Molecular Docking and 3D-Pharmacophore Modeling to Study the Interactions of Chalcone Derivatives with Estrogen Receptor Alpha

 ,
,  ,
,
Abstract
:1. Introduction
2. Results
2.1. Modification of ChalcEA Derivatives
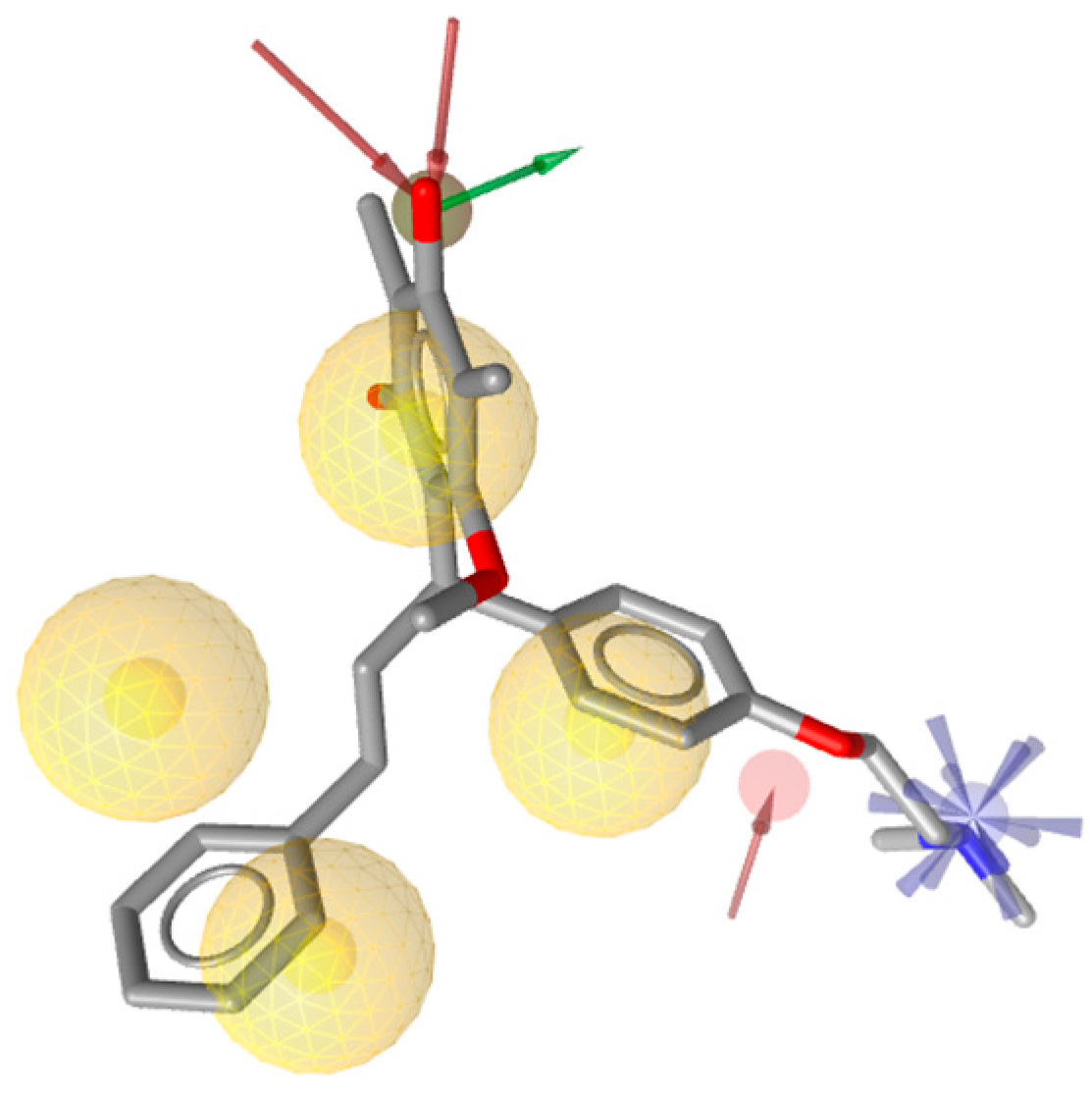
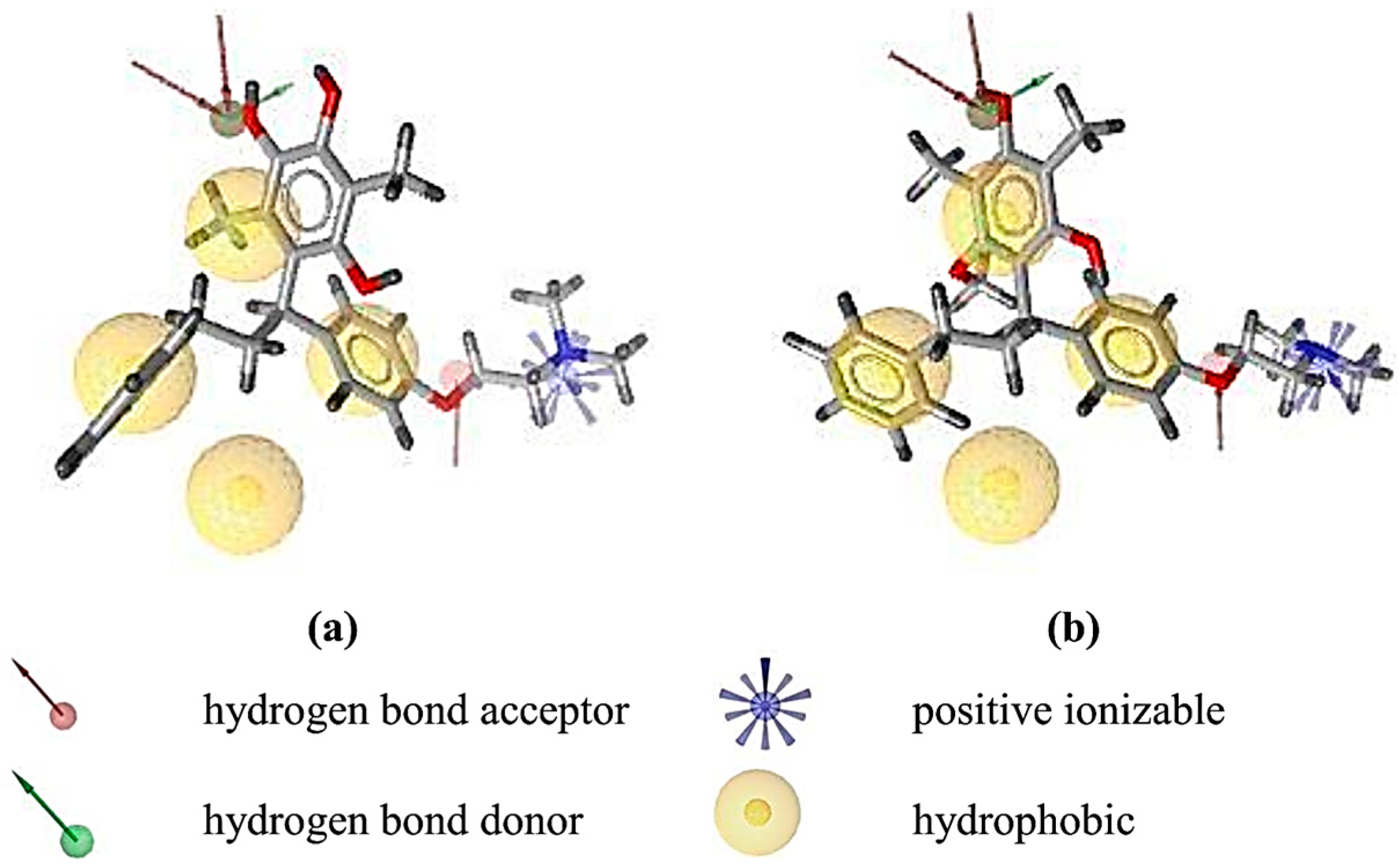
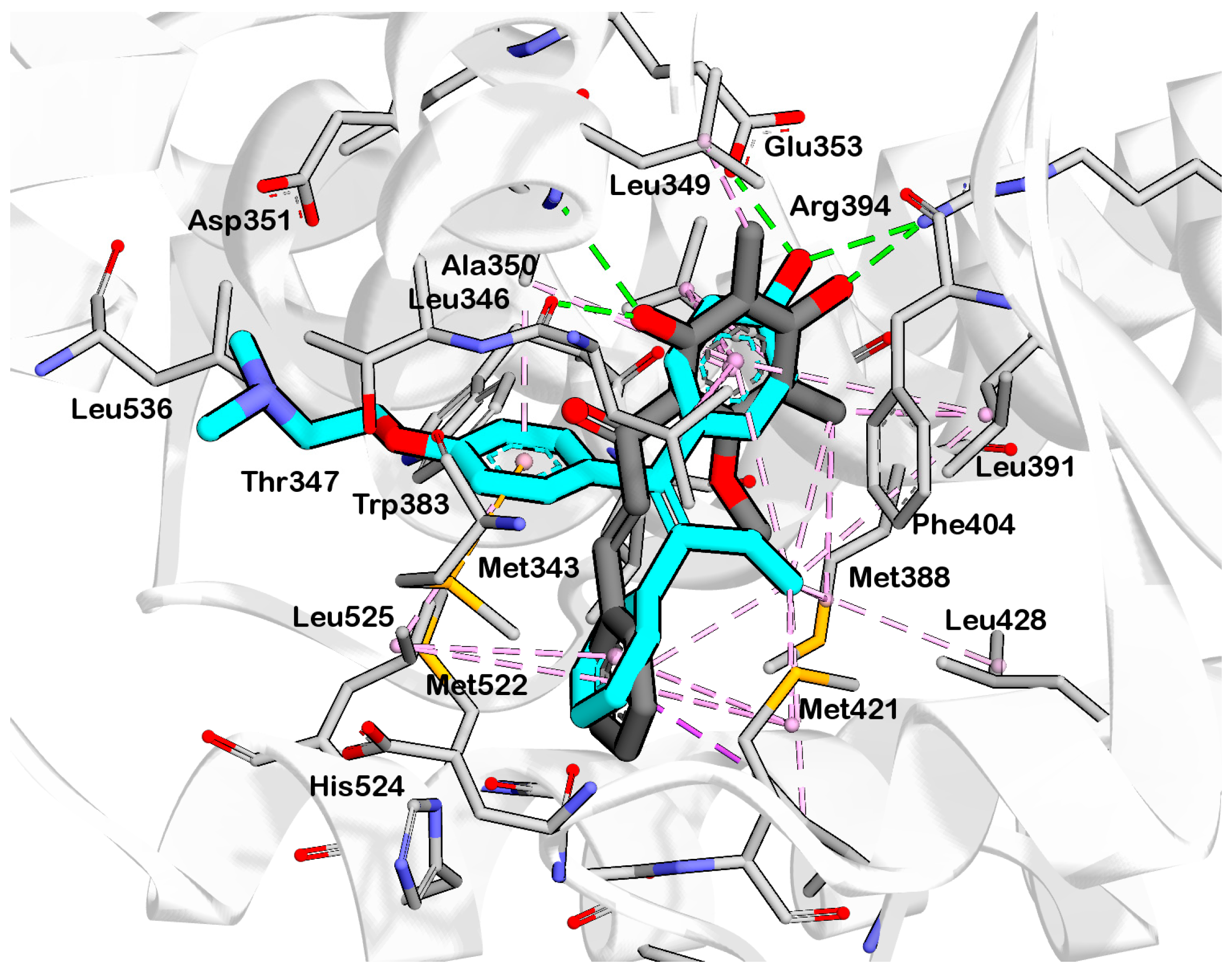
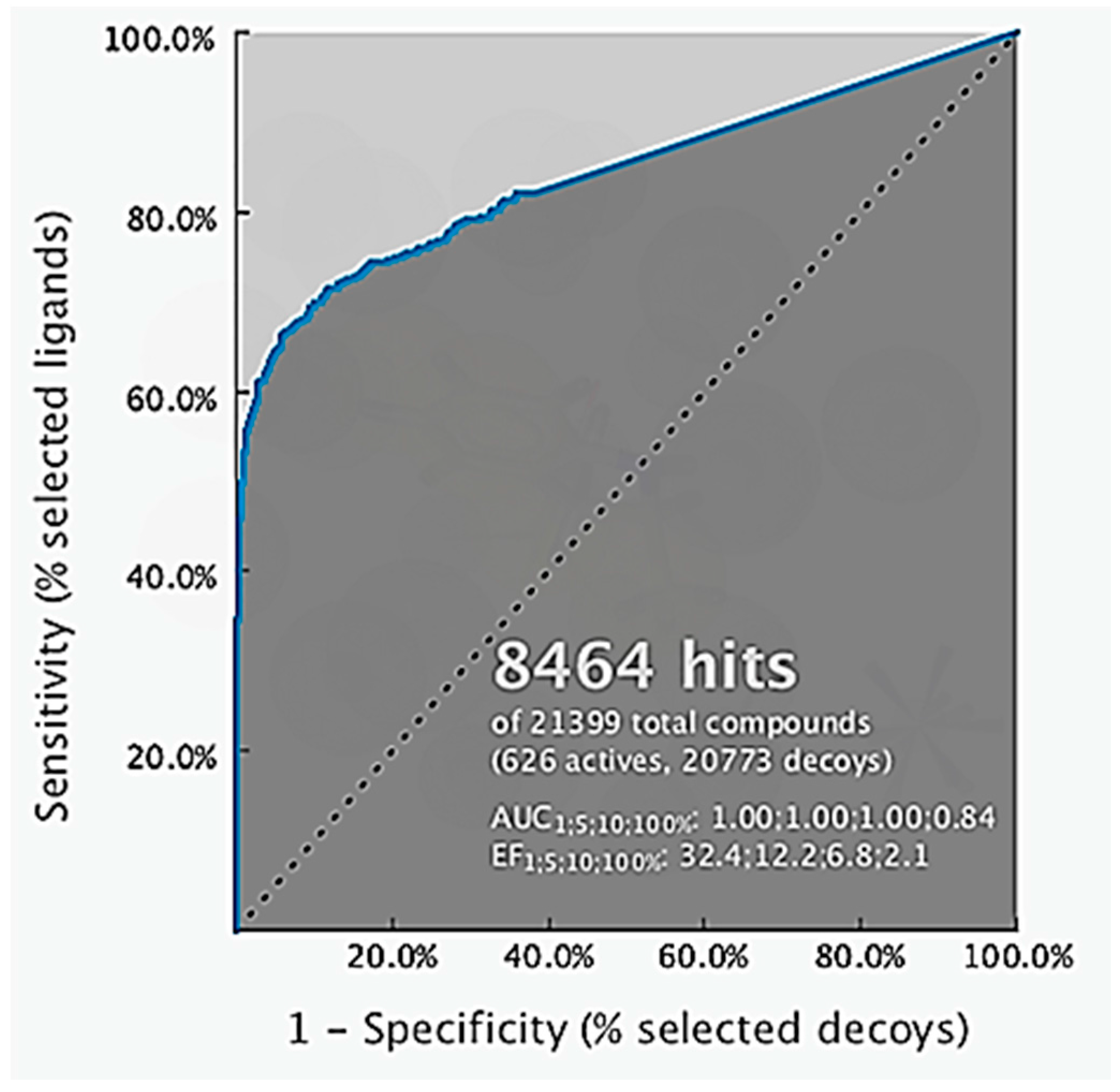
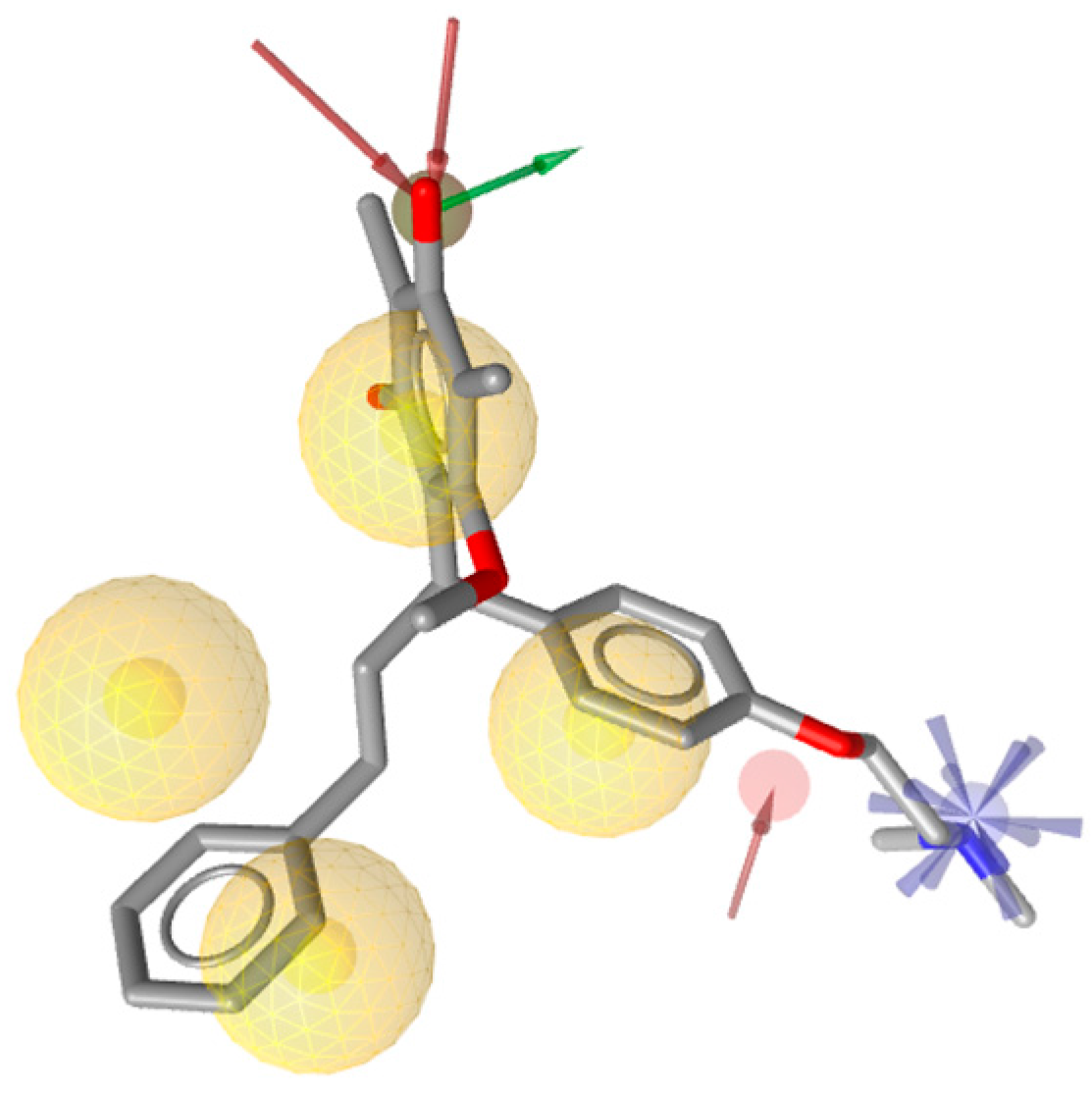
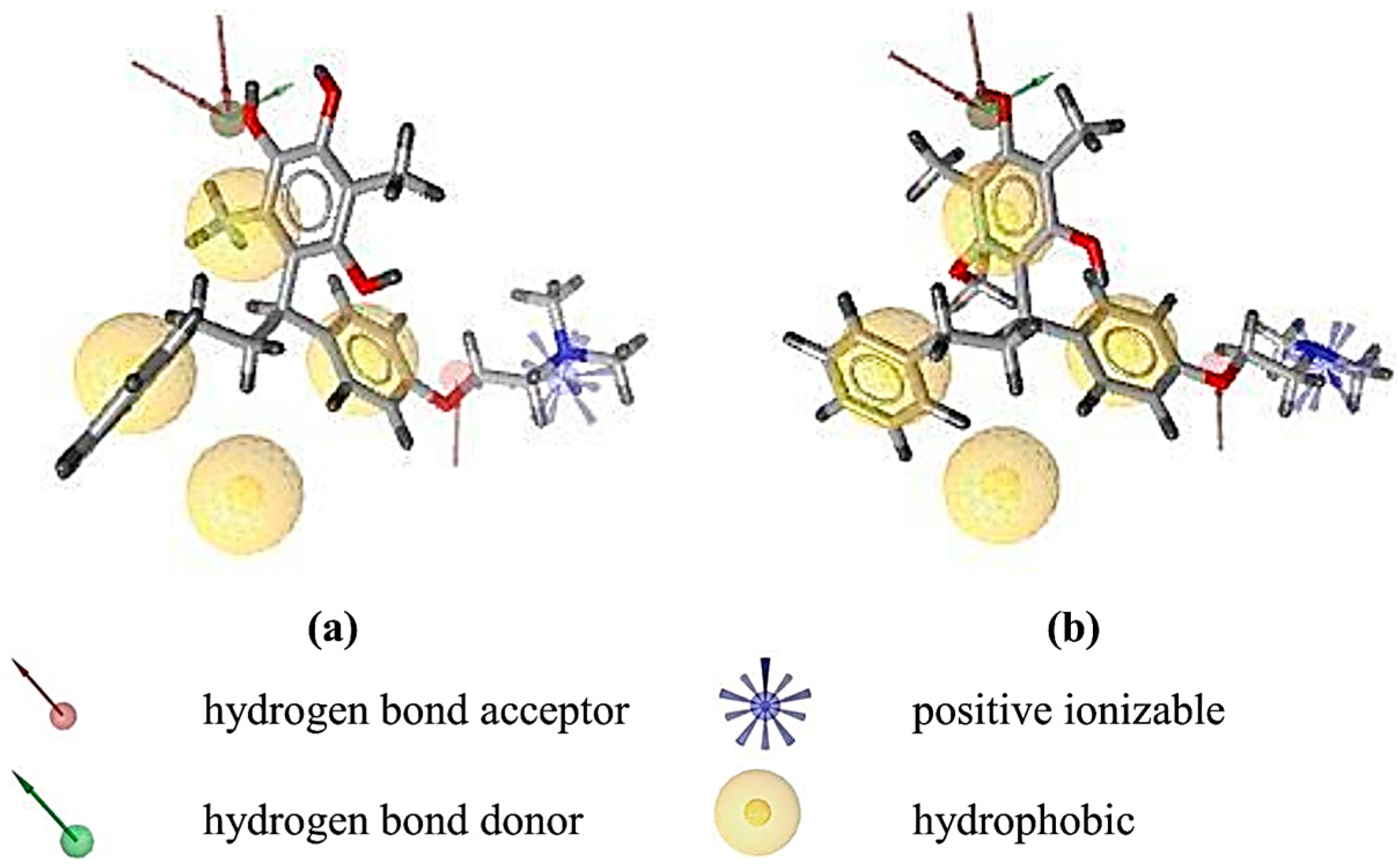
