
Current Status of Radiopharmaceuticals for the Theranostics of Neuroendocrine Neoplasms




Abstract
:1. Introduction
- I
- Despite the fact that sstr2-targeting is well established and clinically accepted, an alternative approach questions the dogma of using radiolabeled somatostatin receptor agonists and demonstrates advantages of antagonists. This is the focus of the respective section covering sstr-targeting.
- II
- The investigation of the clinical relevance of imaging radiopharmaceuticals targeting GLP-1R has already been conducted in humans, and the exploration of the feasibility of respective radiotherapeutic agents is in progress preclinically. The focus of the respective section is the advances and shortcomings in clinical studies as well as research conducted to overcome those shortcomings in the context of radiotheranostics.
- III
- The field of CCK2 receptors is not new, but clinical success is still pending mainly due to a number of limitations of the developed radiolabeled gastrin analogs. These limitations and different approaches to circumvent them are discussed in the CCK2 targeting section, together with the current clinical status in this field.
- IV
- Development of radioligands targeting of GIP-R is a newly emerging and very promising field. The first advances in GIP receptor and respective ligand and radioligand exploration are presented in the corresponding section.
2. Somatostatin Receptor Antagonists
2.1. Development of Radiopharmaceuticals Based on Somatostatin Receptor Antagonists
2.2. In Vitro Human Data
2.3. Clinical Achievements
3. Glucagon-Like Peptide-1 Receptor Targeting
3.1. Clinical Achievements
3.2. Dosimetry and Feasibility of Radiotheranostics
4. Cholecystokinin 2/Gastrin Receptor Targeting
4.1. Clinical Achievements
4.2. Dosimetry and Feasibility of Radiotheranostics
5. Glucose-Dependent Insulinotropic Polypeptide ReceptorTargeting
6. Summary
Conflicts of Interest
Abbreviations
| 5-HTP | 5-Hydroxy-l-tryptophan |
| Ahx | Aminohexanoic acid |
| BFC | Bifunctionalchelator |
| BH | Bolton-Hunter |
| CCK2(R) | Cholecystokinin 2 (Receptor) |
| CB-TE2A | 4,11-Bis(carboxymethyl)-1,4,8,11-tetraazabicyclo[6.6.2]hexadecane |
| CT | Computed tomography |
| DMSA | Dimercaptosuccinic acid |
| DOPA | l-Dihydroxyphenylalanine |
| DOTA | 1,4,7,10-Tetraazacyclododecane-1,4,7,10-tetraacetic acid |
| DTPA | Diethylenetriaminepentaacetic acid |
| EDDA | Ethylenediamine-N,N′-diacetic acid |
| FDA | Food and Drug Administration |
| FDG | Fluorodeoxyglucose |
| GEP-NET | Gastroenderopancreatic neuroendocrine tumors |
| GIP(R) | Glucose-dependent insulinotropic polypeptide (Receptor) |
| GIST | Gastrointestinal stromal tumors |
| GLP-1(R) | Glucagon-like peptide-1 (Receptor) |
| GPCR | G protein-coupled receptors |
| HEK | Human embryonic kidney |
| HYNIC | Hydrazinonicotinamide |
| MIBG | Meta-iodobenzylguanidine |
| MIBI | Methoxyisobutylisonitrile |
| MRI | Magnetic resonance Imaging |
| MTC | Medullary thyroid cancer |
| NENs | Neuroendocrine neoplasms |
| MTD | Maximum tolerated dose |
| NODAGA | 1,4,7-Triazacyclononane,1-glutaric acid-4,7-acetic acid |
| NOTA | 1,4,7-Triazacyclononane-1,4,7-triacetic acid |
| NOTA-MAL | NOTA mono-N-ethylmaleimide |
| PET | Positron emission tomography |
| PRRT | Peptide Receptor Radionuclide Therapy |
| SPECT | Single-photon emission computed tomography |
| SST(R) | Somatostatin (Receptor) |
| VS | Vinylsulfonyl |
References
- Carrasquillo, J.A.; Chen, C.C. Molecular imaging of neuroendocrine tumors. Semin. Oncol. 2010, 37, 662–679. [Google Scholar] [CrossRef] [PubMed]
- Bodei, L.; Sundin, A.; Kidd, M.; Prasad, V.; Modlin, I.M. The status of neuroendocrine tumor imaging: From darkness to light? Neuroendocrinology 2015, 101, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Fani, M. Current and future radiopharmaceuticals in neuroendocrine tumor imaging In Diagnostic and Therapeutic Nuclear Medicine for Neuroendocrine Tumors; Taïeb, D., Pacak, K., Eds.; Springer: Cham, Switzerland, 2017; pp. 141–162. [Google Scholar]
- Oberg, K. Molecular imaging radiotherapy: Theranostics for personalized patient management of neuroendocrine tumors (nets). Theranostics 2012, 2, 448–458. [Google Scholar] [CrossRef] [PubMed]
- Werner, R.A.; Bluemel, C.; Allen-Auerbach, M.S.; Higuchi, T.; Herrmann, K. 68Gallium- and 90Yttrium-/177Lutetium: “Theranostic twins” for diagnosis and treatment of nets. Ann. Nucl. Med. 2015, 29, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Velikyan, I. Radionuclides for imaging and therapy in oncology. In CancerTheranostics; Chen, X., Wong, S., Eds.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 285–325. [Google Scholar]
- Nicolas, G.; Giovacchini, G.; Muller-Brand, J.; Forrer, F. Targeted radiotherapy with radiolabeled somatostatin analogs. Endocrinol. Metab. Clin. N. Am. 2011, 40, 187–204. [Google Scholar] [CrossRef] [PubMed]
- Reubi, J.C. Somatostatin receptor sst1-sst5 expression in normal and neoplastic human tissues using receptor autoradiography with subtype-selective ligands. Eur. J. Nucl. Med. 2001, 28, 836–846. [Google Scholar] [CrossRef] [PubMed]
