Potential of the Other Genetic Information Coded by the Viral RNA Genomes as Antiviral Target




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
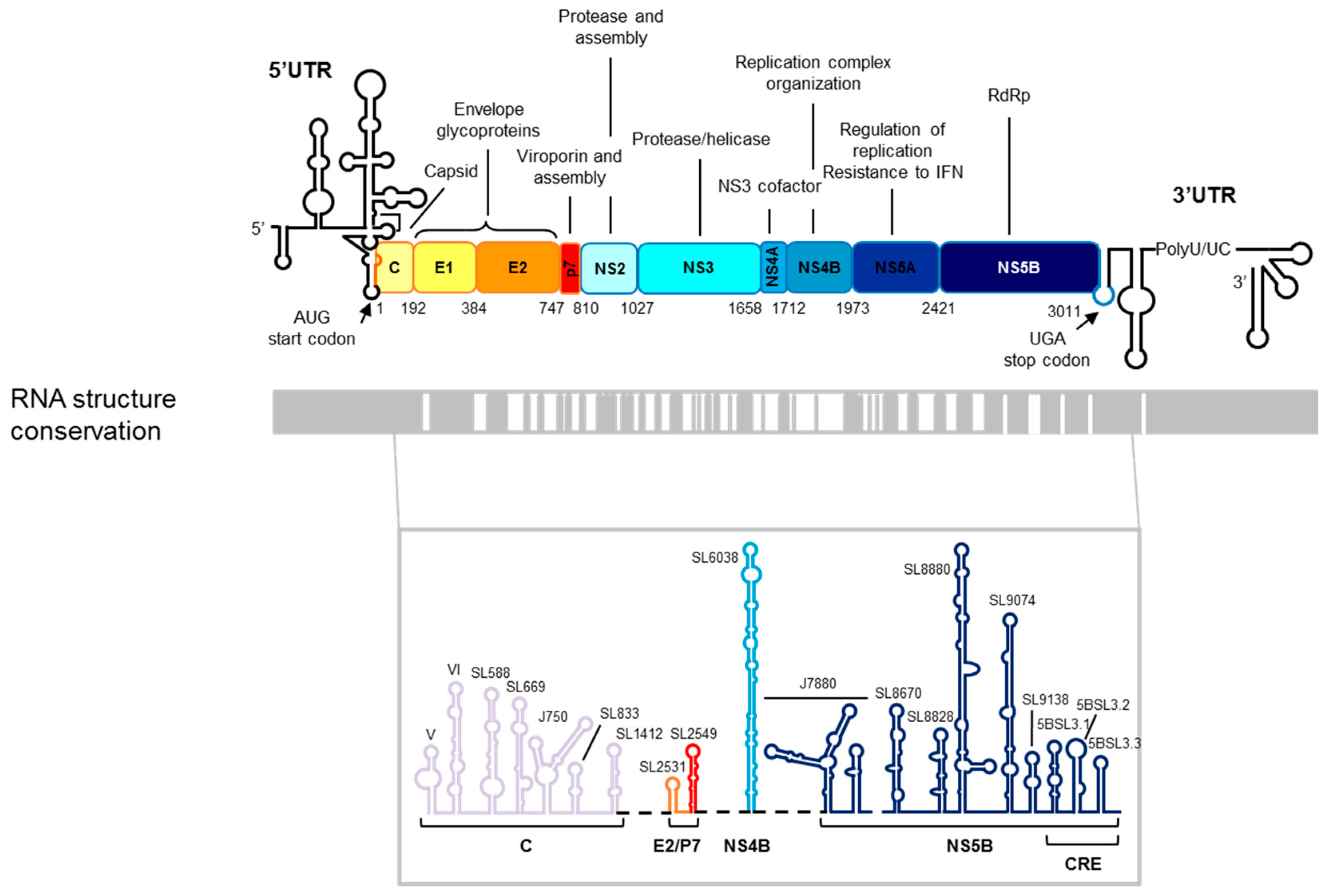
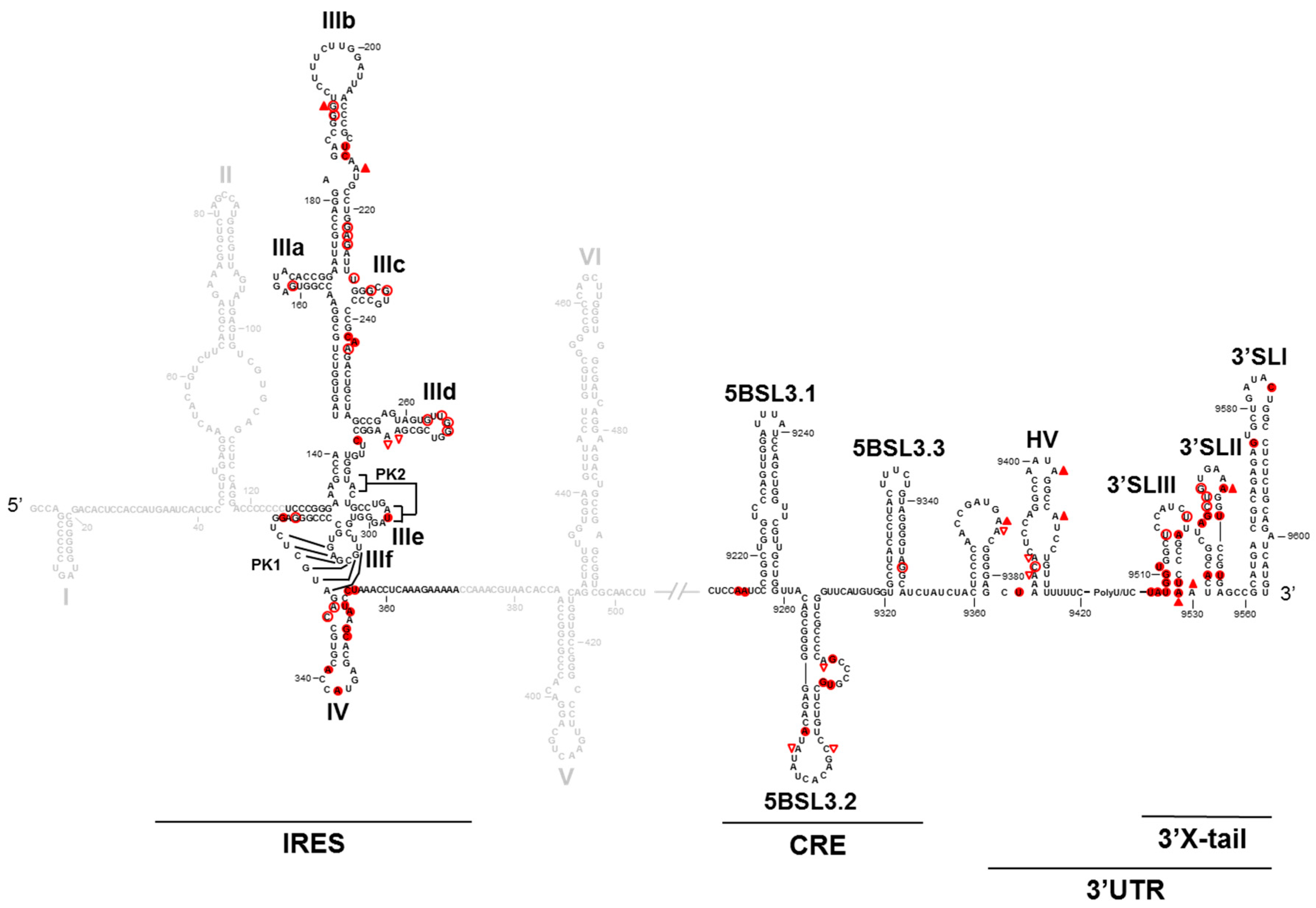
2. HCV Genomic Functional RNA Domains
3. Long-Range Genomic RNA–RNA Interactions
4. Functional Genomic RNA Domains as Therapeutic Targets
4.1. Aptamers
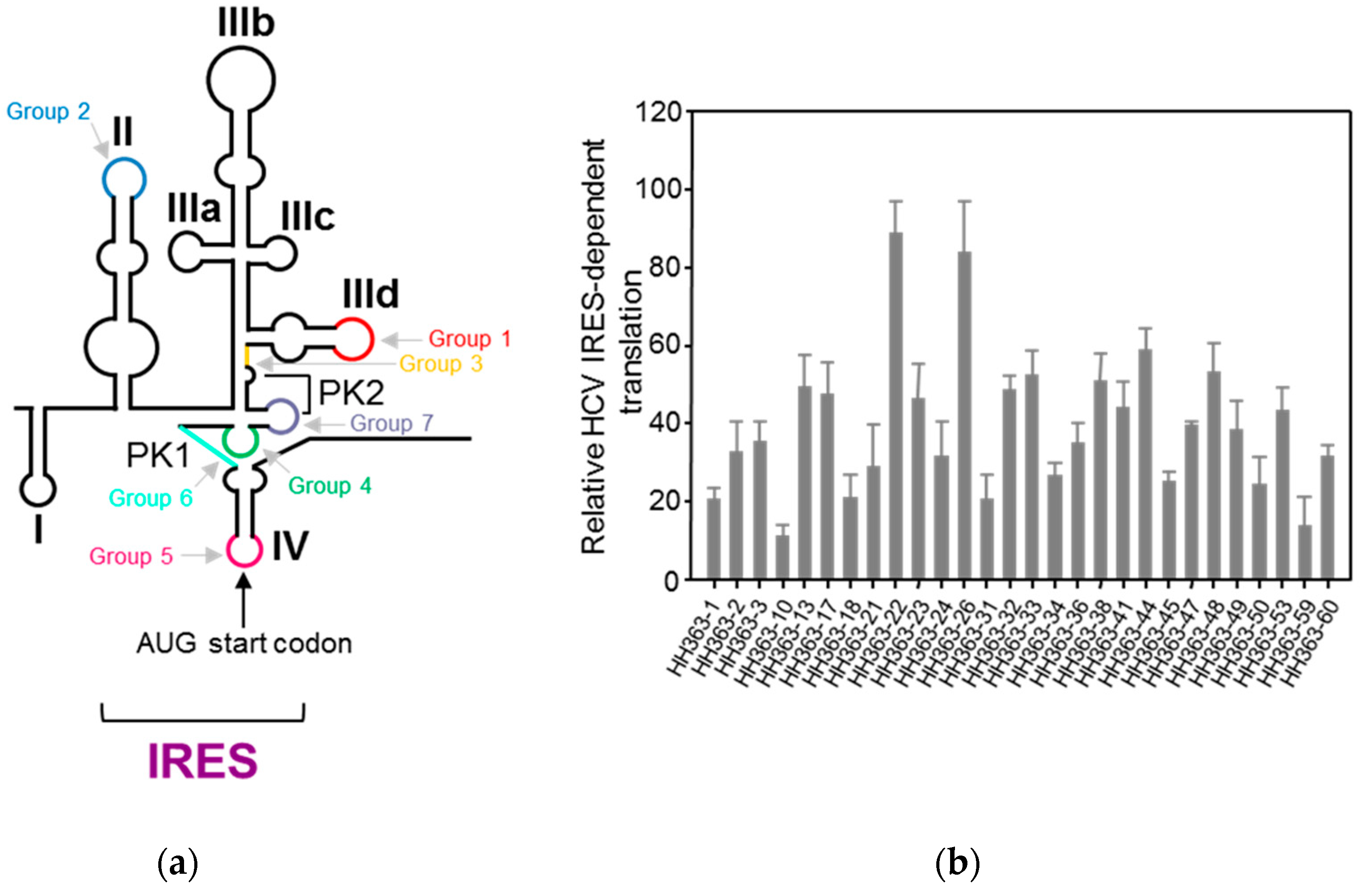
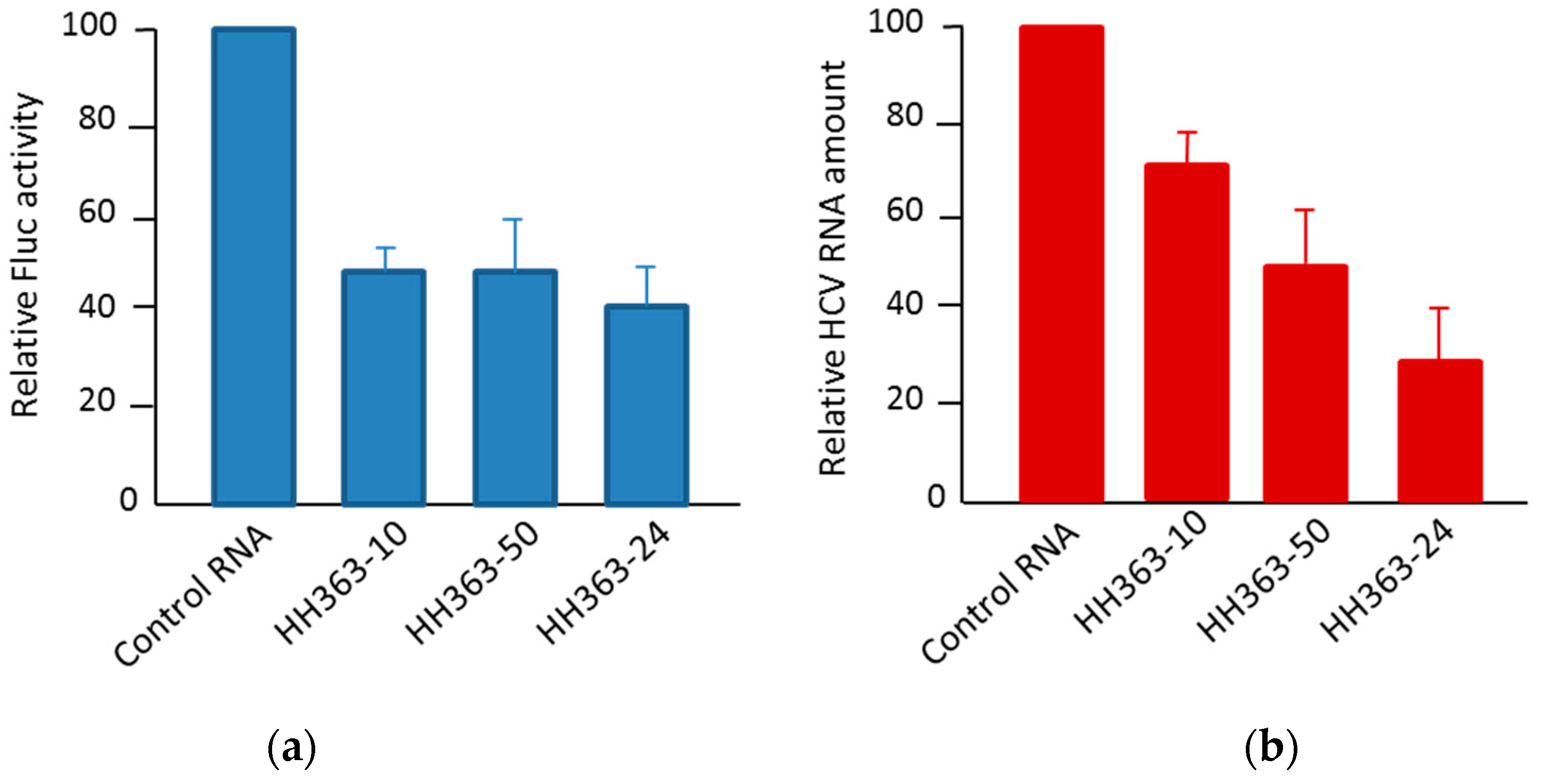
4.1.1. Aptamers Targeting HCV IRES
4.1.2. Anti-HCV CRE Aptamers
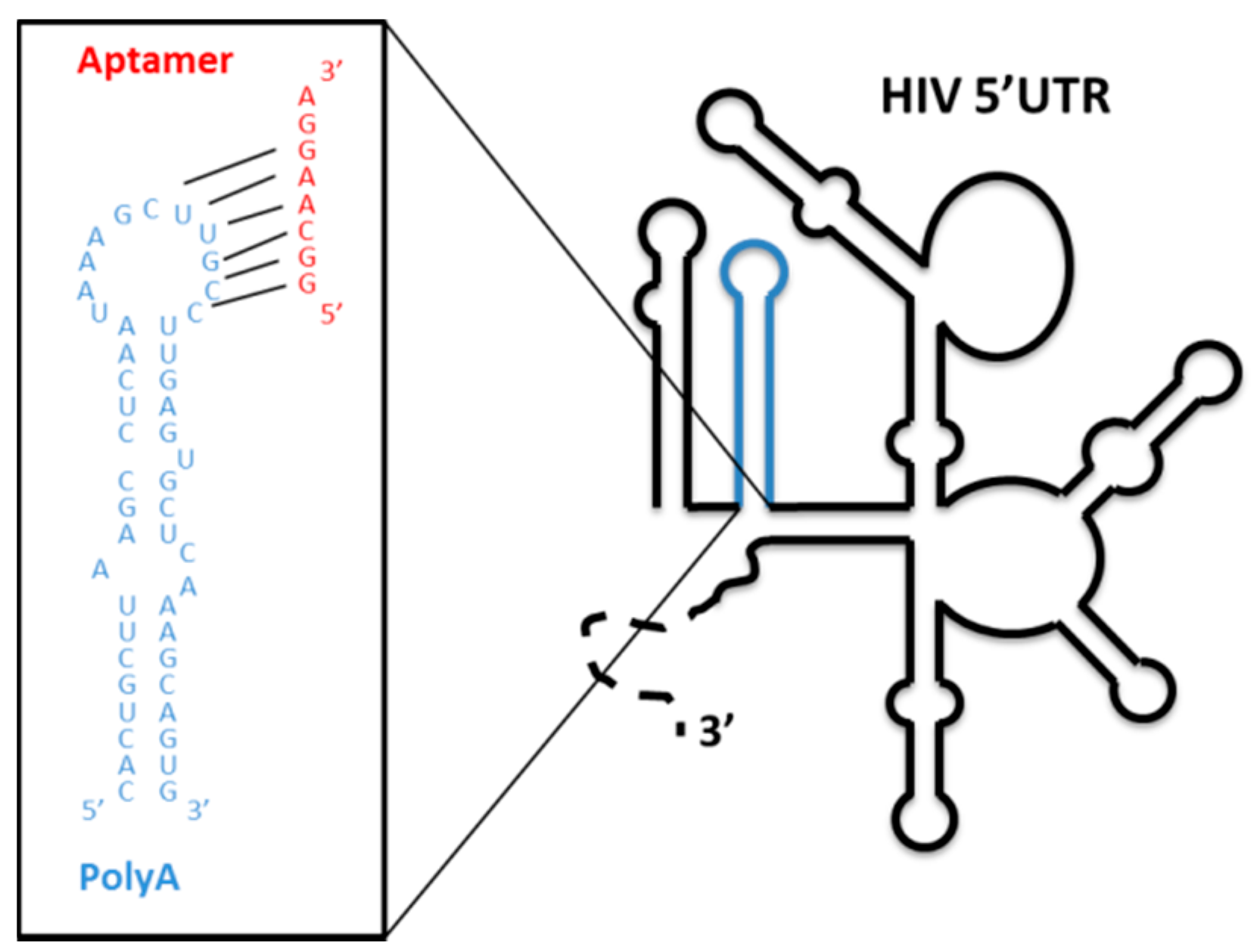
4.1.3. Aptamers Targeting the Structural Elements of the 5′ UTR of the HIV-1 RNA Genome
5. Conclusions
Funding
Conflicts of Interest
References
- Kruger, K.; Grabowski, P.J.; Zaug, A.J.; Sands, J.; Gottschling, D.E.; Cech, T.R. Self-splicing RNA: Autoexcision and autocyclization of the ribosomal RNA intervening sequence of Tetrahymena. Cell 1982, 31, 147–157. [Google Scholar] [CrossRef]
- Guerrier-Takada, C.; Gardiner, K.; Marsh, T.; Pace, N.; Altman, S. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell 1983, 35, 849–857. [Google Scholar] [CrossRef]
- Romero-López, C.; Berzal-Herranz, A. Unmasking the information encoded as structural motifs of viral RNA genomes: A potential antiviral target. Rev. Med. Virol. 2013, 23, 340–354. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, J.E.; Kaminski, A.; Kettinen, H.J.; Grace, K.; Clarke, B.E.; Carroll, A.R.; Rowlands, D.J.; Jackson, R.J. Unique features of internal initiation of hepatitis C virus RNA translation. EMBO J. 1995, 14, 6010–6020. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.H.; Rijnbrand, R.C.; Lemon, S.M. Core protein-coding sequence, but not core protein, modulates the efficiency of cap-independent translation directed by the internal ribosome entry site of hepatitis C virus. J. Virol. 2000, 74, 11347–11358. [Google Scholar] [CrossRef] [PubMed]
- Tsukiyama-Kohara, K.; Iizuka, N.; Kohara, M.; Nomoto, A. Internal ribosome entry site within hepatitis C virus RNA. J. Virol. 1992, 66, 1476–1483. [Google Scholar] [PubMed]