2.2. Structure-Based 3D Pharmacophore Modeling
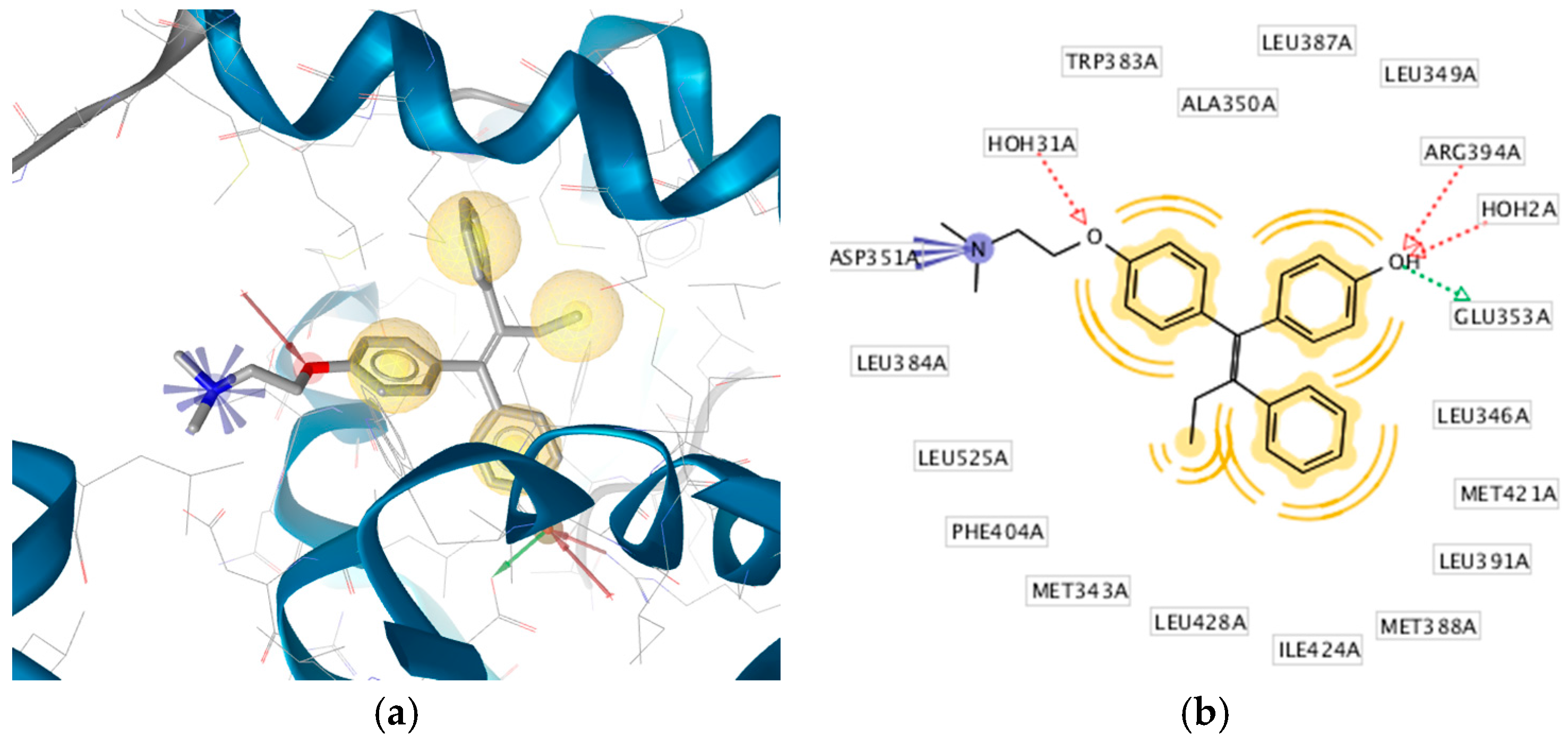
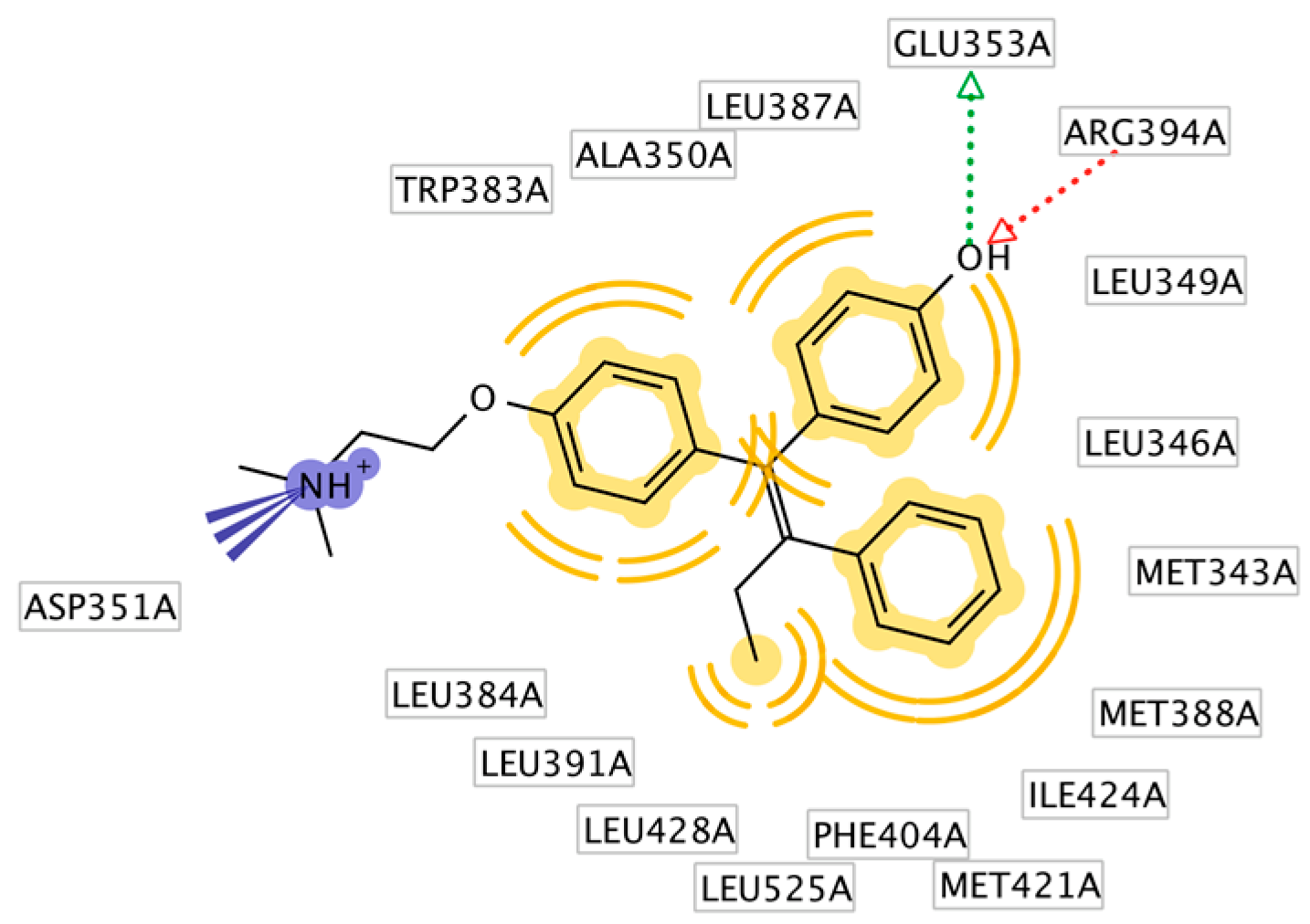
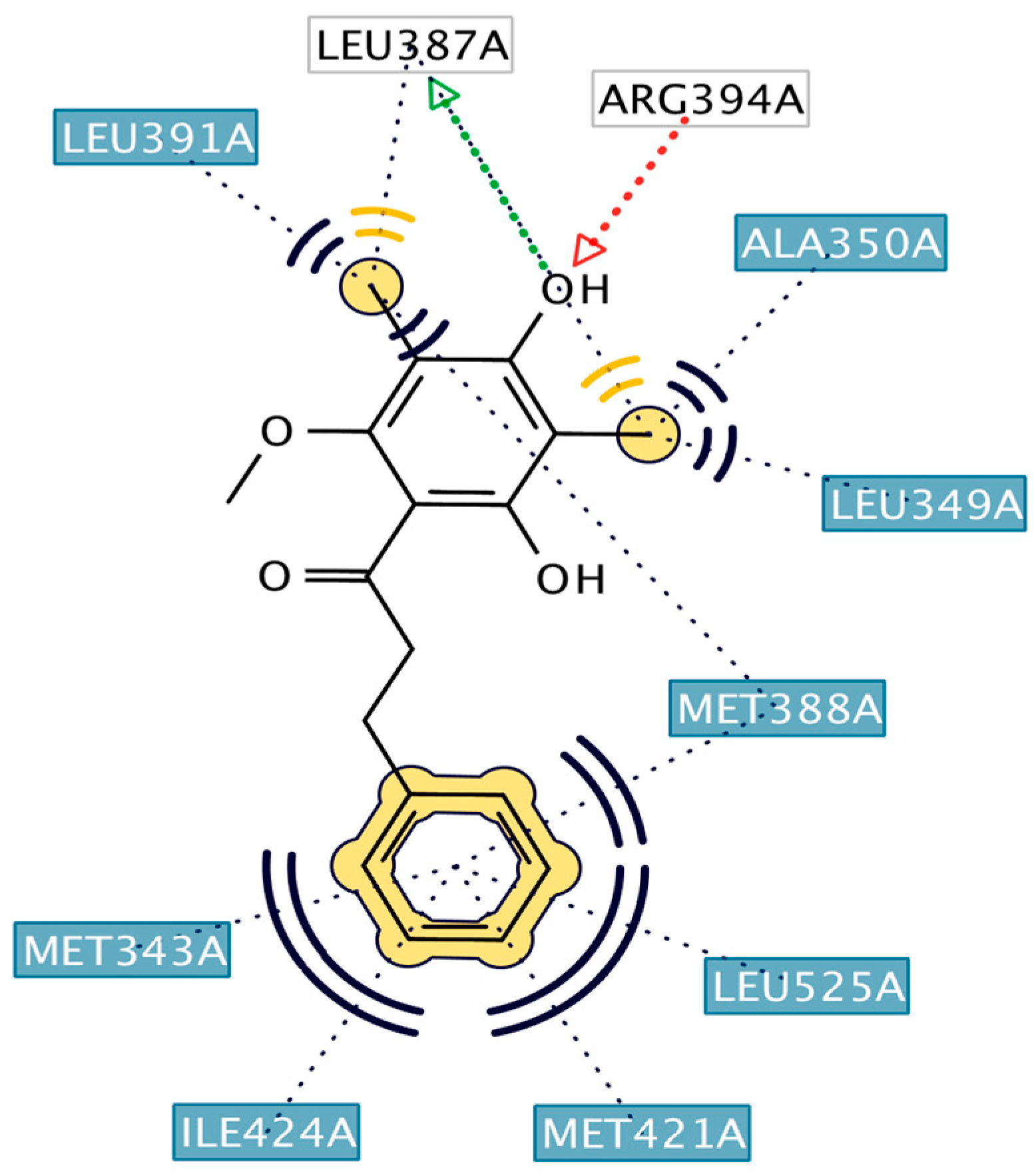
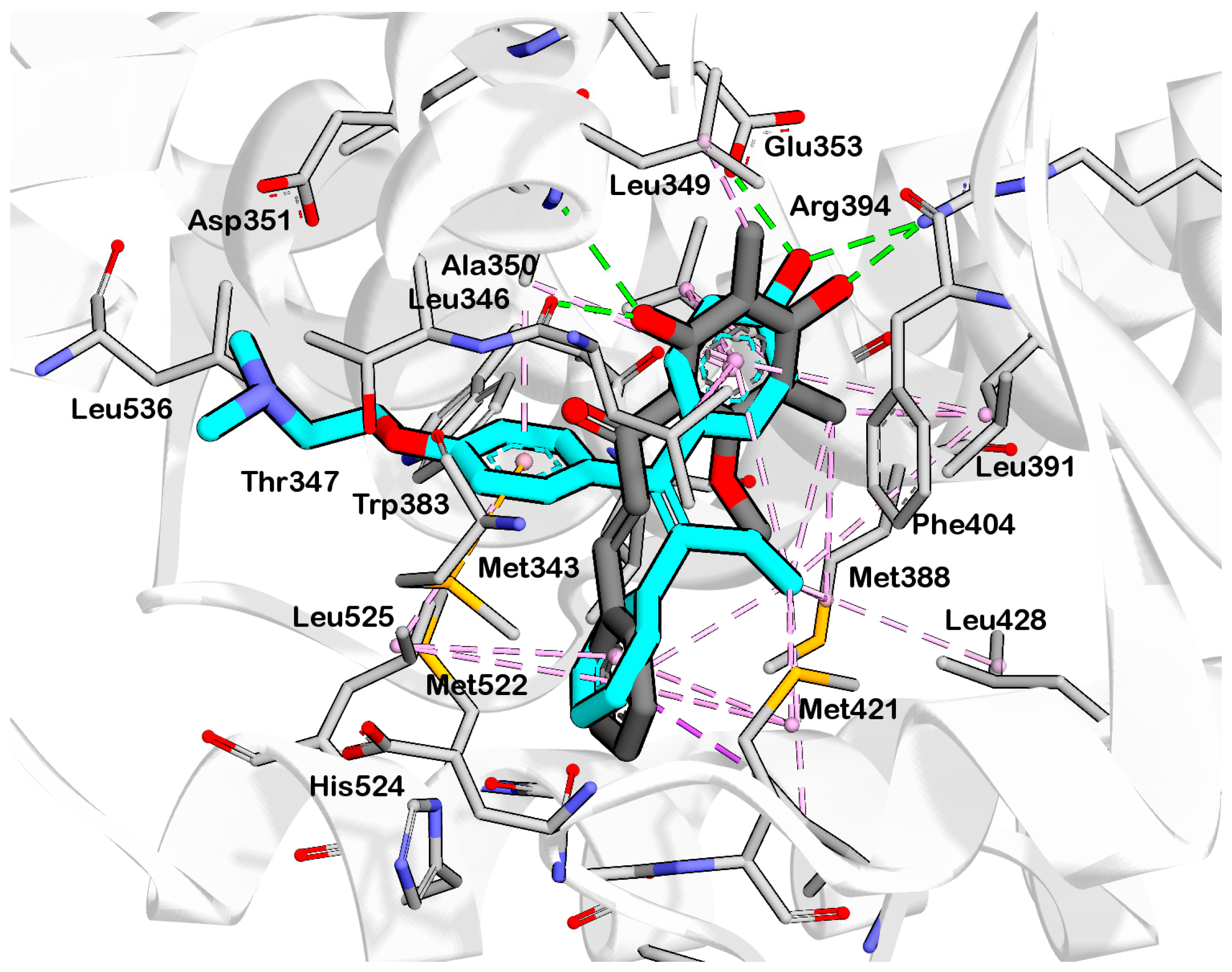