- Kwekkeboom, D.J.; Kam, B.L.; van Essen, M.; Teunissen, J.J.M.; van Eijck, C.H.J.; Valkema, R.; de Jong, M.; de Herder, W.W.; Krenning, E.P. Somatostatin receptor-based imaging and therapy of gastroenteropancreatic neuroendocrine tumors. Endocr. Relat. Cancer 2010, 17, R53–R73. [Google Scholar] [CrossRef] [PubMed]
- Ambrosini, V.; Campana, D.; Tomassetti, P.; Fanti, S. 68Ga-labelled peptides for diagnosis of gastroenteropancreatic net. Eur. J. Nucl. Med. Mol. Imaging 2012, 39 (Suppl. 1), S52–S60. [Google Scholar] [CrossRef] [PubMed]
- Van Essen, M.; Krenning, E.P.; Kam, B.L.; de Jong, M.; Valkema, R.; Kwekkeboom, D.J. Peptide-receptor radionuclide therapy for endocrine tumors. Nat. Rev. Endocrinol. 2009, 5, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Perrin, M.H.; Sutton, S.W.; Cervini, L.A.; Rivier, J.E.; Vale, W.W. Comparison of an agonist, urocortin, and an antagonist, astressin, as radioligands for characterization of corticotropin-releasing factor receptors. J. Pharmacol. Exp. Ther. 1999, 288, 729–734. [Google Scholar] [PubMed]
- Sleight, A.J.; Stam, N.J.; Mutel, V.; Vanderheyden, P.M. Radiolabelling of the human 5-ht2a receptor with an agonist, a partial agonist and an antagonist: Effects on apparent agonist affinities. Biochem. Pharmacol. 1996, 51, 71–76. [Google Scholar] [CrossRef]
- Ginj, M.; Zhang, H.; Waser, B.; Cescato, R.; Wild, D.; Wang, X.; Erchegyi, J.; Rivier, J.; Macke, H.R.; Reubi, J.C. Radiolabeled somatostatin receptor antagonists are preferable to agonists for in vivo peptide receptor targeting of tumors. Proc. Natl. Acad. Sci. USA 2006, 103, 16436–16441. [Google Scholar] [CrossRef] [PubMed]
- Bass, R.T.; Buckwalter, B.L.; Patel, B.P.; Pausch, M.H.; Price, L.A.; Strnad, J.; Hadcock, J.R. Identification and characterization of novel somatostatin antagonists. Mol. Pharmacol. 1996, 50, 709–715. [Google Scholar] [PubMed]
- Reubi, J.C.; Schaer, J.C.; Wenger, S.; Hoeger, C.; Erchegyi, J.; Waser, B.; Rivier, J. Sst3-selective potent peptidic somatostatin receptor antagonists. Proc. Natl. Acad. Sci. USA 2000, 97, 13973–13978. [Google Scholar] [CrossRef] [PubMed]
- Reubi, J.C.; Schär, J.-C.; Waser, B.; Wenger, S.; Heppeler, A.; Schmitt, J.S.; Mäcke, H.R. Affinity profiles for human somatostatin receptor subtypes sst1-sst5 of somatostatin radiotracers selected for scintigraphic and radiotherapeutic use. Eur. J. Nucl. Med. Mol. Imaging 2000, 27, 273–282. [Google Scholar] [CrossRef]
- Wang, X.; Fani, M.; Schulz, S.; Rivier, J.; Reubi, J.C.; Maecke, H.R. Comprehensive evaluation of a somatostatin-based radiolabelled antagonist for diagnostic imaging and radionuclide therapy. Eur. J. Nucl. Med. Mol. Imaging 2012, 39, 1876–1885. [Google Scholar] [CrossRef] [PubMed]
- Wadas, T.J.; Eiblmaier, M.; Zheleznyak, A.; Sherman, C.D.; Ferdani, R.; Liang, K.; Achilefu, S.; Anderson, C.J. Preparation and biological evaluation of 64cu-cb-te2a-sst2-ant, a somatostatin antagonist for pet imaging of somatostatin receptor-positive tumors. J. Nucl. Med. 2008, 49, 1819–1827. [Google Scholar] [CrossRef] [PubMed]
- Cescato, R.; Erchegyi, J.; Waser, B.; Piccand, V.; Maecke, H.R.; Rivier, J.E.; Reubi, J.C. Design and in vitro characterization of highly sst2-selective somatostatin antagonists suitable for radiotargeting. J. Med. Chem. 2008, 51, 4030–4037. [Google Scholar] [CrossRef] [PubMed]
- Hocart, S.J.; Jain, R.; Murphy, W.A.; Taylor, J.E.; Coy, D.H. Highly potent cyclic disulfide antagonists of somatostatin. J. Med. Chem. 1999, 42, 1863–1871. [Google Scholar] [CrossRef] [PubMed]
- Fani, M.; Del Pozzo, L.; Abiraj, K.; Mansi, R.; Tamma, M.L.; Cescato, R.; Waser, B.; Weber, W.A.; Reubi, J.C.; Maecke, H.R. Pet of somatostatin receptor-positive tumors using 64cu- and 68ga-somatostatin antagonists: The chelate makes the difference. J. Nucl. Med. 2011, 52, 1110–1118. [Google Scholar] [CrossRef] [PubMed]
- Fani, M.; Braun, F.; Waser, B.; Beetschen, K.; Cescato, R.; Erchegyi, J.; Rivier, J.E.; Weber, W.A.; Maecke, H.R.; Reubi, J.C. Unexpected sensitivity of sst2 antagonists to n-terminal radiometal modifications. J. Nucl. Med. 2012, 53, 1481–1489. [Google Scholar] [CrossRef] [PubMed]
- Antunes, P.; Ginj, M.; Zhang, H.; Waser, B.; Baum, R.P.; Reubi, J.C.; Maecke, H. Are radiogallium-labelled dota-conjugated somatostatin analogues superior to those labelled with other radiometals? Eur. J. Nucl. Med. Mol. Imaging 2007, 34, 982–993. [Google Scholar] [CrossRef] [PubMed]
- Dalm, S.U.; Nonnekens, J.; Doeswijk, G.N.; de Blois, E.; van Gent, D.C.; Konijnenberg, M.W.; de Jong, M. Comparison of the therapeutic response to treatment with a 177lu-labeled somatostatin receptor agonist and antagonist in preclinical models. J. Nucl. Med. 2016, 57, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, G.; Mansi, R.; Vomstein, S.; Kaufmann, J.; Bouterfa, H.; Maecke, H.; Wild, D.; Fani, M. Wider safety window with radiolabeled somatostatin receptor antagonists over agonists. J. Nucl. Med. 2015, 56, 335. [Google Scholar]
- Nicolas, G.P.; Vomstein, S.; Bouterfa, H.; Maecke, H.R.; Wild, D.; Fani, M. Higher tumour uptake and residence time enhances the therapeutic index of the radiolabeled somatostatin antagonists over the agonists: The influence of the peptide mass. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, S292. [Google Scholar]
