Cysteine Cathepsin Protease Inhibition: An update on its Diagnostic, Prognostic and Therapeutic Potential in Cancer

 ,
, 
Abstract
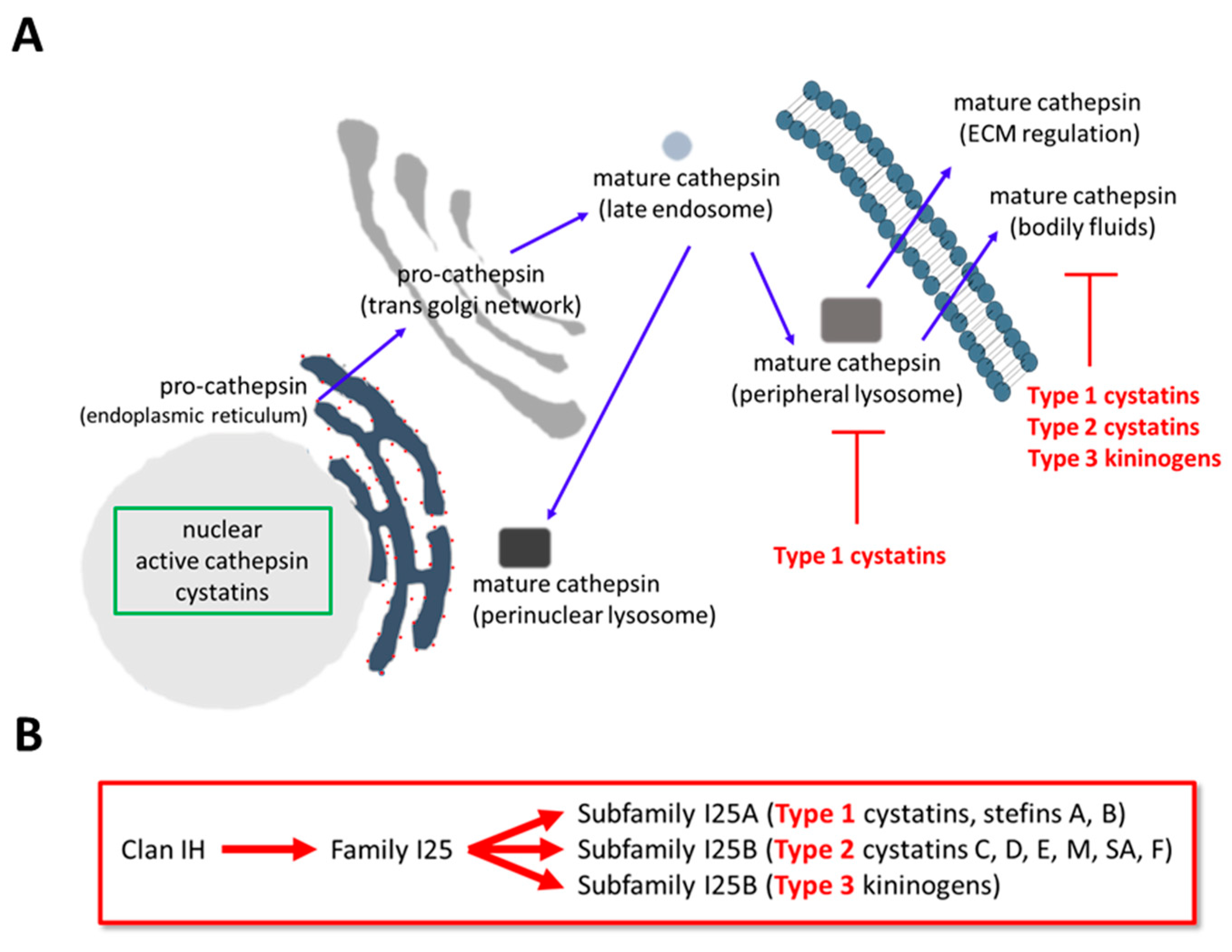
1. Cathepsin Expression and Regulation
2. Transcriptional Regulation of Cystatins in Cancer
3. Cathepsins and Cystatins as Diagnostic Markers for Cancer
4. Cystatins and Cathepsins as Prognostic Markers in Cancer
5. Cathepsin-Derived and -Targeted Therapeutics
6. Clinical Implications and Applications
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Ethical Standards
Abbreviations
| aa | Amino Acids |
| BC | Breast Cancer |
| CC | Colon Carcinoma |
| CRC | Colorectal Cancer |
| ECM | Extracellular Matrix |
| KDa | Kilodaltons |
| LL | Lysosomal Leakage |
| LMP | Lysosomal Membrane Permeabilization |
| PCD | Programmed Cell Death |
| PDAC | Pancreatic Ductal Adenocarcinoma |
| ROC | Receiver Operating Curve Analysis |
References
- Holle, A.W.; Young, J.L.; Spatz, J.P. In vitro cancer cell-ECM interactions inform in vivo cancer treatment. Adv. Drug Deliv. Rev. 2016, 97, 270–279. [Google Scholar] [CrossRef]
- Gocheva, V.; Zeng, W.; Ke, D.; Klimstra, D.; Reinheckel, T.; Peters, C.; Hanahan, D.; Joyce, J.A. Distinct roles for cysteine cathepsin genes in multistage tumorigenesis. Genes Dev. 2006, 20, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Pogorzelska, A.; Zolnowska, B.; Bartoszewski, R. Cysteine cathepsins as a prospective target for anticancer therapies-current progress and prospects. Biochimie 2018, 151, 85–106. [Google Scholar] [CrossRef] [PubMed]
- Kramer, L.; Turk, D.; Turk, B. The Future of Cysteine Cathepsins in Disease Management. Trends Pharmacol. Sci. 2017, 38, 873–898. [Google Scholar] [CrossRef] [PubMed]
- Hamalisto, S.; Jaattela, M. Lysosomes in cancer-living on the edge (of the cell). Curr. Opin. Cell Biol. 2016, 39, 69–76. [Google Scholar] [CrossRef]
- Droga-Mazovec, G.; Bojic, L.; Petelin, A.; Ivanova, S.; Romih, R.; Repnik, U.; Salvesen, G.S.; Stoka, V.; Turk, V.; Turk, B. Cysteine cathepsins trigger caspase-dependent cell death through cleavage of bid and antiapoptotic Bcl-2 homologues. J. Biol. Chem. 2008, 283, 19140–19150. [Google Scholar] [CrossRef]
- Wang, F.; Gomez-Sintes, R.; Boya, P. Lysosomal membrane permeabilization and cell death. Traffic 2018, 19, 918–931. [Google Scholar] [CrossRef]
- Mitrovic, A.; Mirkovic, B.; Sosic, I.; Gobec, S.; Kos, J. Inhibition of endopeptidase and exopeptidase activity of cathepsin B impairs extracellular matrix degradation and tumor invasion. Biol. Chem. 2016, 397, 165–174. [Google Scholar] [CrossRef]
- Poreba, M.; Rut, W.; Vizovisek, M.; Groborz, K.; Kasperkiewicz, P.; Finlay, D.; Vuori, K.; Turk, D.; Turk, B.; Salvesen, G.S.; et al. Selective imaging of cathepsin L in breast cancer by fluorescent activity-based probes. Chem. Sci. 2018, 9, 2113–2129. [Google Scholar] [CrossRef]
- Rut, W.; Kasperkiewicz, P.; Byzia, A.; Poreba, M.; Groborz, K.; Drag, M. Recent advances and concepts in substrate specificity determination of proteases using tailored libraries of fluorogenic substrates with unnatural amino acids. Biol. Chem. 2015, 396, 329–337. [Google Scholar] [CrossRef]
- Ivry, S.L.; Meyer, N.O.; Winter, M.B.; Bohn, M.F.; Knudsen, G.M.; O’Donoghue, A.J.; Craik, C.S. Global substrate specificity profiling of post-translational modifying enzymes. Protein Sci. 2018, 27, 584–594. [Google Scholar] [CrossRef] [PubMed]