- Wang, C.; Sarnow, P.; Siddiqui, A. Translation of human hepatitis C virus RNA in cultured cells is mediated by an internal ribosome-binding mechanism. J. Virol. 1993, 67, 3338–3344. [Google Scholar]
- Kolykhalov, A.A.; Mihalik, K.; Feinstone, S.M.; Rice, C.M. Hepatitis C virus-encoded enzymatic activities and conserved RNA elements in the 3’ nontranslated region are essential for virus replication in vivo. J. Virol. 2000, 74, 2046–2051. [Google Scholar] [CrossRef]
- Friebe, P.; Lohmann, V.; Krieger, N.; Bartenschlager, R. Sequences in the 5’ nontranslated region of hepatitis C virus required for RNA replication. J. Virol. 2001, 75, 12047–12057. [Google Scholar] [CrossRef]
- Friebe, P.; Bartenschlager, R. Genetic analysis of sequences in the 3’ nontranslated region of hepatitis C virus that are important for RNA replication. J. Virol. 2002, 76, 5326–5338. [Google Scholar] [CrossRef]
- Yi, M.; Lemon, S.M. 3’ nontranslated RNA signals required for replication of hepatitis C virus RNA. J. Virol. 2003, 77, 3557–3568. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Lemon, S.M. Structure-function analysis of the 3’ stem-loop of hepatitis C virus genomic RNA and its role in viral RNA replication. RNA 2003, 9, 331–345. [Google Scholar] [CrossRef]
- McCaffrey, A.P.; Ohashi, K.; Meuse, L.; Shen, S.; Lancaster, A.M.; Lukavsky, P.J.; Sarnow, P.; Kay, M.A. Determinants of hepatitis C translational initiation in vitro, in cultured cells and mice. Mol. Ther. 2002, 5, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Bung, C.; Bochkaeva, Z.; Terenin, I.; Zinovkin, R.; Shatsky, I.N.; Niepmann, M. Influence of the hepatitis C virus 3’-untranslated region on IRES-dependent and cap-dependent translation initiation. FEBS Lett. 2010, 584, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Chu, D.; Ren, S.; Hu, S.; Wang, W.G.; Subramanian, A.; Contreras, D.; Kanagavel, V.; Chung, E.; Ko, J.; Amirtham Jacob Appadorai, R.S.; et al. Systematic analysis of enhancer and critical cis-acting RNA elements in the protein-encoding region of the hepatitis C virus genome. J. Virol. 2013, 87, 5678–5696. [Google Scholar] [CrossRef]