- Nicolas, G.P.; Mansi, R.; McDougall; Kaufmann, L.; Bouterfa, H.; Wild, D.; Fani, M. Biodistribution, pharmacokinetics and dosimetry of 177lu-, 90y- and 111in-labeled somatostatin receptor antagonist ops201 in comparison to the agonist 177lu-dota-tate: The mass effect. J. Nucl. Med. 2017, in press. [Google Scholar]
- Cescato, R.; Waser, B.; Fani, M.; Reubi, J.C. Evaluation of 177lu-dota-sst2 antagonist versus 177lu-dota-sst2 agonist binding in human cancers in vitro. J. Nucl. Med. 2011, 52, 1886–1890. [Google Scholar] [CrossRef] [PubMed]
- Reubi, J.C.; Waser, B.; Maecke, H.R.; Rivier, J.E. Highly increased 125i-jr11 antagonist binding in vitro reveals novel indications for sst2 targeting in human cancers. J. Nucl. Med. 2017, 58, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Wild, D.; Fani, M.; Behe, M.; Brink, I.; Rivier, J.E.; Reubi, J.C.; Maecke, H.R.; Weber, W.A. First clinical evidence that imaging with somatostatin receptor antagonists is feasible. J. Nucl. Med. 2011, 52, 1412–1417. [Google Scholar] [CrossRef] [PubMed]
- Wild, D.; Fani, M.; Fischer, R.; Del Pozzo, L.; Kaul, F.; Krebs, S.; Fischer, R.; Rivier, J.E.; Reubi, J.C.; Maecke, H.R.; et al. Comparison of somatostatin receptor agonist and antagonist for peptide receptor radionuclide therapy: A pilot study. J. Nucl. Med. 2014, 55, 1248–1252. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, G.; Schreiter, N.; Kaul, F.; Uiters, J.; Mena, R.; Bouterfa, H.; Maecke, H.; Fani, M.; Wild, D. Pet/ct with the somatostatin receptor antagonist 68ga-ops202 is twice as accurate as with the agonist 68ga-dotatoc for detecting liver metastases: Results of a phase 1/2 study in gastroenteropancreatic net patients. J. Nucl. Med. 2016, 57, 154. [Google Scholar]
- Sandstrom, M.; Velikyan, I.; Garske-Roman, U.; Sorensen, J.; Eriksson, B.; Granberg, D.; Lundqvist, H.; Sundin, A.; Lubberink, M. Comparative biodistribution and radiation dosimetry of 68ga-dotatoc and 68ga-dotatate in patients with neuroendocrine tumors. J. Nucl. Med. 2013, 54, 1755–1759. [Google Scholar] [CrossRef] [PubMed]
- Körner, M.; Christ, E.; Wild, D.; Reubi, J.C. Glucagon-like peptide-1 receptor overexpression in cancer and its impact on clinical applications. Front. Endocrinol. 2012, 3, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korner, M.; Stockli, M.; Waser, B.; Reubi, J.C. Glp-1 receptor expression in human tumors and human normal tissues: Potential for in vivo targeting. J. Nucl. Med. 2007, 48, 736–743. [Google Scholar] [CrossRef] [PubMed]
- Reubi, J.C.; Waser, B. Concomitant expression of several peptide receptors in neuroendocrine tumours: Molecular basis for in vivo multireceptor tumour targeting. Eur. J. Nucl. Med. Mol. Imaging 2003, V30, 781–793. [Google Scholar] [CrossRef] [PubMed]
- De Herder, W.W.; Niederle, B.; Scoazec, J.Y.; Pauwels, S.; Klöppel, G.; Falconi, M.; Kwekkeboom, D.J.; Öberg, K.; Eriksson, B.; Wiedenmann, B.; et al. Well-differentiated pancreatic tumor/carcinoma: Insulinoma. Neuroendocrinology 2006, 84, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Soga, J.; Yakuwa, Y. Pancreatic endocrinomas: A statistical analysis of 1857 cases. J. Hepato-Biliary-Pancreat. Surg. 1994, 1, 522–529. [Google Scholar] [CrossRef]
- Öberg, K.; Eriksson, B. Endocrine tumours of the pancreas. Best Pract. Res. Clin. Gastroenterol. 2005, 19, 753–781. [Google Scholar] [CrossRef] [PubMed]
- Aisha, S.; Lubna, M.Z.; Huque, N. Recurrent insulinoma—Rare among the rarities. J. Coll. Phys. Surg. Pak. 2007, 17, 364–366. [Google Scholar]
- Krenning, E.P.; Kwekkeboom, D.J.; Reubi, J.C.; van Hagen, P.M.; van Eijck, C.H.J.; Oei, H.Y.; Lamberts, S.W.J. 111in-octreotide scintigraphy in oncology. Metabolism 1992, 41, 83–86. [Google Scholar] [CrossRef]
- Krenning, E.P.; Kwekkeboom, D.J.; Bakker, W.H.; Breeman, W.A.; Kooij, P.P.; Oei, H.Y.; van Hagen, M.; Postema, P.T.; de Jong, M.; Reubi, J.C.; et al. Somatostatin receptor scintigraphy with [111in-dtpa-d-phe1]- and [123i-tyr3]-octreotide: The rotterdam experience with more than 1000 patients. Eur. J. Nucl. Med. 1993, 20, 716–731. [Google Scholar] [CrossRef] [PubMed]
- Reubi, J.C. Peptide receptors as molecular targets for cancer diagnosis and therapy. Endocr. Rev. 2003, 24, 389–427. [Google Scholar] [CrossRef] [PubMed]
- Wild, D.; Christ, E.; Caplin, M.E.; Kurzawinski, T.R.; Forrer, F.; Brandle, M.; Seufert, J.; Weber, W.A.; Bomanji, J.; Perren, A.; et al. Glucagon-like peptide-1 versus somatostatin receptor targeting reveals 2 distinct forms of malignant insulinomas. J. Nucl. Med. 2011, 52, 1073–1078. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, O.; Velikyan, I.; Selvaraju, R.K.; Kandeel, F.; Johansson, L.; Antoni, G.; Eriksson, B.; Sörensen, J.; Korsgren, O. Detection of metastatic insulinoma by positron emission tomography with [68ga]exendin-4-a case report. J. Clin. Endocrinol. Metab. 2014, 99, 1519–1524. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Pan, Q.; Yao, S.; Yu, M.; Wu, W.; Xue, H.; Kiesewetter, D.O.; Zhu, Z.; Li, F.; Zhao, Y.; et al. Glucagon-like peptide-1 receptor pet/ct with 68ga-nota-exendin-4 for detecting localized insulinoma: A prospective cohort study. J. Nucl. Med. 2016, 57, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Sowa-Staszczak, A.; Trofimiuk-Muldner, M.; Stefanska, A.; Tomaszuk, M.; Buziak-Bereza, M.; Gilis-Januszewska, A.; Jabrocka-Hybel, A.; Glowa, B.; Malecki, M.; Bednarczuk, T.; et al. 99mtc labeled glucagon-like peptide-1-analogue (99mtc-glp1) scintigraphy in the management of patients with occult insulinoma. PLoS ONE 2016, 11, e0160714. [Google Scholar] [CrossRef] [PubMed]
