NUC041, a Prodrug of the DNA Methytransferase Inhibitor 5-aza-2′,2′-Difluorodeoxycytidine (NUC013), Leads to Tumor Regression in a Model of Non-Small Cell Lung Cancer

Abstract
1. Introduction
2. Results
2.1. In Vitro Comparison of 5-azaC or NUC013 with Its Respective TMS Prodrug
2.2. In Vivo Studies with NUC041 Formulated in a Lipid Nano-Emulsion
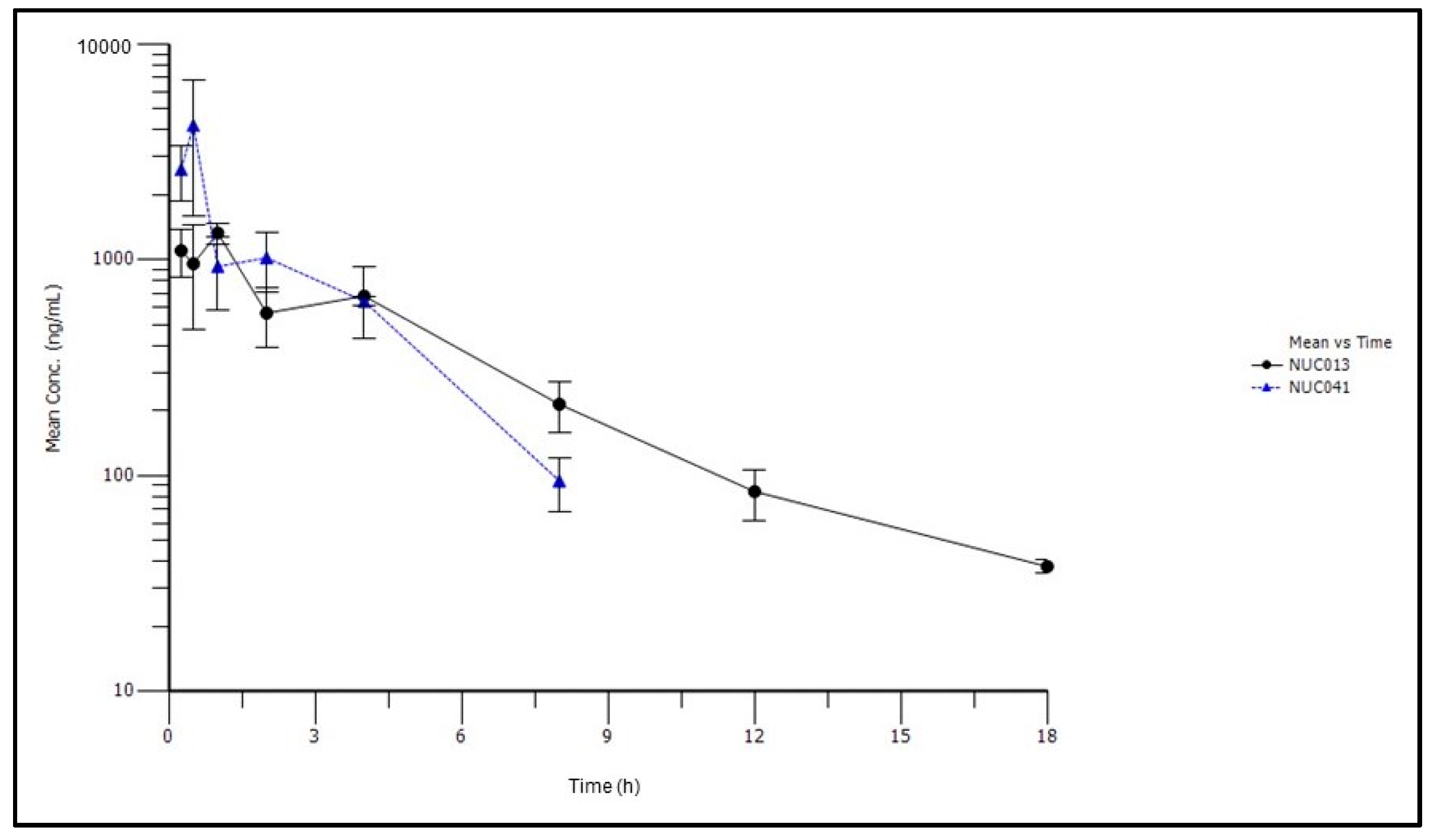
2.3. Pharmacokinetic Studies of NUC041 Formulated in PEG-Phospholipid Depot
2.4. Tolerability Studies of NUC041 Formulated in PPD
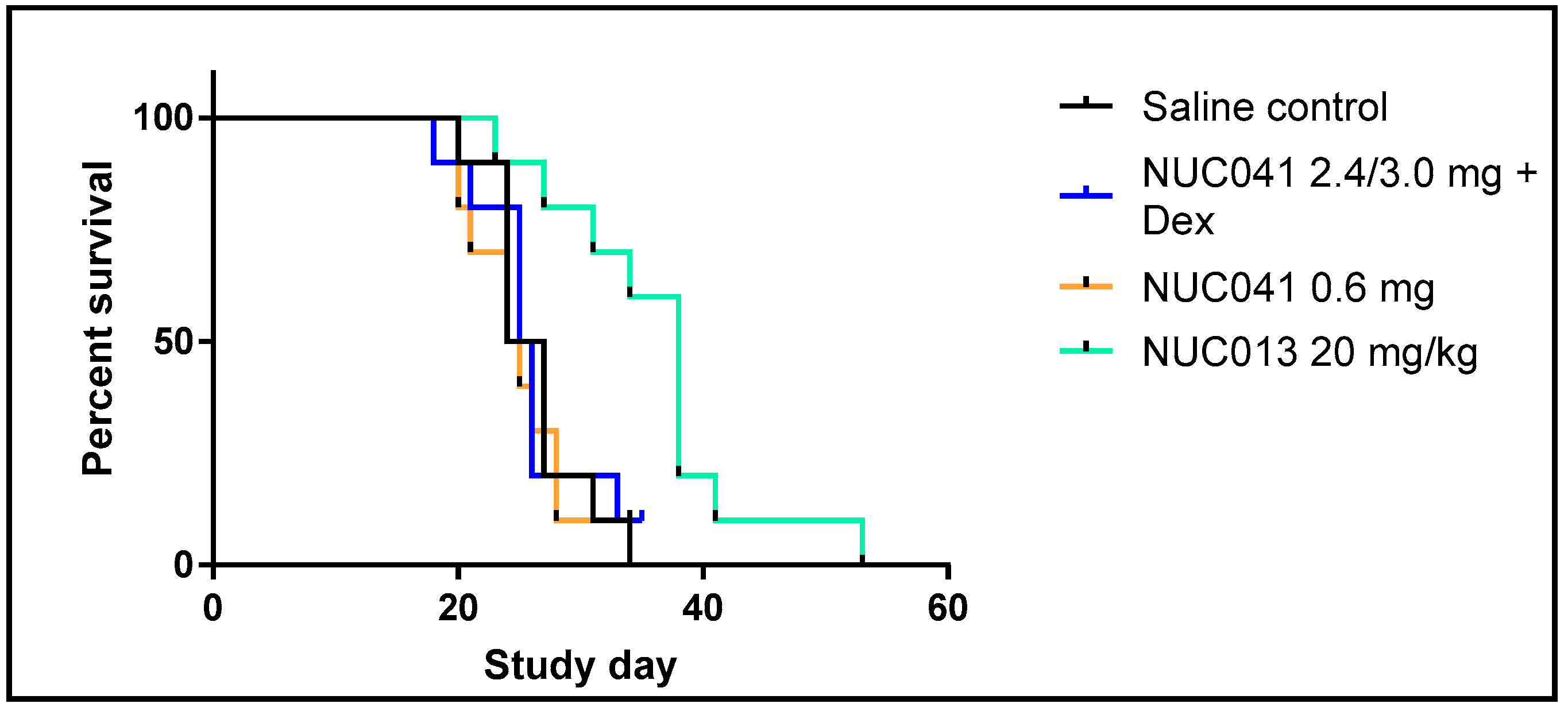
2.5. Human NSCLC NCI-H460 Xenograft Treated with NUC013 and NUC041 in PPD
2.6. Tumor Histology
3. Discussion
4. Materials and Methods
4.1. NUC041 Synthesis
4.2. In Vitro Activity
4.3. Pharmacokinetic Studies
4.4. Tumor Xenograft Studies
4.5. Formulations
4.6. Histology
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Feinberg, A.P. The key role of epigenetics in human disease prevention and mitigation. N. Engl. J. Med. 2018, 378, 1323–1334. [Google Scholar] [CrossRef] [PubMed]