- Davis, M.; Sagan, S.M.; Pezacki, J.P.; Evans, D.J.; Simmonds, P. Bioinformatic and physical characterizations of genome-scale ordered RNA structure in mammalian RNA viruses. J. Virol. 2008, 82, 11824–11836. [Google Scholar] [CrossRef] [PubMed]
- Diviney, S.; Tuplin, A.; Struthers, M.; Armstrong, V.; Elliott, R.M.; Simmonds, P.; Evans, D.J. A hepatitis C virus cis-acting replication element forms a long-range RNA-RNA interaction with upstream RNA sequences in NS5B. J. Virol. 2008, 82, 9008–9022. [Google Scholar] [CrossRef] [PubMed]
- Fricke, M.; Dunnes, N.; Zayas, M.; Bartenschlager, R.; Niepmann, M.; Marz, M. Conserved RNA secondary structures and long-range interactions in hepatitis C viruses. RNA 2015, 21, 1219–1232. [Google Scholar] [CrossRef]
- Mauger, D.M.; Golden, M.; Yamane, D.; Williford, S.; Lemon, S.M.; Martin, D.P.; Weeks, K.M. Functionally conserved architecture of hepatitis C virus RNA genomes. Proc. Natl. Acad. Sci. USA 2015, 112, 3692–3697. [Google Scholar] [CrossRef] [PubMed]
- McMullan, L.K.; Grakoui, A.; Evans, M.J.; Mihalik, K.; Puig, M.; Branch, A.D.; Feinstone, S.M.; Rice, C.M. Evidence for a functional RNA element in the hepatitis C virus core gene. Proc. Natl. Acad. Sci. USA 2007, 104, 2879–2884. [Google Scholar] [CrossRef] [PubMed]
- Pirakitikulr, N.; Kohlway, A.; Lindenbach, B.D.; Pyle, A.M. The Coding Region of the HCV Genome Contains a Network of Regulatory RNA Structures. Mol. Cell 2016, 62, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Tuplin, A.; Wood, J.; Evans, D.J.; Patel, A.H.; Simmonds, P. Thermodynamic and phylogenetic prediction of RNA secondary structures in the coding region of hepatitis C virus. RNA 2002, 8, 824–841. [Google Scholar] [CrossRef] [PubMed]
- Tuplin, A.; Evans, D.J.; Simmonds, P. Detailed mapping of RNA secondary structures in core and NS5B-encoding region sequences of hepatitis C virus by RNase cleavage and novel bioinformatic prediction methods. J. Gen. Virol. 2004, 85, 3037–3047. [Google Scholar] [CrossRef]
- Lee, H.; Shin, H.; Wimmer, E.; Paul, A.V. cis-acting RNA signals in the NS5B C-terminal coding sequence of the hepatitis C virus genome. J. Virol. 2004, 78, 10865–10877. [Google Scholar] [CrossRef] [PubMed]