- Okabayashi, T.; Shima, Y.; Sumiyoshi, T.; Kozuki, A.; Ito, S.; Ogawa, Y.; Kobayashi, M.; Hanazaki, K. Diagnosis and management of insulinoma. World J. Gastroenterol. 2013, 19, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Gotthardt, M.; Lalyko, G.; van Eerd-Vismale, J.; Keil, B.; Schurrat, T.; Hower, M.; Laverman, P.; Behr, T.M.; Boerman, O.C.; Göke, B.; et al. A new technique for in vivo imaging of specific glp-1 binding sites: First results in small rodents. Regul. Pept. 2006, 137, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Wild, D.; Behe, M.; Wicki, A.; Storch, D.; Waser, B.; Gotthardt, M.; Keil, B.; Christofori, G.; Reubi, J.C.; Macke, H.R. [lys40(ahx-dtpa-111in)nh2]exendin-4, a very promising ligand for glucagon-like peptide-1 (glp-1) receptor targeting. J. Nucl. Med. 2006, 47, 2025–2033. [Google Scholar] [PubMed]
- Gotthardt, M.; Fischer, M.; Naeher, I.; Holz, J.B.; Jungclas, H.; Fritsch, H.W.; Behe, M.; Goke, B.; Joseph, K.; Behr, T.M. Use of the incretin hormone glucagon-like peptide-1 (glp-1) for the detection of insulinomas: Initial experimental results. Eur. J. Nucl. Med. Mol. Imaging 2002, 29, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Wild, D.; Macke, H.; Christ, E.; Gloor, B.; Reubi, J.C. Glucagon-like peptide 1-receptor scans to localize occult insulinomas. New Engl. J. Med. 2008, 359, 766–768. [Google Scholar] [CrossRef] [PubMed]
- Sowa-Staszczak, A.; Pach, D.; Mikolajczak, R.; Macke, H.; Jabrocka-Hybel, A.; Stefanska, A.; Tomaszuk, M.; Janota, B.; Gilis-Januszewska, A.; Malecki, M.; et al. Glucagon-like peptide-1 receptor imaging with [lys40(ahx-hynic-99mtc/edda)nh2]-exendin-4 for the detection of insulinoma. Eur. J. Nucl. Med. Mol. Imaging 2013, 40, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Christ, E.; Wild, D.; Forrer, F.; Brandle, M.; Sahli, R.; Clerici, T.; Gloor, B.; Martius, F.; Maecke, H.; Reubi, J.C. Glucagon-like peptide-1 receptor imaging for localization of insulinomas. J. Clin. Endocrinol. Metab. 2009, 94, 4398–4405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brom, M.; Joosten, L.; Oyen, W.J.G.; Gotthardt, M.; Boerman, O.C. Radiolabelled glp-1 analogues for in vivo targeting of insulinomas. Contrast Media Mol. Imaging 2012, 7, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Brom, M.; Oyen, W.J.; Joosten, L.; Gotthardt, M.; Boerman, O.C. 68ga-labelled exendin-3, a new agent for the detection of insulinomas with pet. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 1345–1355. [Google Scholar] [CrossRef] [PubMed]
- Wild, D.; Wicki, A.; Mansi, R.; Behe, M.; Keil, B.; Bernhardt, P.; Christofori, G.; Ell, P.J.; Macke, H.R. Exendin-4-based radiopharmaceuticals for glucagonlike peptide-1 receptor pet/ct and spect/ct. J. Nucl. Med. 2010, 51, 1059–1067. [Google Scholar] [CrossRef] [PubMed]
- Selvaraju, R.K.; Velikyan, I.; Asplund, V.; Johansson, L.; Wu, Z.; Todorov, I.; Shively, J.; Kandeel, F.; Eriksson, B.; Korsgren, O.; et al. Pre-clinical evaluation of [68ga]ga-do3a-vs-cys40-exendin-4 for imaging of insulinoma. Nucl. Med. Biol. 2014, 41, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Liu, S.; Nair, I.; Omori, K.; Scott, S.; Todorov, I.; Shively, J.E.; Conti, P.S.; Li, Z.; Kandeel, F. (64)cu labeled sarcophagine exendin-4 for micropet imaging of glucagon like peptide-1 receptor expression. Theranostics 2014, 4, 770–777. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lim, K.; Normandin, M.; Zhao, X.; Cline, G.W.; Ding, Y.S. Synthesis and evaluation of [18f]exendin (9-39) as a potential biomarker to measure pancreatic beta-cell mass. Nucl. Med. Biol. 2012, 39, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Niu, G.; Yang, M.; Quan, Q.; Ma, Y.; Murage, E.N.; Ahn, J.M.; Kiesewetter, D.O.; Chen, X. Pet of insulinoma using (1)(8)f-fbem-em3106b, a new glp-1 analogue. Mol. Pharm. 2011, 8, 1775–1782. [Google Scholar] [CrossRef] [PubMed]
- Kiesewetter, D.O.; Gao, H.; Ma, Y.; Niu, G.; Quan, Q.; Guo, N.; Chen, X. 18f-radiolabeled analogs of exendin-4 for pet imaging of glp-1 in insulinoma. Eur. J. Nucl. Med. Mol. Imaging 2012, 39, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Yu, M.; Pan, Q.; Wu, W.; Zhang, T.; Kiesewetter, D.O.; Zhu, Z.; Li, F.; Chen, X.; Zhao, Y. 68ga-nota-exendin-4 pet/ct in detection of occult insulinoma and evaluation of physiological uptake. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 531–532. [Google Scholar] [CrossRef] [PubMed]
- Christ, E.; Wild, D.; Antwi, K.; Waser, B.; Fani, M.; Schwanda, S.; Heye, T.; Schmid, C.; Baer, H.U.; Perren, A.; et al. Preoperative localization of adult nesidioblastosis using 68ga-dota-exendin-4-pet/ct. Endocrine 2015, 50, 821–823. [Google Scholar] [CrossRef] [PubMed]