- Gnyszka, A.; Jastrzebski, Z.; Flis, S. DNA methyltransferase inhibitors and their emerging role in epigenetic therapy of cancer. Anticancer Res. 2013, 33, 2989–2996. [Google Scholar] [PubMed]
- Holleran, J.L.; Beumer, J.H.; McCormick, D.L.; Johnson, W.D.; Newman, E.M.; Doroshow, J.H.; Kummar, S.; Covey, J.M.; Davis, M.; Eiseman, J.L. Oral and intravenous pharmacokinetics of 5-fluoro-2’-deoxycytidine and THU in cynomolgus monkeys and humans. Cancer Chemother. Pharmacolol. 2015, 76, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Thottassery, J.V.; Sambandam, V.; Allan, P.W.; Maddry, J.A.; Maxuitenko, Y.Y.; Tiwari, K.; Hollingshead, M.; Parker, W.B. Novel DNA methyltransferase 1 (DNMT1) depleting anticancer nucleosides, 4′-thio-2′-deoxycytidine and 5-aza-4′-thio-2′-deoxycytidine. Cancer Chemother. Pharmacol. 2014, 74, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Highlights of Prescribing Information. Dacogen® (Decitabine) for Injection. Available online: https://www.otsuka-us.com/media/static/DACOGEN-PI.pdf (accessed on 7 February 2018).