- You, S.; Stump, D.D.; Branch, A.D.; Rice, C.M. A cis-acting replication element in the sequence encoding the NS5B RNA-dependent RNA polymerase is required for hepatitis C virus RNA replication. J. Virol. 2004, 78, 1352–1366. [Google Scholar] [CrossRef]
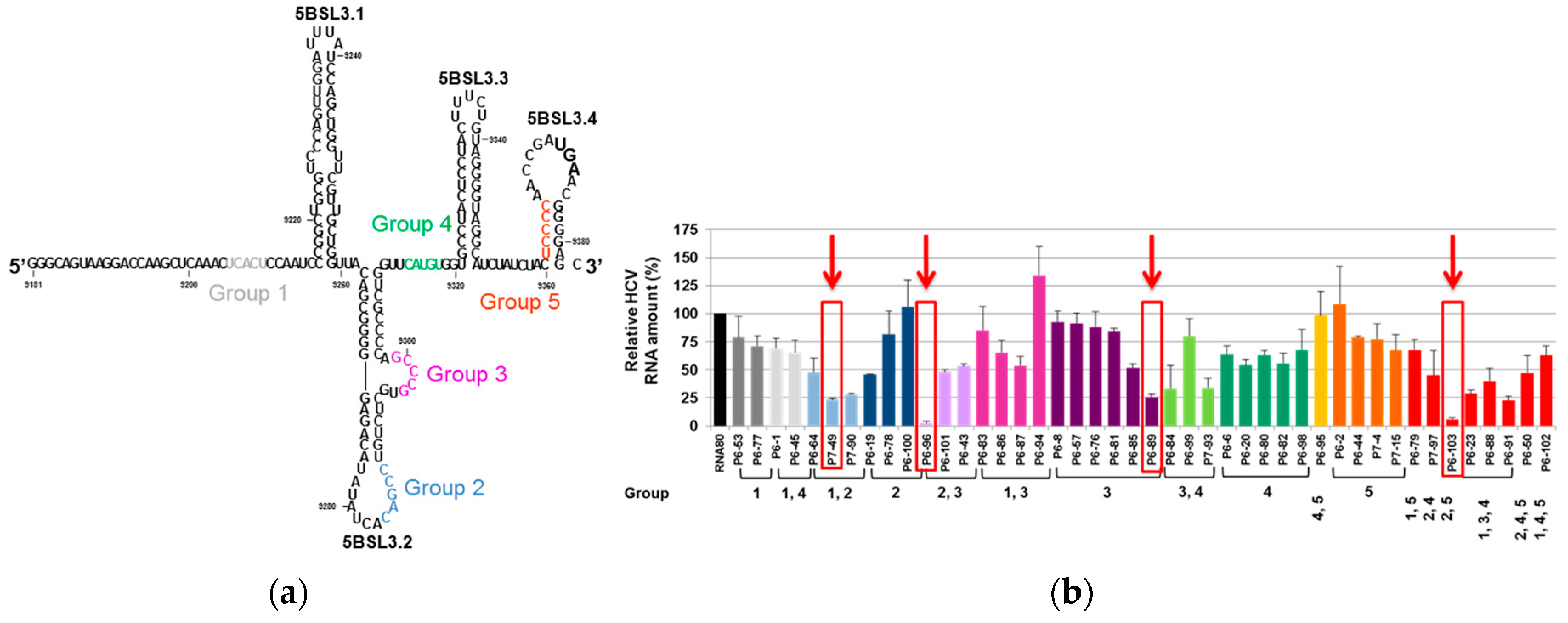
- Romero-López, C.; Berzal-Herranz, A. The functional RNA domain 5BSL3.2 within the NS5B coding sequence influences hepatitis C virus IRES-mediated translation. Cell Mol. Life Sci. 2012, 69, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Romero-López, C.; Berzal-Herranz, A. The 5BSL3.2 Functional RNA Domain Connects Distant Regions in the Hepatitis C Virus Genome. Front. Microbiol. 2017, 8, 2093. [Google Scholar] [CrossRef]
- Bradrick, S.S.; Walters, R.W.; Gromeier, M. The hepatitis C virus 3’-untranslated region or a poly(A) tract promote efficient translation subsequent to the initiation phase. Nucleic Acids Res. 2006, 34, 1293–1303. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Friebe, P.; Tzima, E.; Junemann, C.; Bartenschlager, R.; Niepmann, M. The hepatitis C virus RNA 3’-untranslated region strongly enhances translation directed by the internal ribosome entry site. J. Virol. 2006, 80, 11579–11588. [Google Scholar] [CrossRef] [PubMed]
- Lourenco, S.; Costa, F.; Debarges, B.; Andrieu, T.; Cahour, A. Hepatitis C virus internal ribosome entry site-mediated translation is stimulated by cis-acting RNA elements and trans-acting viral factors. FEBS J. 2008, 275, 4179–4197. [Google Scholar] [CrossRef]
- Romero-López, C.; Berzal-Herranz, A. A long-range RNA-RNA interaction between the 5’ and 3’ ends of the HCV genome. RNA 2009, 15, 1740–1752. [Google Scholar] [CrossRef] [PubMed]
- Romero-López, C.; Barroso-Deljesus, A.; García-Sacristán, A.; Briones, C.; Berzal-Herranz, A. End-to-end crosstalk within the hepatitis C virus genome mediates the conformational switch of the 3’X-tail region. Nucleic Acids Res. 2014, 42, 567–582. [Google Scholar] [CrossRef] [PubMed]
- Friebe, P.; Boudet, J.; Simorre, J.P.; Bartenschlager, R. Kissing-loop interaction in the 3’ end of the hepatitis C virus genome essential for RNA replication. J. Virol. 2005, 79, 380–392. [Google Scholar] [CrossRef] [PubMed]