- Jodal, A.; Lankat-Buttgereit, B.; Brom, M.; Schibli, R.; Behe, M. A comparison of three (67/68)ga-labelled exendin-4 derivatives for beta-cell imaging on the glp-1 receptor: The influence of the conjugation site of nodaga as chelator. EJNMMI Res. 2014, 4, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selvaraju, R.K.; Velikyan, I.; Johansson, L.; Wu, Z.; Todorov, I.; Shively, J.; Kandeel, F.; Korsgren, O.; Eriksson, O. In vivo imaging of the glucagonlike peptide 1 receptor in the pancreas with 68ga-labeled do3a-exendin-4. J. Nucl. Med. 2013, 54, 1458–1463. [Google Scholar] [CrossRef] [PubMed]
- Kirsi, M.; Yim, C.B.; Veronica, F.; Tamiko, I.; Viki-Veikko, E.; Johan, R.; Jori, J.; Tiina, S.; Tuula, T.; Marko, T.; et al. 64cu- and 68ga-labelled [nle14,lys 40(ahx-nodaga)nh2]-exendin-4 for pancreatic beta cell imaging in rats. Mol. Imaging Biol. 2014, 16, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Todorov, I.; Li, L.; Bading, J.R.; Li, Z.; Nair, I.; Ishiyama, K.; Colcher, D.; Conti, P.E.; Fraser, S.E.; et al. In vivo imaging of transplanted islets with 64cu-do3a-vs-cys40-exendin-4 by targeting glp-1 receptor. Bioconjug.Chem. 2011, 22, 1587–1594. [Google Scholar] [CrossRef] [PubMed]
- Kiesewetter, D.O.; Guo, N.; Guo, J.; Gao, H.; Zhu, L.; Ma, Y.; Niu, G.; Chen, X. Evaluation of an [(18)f]alf-nota analog of exendin-4 for imaging of glp-1 receptor in insulinoma. Theranostics 2012, 2, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Kiesewetter, D.O.; Guo, J.; Sun, Z.; Zhang, X.; Zhu, L.; Niu, G.; Ma, Y.; Lang, L.; Chen, X. Development of a new thiol site-specific prosthetic group and its conjugation with [cys40]-exendin-4 for in vivo targeting of insulinomas. Bioconjug. Chem. 2013, 24, 1191–1200. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Yan, X.; Wu, C.; Niu, G.; Ma, Y.; Jacobson, O.; Shen, B.; Kiesewetter, D.O.; Chen, X. One-pot two-step radiosynthesis of a new (18)f-labeled thiol reactive prosthetic group and its conjugate for insulinoma imaging. Mol. Pharm. 2014, 11, 3875–3884. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Liang, S.; Liu, S.; Pan, Y.; Cheng, D.; Zhang, Y. 18f-radiolabeled glp-1 analog exendin-4 for pet/ct imaging of insulinoma in small animals. Nucl. Med. Commun. 2013, 34, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Liu, S.; Hassink, M.; Nair, I.; Park, R.; Li, L.; Todorov, I.; Fox, J.M.; Li, Z.; Shively, J.E.; et al. Development and evaluation of 18f-ttco-cys40-exendin-4: A pet probe for imaging transplanted islets. J. Nucl. Med. 2013, 54, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Bauman, A.; Valverde, I.E.; Fischer, C.A.; Vomstein, S.; Mindt, T.L. Development of 68ga- and 89zr-labeled exendin-4 as potential radiotracers for the imaging of insulinomas by pet. J. Nucl. Med. 2015, 56, 1569–1574. [Google Scholar] [CrossRef] [PubMed]
- Janota, B.; Karczmarczyk, U.; Laszuk, E.; Garnuszek, P.; Mikolajczak, R. Oxidation of methionine—Is it limiting the diagnostic properties of 99mtc-labeled exendin-4, a glucagon-like peptide-1 receptor agonist? Nucl. Med. Rev. Cent. East. Eur. 2016, 19, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Espes, D.; Selvaraju, R.; Velikyan, I.; Krajcovic, M.; Carlsson, P.O.; Eriksson, O. Quantification of beta-cell mass in intramuscular islet grafts using radiolabeled exendin-4. Transpl. Direct 2016, 2, e93. [Google Scholar] [CrossRef] [PubMed]
- Christ, E.; Wild, D.; Ederer, S.; Béhé, M.; Nicolas, G.; Caplin, M.E.; Brändle, M.; Clerici, T.; Fischli, S.; Stettler, C.; et al. Glucagon-like peptide-1 receptor imaging for the localisation of insulinomas: A prospective multicentre imaging study. Lancet Diabetes Endocrinol. 2013, 1, 115–122. [Google Scholar] [CrossRef]
- Pach, D.; Sowa-Staszczak, A.; Jabrocka-Hybel, A.; Stefanska, A.; Tomaszuk, M.; Mikolajczak, R.; Janota, B.; Trofimiuk-Muldner, M.; Przybylik-Mazurek, E.; Hubalewska-Dydejczyk, A. Glucagon-like peptide-1 receptor imaging with [lys (40) (ahx-hynic-(99 m) tc/edda)nh 2]-exendin-4 for the diagnosis of recurrence or dissemination of medullary thyroid cancer: A preliminary report. Int. J. Endocrinol. 2013, 2013, 384508. [Google Scholar] [CrossRef] [PubMed]
- Velikyan, I. Prospective of 68ga-radiopharmaceutical development. Theranostics 2014, 4, 47–80. [Google Scholar] [CrossRef] [PubMed]
- Nalin, L.; Selvaraju, R.K.; Velikyan, I.; Berglund, M.; Andreasson, S.; Wikstrand, A.; Ryden, A.; Lubberink, M.; Kandeel, F.; Nyman, G.; et al. Positron emission tomography imaging of the glucagon-like peptide-1 receptor in healthy and streptozotocin-induced diabetic pigs. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 1800–1810. [Google Scholar] [CrossRef] [PubMed]
- Ryden, A.; Nyman, G.; Nalin, L.; Andreasson, S.; Velikyan, I.; Korsgren, O.; Eriksson, O.; Jensen-Waern, M. Corrigendum to “cardiovascular side-effects and insulin secretion after intravenous administration of radiolabeled exendin-4 in pigs” [nucl med biol 43 (2016) 397–402]. Nucl. Med. Biol. 2016, 43, 742. [Google Scholar] [CrossRef] [PubMed]
- Selvaraju, R.; Bulenga, T.N.; Espes, D.; Lubberink, M.; Sörensen, J.; Eriksson, B.; Estrada, S.; Velikyan, I.; Eriksson, O. Dosimetry of [68ga]ga-do3a-vs-cys40-exendin-4 in rodents, pigs, non-human primates and human - repeated scanning in human is possible. Am. J. Nucl. Med. Mol. Imaging 2015, 5, 259–269. [Google Scholar] [PubMed]