- Karahoca, M.; Momparler, R.L. Pharmacokinetic and pharmacodynamic analysis of 5-aza-2′-deoxycytidine (decitabine) in the design of its dose-schedule for cancer therapy. Clin. Epigenet. 2013, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Samlowski, W.E.; Leachman, S.A.; Wade, M.; Cassidy, P.; Porter-Gill, P.; Busby, L.; Wheeler, R.; Boucher, K.; Fitzpatrick, F.; Jones, D.A.; et al. Evaluation of a 7-day continuous intravenous infusion of decitabine: Inhibition of promoter-specific and global genomic DNA methylation. J. Clin. Oncol. 2005, 23, 3897–3905. [Google Scholar] [CrossRef] [PubMed]
- Scandura, J.M.; Roboz, G.J.; Moh, M.; Morawa, E.; Brenet, F.; Bose, J.R.; Villegas, L.; Gergis, U.S.; Mayer, S.A.; Ippoliti, C.M.; et al. Phase 1 study of epigenetic priming with decitabine prior to standard induction chemotherapy for patients with AML. Blood 2011, 118, 1472–1480. [Google Scholar] [CrossRef] [PubMed]
- Daifuku, R.; Hu, Z.; Saunthararajah, Y. 5-aza-2′,2′-difluoro deoxycytidine (NUC013): A novel nucleoside DNA methyl transferase inhibitor and ribonucleotide reductase inhibitor for the treatment of cancer. Pharmaceuticals 2017, 10, 65. [Google Scholar] [CrossRef] [PubMed]
- Stresemann, C.; Lyko, F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int. J. Cancer 2008, 123, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Utoh, M.; Sawada, N.; Nishida, M.; Fukase, Y.; Sekiguchi, F.; Ishitsuka, H. Tumor selective delivery of 5-fluorouracil by capecitabine, a new oral fluoropyrimidine carbamate, in human cancer xenografts. Biochem. Pharmacol. 1998, 55, 1091–1097. [Google Scholar] [CrossRef]
- Harris, K.S.; Brabant, W.; Styrchak, S.; Gall, A.; Daifuku, R. KP-1212/1461, a nucleoside designed for the treatment of HIV by viral mutagenesis. Antivir. Res. 2005, 67, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Montana, J.G.; Bains, W. Silicon Compounds Useful in Cancer Therapy. WO 2004/050666, 17 June 2004. [Google Scholar]
- Mahkam, M.; Assadi, M.G.; Golipour, N. pH-sensitive hydrogel containing acetaminophen silyl ethers for colon-specific drug delivery. Des. Monomers Polym. 2006, 9, 607–615. [Google Scholar] [CrossRef]
- Assadi, M.G.; Golipour, N. Synthesis and characterization of methyl salicylate and acetaminophen silyl ether candidates for prodrugs. Main Group Chem. 2006, 5, 179–190. [Google Scholar] [CrossRef]
- National Research Council. Spacecraft Maximum Allowable Concentrations for Selected Airborne Contaminants. Volume 1. Available online: https://www.nap.edu/read/9062/chapter/13 (accessed on 9 January 2018).
