudell, D.L.; Elford, H.L.; Pardee, T.S. The Efficacy of the Ribonucleotide Reductase Inhibitor Didox in Preclinical Models of AML. PLoS ONE 2014, 9, e112619. [Google Scholar] [CrossRef]
- Carmichael, J.; Cantwell, B.; Mannix, K.; Veale, D.; Elford, H.; Blackie, R.; Kerr, D.; Kaye, S.; Harris, A. A phase I and pharmacokinetic study of didox administered by 36 hour infusion. Br. J. Cancer 1990, 61, 447–450. [Google Scholar] [CrossRef]
- Veale, D.; Carmichael, J.; Cantwell, B.; Elford, H.; Blackie, R.; Kerr, D.; Kaye, S.; Harris, A. A phase 1 and pharmacokinetic study of didox: A ribonucleotide reductase inhibitor. Br. J. Cancer 1988, 58, 70–72. [Google Scholar] [CrossRef]
- Fritzer-Szekeres, M.; Novotný, L.; Vachálková, A.; Findenig, G.; Elford, H.L.; Szekeres, T. Iron binding capacity of didox (3,4-dihydroxybenzohydroxamic acid) and amidox (3,4-dihydroxybenzamidoxime) new inhibitors of the enzyme ribonucleotide reductase. Life Sci. 1997, 61, 2231–2237. [Google Scholar] [CrossRef]
- Fanzani, A.; Poli, M. Iron, Oxidative Damage and Ferroptosis in Rhabdomyosarcoma. Int. J. Mol. Sci. 2017, 18, 1718. [Google Scholar] [CrossRef] [PubMed]
- Torti, S.V.; Torti, F.M. Iron and cancer: More ore to be mined. Nat. Rev. Cancer 2013, 13, 342–355. [Google Scholar] [CrossRef] [PubMed]
- Blatt, J.; Stitely, S. Antineuroblastoma activity of desferoxamine in human cell lines. Cancer Res. 1987, 47, 1749–1750. [Google Scholar] [PubMed]
- Reddel, R.R.; Hedley, D.W.; Sutherland, R.L. Cell cycle effects of iron depletion on T-47D human breast cancer cells. Exp. Cell Res. 1985, 161, 277–284. [Google Scholar] [CrossRef]
- Richardson, D.R.; Milnes, K. The potential of iron chelators of the pyridoxal isonicotinoyl hydrazone class as effective antiproliferative agents II: The mechanism of action of ligands derived from salicylaldehyde benzoyl hydrazone and 2-hydroxy-1-naphthylaldehyde benzoyl hydrazone. Blood 1997, 89, 3025–3038. [Google Scholar] [PubMed]
- Richardson, D.R.; Tran, E.H.; Ponka, P. The potential of iron chelators of the pyridoxal isonicotinoyl hydrazone class as effective antiproliferative agents. Blood 1995, 86, 4295–4306. [Google Scholar]
- Cazzola, M.; Bergamaschi, G.; Dezza, L.; Arosio, P. Manipulations of cellular iron-metabolism for modulating normal and malignant-cell proliferation—Achievements and prospects. Blood 1990, 75, 1903–1919. [Google Scholar]
- Lui, G.Y.; Kovacevic, Z.; Richardson, V.; Merlot, A.M.; Kalinowski, D.S.; Richardson, D.R. Targeting cancer by binding iron: Dissecting cellular signaling pathways. Oncotarget 2015, 6, 18748–18779. [Google Scholar] [CrossRef]
- Bogdan, A.R.; Miyazawa, M.; Hashimoto, K.; Tsuji, Y. Regulators of Iron Homeostasis: New Players in Metabolism, Cell Death, and Disease. Trends Biochem. Sci. 2016, 41, 274–286. [Google Scholar] [CrossRef]
- Hatcher, H.C.; Singh, R.N.; Torti, F.M.; Torti, S.V. Synthetic and natural iron chelators: Therapeutic potential and clinical use. Future Med. Chem. 2009, 1, 1643–1670. [Google Scholar] [CrossRef] [PubMed]
- Epsztejn, S.; Breuer, W.; Cabantchik, Z.I. Iron Acquired from Transferrin by K562 Cells Is Delivered into a Cytoplasmic Pool of Chelatable Iron(II). J. Biol. Chem. 1995, 270, 24209–24215. [Google Scholar]
- Šebestík, J.; Šafařík, M.; Bouř, P. Ferric Complexes of 3-Hydroxy-4-pyridinones Characterized by Density Functional Theory and Raman and UV–vis Spectroscopies. Inorg. Chem. 2012, 51, 4473–4481. [Google Scholar] [CrossRef] [PubMed]
- Yarbro, J.W. Mechanism of action of hydroxyurea. Semin. Oncol. 1992, 19, 1–10. [Google Scholar] [PubMed]
- Andrews, S.C.; Robinson, A.K.; Rodriguez-Quinones, F. Bacterial iron homeostasis. FEMS Microbiol. Rev. 2003, 27, 215–237. [Google Scholar] [CrossRef]
- Schlachterman, A.; Craft, W.W., Jr.; Hilgenfeldt, E.; Mitra, A.; Cabrera, R. Current and future treatments for hepatocellular carcinoma. World J. Gastroenterol. 2015, 21, 8478–8491. [Google Scholar] [CrossRef] [PubMed]
- Agoro, R.; Benmerzoug, S.; Rose, S.; Bouyer, M.; Gozzelino, R.; Quesniaux, V.F.J.; Mura, C.; Garcia, I.; Ryffel, B. An Iron-Rich Diet Decreases the Mycobacterial Burden and Correlates with Hepcidin Upregulation, Lower Levels of Proinflammatory Mediators, and Increased T-Cell Recruitment in a Model of Mycobacterium bovis Bacille Calmette-Guerin Infection. J. Infect. Dis. 2017, 216, 907–918. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asperti, M.; Cantamessa, L.; Ghidinelli, S.; Gryzik, M.; Denardo, A.; Giacomini, A.; Longhi, G.; Fanzani, A.; Arosio, P.; Poli, M. The Antitumor Didox Acts as an Iron Chelator in Hepatocellular Carcinoma Cells. Pharmaceuticals 2019, 12, 129. https://doi.org/10.3390/ph12030129
Asperti M, Cantamessa L, Ghidinelli S, Gryzik M, Denardo A, Giacomini A, Longhi G, Fanzani A, Arosio P, Poli M. The Antitumor Didox Acts as an Iron Chelator in Hepatocellular Carcinoma Cells. Pharmaceuticals. 2019; 12(3):129. https://doi.org/10.3390/ph12030129
Chicago/Turabian StyleAsperti, Michela, Luca Cantamessa, Simone Ghidinelli, Magdalena Gryzik, Andrea Denardo, Arianna Giacomini, Giovanna Longhi, Alessandro Fanzani, Paolo Arosio, and Maura Poli. 2019. "The Antitumor Didox Acts as an Iron Chelator in Hepatocellular Carcinoma Cells" Pharmaceuticals 12, no. 3: 129. https://doi.org/10.3390/ph12030129
APA StyleAsperti, M., Cantamessa, L., Ghidinelli, S., Gryzik, M., Denardo, A., Giacomini, A., Longhi, G., Fanzani, A., Arosio, P., & Poli, M. (2019). The Antitumor Didox Acts as an Iron Chelator in Hepatocellular Carcinoma Cells. Pharmaceuticals, 12(3), 129. https://doi.org/10.3390/ph12030129