- Cantero-Camacho, A.; Fan, L.; Wang, Y.X.; Gallego, J. Three-dimensional structure of the 3’X-tail of hepatitis C virus RNA in monomeric and dimeric states. RNA 2017, 23, 1465–1476. [Google Scholar] [CrossRef] [PubMed]
- Tuplin, A.; Struthers, M.; Simmonds, P.; Evans, D.J. A twist in the tail: SHAPE mapping of long-range interactions and structural rearrangements of RNA elements involved in HCV replication. Nucleic Acids Res. 2012, 40, 6908–6921. [Google Scholar] [CrossRef] [PubMed]
- Shetty, S.; Stefanovic, S.; Mihailescu, M.R. Hepatitis C virus RNA: Molecular switches mediated by long-range RNA-RNA interactions? Nucleic Acids Res. 2013, 41, 2526–2540. [Google Scholar] [CrossRef]
- Kim, Y.K.; Lee, S.H.; Kim, C.S.; Seol, S.K.; Jang, S.K. Long-range RNA-RNA interaction between the 5’ nontranslated region and the core-coding sequences of hepatitis C virus modulates the IRES- dependent translation. RNA 2003, 9, 599–606. [Google Scholar] [CrossRef]
- Beguiristain, N.; Robertson, H.D.; Gomez, J. RNase III cleavage demonstrates a long range RNA: RNA duplex element flanking the hepatitis C virus internal ribosome entry site. Nucleic Acids Res. 2005, 33, 5250–5261. [Google Scholar] [CrossRef]
- Diaz-Toledano, R.; Ariza-Mateos, A.; Birk, A.; Martinez-Garcia, B.; Gomez, J. In vitro characterization of a miR-122-sensitive double-helical switch element in the 5’ region of hepatitis C virus RNA. Nucleic Acids Res. 2009, 37, 5498–5510. [Google Scholar] [CrossRef]
- Ariza-Mateos, A.; Diaz-Toledano, R.; Block, T.M.; Prieto-Vega, S.; Birk, A.; Gomez, J. Geneticin Stabilizes the Open Conformation of the 5’ Region of Hepatitis C Virus RNA and Inhibits Viral Replication. Antimicrob. Agents Chemother. 2016, 60, 925–935. [Google Scholar] [CrossRef]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef]
- Henke, J.I.; Goergen, D.; Zheng, J.; Song, Y.; Schuttler, C.G.; Fehr, C.; Junemann, C.; Niepmann, M. microRNA-122 stimulates translation of hepatitis C virus RNA. EMBO J. 2008, 27, 3300–3310. [Google Scholar] [CrossRef] [PubMed]
- Romero-López, C.; Berzal-Herranz, A. Structure-function relationship in viral RNA genomes. The case of HCV. World J. Med. Genet. 2014, 4, 6–18. [Google Scholar] [CrossRef]