- Kasperkiewicz, P.; Poreba, M.; Groborz, K.; Drag, M. Emerging challenges in the design of selective substrates, inhibitors and activity-based probes for indistinguishable proteases. FEBS J. 2017, 284, 1518–1539. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, L.; Wei, L.; Gao, X.; Tang, L.I.; Gong, W.; Min, N.A.; Zhang, L.I.; Yuan, Y. Knockdown of cathepsin L sensitizes ovarian cancer cells to chemotherapy. Oncol. Lett. 2016, 11, 4235–4239. [Google Scholar] [CrossRef] [PubMed]
- Han, M.L.; Zhao, Y.F.; Tan, C.H.; Xiong, Y.J.; Wang, W.J.; Wu, F.; Fei, Y.; Wang, L.; Liang, Z.Q. Cathepsin L upregulation-induced EMT phenotype is associated with the acquisition of cisplatin or paclitaxel resistance in A549 cells. Acta Pharmacol. Sin. 2016, 37, 1606–1622. [Google Scholar] [CrossRef] [PubMed]
- Sui, H.; Shi, C.; Yan, Z.; Wu, M. Overexpression of Cathepsin L is associated with chemoresistance and invasion of epithelial ovarian cancer. Oncotarget 2016, 7, 45995–46001. [Google Scholar] [CrossRef] [PubMed]
- Ishida, Y.; Nayak, S.; Mindell, J.A.; Grabe, M. A model of lysosomal pH regulation. J. Gen. Physiol. 2013, 141, 705–720. [Google Scholar] [CrossRef] [PubMed]
- Van der Stappen, J.W.; Williams, A.C.; Maciewicz, R.A.; Paraskeva, C. Activation of cathepsin B, secreted by a colorectal cancer cell line requires low pH and is mediated by cathepsin D. Int. J. Cancer 1996, 67, 547–554. [Google Scholar] [CrossRef]
- Verma, S.; Dixit, R.; Pandey, K.C. Cysteine Proteases: Modes of Activation and Future Prospects as Pharmacological Targets. Front. Pharmacol. 2016, 7, 107. [Google Scholar] [CrossRef]
- Vray, B.; Hartmann, S.; Hoebeke, J. Immunomodulatory properties of cystatins. Cell. Mol. Life Sci. 2002, 59, 1503–1512. [Google Scholar] [CrossRef] [PubMed]
- Ochieng, J.; Chaudhuri, G. Cystatin superfamily. J. Health Care Poor Underserv. 2010, 21, 51–70. [Google Scholar] [CrossRef] [PubMed]
- Turk, V.; Stoka, V.; Vasiljeva, O.; Renko, M.; Sun, T.; Turk, B.; Turk, D. Cysteine cathepsins: From structure, function and regulation to new frontiers. Biochim. Biophys. Acta 2012, 1824, 68–88. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, J.L.; Skepper, J.N.; McNair, R.; Kasama, T.; Gupta, K.; Weissberg, P.L.; Jahnen-Dechent, W.; Shanahan, C.M. Multifunctional roles for serum protein fetuin-a in inhibition of human vascular smooth muscle cell calcification. J. Am. Soc. Nephrol. 2005, 16, 2920–2930. [Google Scholar] [CrossRef] [PubMed]
- Kordis, D.; Turk, V. Phylogenomic analysis of the cystatin superfamily in eukaryotes and prokaryotes. BMC Evol. Biol. 2009, 9, 266. [Google Scholar] [CrossRef] [PubMed]
- Turk, V.; Bode, W. The cystatins: Protein inhibitors of cysteine proteinases. FEBS Lett. 1991, 285, 213–219. [Google Scholar] [CrossRef]
- Abrahamson, M.; Barrett, A.J.; Salvesen, G.; Grubb, A. Isolation of six cysteine proteinase inhibitors from human urine. Their physicochemical and enzyme kinetic properties and concentrations in biological fluids. J. Biol. Chem. 1986, 261, 11282–11289. [Google Scholar] [PubMed]
- Ni, J.; Fernandez, M.A.; Danielsson, L.; Chillakuru, R.A.; Zhang, J.; Grubb, A.; Su, J.; Gentz, R.; Abrahamson, M. Cystatin F is a glycosylated human low molecular weight cysteine proteinase inhibitor. J. Biol. Chem. 1998, 273, 24797–24804. [Google Scholar] [CrossRef] [PubMed]