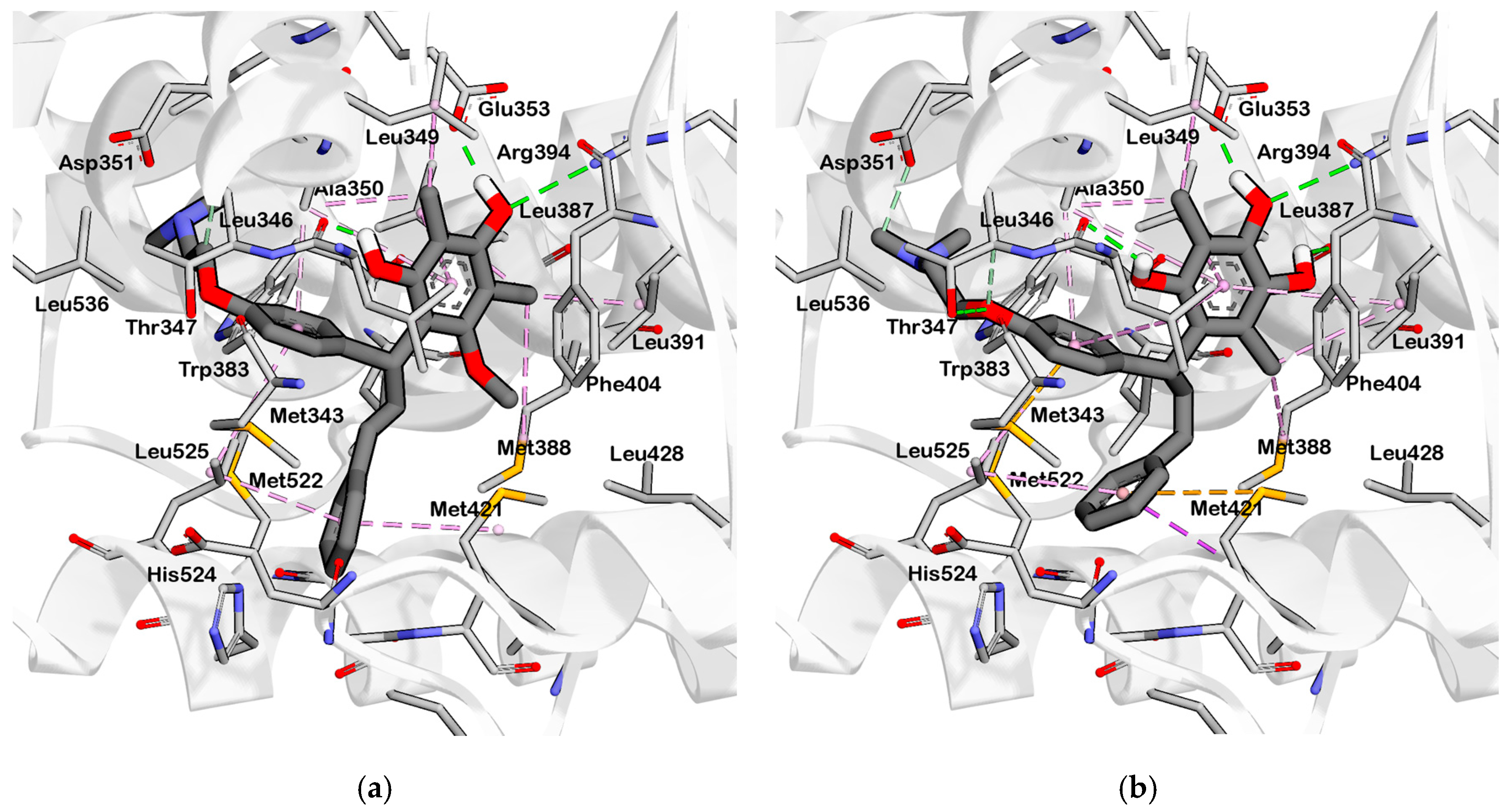
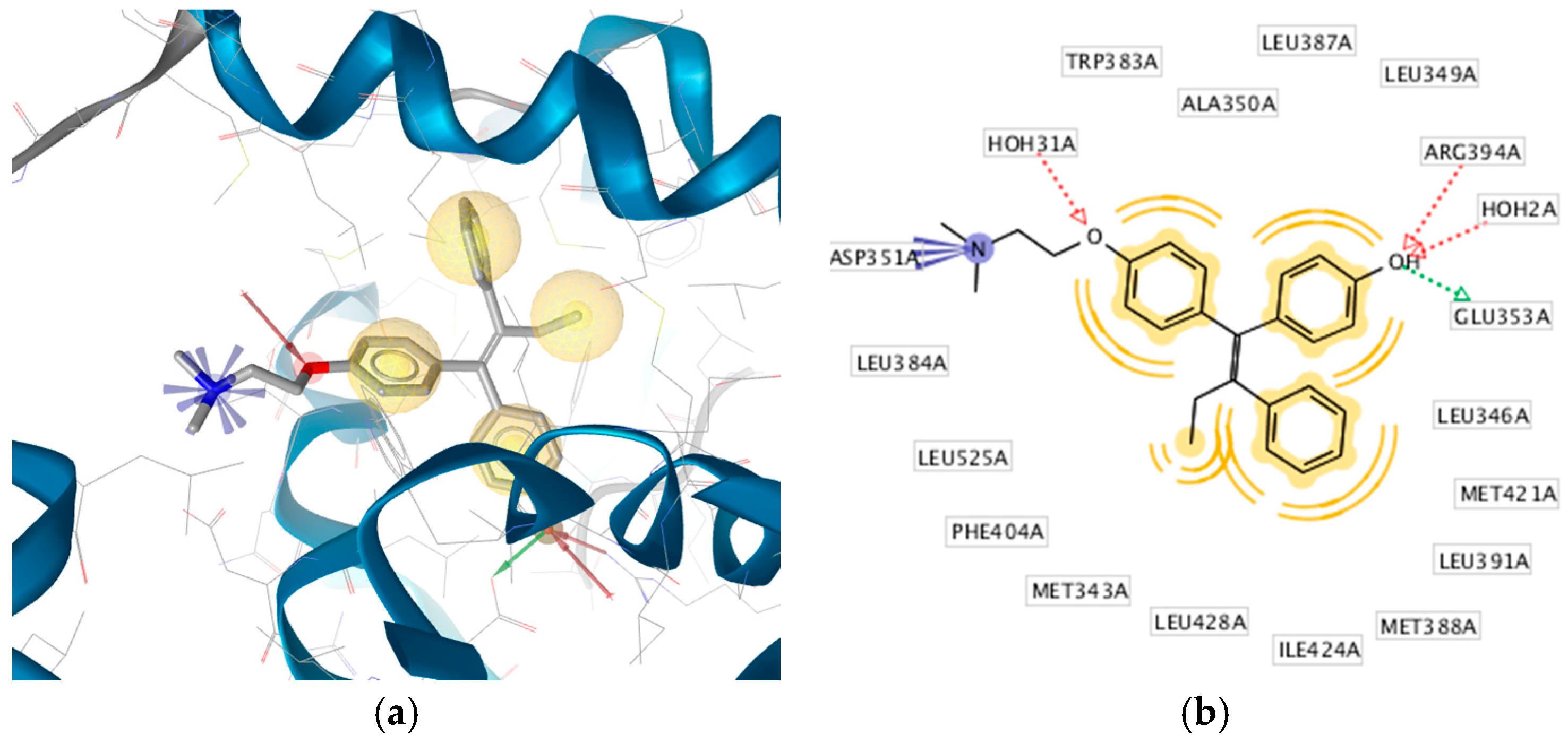
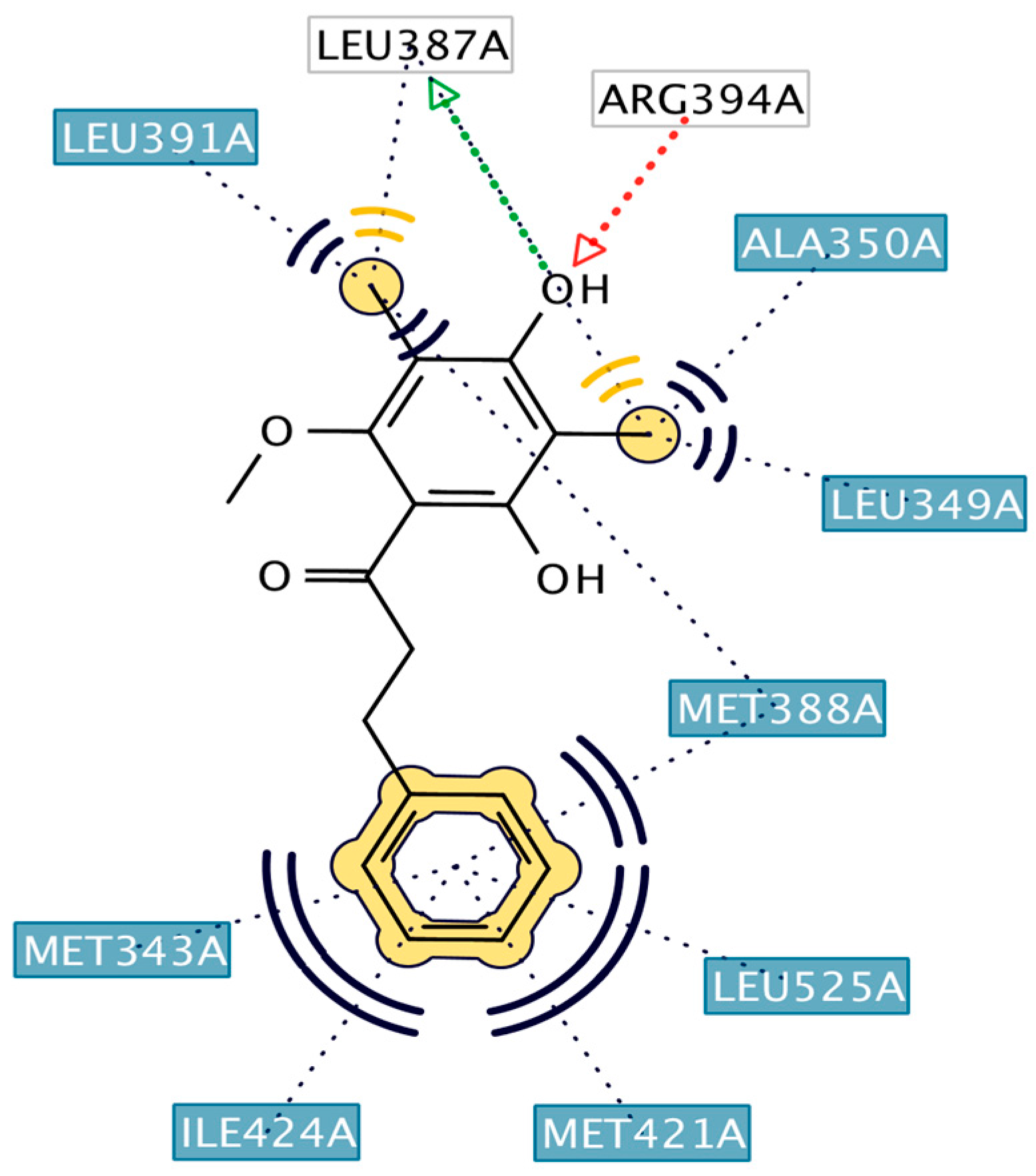
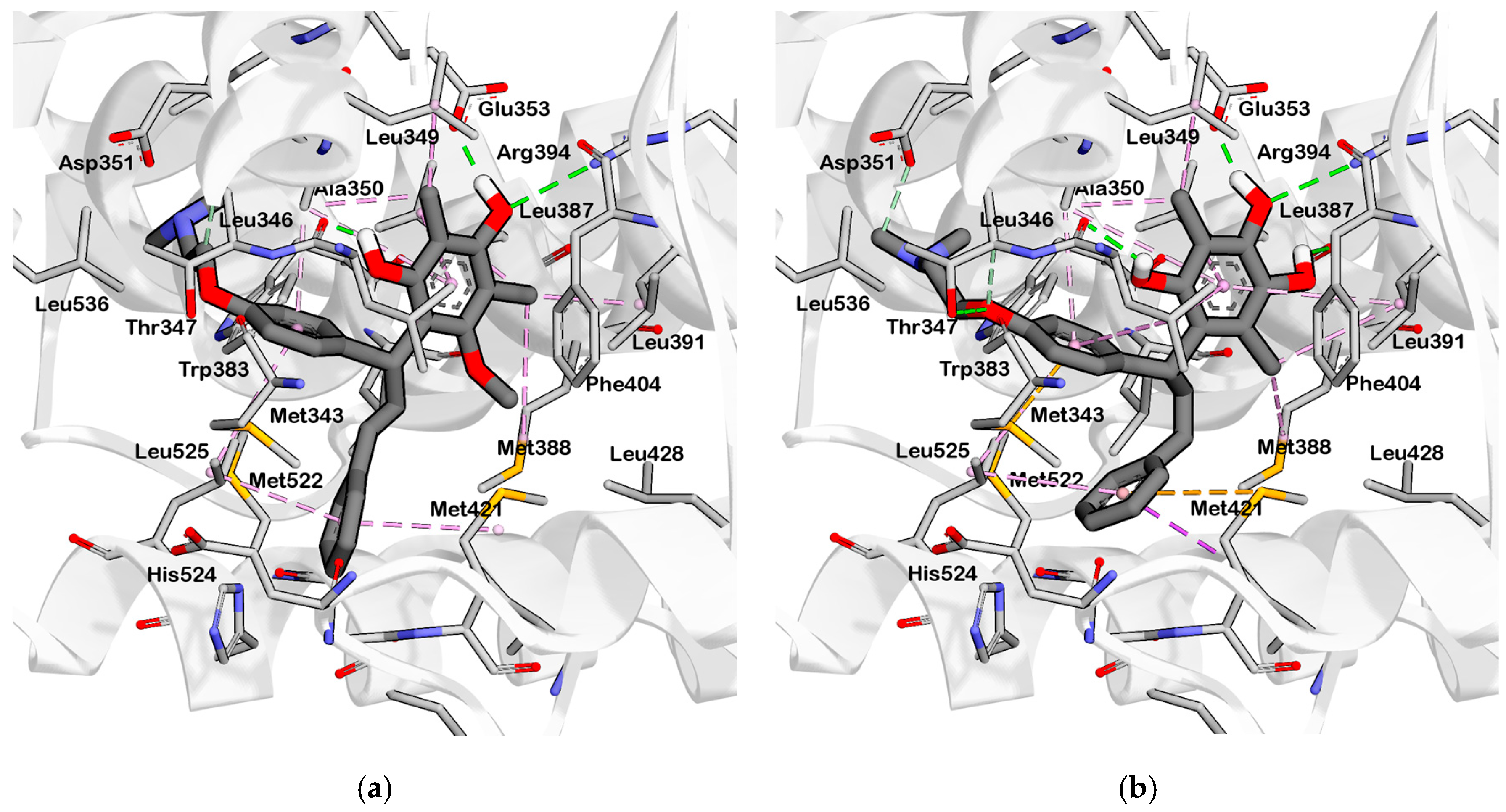
2.3. Interpretation of Molecular Docking Results and Pharmacophore Modeling