- Szebeni, J. Complement activation-related pseudoallergy: A new class of drug-induced acute immune toxicity. Toxicology 2005, 216, 106–121. [Google Scholar] [CrossRef] [PubMed]
- Szebeni, J.; Barenholz, Y. Adverse Immune Effects of Liposomes: Complement Activation, Immunogenicity and Immune Suppression. Available online: http://seroscience.com/wp-content/uploads/2014/10/2009_Liposomes.pdf (accessed on 26 January 2018).
- Jang, H.J.; Shin, C.Y.; Kim, K.B. Safety of polyethylene glycol (PEG) compounds for cosmetic use. Toxicol Res. 2015, 31, 105–136. [Google Scholar] [CrossRef] [PubMed]
- Butani, L.; Calogiuri, G. Hypersensitivity reactions in patients receiving hemodialysis. Ann. Allergy Asthma Immunol. 2017, 118, 680–684. [Google Scholar] [CrossRef] [PubMed]
- Marina, N.M.; Cochrane, D.; Harney, E.; Zomorodi, K.; Blaney, S.; Winick, N.; Bernstein, M.; Link, M.P. Dose escalation and pharmacokinetics of pegylated liposomal doxorubicin (Doxil) in children with solid tumors: A pediatric oncology group study. Clin. Cancer Res. 2002, 8, 413–418. [Google Scholar] [PubMed]
- Krakauer, T.; Buckley, M. Dexamethasone attenuates staphylococcal enterotoxin B-induced hypothermic response and protects mice from superantigen-induced toxic shock. Antimicrob. Agents Chemother. 2006, 50, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Daifuku, R.; Koratich, M.; Stackhouse, M. Vitamin E phosphate nucleosides prodrugs: A platform for intracellular delivery of monophosphorylated nucleosides. Pharmaceuticals 2018, 11, 16. [Google Scholar] [CrossRef] [PubMed]
- Salamon, J.; Hoffmann, T.; Elies, E.; Peldschus, K.; Johansen, J.S.; Lüers, G.; Schumacher, U.; Wicklein, D. Antibody directed against human YKL-40 increases tumor volume in a human melanoma xenograft model in Scid mice. PLoS ONE 2014, 9, e95822. [Google Scholar] [CrossRef] [PubMed]
- Chiou, V.L.; Burotto, M. Pseudoprogression and immune-related response in solid tumors. J. Clin. Oncol. 2015, 33, 3541–3543. [Google Scholar] [CrossRef] [PubMed]
- Jewell, R.; Elliott, F.; Laye, J.; Nsengimana, J.; Davies, J.; Walker, C.; Conway, C.; Mitra, A.; Harland, M.; Cook, M.G.; et al. The clinicopathological and gene expression patterns associated with ulceration of primary melanoma. Pigment Cell Melanoma Res. 2015, 28, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Al-Salihi, M.; Yu, M.; Burnett, D.M.; Alexander, A.; Samlowski, W.E.; Fitzpatrick, F.A. The depletion of DNA methyltransferase-1 and the epigenetic effects of 5-aza-2′deoxycytidine (decitabine) are differentially regulated by cell cycle progression. Epigenetics 2011, 6, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, E.A.; Choy, G.; Redkar, S.; Taverna, P.; Azab, M.; Karpf, A.R. SGI 110: DNA methyltransferase inhibitor oncolytic. Drugs Future 2013, 38, 535–543. [Google Scholar] [PubMed]
- Yoo, C.B.; Jeong, S.; Egger, G.; Liang, G.; Phiasivongsa, P.; Tang, C.; Redkar, S.; Jones, P.A. Delivery of 5-aza-2′-deoxycytidine to cells using oligodeoxynucleotides. Cancer Res. 2007, 67, 6400–6408. [Google Scholar] [CrossRef] [PubMed]
- Issa, J.J.; Roboz, G.; Rizzieri, D.; Jabbour, E.; Stock, W.; O’Connell, C.; Yee, K.; Tibes, R.; Griffiths, E.A.; Walsh, K.; et al. Safety and tolerability of guadecitabine (SGI-110) in patients with myelodysplastic syndrome and acute myeloid leukaemia: A multicentre, randomised, dose-escalation phase 1 study. Lancet Oncol. 2015, 16, 1099–1110. [Google Scholar] [CrossRef]
- Yi, L.; Sun, Y.; Levine, A. Selected drugs that inhibit DNA methylation can preferentially kill p53 deficient cells. Oncotarget 2014, 5, 8924–8936. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zeng, S.X.; Lu, H. Targeting p53-MDM2-MDMX loop for cancer therapy. Subcell Biochem. 2014, 85, 281–319. [Google Scholar] [PubMed]