- Turk, V.; Stoka, V.; Turk, D. Cystatins: Biochemical and structural properties, and medical relevance. Front. Biosci. 2008, 13, 5406–5420. [Google Scholar] [CrossRef]
- Bode, W.; Engh, R.; Musil, D.; Thiele, U.; Huber, R.; Karshikov, A.; Brzin, J.; Kos, J.; Turk, V. The 2.0 A X-ray crystal structure of chicken egg white cystatin and its possible mode of interaction with cysteine proteinases. EMBO J. 1988, 7, 2593–2599. [Google Scholar] [CrossRef]
- Jerala, R.; Trstenjak, M.; Lenarcic, B.; Turk, V. Cloning a synthetic gene for human stefin B and its expression in E. coli. FEBS Lett. 1988, 239, 41–44. [Google Scholar] [CrossRef]
- Renko, M.; Pozgan, U.; Majera, D.; Turk, D. Stefin A displaces the occluding loop of cathepsin B only by as much as required to bind to the active site cleft. FEBS J. 2010, 277, 4338–4345. [Google Scholar] [CrossRef]
- Naudin, C.; Lecaille, F.; Chowdhury, S.; Krupa, J.C.; Purisima, E.; Mort, J.S.; Lalmanach, G. The occluding loop of cathepsin B prevents its effective inhibition by human kininogens. J. Mol. Biol. 2010, 400, 1022–1035. [Google Scholar] [CrossRef]
- Mihelic, M.; Teuscher, C.; Turk, V.; Turk, D. Mouse stefins A1 and A2 (Stfa1 and Stfa2) differentiate between papain-like endo- and exopeptidases. FEBS Lett. 2006, 580, 4195–4199. [Google Scholar] [CrossRef]
- Choe, Y.; Leonetti, F.; Greenbaum, D.C.; Lecaille, F.; Bogyo, M.; Bromme, D.; Ellman, J.A.; Craik, C.S. Substrate profiling of cysteine proteases using a combinatorial peptide library identifies functionally unique specificities. J. Biol. Chem. 2006, 281, 12824–12832. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, J.; Gilberg, E.; Loser, R.; Bajorath, J.; Bartz, U.; Gutschow, M. Cathepsin B: Active site mapping with peptidic substrates and inhibitors. Bioorg. Med. Chem. 2019, 27, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Mori, J.; Tanikawa, C.; Funauchi, Y.; Lo, P.H.; Nakamura, Y.; Matsuda, K. Cystatin C as a p53-inducible apoptotic mediator that regulates cathepsin L activity. Cancer Sci. 2016, 107, 298–306. [Google Scholar] [CrossRef]
- Maier, O.; Galan, D.L.; Wodrich, H.; Wiethoff, C.M. An N-terminal domain of adenovirus protein VI fragments membranes by inducing positive membrane curvature. Virology 2010, 402, 11–19. [Google Scholar] [CrossRef]
- Ma, Y.; Chen, Y.; Li, Y.; Grun, K.; Berndt, A.; Zhou, Z.; Petersen, I. Cystatin A suppresses tumor cell growth through inhibiting epithelial to mesenchymal transition in human lung cancer. Oncotarget 2018, 9, 14084–14098. [Google Scholar] [CrossRef]
- Tamhane, T.; Lllukkumbura, R.; Lu, S.; Maelandsmo, G.M.; Haugen, M.H.; Brix, K. Nuclear cathepsin L activity is required for cell cycle progression of colorectal carcinoma cells. Biochimie 2016, 122, 208–218. [Google Scholar] [CrossRef]
- Ferrer-Mayorga, G.; Alvarez-Diaz, S.; Valle, N.; De Las Rivas, J.; Mendes, M.; Barderas, R.; Canals, F.; Tapia, O.; Casal, J.I.; Lafarga, M.; et al. Cystatin D locates in the nucleus at sites of active transcription and modulates gene and protein expression. J. Biol. Chem. 2015, 290, 26533–26548. [Google Scholar] [CrossRef]
- Goulet, B.; Baruch, A.; Moon, N.S.; Poirier, M.; Sansregret, L.L.; Erickson, A.; Bogyo, M.; Nepveu, A. A cathepsin L isoform that is devoid of a signal peptide localizes to the nucleus in S phase and processes the CDP/Cux transcription factor. Mol. Cell 2004, 14, 207–219. [Google Scholar] [CrossRef]