3. Discussion
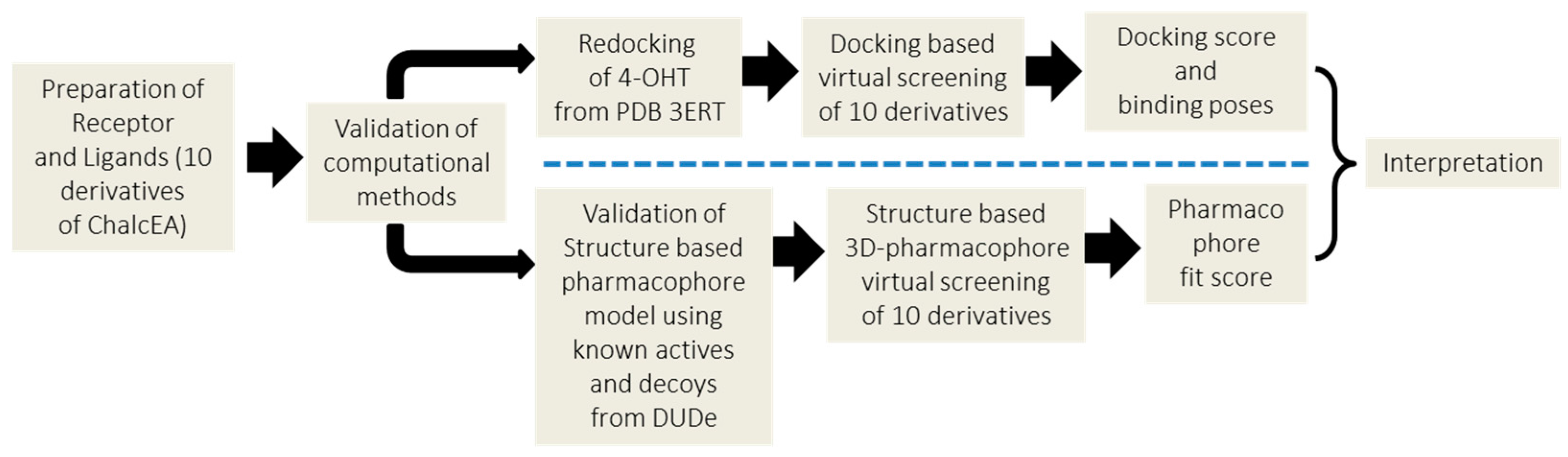
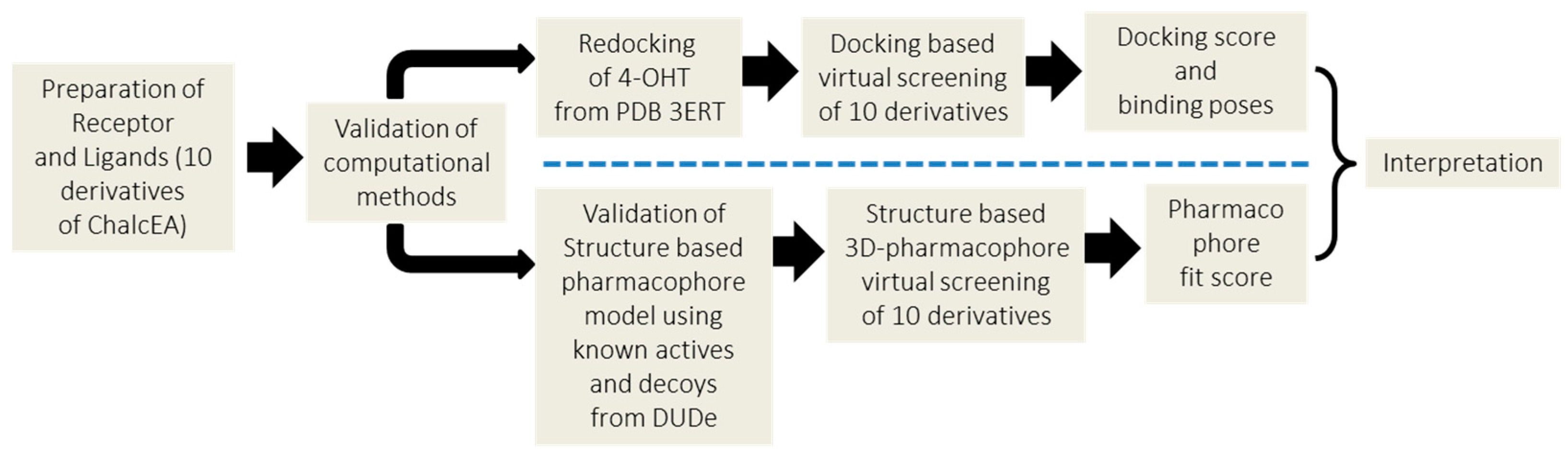
4. Materials and Methods
4.1. Molecular Docking Simulation
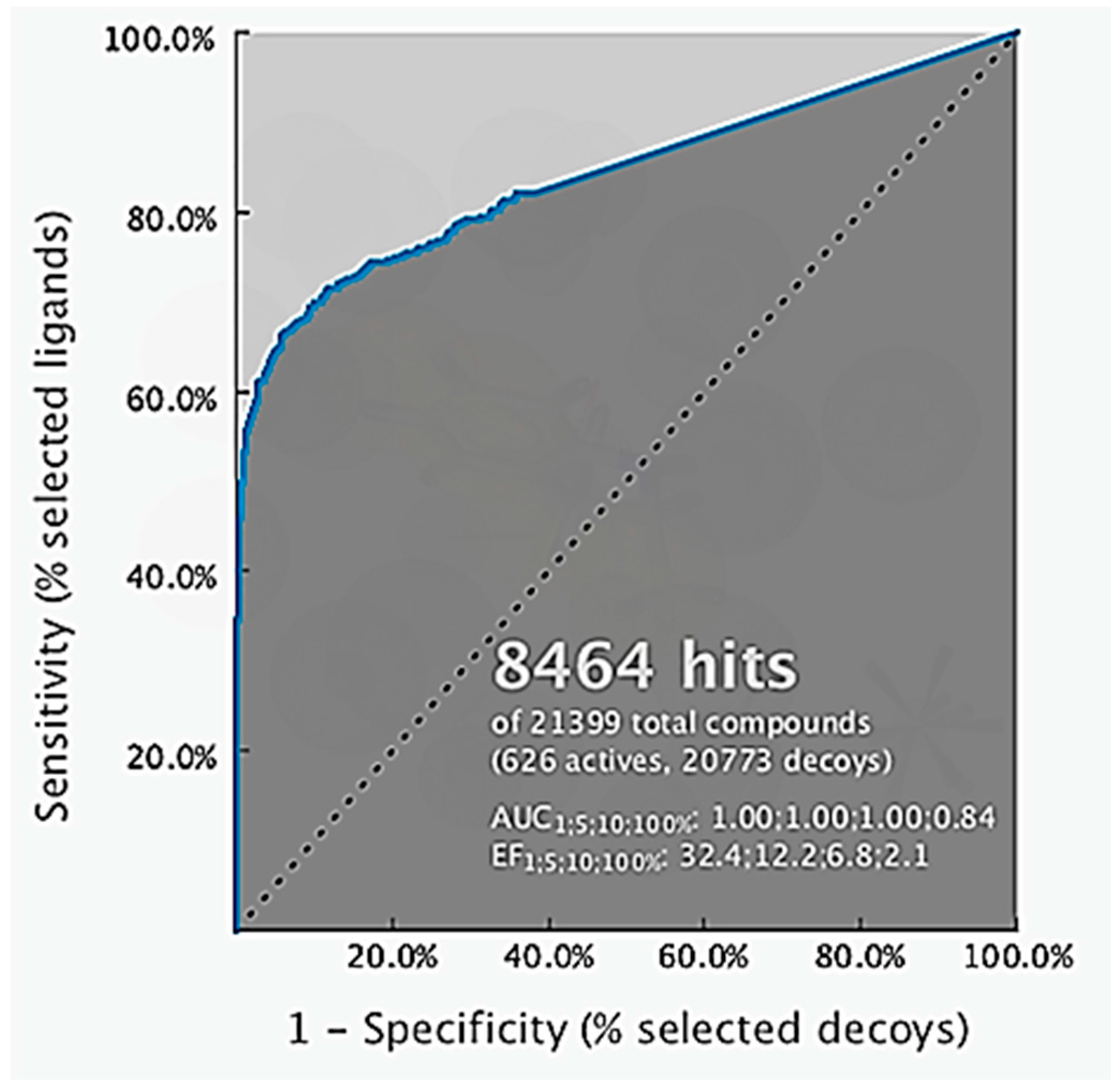
4.2. Structure-Based 3D-Pharmacophore Modeling
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, F.; Harbeck, N.; Fallowfield, L.; Kyriakides, S.; Senkus, E.; Group, E.G.W. Locally recurrent or metastatic breast cancer: Esmo clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2012, 23, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Budijanto, D. Infodatin-Breast Cancer. Available online: http://tinyurl.com/ycx6sgxh (accessed on 11 June 2015).