- Romero-López, C.; Barroso-Deljesus, A.; García-Sacristán, A.; Briones, C.; Berzal-Herranz, A. The folding of the hepatitis C virus internal ribosome entry site depends on the 3’-end of the viral genome. Nucleic Acids Res. 2012, 40, 11697–11713. [Google Scholar] [CrossRef] [PubMed]
- Cristofari, G.; Ivanyi-Nagy, R.; Gabus, C.; Boulant, S.; Lavergne, J.P.; Penin, F.; Darlix, J.L. The hepatitis C virus Core protein is a potent nucleic acid chaperone that directs dimerization of the viral (+) strand RNA in vitro. Nucleic Acids Res. 2004, 32, 2623–2631. [Google Scholar] [CrossRef] [PubMed]
- Ivanyi-Nagy, R.; Kanevsky, I.; Gabus, C.; Lavergne, J.P.; Ficheux, D.; Penin, F.; Fosse, P.; Darlix, J.L. Analysis of hepatitis C virus RNA dimerization and core-RNA interactions. Nucleic Acids Res. 2006, 34, 2618–2633. [Google Scholar] [CrossRef] [PubMed]
- Blight, K.J.; Rice, C.M. Secondary structure determination of the conserved 98-base sequence at the 3’ terminus of hepatitis C virus genome RNA. J. Virol. 1997, 71, 7345–7352. [Google Scholar]
- Tanaka, T.; Kato, N.; Cho, M.J.; Sugiyama, K.; Shimotohno, K. Structure of the 3’ terminus of the hepatitis C virus genome. J. Virol. 1996, 70, 3307–3312. [Google Scholar]
- Romero-López, C.; Barroso-delJesus, A.; Berzal-Herranz, A. The chaperone-like activity of the hepatitis C virus IRES and CRE elements regulates genome dimerization. Sci Rep. 2017, 7. [Google Scholar] [CrossRef]
- Reyes-Darias, J.A.; Sanchez-Luque, F.J.; Berzal-Herranz, A. Inhibition of HIV-1 replication by RNA-based strategies. Curr. HIV Res. 2008, 6, 500–514. [Google Scholar] [CrossRef]
- Poolsup, S.; Kim, C.Y. Therapeutic applications of synthetic nucleic acid aptamers. Curr. Opin. Biotechnol. 2017, 48, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman, J.E.; Davis, M.E. Clinical experiences with systemically administered siRNA-based therapeutics in cancer. Nat. Rev. Drug Discov. 2015, 14, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.S.; Szostak, J.W. In vitro selection of functional nucleic acids. Annu. Rev. Biochem. 1999, 68, 611–647. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, M.; Ellington, A.D. Selection of fluorescent aptamer beacons that light up in the presence of zinc. Anal. Bioanal. Chem. 2008, 390, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Raddatz, M.S.; Dolf, A.; Endl, E.; Knolle, P.; Famulok, M.; Mayer, G. Enrichment of cell-targeting and population-specific aptamers by fluorescence-activated cell sorting. Angew. Chem. Int. Ed. Engl. 2008, 47, 5190–5193. [Google Scholar] [CrossRef]
- Marton, S.; Reyes-Darias, J.A.; Sanchez-Luque, F.J.; Romero-Lopez, C.; Berzal-Herranz, A. In vitro and ex vivo selection procedures for identifying potentially therapeutic DNA and RNA molecules. Molecules 2010, 15, 4610–4638. [Google Scholar] [CrossRef]
- Kumar, P.K. Monitoring Intact Viruses Using Aptamers. Biosensors (Basel) 2016, 6, 40. [Google Scholar] [CrossRef]
- Romero-López, C.; Barroso-delJesus, A.; Puerta-Fernández, E.; Berzal-Herranz, A. Interfering with hepatitis C virus IRES activity using RNA molecules identified by a novel in vitro selection method. Biol. Chem. 2005, 386, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Romero-López, C.; Díaz-González, R.; Berzal-Herranz, A. Inhibition of hepatitis C virus internal ribosome entry site-mediated translation by an RNA targeting the conserved IIIf domain. Cell Mol. Life Sci. 2007, 64, 2994–3006. [Google Scholar] [CrossRef] [PubMed]
- Romero-López, C.; Díaz-González, R.; Barroso-delJesus, A.; Berzal-Herranz, A. Inhibition of hepatitis C virus replication and internal ribosome entry site-dependent translation by an RNA molecule. J. Gen. Virol. 2009, 90, 1659–1669. [Google Scholar] [CrossRef] [PubMed]
- Romero-López, C.; Berzal-Herranz, B.; Gómez, J.; Berzal-Herranz, A. An engineered inhibitor RNA that efficiently interferes with hepatitis C virus translation and replication. Antiviral Res. 2012, 94, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Romero-López, C.; Lahlali, T.; Berzal-Herranz, B.; Berzal-Herranz, A. Development of Optimized Inhibitor RNAs Allowing Multisite-Targeting of the HCV Genome. Molecules 2017, 22, 861. [Google Scholar] [CrossRef] [PubMed]