- Abudula, A.; Rommerskirch, W.; Weber, E.; Gunther, D.; Wiederanders, B. Splice variants of human cathepsin L mRNA show different expression rates. Biol. Chem. 2001, 382, 1583–1591. [Google Scholar] [CrossRef] [PubMed]
- Sloane, B.F.; Yan, S.; Podgorski, I.; Linebaugh, B.E.; Cher, M.L.; Mai, J.; Cavallo-Medved, D.; Sameni, M.; Dosescu, J.; Moin, K. Cathepsin B and tumor proteolysis: Contribution of the tumor microenvironment. Semin. Cancer Biol. 2005, 15, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Tan, G.; Liu, Q.; Tang, X.; Kang, T.; Li, Y.; Lu, J.; Zhao, X.; Tang, F. Diagnostic values of serum cathepsin B and D in patients with nasopharyngeal carcinoma. BMC Cancer 2016, 16, 241. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.L.; Liu, D.; Cheng, K.; Liu, Y.J.; Xing, S.; Chi, P.D.; Liu, X.H.; Xue, N.; Lai, Y.Z.; Guo, L.; et al. Evaluating the diagnostic and prognostic value of circulating cathepsin S in gastric cancer. Oncotarget 2016, 7, 28124–28138. [Google Scholar] [CrossRef] [PubMed]
- Kehlet, S.N.; Bager, C.L.; Willumsen, N.; Dasgupta, B.; Brodmerkel, C.; Curran, M.; Brix, S.; Leeming, D.J.; Karsdal, M.A. Cathepsin-S degraded decorin are elevated in fibrotic lung disorders—Development and biological validation of a new serum biomarker. BMC Pulm. Med. 2017, 17, 110. [Google Scholar] [CrossRef] [PubMed]
- Komura, T.; Takabatake, H.; Harada, K.; Yamato, M.; Miyazawa, M.; Yoshida, K.; Honda, M.; Wada, T.; Kitagawa, H.; Ohta, T.; et al. Clinical features of cystatin A expression in patients with pancreatic ductal adenocarcinoma. Cancer Sci. 2017, 108, 2122–2129. [Google Scholar] [CrossRef]
- Tokarzewicz, A.; Guszcz, T.; Onopiuk, A.; Kozlowski, R.; Gorodkiewicz, E. Utility of cystatin C as a potential bladder tumor biomarker confirmed by surface plasmon resonance technique. Indian J. Med. Res. 2018, 147, 46–50. [Google Scholar] [CrossRef]
- Kos, J.; Krasovec, M.; Cimerman, N.; Nielsen, H.J.; Christensen, I.J.; Brunner, N. Cysteine proteinase inhibitors stefin A, stefin B, and cystatin C in sera from patients with colorectal cancer: Relation to prognosis. Clin. Cancer Res. 2000, 6, 505–511. [Google Scholar]
- Kothapalli, R.; Bailey, R.D.; Kusmartseva, I.; Mane, S.; Epling-Burnette, P.K.; Loughran, T.P., Jr. Constitutive expression of cytotoxic proteases and down-regulation of protease inhibitors in LGL leukemia. Int. J. Oncol. 2003, 22, 33–39. [Google Scholar] [CrossRef]
- Zhang, J.; He, P.; Zhong, Q.; Li, K.; Chen, D.; Lin, Q.; Liu, W. Increasing Cystatin C and Cathepsin B in Serum of Colorectal Cancer Patients. Clin. Lab. 2017, 63, 365–371. [Google Scholar] [CrossRef]
- Yan, Y.; Zhou, K.; Wang, L.; Wang, F.; Chen, X.; Fan, Q. Clinical significance of serum cathepsin B and cystatin C levels and their ratio in the prognosis of patients with esophageal cancer. Onco Targets Ther. 2017, 10, 1947–1954. [Google Scholar] [CrossRef]