- Levin, E.R. Integration of the extranuclear and nuclear actions of estrogen. Mol. Endocrinol. 2005, 19, 1951–1959. [Google Scholar] [CrossRef] [PubMed]
- Sanyakamdhorn, S.; Agudelo, D.; Bekale, L.; Tajmir-Riahi, H.A. Targeted conjugation of breast anticancer drug tamoxifen and its metabolites with synthetic polymers. Coll. Surf. B: Biointerfaces 2016, 145, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Chang, M. Tamoxifen resistance in breast cancer. Biomol. Ther. 2012, 20, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Subarnas, A.; Diantini, A.; Abdulah, R.; Zuhrotun, A.; Hadisaputri, Y.E.; Puspitasari, I.M.; Yamazaki, C.; Kuwano, H.; Koyama, H. Apoptosis induced in mcf-7 human breast cancer cells by 2′,4′-dihydroxy-6-methoxy-3,5-dimethylchalcone isolated from eugenia aquea burm f. Leaves. Oncol. Lett. 2015, 9, 2303–2306. [Google Scholar] [CrossRef] [PubMed]
- Cohen, L.H.; Remley, M.J.; Raunig, D.; Vaz, A.D. In vitro drug interactions of cytochrome p450: An evaluation of fluorogenic to conventional substrates. Drug Metab. Dispos. 2003, 31, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.Y.; Zhang, H.X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Guner, O.F.; Bowen, J.P. Setting the record straight: The origin of the pharmacophore concept. J. Chem. Inf. Model. 2014, 54, 1269–1283. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Chen, Y.P. Structure-based drug design to augment hit discovery. Drug Discov. Today 2011, 16, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Smyth, M.S.; Martin, J.H. X-ray crystallography. Mol. Pathol. 2000, 53, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Muchtaridi, M.; Yusuf, M.; Diantini, A.; Choi, S.B.; Al-Najjar, B.O.; Manurung, J.V.; Subarnas, A.; Achmad, T.H.; Wardhani, S.R.; Wahab, H.A. Potential activity of fevicordin-a from phaleria macrocarpa (Scheff) boerl. Seeds as estrogen receptor antagonist based on cytotoxicity and molecular modelling studies. Int. J. Mol. Sci. 2014, 15, 7225–7249. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Wolber, G.; Langer, T. Ligandscout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Kirchmair, J.; Distinto, S.; Markt, P.; Schuster, D.; Spitzer, G.M.; Liedl, K.R.; Wolber, G. How to optimize shape-based virtual screening: Choosing the right query and including chemical information. J. Chem. Inf. Model. 2009, 49, 678–692. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, Y.; Ai, C.; Wang, Y. In silico prediction of estrogen receptor subtype binding affinity and selectivity using statistical methods and molecular docking with 2-arylnaphthalenes and 2-arylquinolines. Int. J. Mol. Sci. 2010, 11, 3434. [Google Scholar] [CrossRef] [PubMed]
- Vedavalli, S.; Srinivasan, T.; Suhitha, S.; Velmurugan, D.; Kandaswamy, M. Molecular docking study of chalcone derivative with human estrogen receptor as target protein and its anti-cancer activity against hepg2 cells. Int. J. Pharm. Tech. Res. 2014, 6, 1580–1583. [Google Scholar]
- Jensen, E.V.; Jordan, V.C. The estrogen receptor: A Model for Molecular Medicine. Clin. Cancer Res. 2003, 9, 1980–1989. [Google Scholar] [PubMed]
- Cano, A.; Hermenegildo, C. The endometrial effects of serms. Hum. Reprod. Update 2000, 6, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Shiau, A.K.; Barstad, D.; Loria, P.M.; Cheng, L.; Kushner, P.J.; Agard, D.A.; Greene, G.L. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 1998, 95, 927–937. [Google Scholar] [CrossRef]
- Scott, J.; Weiner, S.J.; Kollman, P.A.; Case, D.A.; Singh, U.C.; Ghio, C.; Alagona, G.; Profeta, S.; Weiner, P. A new force field for molecular mechanical simulation of nucleic acids and proteins. J. Am. Chem. Soc. 1984, 106, 765–784. [Google Scholar]
- Gasteiger, J.; Marsili, M. Iterative partial equalization of orbital electronegativity—A rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Ikram, N.K.K.; Durrant, J.D.; Muchtaridi, M.; Zalaludin, A.S.; Purwitasari, N.; Mohamed, N.; Rahim, A.S.A.; Lam, C.K.; Normi, Y.M.; Rahman, N.A. A virtual screening approach for identifying plants with anti h5n1 neuraminidase activity. J. Chem. Inf. Model. 2015, 55, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ChalcEA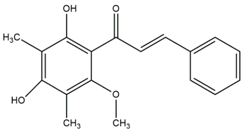 | ChalcEA Derivatives (HNS1-10)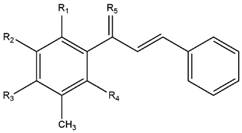 | |||||
|---|---|---|---|---|---|---|
| No. | Molecule Name | R1 | R2 | R3 | R4 | R5 |
| 1 | HNS1 | OCH3 | H | H | H | - |
| 2 | HNS2 | H | H | H | H | 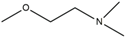 |
| 3 | HNS3 | H | H | H | H |  |
| 4 | HNS4 | H | H | H | H |  |
| 5 | HNS5 | H | H | H | H |  |
| 6 | HNS6 | H | H | H | H | - |
| 7 | HNS7 | CH3 | H | - | H | 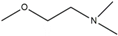 |
| 8 | HNS8 | H | H | H | H | 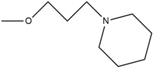 |
| 9 | HNS9 | CH3 | OH | H | OH | 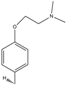 |
| 10 | HNS10 | - | H | - | H | 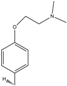 |
| No. | Derivative Name | Molecular Weight | Log P | Number of HB Donors | Number of HB Acceptors |
|---|---|---|---|---|---|
| 1 | HNS1 | 284.355 | 3.60 | 1 | 3 |
| 2 | HNS2 | 374.501 | 2.27 | 3 | 4 |
| 3 | HNS3 | 328.408 | 4.03 | 2 | 4 |
| 4 | HNS4 | 330.484 | 3.07 | 3 | 3 |
| 5 | HNS5 | 330.424 | 3.82 | 3 | 4 |
| 6 | HNS6 | 399.531 | 4.22 | 2 | 4 |
| 7 | HNS7 | 372.529 | 2.88 | 2 | 3 |
| 8 | HNS8 | 427.585 | 5.00 | 2 | 4 |
| 9 | HNS9 | 436.572 | 3.75 | 4 | 4 |
| 10 | HNS10 | 450.599 | 4.05 | 3 | 4 |
| No. | Molecule Name | Chemical Formula | ∆G (kcal/mol) | Number in Cluster | Calculated Ki (nM) | Interactions with Amino Acids | |
|---|---|---|---|---|---|---|---|
| Hydrogen Bond | van der Waals (Hydrophobic) | ||||||
| 1 | HNS1 | C18H18O3 | −9.30 | 35 | 153.41 | Glu353, Arg394 | Leu525, Met421, Leu387, Ala350, Leu349 |
| 2 | HNS2 | C22H29NO4 | −9.25 | 29 | 66.92 | Leu346, Leu387 | Leu391, Met388, Leu384, Leu525, Met421, Met343, Asp351, Thr347 |
| 3 | HNS3 | C22H29NO4 | −9.01 | 64 | 249.32 | Gly420 | Gly521, Val418, Leu346, Met343, Met421, Leu525, Ala350, Thr347, Trp383, Leu387, Leu391, Asp351, Leu348 |
| 4 | HNS4 | C20H23NO3 | −9.26 | 53 | 162.64 | Thr347, Leu346, Glu353, Arg394 | Leu349, Ala350, Leu387, Leu391, Met388, Met421, His524, Leu525 |
| 5 | HNS5 | C20H24O4 | −9.44 | 75 | 120.52 | Thr347, Leu346, Glu353, Arg394 | Leu387, Ala350, Leu349, Leu391, Met388, Met421, Leu525 |
| 6 | HNS6 | C24H31NO4 | −9.76 | 30 | 70.51 | Leu346, Glu353, Arg394 | Leu387, Leu349, Leu391, Ala350, Leu525 |
| 7 | HNS7 | C23H31NO3 | −9.93 | 49 | 52.67 | Arg394, Leu387, Thr347 | Leu346, Leu349, Ala350, Asp351, Leu525, Met421, Met388, Leu391 |
| 8 | HNS8 | C26H35NO4 | −10.96 | 43 | 9.27 | Glu353 | Met421, Leu525, Met388, Leu391, Leu346, Leu349, Leu387, Ala350, Thr347, Trp383 |
| 9 | HNS9 | C27H31NO4 | −12.15 | 83 | 1.25 | Leu346, Glu353, Arg394, Leu387 | Met421, Leu525, Met343, Asp351, Thr347, Ala350, Leu349, Leu391, Leu384, Met388 |
| 10 | HNS10 | C28H33NO4 | −12.33 | 42 | 0.91 | Leu346, Glu353, Arg394 | Met421, Leu525, Thr347, Ala350, Leu387, Leu349, Met388, Leu391 |
| No. | Compound | Pharmacophore-Fit Score | Docking Score (kcal/mol) |
|---|---|---|---|
| 1 | ChalcEA | 45.90 | −8.23 |
| 2 | HNS1 | 46.76 | −9.30 |
| 3 | HNS2 | 46.62 | −9.25 |
| 4 | HNS3 | 56.57 | −9.01 |
| 5 | HNS4 | 46.61 | −9.26 |
| 6 | HNS5 | 56.68 | −9.44 |
| 7 | HNS6 | 55.33 | −9.76 |
| 8 | HNS7 | 55.00 | −9.93 |
| 9 | HNS8 | 56.20 | −10.96 |
| 10 | HNS9 | 66.50 | −12.15 |
| 11 | HNS10 | 67.07 | −12.33 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muchtaridi, M.; Syahidah, H.N.; Subarnas, A.; Yusuf, M.; Bryant, S.D.; Langer, T. Molecular Docking and 3D-Pharmacophore Modeling to Study the Interactions of Chalcone Derivatives with Estrogen Receptor Alpha. Pharmaceuticals 2017, 10, 81. https://doi.org/10.3390/ph10040081
Muchtaridi M, Syahidah HN, Subarnas A, Yusuf M, Bryant SD, Langer T. Molecular Docking and 3D-Pharmacophore Modeling to Study the Interactions of Chalcone Derivatives with Estrogen Receptor Alpha. Pharmaceuticals. 2017; 10(4):81. https://doi.org/10.3390/ph10040081
Chicago/Turabian StyleMuchtaridi, Muchtaridi, Hasna Nur Syahidah, Anas Subarnas, Muhammad Yusuf, Sharon D. Bryant, and Thierry Langer. 2017. "Molecular Docking and 3D-Pharmacophore Modeling to Study the Interactions of Chalcone Derivatives with Estrogen Receptor Alpha" Pharmaceuticals 10, no. 4: 81. https://doi.org/10.3390/ph10040081
APA StyleMuchtaridi, M., Syahidah, H. N., Subarnas, A., Yusuf, M., Bryant, S. D., & Langer, T. (2017). Molecular Docking and 3D-Pharmacophore Modeling to Study the Interactions of Chalcone Derivatives with Estrogen Receptor Alpha. Pharmaceuticals, 10(4), 81. https://doi.org/10.3390/ph10040081