- Velikyan, I.; Bulenga, T.N.; Selvaraju, K.R.; Lubberink, M.; Espes, D.; Rosenstrom, U.; Eriksson, O. Dosimetry of [177lu]-do3a-vs-cys40-exendin-4—Impact on the feasibility of insulinoma internal radiotherapy. Am. J. Nucl. Med. Mol. Imaging 2015, 5, 109–126. [Google Scholar] [PubMed]
- Rylova, S.N.; Waser, B.; Del Pozzo, L.; Tonnesmann, R.; Mansi, R.; Meyer, P.T.; Reubi, J.C.; Maecke, H.R. Approaches to improve the pharmacokinetics of radiolabeled glucagon-like peptide-1 receptor ligands using antagonistic tracers. J. Nucl. Med. 2016, 57, 1282–1288. [Google Scholar] [CrossRef] [PubMed]
- Mukai, E.; Toyoda, K.; Kimura, H.; Kawashima, H.; Fujimoto, H.; Ueda, M.; Temma, T.; Hirao, K.; Nagakawa, K.; Saji, H.; et al. Glp-1 receptor antagonist as a potential probe for pancreatic β-cell imaging. Biochem. Biophys. Res. Commun. 2009, 389, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Waser, B.; Reubi, J.C. Radiolabelled glp-1 receptor antagonist binds to glp-1 receptor-expressing human tissues. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 1166–1171. [Google Scholar] [CrossRef] [PubMed]
- Gotthardt, M.; van Eerd-Vismale, J.; Oyen, W.J.; de Jong, M.; Zhang, H.; Rolleman, E.; Maecke, H.R.; Béhé, M.; Boerman, O. Indication for different mechanisms of kidney uptake of radiolabeled peptides. J. Nucl. Med. 2007, 48, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Laeppchen, T.; Toennesmann, R.; Meyer, P.; Maecke, H.; Rylova, S. Radioiodinated exendin-4 demonstrates superior pharmacokinetics compared to radiometalated analogs. J. Nucl. Med. 2016, 57, 1388. [Google Scholar]
- Wicki, A.; Wild, D.; Storch, D.; Seemayer, C.; Gotthardt, M.; Behe, M.; Kneifel, S.; Mihatsch, M.J.; Reubi, J.C.; Macke, H.R.; et al. [lys40(ahx-dtpa-111in)nh2]-exendin-4 is a highly efficient radiotherapeutic for glucagon-like peptide-1 receptor-targeted therapy for insulinoma. Clin. Cancer Res. 2007, 13, 3696–3705. [Google Scholar] [CrossRef] [PubMed]
- Reubi, J.C.; Schaer, J.C.; Waser, B. Cholecystokinin(cck)-a and cck-b/gastrin receptors in human tumors. Cancer Res. 1997, 57, 1377–1386. [Google Scholar] [PubMed]
- Reubi, J.C.; Waser, B. Unexpected high incidence of cholecystokinin-b/gastrin receptors in human medullary thyroid carcinomas. Int. J. Cancer 1996, 67, 644–647. [Google Scholar] [CrossRef]
- Kebebew, E.; Kikuchi, S.; Duh, Q.Y.; Clark, O.H. Long-term results of reoperation and localizing studies in patients with persistent or recurrent medullary thyroid cancer. Arch. Surg. 2000, 135, 895–901. [Google Scholar] [CrossRef] [PubMed]
- Pacini, F.; Castagna, M.G.; Cipri, C.; Schlumberger, M. Medullary thyroid carcinoma. Clin. Oncol. (R. Coll. Radiol.) 2010, 22, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Treglia, G.; Aktolun, C.; Chiti, A.; Frangos, S.; Giovanella, L.; Hoffmann, M.; Iakovou, I.; Mihailovic, J.; Krause, B.J.; Langsteger, W.; et al. The 2015 revised american thyroid association guidelines for the management of medullary thyroid carcinoma: The “evidence-based” refusal to endorse them by eanm due to the “not evidence-based” marginalization of the role of nuclear medicine. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 1486–1490. [Google Scholar] [CrossRef] [PubMed]
- Wells, S.A., Jr.; Asa, S.L.; Dralle, H.; Elisei, R.; Evans, D.B.; Gagel, R.F.; Lee, N.; Machens, A.; Moley, J.F.; Pacini, F.; et al. Revised american thyroid association guidelines for the management of medullary thyroid carcinoma. Thyroid 2015, 25, 567–610. [Google Scholar] [CrossRef] [PubMed]
- Moley, J.F. Medullary thyroid cancer. Surg. Clin. N. Am. 1995, 75, 405–420. [Google Scholar] [CrossRef]
- Gotthardt, M.; Behe, M.P.; Beuter, D.; Battmann, A.; Bauhofer, A.; Schurrat, T.; Schipper, M.; Pollum, H.; Oyen, W.J.; Behr, T.M. Improved tumour detection by gastrin receptor scintigraphy in patients with metastasised medullary thyroid carcinoma. Eur. J. Nucl. Med. Mol. Imaging 2006, 33, 1273–1279. [Google Scholar] [CrossRef] [PubMed]
- Gotthardt, M.; Behe, M.P.; Grass, J.; Bauhofer, A.; Rinke, A.; Schipper, M.L.; Kalinowski, M.; Arnold, R.; Oyen, W.J.; Behr, T.M. Added value of gastrin receptor scintigraphy in comparison to somatostatin receptor scintigraphy in patients with carcinoids and other neuroendocrine tumours. Endocr. Relat. Cancer 2006, 13, 1203–1211. [Google Scholar] [CrossRef] [PubMed]
- Behr, T.M.; Gratz, S.; Markus, P.M.; Dunn, R.M.; Hufner, M.; Schauer, A.; Fischer, M.; Munz, D.L.; Becker, H.; Becker, W. Anti-carcinoembryonic antigen antibodies versus somatostatin analogs in the detection of metastatic medullary thyroid carcinoma: Are carcinoembryonic antigen and somatostatin receptor expression prognostic factors? Cancer 1997, 80, 2436–2457. [Google Scholar] [CrossRef]
- De Jong, M.; Bakker, W.H.; Bernard, B.F.; Valkema, R.; Kwekkeboom, D.J.; Reubi, J.C.; Srinivasan, A.; Schmidt, M.; Krenning, E.P. Preclinical and initial clinical evaluation of 111in-labeled nonsulfated cck8 analog: A peptide for cck-b receptor-targeted scintigraphy and radionuclide therapy. J. Nucl. Med. 1999, 40, 2081–2087. [Google Scholar] [PubMed]