- Ben-Nun, Y.; Fichman, G.; Adler-Abramovich, L.; Turk, B.; Gazit, E.; Blum, G. Cathepsin nanofiber substrates as potential agents for targeted drug delivery. J. Control. Release 2017, 257, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Blum, G.; Mullins, S.R.; Keren, K.; Fonovic, M.; Jedeszko, C.; Rice, M.J.; Sloane, B.F.; Bogyo, M. Dynamic imaging of protease activity with fluorescently quenched activity-based probes. Nat. Chem. Biol. 2005, 1, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Blum, G.; von Degenfeld, G.; Merchant, M.J.; Blau, H.M.; Bogyo, M. Noninvasive optical imaging of cysteine protease activity using fluorescently quenched activity-based probes. Nat. Chem. Biol. 2007, 3, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Blum, G.; Weimer, R.M.; Edgington, L.E.; Adams, W.; Bogyo, M. Comparative assessment of substrates and activity based probes as tools for non-invasive optical imaging of cysteine protease activity. PLoS ONE 2009, 4, e6374. [Google Scholar] [CrossRef] [PubMed]
- Ben-Aderet, L.; Merquiol, E.; Fahham, D.; Kumar, A.; Reich, E.; Ben-Nun, Y.; Kandel, L.; Haze, A.; Liebergall, M.; Kosinska, M.K.; et al. Detecting cathepsin activity in human osteoarthritis via activity-based probes. Arthritis Res. Ther. 2015, 17, 69. [Google Scholar] [CrossRef] [PubMed]
- Oresic Bender, K.; Ofori, L.; van der Linden, W.A.; Mock, E.D.; Datta, G.K.; Chowdhury, S.; Li, H.; Segal, E.; Sanchez Lopez, M.; Ellman, J.A.; et al. Design of a highly selective quenched activity-based probe and its application in dual color imaging studies of cathepsin S activity localization. J. Am. Chem. Soc. 2015, 137, 4771–4777. [Google Scholar] [CrossRef] [PubMed]
- Withana, N.P.; Ma, X.; McGuire, H.M.; Verdoes, M.; van der Linden, W.A.; Ofori, L.O.; Zhang, R.; Li, H.; Sanman, L.E.; Wei, K.; et al. Non-invasive Imaging of Idiopathic Pulmonary Fibrosis Using Cathepsin Protease Probes. Sci. Rep. 2016, 6, 19755. [Google Scholar] [CrossRef]
- Abd-Elrahman, I.; Kosuge, H.; Wises Sadan, T.; Ben-Nun, Y.; Meir, K.; Rubinstein, C.; Bogyo, M.; McConnell, M.V.; Blum, G. Cathepsin Activity-Based Probes and Inhibitor for Preclinical Atherosclerosis Imaging and Macrophage Depletion. PLoS ONE 2016, 11, e0160522. [Google Scholar] [CrossRef] [PubMed]
- Edgington-Mitchell, L.E.; Bogyo, M.; Verdoes, M. Live Cell Imaging and Profiling of Cysteine Cathepsin Activity Using a Quenched Activity-Based Probe. Methods Mol. Biol. 2017, 1491, 145–159. [Google Scholar] [CrossRef]
- Saranya, G.; Joseph, M.M.; Karunakaran, V.; Nair, J.B.; Saritha, V.N.; Veena, V.S.; Sujathan, K.; Ajayaghosh, A.; Maiti, K.K. Enzyme-Driven Switchable Fluorescence-SERS Diagnostic Nanococktail for the Multiplex Detection of Lung Cancer Biomarkers. ACS Appl. Mater. Interfaces 2018, 10, 38807–38818. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.Y.; Chen, Z.W.; Lin, Z.P.; Lin, L.B.; Yang, X.M.; Xu, L.Y.; Xie, Q. Tissue Levels of Stefin A and Stefin B in Hepatocellular Carcinoma. Anat. Rec. 2016, 299, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Wang, C.; Song, X.; Liu, H.; Li, X.; Zhang, Y. Elevated Cathepsin K potentiates metastasis of epithelial ovarian cancer. Histol. Histopathol. 2018, 33, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Breznik, B.; Limbaeck Stokin, C.; Kos, J.; Khurshed, M.; Hira, V.V.V.; Bosnjak, R.; Lah, T.T.; Van Noorden, C.J.F. Cysteine cathepsins B, X and K expression in peri-arteriolar glioblastoma stem cell niches. J. Mol. Histol. 2018, 49, 481–497. [Google Scholar] [CrossRef] [PubMed]