- Liu, K.; Zhan, M.; Zheng, P. Loss of p73 expression in six non-small cell lung cancer cell lines is associated with 5’CpG island methylation. Exp. Mol. Pathol. 2008, 84, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Izumi, H.; Inoue, J.; Yokoi, S.; Hosoda, H.; Shibata, T.; Sunamori, M.; Hirohashi, S.; Inazawa, J.; Imoto, I. Frequent silencing of DBC1 is by genetic or epigenetic mechanisms in non-small cell lung cancers. Hum. Mol. Genet. 2005, 14, 997–1007. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sigalotti, L.; Fratta, E.; Coral, S.; Maio, M. Epigenetic drugs as immunomodulators for combination therapies in solid tumors. Pharmacol. Ther. 2014, 142, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, P.; Paluch, B.E.; Matsuzaki, J.; James, S.R.; Collamat-Lai, G.; Blagitko-Dorfs, N.; Ford, L.A.; Naqash, R.; Lübbert, M.; Karpf, A.R.; et al. Induction of cancer testis antigen expression in circulating acute myeloid leukemia blasts following hypomethylating agent monotherapy. Oncotarget 2016, 7, 12840–12856. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.; Li, M.; Wang, R.; Xiao, Q.; Wang, J.; Li, M.; He, D.; Xiao, X. Knockdown of CABYR-a/b increases chemosensitivity of human non-small cell lung cancer cells through inactivation of Akt. Mol. Cancer Res. 2014, 12, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Belizário, J.E. Immunodeficient mouse models: An overview. Open Immunol. J. 2009, 2, 79–85. [Google Scholar] [CrossRef]
- Yeadon, J. Immunodeficient Mice for Cancer Studies: Which Host Strain Should I Use? Available online: https://www.jax.org/news-and-insights/jax-blog/2013/july/which-host-strain-should-i-use (accessed on 28 February 2018).
- Li, X.; Zhang, Y.; Chen, M.; Mei, Q.; Liu, Y.; Feng, K.; Jia, H.; Dong, L.; Shi, L.; Liu, L.; Nie, J.; Han, W. Increased IFNγ+ T cells are responsible for the clinical responses of low-dose DNA-demethylating agent decitabine antitumor therapy. Clin. Cancer Res. 2017, 23, 6031–6043. [Google Scholar] [CrossRef] [PubMed]
- Collin, R.; St-Pierre, C.; Guilbault, L.; Mullins-Dansereau, V.; Policheni, A.; Guimont-Desrochers, F.; Pelletier, A.N.; Gray, D.H.; Drobetsky, E.; Perreault, C.; et al. An unbiased linkage approach reveals that the p53 pathway is coupled to NK cell maturation. J. Immunol. 2017, 199, 1490–1504. [Google Scholar] [CrossRef] [PubMed]
- Textor, S.; Fiegler, N.; Arnold, A.; Porgador, A.; Hofmann, T.G.; Cerwenka, A. Human NK cells are alerted to induction of p53 in cancer cells by upregulation of the NKG2D ligands ULBP1 and ULBP2. Cancer Res. 2011, 71, 5998–6009. [Google Scholar] [CrossRef] [PubMed]
- Iannello, A.; Raulet, D.H. Immunosurveillance of senescent cancer cells by natural killer cells. Oncoimmunology 2014, 3, e27616. [Google Scholar] [CrossRef] [PubMed]
- Rock, K.L.; Kono, H. The inflammatory response to cell death. Annu. Rev. Pathol. 2008, 3, 99–126. [Google Scholar] [CrossRef] [PubMed]
- Baurain, J.; Stas, M.; Hammouch, F.; Gillain, A.; Feyens, A.; Van Baren, N.; Tromme, I.; Van Wijck, R.; Garmyn, M.; Coulie, P.G. Association of primary melanoma ulceration and clinical benefit of adjuvant vaccination with tumor-specific antigenic peptides. J. Clin. Oncol. 2009, 27, 3022. [Google Scholar] [CrossRef]
- Eggermont, A.M.; Suciu, S.; Testori, A.; Santinami, M.; Kruit, W.H.; Marsden, J.; Punt, C.J.; Salès, F.; Dummer, R.; Robert, C.; et al. Long term results of the randomized phase III trial EORTC 18991 of adjuvant therapy with pegylated interferon alfa-2B versus observation in resected stage III melanoma. J. Clin. Oncol. 2012, 30, 3810–3818. [Google Scholar] [CrossRef] [PubMed]
- Boylan, J.C.; Nail, S.L. Parenteral Products. Modern Pharmaceutics Volume 1. Basic Principles and Systems. Fifth Edition. Available online: https://books.google.es/books?id=X9_qBgAAQBAJ&pg=PA565&dq=parenteral+products++boylan+nail&hl=es&sa=X&ved=0ahUKEwjH35yf0pbZAhXDPhQKHUXuDhcQ6AEIUjAG#v=onepage&q=parenteral%20products%20%20boylan%20nail&f=false (accessed on 8 February 2018).