- Kwekkeboom, D.J.; Bakker, W.H.; Kooij, P.P.; Erion, J.; Srinivasan, A.; de Jong, M.; Reubi, J.C.; Krenning, E.P. Cholecystokinin receptor imaging using an octapeptide dtpa-cck analogue in patients with medullary thyroid carcinoma. Eur. J. Nucl. Med. 2000, 27, 1312–1317. [Google Scholar] [CrossRef] [PubMed]
- Behr, T.M.; Behe, M.; Angerstein, C.; Gratz, S.; Mach, R.; Hagemann, L.; Jenner, N.; Stiehler, M.; Frank-Raue, K.; Raue, F.; et al. Cholecystokinin-b/gastrin receptor binding peptides: Preclinical development and evaluation of their diagnostic and therapeutic potential. Clin. Cancer Res. 1999, 5, 3124s–3138s. [Google Scholar] [CrossRef] [PubMed]
- Behr, T.M.; Jenner, N.; Radetzky, S.; Behe, M.; Gratz, S.; Yucekent, S.; Raue, F.; Becker, W. Targeting of cholecystokinin-b/gastrin receptors in vivo: Preclinical and initial clinical evaluation of the diagnostic and therapeutic potential of radiolabelled gastrin. Eur. J. Nucl. Med. 1998, 25, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Behr, T.M.; Jenner, N.; Behe, M.; Angerstein, C.; Gratz, S.; Raue, F.; Becker, W. Radiolabeled peptides for targeting cholecystokinin-b/gastrin receptor-expressing tumors. J. Nucl. Med. 1999, 40, 1029–1044. [Google Scholar] [PubMed]
- Behe, M.; Becker, W.; Gotthardt, M.; Angerstein, C.; Behr, T.M. Improved kinetic stability of dtpa- dglu as compared with conventional monofunctional dtpa in chelating indium and yttrium: Preclinical and initial clinical evaluation of radiometal labelled minigastrin derivatives. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 1140–1146. [Google Scholar] [CrossRef] [PubMed]
- Behe, M.; Behr, T.M. Cholecystokinin-b (cck-b)/gastrin receptor targeting peptides for staging and therapy of medullary thyroid cancer and other cck-b receptor expressing malignancies. Biopolymers 2002, 66, 399–418. [Google Scholar] [CrossRef] [PubMed]
- Reubi, J.C.; Waser, B.; Schaer, J.C.; Laederach, U.; Erion, J.; Srinivasan, A.; Schmidt, M.A.; Bugaj, J.E. Unsulfated dtpa- and dota-cck analogs as specific high-affinity ligands for cck-b receptor-expressing human and rat tissues in vitro and in vivo. Eur. J. Nucl. Med. 1998, 25, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Breeman, W.A.; Froberg, A.C.; de Blois, E.; van Gameren, A.; Melis, M.; de Jong, M.; Maina, T.; Nock, B.A.; Erion, J.L.; Macke, H.R.; et al. Optimised labeling, preclinical and initial clinical aspects of cck-2 receptor-targeting with 3 radiolabeled peptides. Nucl. Med. Biol. 2008, 35, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Froberg, A.C.; de Jong, M.; Nock, B.A.; Breeman, W.A.; Erion, J.L.; Maina, T.; Verdijsseldonck, M.; de Herder, W.W.; van der Lugt, A.; Kooij, P.P.; et al. Comparison of three radiolabelled peptide analogues for cck-2 receptor scintigraphy in medullary thyroid carcinoma. Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 1265–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Good, S.; Walter, M.A.; Waser, B.; Wang, X.; Muller-Brand, J.; Behe, M.P.; Reubi, J.C.; Maecke, H.R. Macrocyclic chelator-coupled gastrin-based radiopharmaceuticals for targeting of gastrin receptor-expressing tumours. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 1868–1877. [Google Scholar] [CrossRef] [PubMed]
- Nock, B.A.; Kaloudi, A.; Lymperis, E.; Giarika, A.; Kulkarni, H.R.; Klette, I.; Singh, A.; Krenning, E.P.; de Jong, M.; Maina, T.; et al. Theranostic perspectives in prostate cancer with the gastrin-releasing peptide receptor antagonist neobomb1: Preclinical and first clinical results. J. Nucl. Med. 2017, 58, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Mather, S.J.; McKenzie, A.J.; Sosabowski, J.K.; Morris, T.M.; Ellison, D.; Watson, S.A. Selection of radiolabeled gastrin analogs for peptide receptor-targeted radionuclide therapy. J. Nucl. Med. 2007, 48, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Von Guggenberg, E.; Rangger, C.; Sosabowski, J.; Laverman, P.; Reubi, J.C.; Virgolini, I.J.; Decristoforo, C. Preclinical evaluation of radiolabeled dota-derivatized cyclic minigastrin analogs for targeting cholecystokinin receptor expressing malignancies. Mol. Imaging Biol. 2012, 14, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Sosabowski, J.K.; Matzow, T.; Foster, J.M.; Finucane, C.; Ellison, D.; Watson, S.A.; Mather, S.J. Targeting of cck-2 receptor-expressing tumors using a radiolabeled divalent gastrin peptide. J. Nucl. Med. 2009, 50, 2082–2089. [Google Scholar] [CrossRef] [PubMed]
- Kolenc-Peitl, P.; Mansi, R.; Tamma, M.; Gmeiner-Stopar, T.; Sollner-Dolenc, M.; Waser, B.; Baum, R.P.; Reubi, J.C.; Maecke, H.R. Highly improved metabolic stability and pharmacokinetics of indium-111-dota-gastrin conjugates for targeting of the gastrin receptor. J. Med. Chem. 2011, 54, 2602–2609. [Google Scholar] [CrossRef] [PubMed]
- Kolenc Peitl, P.; Tamma, M.; Kroselj, M.; Braun, F.; Waser, B.; Reubi, J.C.; Sollner Dolenc, M.; Maecke, H.R.; Mansi, R. Stereochemistry of amino acid spacers determines the pharmacokinetics of (111)in-dota-minigastrin analogues for targeting the cck2/gastrin receptor. Bioconjug. Chem. 2015, 26, 1113–1119. [Google Scholar] [CrossRef] [PubMed]