- Bach, A.S.; Derocq, D.; Laurent-Matha, V.; Montcourrier, P.; Sebti, S.; Orsetti, B.; Theillet, C.; Gongora, C.; Pattingre, S.; Ibing, E.; et al. Nuclear cathepsin D enhances TRPS1 transcriptional repressor function to regulate cell cycle progression and transformation in human breast cancer cells. Oncotarget 2015, 6, 28084–28103. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Niu, H.; Hu, Z.; Zhu, M.; Wang, L.; Han, L.; Qian, L.; Tian, K.; Yuan, H.; Lou, H. Targeting the lysosome by an aminomethylated Riccardin D triggers DNA damage through cathepsin B-mediated degradation of BRCA1. J. Cell. Mol. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Riccio, M.; Di Giaimo, R.; Pianetti, S.; Palmieri, P.P.; Melli, M.; Santi, S. Nuclear localization of cystatin B, the cathepsin inhibitor implicated in myoclonus epilepsy (EPM1). Exp. Cell Res. 2001, 262, 84–94. [Google Scholar] [CrossRef]
- Laurent-Matha, V.; Huesgen, P.F.; Masson, O.; Derocq, D.; Prebois, C.; Gary-Bobo, M.; Lecaille, F.; Rebiere, B.; Meurice, G.; Orear, C.; et al. Proteolysis of cystatin C by cathepsin D in the breast cancer microenvironment. FASEB J. 2012, 26, 5172–5181. [Google Scholar] [CrossRef]
- Guerra, E.; Cimadamore, A.; Simeone, P.; Vacca, G.; Lattanzio, R.; Botti, G.; Gatta, V.; D’Aurora, M.; Simionati, B.; Piantelli, M.; et al. p53, cathepsin D, Bcl-2 are joint prognostic indicators of breast cancer metastatic spreading. BMC Cancer 2016, 16, 649. [Google Scholar] [CrossRef]
- Crown, J.; O’Shaughnessy, J.; Gullo, G. Emerging targeted therapies in triple-negative breast cancer. Ann. Oncol. 2012, 23 (Suppl. S6), vi56–vi65. [Google Scholar] [CrossRef]
- Hudis, C.A.; Gianni, L. Triple-negative breast cancer: An unmet medical need. Oncologist 2011, 16 (Suppl. S1), 1–11. [Google Scholar] [CrossRef]
- Guo, K.; Chen, Q.; He, X.; Yao, K.; Li, Z.; Liu, Z.; Chen, J.; Liu, Z.; Guo, C.; Lu, J.; et al. Expression and significance of Cystatin-C in clear cell renal cell carcinoma. Biomed. Pharmacother. 2018, 107, 1237–1245. [Google Scholar] [CrossRef]
- Sun, T.; Jiang, D.; Zhang, L.; Su, Q.; Mao, W.; Jiang, C. Expression profile of cathepsins indicates the potential of cathepsins B and D as prognostic factors in breast cancer patients. Oncol. Lett. 2016, 11, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Carmona, S.; Sukhumalchandra, P.; Roszik, J.; Philips, A.; Perakis, A.A.; Kerros, C.; Zhang, M.; Qiao, N.; John, L.S.S.; et al. Cathepsin G Is Expressed by Acute Lymphoblastic Leukemia and Is a Potential Immunotherapeutic Target. Front. Immunol. 2017, 8, 1975. [Google Scholar] [CrossRef] [PubMed]
- Pandey, G.; Bakhshi, S.; Thakur, B.; Jain, P.; Chauhan, S.S. Prognostic significance of cathepsin L expression in pediatric acute myeloid leukemia. Leuk. Lymphoma 2018, 59, 2175–2187. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Qian, X.; Song, B.; An, X.; Cai, T.; Zuo, Z.; Ding, D.; Lu, Y.; Li, H. Integrated bioinformatics analysis reveals that the expression of cathepsin S is associated with lymph node metastasis and poor prognosis in papillary thyroid cancer. Oncol. Rep. 2018, 40, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Kos, J.; Mitrovic, A.; Mirkovic, B. The current stage of cathepsin B inhibitors as potential anticancer agents. Future Med. Chem. 2014, 6, 1355–1371. [Google Scholar] [CrossRef] [PubMed]
- Reiser, J.; Adair, B.; Reinheckel, T. Specialized roles for cysteine cathepsins in health and disease. J. Clin. Investig. 2010, 120, 3421–3431. [Google Scholar] [CrossRef] [PubMed]