- Romero-López, C.; Ríos-Marco, P.; Berzal-Herranz, B.; Berzal-Herranz, A. The HCV genome domains 5BSL3.1 and 5BSL3.3 act as managers of translation. Sci. Rep. 2018, 8, 16101. [Google Scholar] [CrossRef] [PubMed]
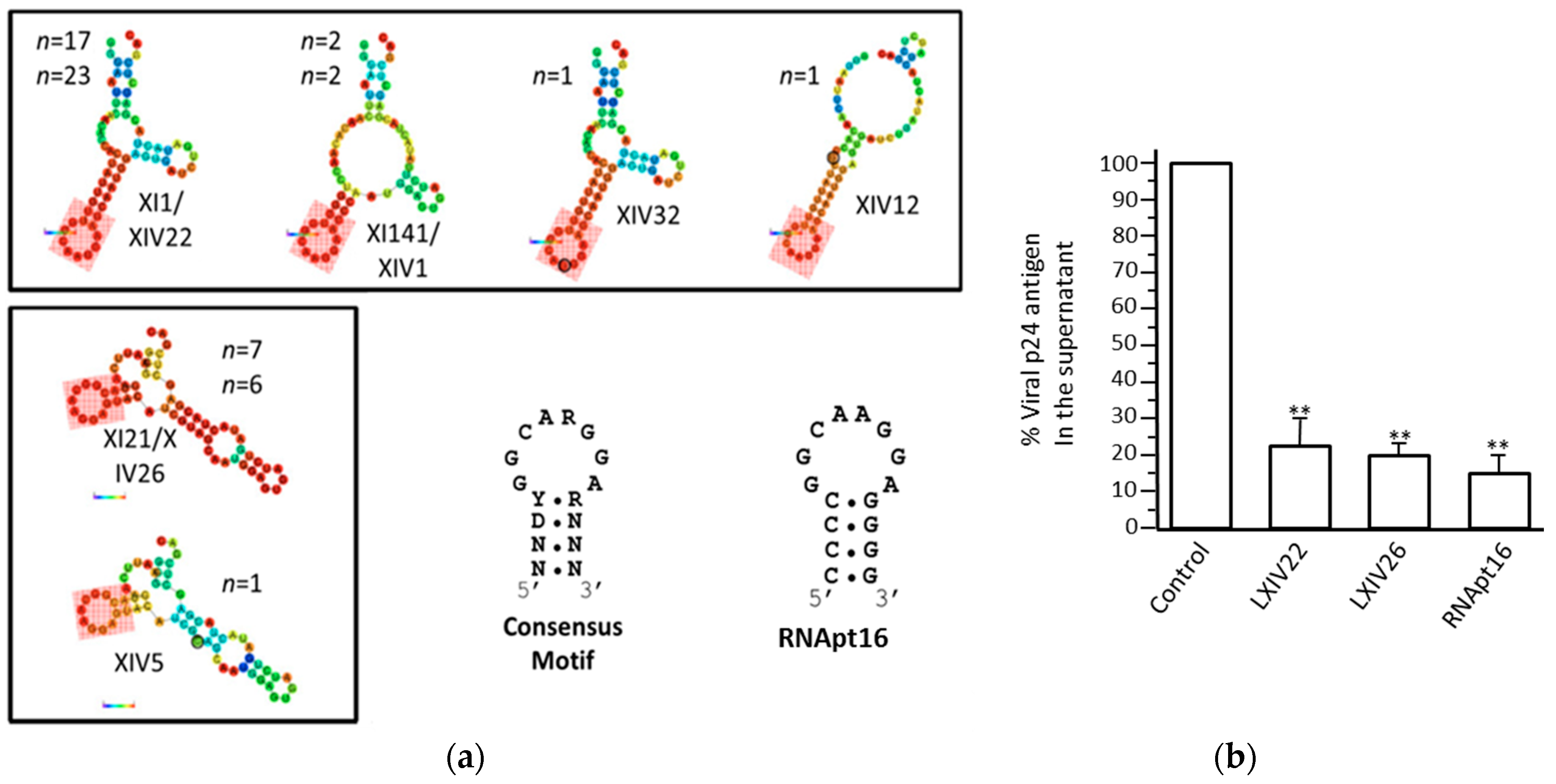
- Marton, S.; Berzal-Herranz, B.; Garmendia, E.; Cueto, F.J.; Berzal-Herranz, A. Anti-HCV RNA Aptamers Targeting the Genomic cis-Acting Replication Element. Pharmaceuticals (Basel) 2012, 5, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Sanlés, A.; Berzal-Herranz, B.; González-Matamala, R.; Ríos-Marco, P.; Romero-López, C.; Berzal-Herranz, A. RNA Aptamers as Molecular Tools to Study the Functionality of the Hepatitis C Virus CRE Region. Molecules 2015, 20, 16030–16047. [Google Scholar] [CrossRef]
- Marton, S.; Romero-Lopez, C.; Berzal-Herranz, A. RNA aptamer-mediated interference of HCV replication by targeting the CRE-5BSL3.2 domain. J. Viral. Hepat. 2013, 20, 103–112. [Google Scholar] [CrossRef]
- Sánchez-Luque, F.J.; Stich, M.; Manrubia, S.; Briones, C.; Berzal-Herranz, A. Efficient HIV-1 inhibition by a 16 nt-long RNA aptamer designed by combining in vitro selection and in silico optimisation strategies. Sci. Rep. 2014, 4, 6242. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berzal-Herranz, A.; Romero-López, C.; Berzal-Herranz, B.; Ramos-Lorente, S. Potential of the Other Genetic Information Coded by the Viral RNA Genomes as Antiviral Target. Pharmaceuticals 2019, 12, 38. https://doi.org/10.3390/ph12010038
Berzal-Herranz A, Romero-López C, Berzal-Herranz B, Ramos-Lorente S. Potential of the Other Genetic Information Coded by the Viral RNA Genomes as Antiviral Target. Pharmaceuticals. 2019; 12(1):38. https://doi.org/10.3390/ph12010038
Chicago/Turabian StyleBerzal-Herranz, Alfredo, Cristina Romero-López, Beatriz Berzal-Herranz, and Sara Ramos-Lorente. 2019. "Potential of the Other Genetic Information Coded by the Viral RNA Genomes as Antiviral Target" Pharmaceuticals 12, no. 1: 38. https://doi.org/10.3390/ph12010038
APA StyleBerzal-Herranz, A., Romero-López, C., Berzal-Herranz, B., & Ramos-Lorente, S. (2019). Potential of the Other Genetic Information Coded by the Viral RNA Genomes as Antiviral Target. Pharmaceuticals, 12(1), 38. https://doi.org/10.3390/ph12010038