- Bozzuto, G.; Molinari, A. Liposomes as nanomedical devices. Int. J. Nanomed. 2015, 10, 975–999. [Google Scholar] [CrossRef] [PubMed]
- Doxil (Doxorubicin Hydrochloride Liposome Injection) for Intravenous Use. Highlights of Prescribing Information. Available online: https://www.doxil.com/shared/product/doxil/doxil-prescribing-information.pdf (accessed on 8 February 2018).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | NSCLC NCI-H460 GI50 (µM) | Colon Cancer HCT-116 GI50 (µM) |
|---|---|---|
| NUC013 | 1.57 | 2.58 |
| NUC041 | 1.61 | 2.80 |
| Analyte | Cmax (ng/mL) | Tmax (min) | T1/2 (min) | AUClast (h·ng/mL) | AUCinf (h·ng/mL) | Cl (mL/min/kg) | Vss (mL/kg) |
|---|---|---|---|---|---|---|---|
| NUC041 | 2803 | 3 | 11 | 580.4 | 590.6 | 423 | 4431 |
| NUC013 | 745 | 15 | 15 | 464.0 | 468.0 | NA | NA |
| Analyte | Cmax (ng/mL) | Tmax (h) | T1/2 (h) | AUClast (h·ng/mL) | AUCinf (h·ng/mL) | MRT (h) | Vz/F (mL/kg) |
|---|---|---|---|---|---|---|---|
| NUC041 | 4210 | 0.5 | 1.7 | 6030 | 6261 | 2.6 | 1172 |
| NUC013 | 1333 | 1 | 3.4 | 5629 | 5813 | 5.1 | NA |
| Mouse Identifier | NUC041 Dose & Regimen | Study Day of Scheduled Euthanasia | Tumor Volume (mm3)/Ulceration | Histopathology |
|---|---|---|---|---|
| 1 | 2.4/3.0 mg/mouse qwk + dexamethasone | 35 | 2746/no ulceration | 30–40% necrosis |
| 2 | 0.6 mg/mouse qod | 34 | 3035/no ulceration | 50–60% necrosis |
| 3 | 0.6 mg/mouse qod | 34 | 1960/ulceration | 10% necrosis * |
| 4 | 0.6 mg /mouse qod | 34 | 936/ulceration | 5% necrosis * |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daifuku, R.; Grimes, S.; Stackhouse, M. NUC041, a Prodrug of the DNA Methytransferase Inhibitor 5-aza-2′,2′-Difluorodeoxycytidine (NUC013), Leads to Tumor Regression in a Model of Non-Small Cell Lung Cancer. Pharmaceuticals 2018, 11, 36. https://doi.org/10.3390/ph11020036
Daifuku R, Grimes S, Stackhouse M. NUC041, a Prodrug of the DNA Methytransferase Inhibitor 5-aza-2′,2′-Difluorodeoxycytidine (NUC013), Leads to Tumor Regression in a Model of Non-Small Cell Lung Cancer. Pharmaceuticals. 2018; 11(2):36. https://doi.org/10.3390/ph11020036
Chicago/Turabian StyleDaifuku, Richard, Sheila Grimes, and Murray Stackhouse. 2018. "NUC041, a Prodrug of the DNA Methytransferase Inhibitor 5-aza-2′,2′-Difluorodeoxycytidine (NUC013), Leads to Tumor Regression in a Model of Non-Small Cell Lung Cancer" Pharmaceuticals 11, no. 2: 36. https://doi.org/10.3390/ph11020036
APA StyleDaifuku, R., Grimes, S., & Stackhouse, M. (2018). NUC041, a Prodrug of the DNA Methytransferase Inhibitor 5-aza-2′,2′-Difluorodeoxycytidine (NUC013), Leads to Tumor Regression in a Model of Non-Small Cell Lung Cancer. Pharmaceuticals, 11(2), 36. https://doi.org/10.3390/ph11020036