- Pulz, L.H.; Strefezzi, R.F. Proteases as prognostic markers in human and canine cancers. Vet. Comp. Oncol. 2017, 15, 669–683. [Google Scholar] [CrossRef] [PubMed]
- Sadaghiani, L.; Wilson, M.A.; Wilson, N.H. Effect of selected mouthwashes on the surface roughness of resin modified glass-ionomer restorative materials. Dent. Mater. 2007, 23, 325–334. [Google Scholar] [CrossRef]
- Li, Y.Y.; Fang, J.; Ao, G.Z. Cathepsin B and L inhibitors: A patent review (2010–present). Expert Opin. Ther. Pat. 2017, 27, 643–656. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Iioka, H.; Kojima, C.; Ogawa, M.; Kondo, E. Peptide-based tumor inhibitor encoding mitochondrial p14(ARF) is highly efficacious to diverse tumors. Cancer Sci. 2016, 107, 1290–1301. [Google Scholar] [CrossRef] [PubMed]
- Kramer, L.; Renko, M.; Zavrsnik, J.; Turk, D.; Seeger, M.A.; Vasiljeva, O.; Grutter, M.G.; Turk, V.; Turk, B. Non-invasive in vivo imaging of tumor-associated cathepsin B by a highly selective inhibitory DARPin. Theranostics 2017, 7, 2806–2821. [Google Scholar] [CrossRef] [PubMed]
- Raghav, N.; Singh, M. SAR studies of some acetophenone phenylhydrazone based pyrazole derivatives as anticathepsin agents. Bioorg. Chem. 2017, 75, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Li, S.; Wang, X.; Zhang, Y.; Sun, Y.; Wang, Y.; Wang, X.; He, B.; Dai, W.; Zhang, H.; et al. A comparative study of the antitumor efficacy of peptide-doxorubicin conjugates with different linkers. J. Control. Release 2018, 275, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Shim, M.K.; Park, J.; Yoon, H.Y.; Lee, S.; Um, W.; Kim, J.H.; Kang, S.W.; Seo, J.W.; Hyun, S.W.; Park, J.H.; et al. Carrier-free nanoparticles of cathepsin B-cleavable peptide-conjugated doxorubicin prodrug for cancer targeting therapy. J. Control. Release 2018. [Google Scholar] [CrossRef] [PubMed]
- Anantaraju, H.S.; Battu, M.B.; Viswanadha, S.; Sriram, D.; Yogeeswari, P. Cathepsin D inhibitors as potential therapeutics for breast cancer treatment: Molecular docking and bioevaluation against triple-negative and triple-positive breast cancers. Mol. Divers. 2016, 20, 521–535. [Google Scholar] [CrossRef] [PubMed]
- Talieri, M.; Papadopoulou, S.; Scorilas, A.; Xynopoulos, D.; Arnogianaki, N.; Plataniotis, G.; Yotis, J.; Agnanti, N. Cathepsin B and cathepsin D expression in the progression of colorectal adenoma to carcinoma. Cancer Lett. 2004, 205, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Lah, T.T.; Cercek, M.; Blejec, A.; Kos, J.; Gorodetsky, E.; Somers, R.; Daskal, I. Cathepsin B, a prognostic indicator in lymph node-negative breast carcinoma patients: Comparison with cathepsin D, cathepsin L, and other clinical indicators. Clin. Cancer Res. 2000, 6, 578–584. [Google Scholar] [PubMed]
- Palermo, C.; Joyce, J.A. Cysteine cathepsin proteases as pharmacological targets in cancer. Trends Pharmacol. Sci. 2008, 29, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Saftig, P.; Klumperman, J. Lysosome biogenesis and lysosomal membrane proteins: Trafficking meets function. Nat. Rev. Mol. Cell Biol. 2009, 10, 623–635. [Google Scholar] [CrossRef]
- Yuan, L.; Sheng, L.; He, W.; Zou, C.; Hu, B.; Liu, J.; Ge, W.; Liu, Y.; Wang, J.; Ma, E. Discovery of novel cathepsin inhibitors with potent anti-metastatic effects in breast cancer cells. Bioorg. Chem. 2018, 81, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Tawara, K.; Oxford, J.T.; Jorcyk, C.L. Clinical significance of interleukin (IL)-6 in cancer metastasis to bone: Potential of anti-IL-6 therapies. Cancer Manag. Res. 2011, 3, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Freeman, C.L.; Sehn, L. Anti-CD20 Directed Therapy of B Cell Lymphomas: Are New Agents Really Better? Curr. Oncol. Rep. 2018, 20, 103. [Google Scholar] [CrossRef] [PubMed]
- Storm van’s Gravesande, K.; Layne, M.D.; Ye, Q.; Le, L.; Baron, R.M.; Perrella, M.A.; Santambrogio, L.; Silverman, E.S.; Riese, R.J. IFN regulatory factor-1 regulates IFN-gamma-dependent cathepsin S expression. J. Immunol. 2002, 168, 4488–4494. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.A.; El-Ghonaimy, E.A.; Hassan, H.; Mahana, N.; Mahmoud, M.A.; El-Mamlouk, T.; El-Shinawi, M.; Mohamed, M.M. Hormonal-receptor positive breast cancer: IL-6 augments invasion and lymph node metastasis via stimulating cathepsin B expression. J. Adv. Res. 2016, 7, 661–670. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Cathepsin | Cystatin |
|---|---|
| Cysteine- | - |
| B | A, B, C, S |
| C | S, F |
| F | F |
| L | A, B, C, D, E, M, F |
| H | A, B, C, D, F |
| K | F |
| S | B, C, D, F |
| V | E, M, F |
| Serine- | |
| A | - |
| G | - |
| Aspartate- | |
| D | C |
| E | - |
| Cathepsin | mRNA | Protein | ELISA | Biopsy | Serum | Cancer [Reference] |
|---|---|---|---|---|---|---|
| S | X | X | X | Gastric [44] | ||
| B/D-Stef | X | X | X | Hepatocarcinoma [62] | ||
| B/D-Cys | X | X | Colorectal [50] | |||
| K | X | X | Ovarian [63] | |||
| Cys A | X | X | X | X | Pancreatic [46] | |
| B/Cys C | X | X | X | Esophageal [43] | ||
| X | X | X | X | Glioblastoma [64] |
| Theranostic | Cathepsin Specificity | Cathepsin Labelling | Cell Imaging | In Vivo Imaging | Reference |
|---|---|---|---|---|---|
| ABP-GB123 | B, S, L | NA | NA | NA | [55,58] |
| ABP-BMV101 | B, S, L | Y | Y | Y | [57] |
| qABP-BMV109 | B, S, L, X | Y | Y | NA | [56,59] |
| qABP-GB137 | B, L | NA | NA | Y | [52] |
| qABP-YBN1-8 | B, S, L | Y | Y | Y | [51] |
| qABP-BMV083 | B, S, L | Y | NA | NA | [56] |
| qABP-BMV117 | S | Y | NA | NA | [56] |
| qABP-BMV157 | S | Y | Y | Y | [56] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soond, S.M.; Kozhevnikova, M.V.; Townsend, P.A.; Zamyatnin, A.A., Jr. Cysteine Cathepsin Protease Inhibition: An update on its Diagnostic, Prognostic and Therapeutic Potential in Cancer. Pharmaceuticals 2019, 12, 87. https://doi.org/10.3390/ph12020087
Soond SM, Kozhevnikova MV, Townsend PA, Zamyatnin AA Jr. Cysteine Cathepsin Protease Inhibition: An update on its Diagnostic, Prognostic and Therapeutic Potential in Cancer. Pharmaceuticals. 2019; 12(2):87. https://doi.org/10.3390/ph12020087
Chicago/Turabian StyleSoond, Surinder M., Maria V. Kozhevnikova, Paul A. Townsend, and Andrey A. Zamyatnin, Jr. 2019. "Cysteine Cathepsin Protease Inhibition: An update on its Diagnostic, Prognostic and Therapeutic Potential in Cancer" Pharmaceuticals 12, no. 2: 87. https://doi.org/10.3390/ph12020087
APA StyleSoond, S. M., Kozhevnikova, M. V., Townsend, P. A., & Zamyatnin, A. A., Jr. (2019). Cysteine Cathepsin Protease Inhibition: An update on its Diagnostic, Prognostic and Therapeutic Potential in Cancer. Pharmaceuticals, 12(2), 87. https://doi.org/10.3390/ph12020087