- Maina, T.; Konijnenberg, M.W.; KolencPeitl, P.; Garnuszek, P.; Nock, B.A.; Kaloudi, A.; Kroselj, M.; Zaletel, K.; Maecke, H.; Mansi, R.; et al. Preclinical pharmacokinetics, biodistribution, radiation dosimetry and toxicity studies required for regulatory approval of a phase i clinical trial with (111)in-cp04 in medullary thyroid carcinoma patients. Eur. J. Pharm.Sci. 2016, 91, 236–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroselj, M.; Mansi, R.; Reubi, J.; Maecke, H.; Peitl, P.K. Comparison of dota-coupled minigastrin analogues and corresponding nle congeners. Eur. J. Nucl. Med. Mol. Imaging 2012, 39 (Suppl. 1), S533–S534. [Google Scholar]
- Aloj, L.; Aurilio, M.; Rinaldi, V.; D'Ambrosio, L.; Tesauro, D.; Peitl, P.K.; Maina, T.; Mansi, R.; von Guggenberg, E.; Joosten, L.; et al. Comparison of the binding and internalization properties of 12 dota-coupled and (1)(1)(1)in-labelled cck2/gastrin receptor binding peptides: A collaborative project under cost action bm0607. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 1417–1425. [Google Scholar] [CrossRef] [PubMed]
- Laverman, P.; Joosten, L.; Eek, A.; Roosenburg, S.; Peitl, P.K.; Maina, T.; Macke, H.; Aloj, L.; von Guggenberg, E.; Sosabowski, J.K.; et al. Comparative biodistribution of 12 (1)(1)(1)in-labelled gastrin/cck2 receptor-targeting peptides. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 1410–1416. [Google Scholar] [CrossRef] [PubMed]
- Ocak, M.; Helbok, A.; Rangger, C.; Peitl, P.K.; Nock, B.A.; Morelli, G.; Eek, A.; Sosabowski, J.K.; Breeman, W.A.; Reubi, J.C.; et al. Comparison of biological stability and metabolism of cck2 receptor targeting peptides, a collaborative project under cost bm0607. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 1426–1435. [Google Scholar] [CrossRef] [PubMed]
- Behe, M.; Kluge, G.; Becker, W.; Gotthardt, M.; Behr, T.M. Use of polyglutamic acids to reduce uptake of radiometal-labeled minigastrin in the kidneys. J. Nucl. Med. 2005, 46, 1012–1015. [Google Scholar] [PubMed]
- Konijnenberg, M.; Breeman, W.; de Blois, E.; Chan, H.; Boerman, O.; Laverman, P.; Kolenc-Peitl, P.; Melis, M.; de Jong, M. Therapeutic application of cck2r-targeting pp-f11: Influence of particle range, activity and peptide amount. EJNMMI Res. 2014, 4, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roosenburg, S.; Laverman, P.; Joosten, L.; Cooper, M.S.; Kolenc Peitl, P.; Foster, J.; Hudson, C.; Leyton, J.; Burnet, J.; Oyen, W.; et al. Pet and spect imaging of a radiolabeled minigastrin analog conjugated with dota, nota and nodaga and labeled with cu, ga and in. Mol. Pharm. 2014, 11, 3930–3937. [Google Scholar] [CrossRef] [PubMed]
- Kunikowska, J.; Ziemnicka, K.; Pawlak, D.; Ruchala, M.; Kolasa, A.; Janicka-Jedynska, M.; Wozniak, A.; Mikolajczak, R.; Krolicki, L. Medullary thyroid carcinoma—Pet/ct imaging with 68ga-labelled gastrin and somatostatin analogues. Endokrynol. Polska 2016, 67, 68–71. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, D.; Rangger, C.; Kolenc Peitl, P.; Garnuszek, P.; Maurin, M.; Ihli, L.; Kroselj, M.; Maina, T.; Maecke, H.; Erba, P.; et al. From preclinical development to clinical application: Kit formulation for radiolabelling the minigastrin analogue cp04 with in-111 for a first-in-human clinical trial. Eur. J. Pharm. Sci. 2016, 85, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Behr, T.M.; Behe, M.P. Cholecystokinin-b/gastrin receptor-targeting peptides for staging and therapy of medullary thyroid cancer and other cholecystokinin-b receptor-expressing malignancies. Semin. Nucl. Med. 2002, 32, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Wessels, B.W.; Konijnenberg, M.W.; Dale, R.G.; Breitz, H.B.; Cremonesi, M.; Meredith, R.F.; Green, A.J.; Bouchet, L.G.; Brill, A.B.; Bolch, W.E.; et al. Mird pamphlet no. 20: The effect of model assumptions on kidney dosimetry and response—Implications for radionuclide therapy. J. Nucl. Med. 2008, 49, 1884–1899. [Google Scholar] [CrossRef] [PubMed]
- Mayo, K.E.; Miller, L.J.; Bataille, D.; Dalle, S.; Goke, B.; Thorens, B.; Drucker, D.J. International union of pharmacology. Xxxv. The glucagon receptor family. Pharmacol. Rev. 2003, 55, 167–194. [Google Scholar] [CrossRef] [PubMed]
- Waser, B.; Rehmann, R.; Sanchez, C.; Fourmy, D.; Reubi, J.C. Glucose-dependent insulinotropic polypeptide receptors in most gastroenteropancreatic and bronchial neuroendocrine tumors. J. Clin. Endocrinol. Metab. 2012, 97, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Yaqub, T.; Tikhonova, I.G.; Lattig, J.; Magnan, R.; Laval, M.; Escrieut, C.; Boulegue, C.; Hewage, C.; Fourmy, D. Identification of determinants of glucose-dependent insulinotropic polypeptide receptor that interact with n-terminal biologically active region of the natural ligand. Mol. Pharmacol. 2010, 77, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Gourni, E.; Waser, B.; Clerc, P.; Fourmy, D.; Reubi, J.C.; Maecke, H.R. The glucose-dependent insulinotropic polypeptide receptor: A novel target for neuroendocrine tumor imaging-first preclinical studies. J. Nucl. Med. 2014, 55, 976–982. [Google Scholar] [CrossRef] [PubMed]
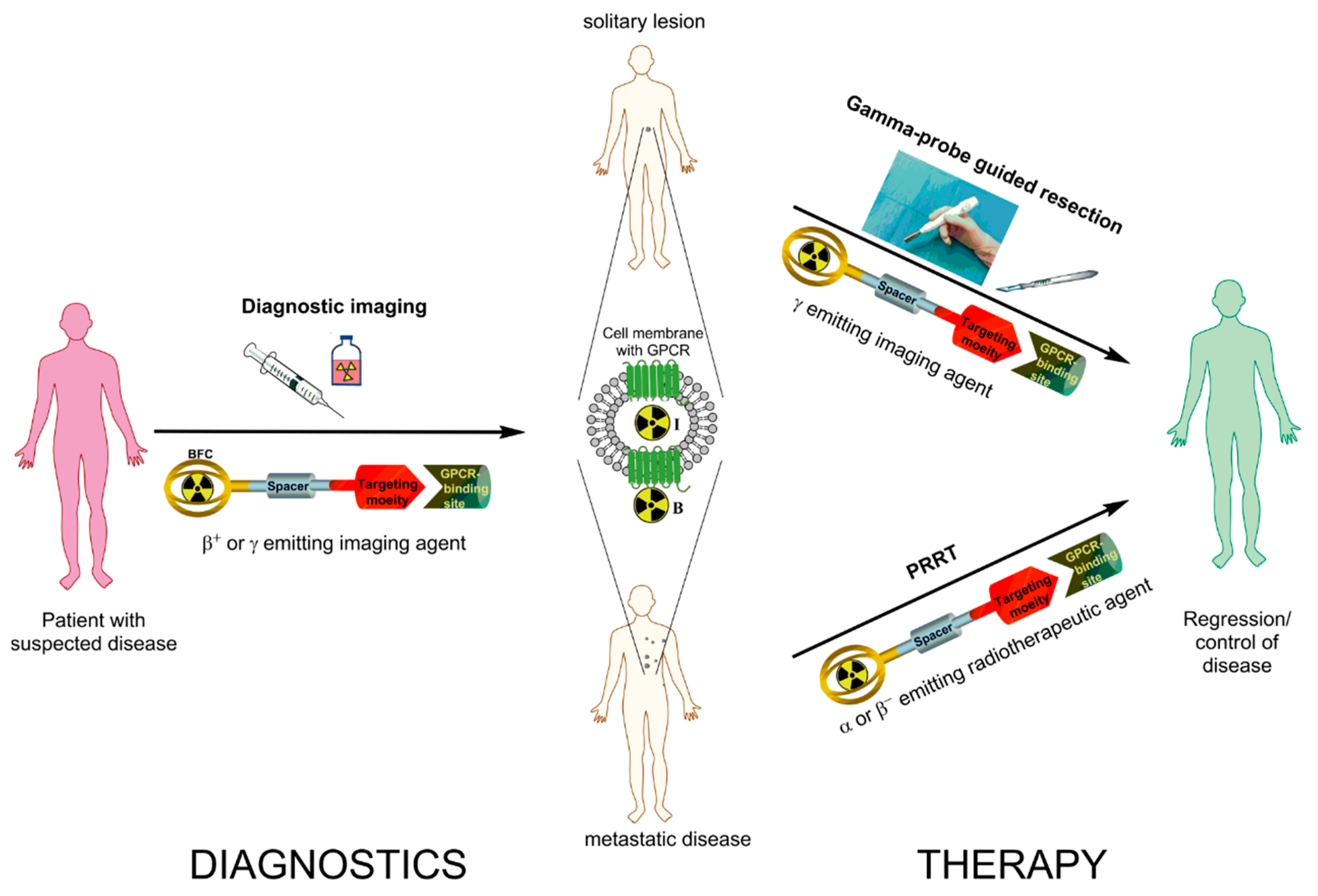
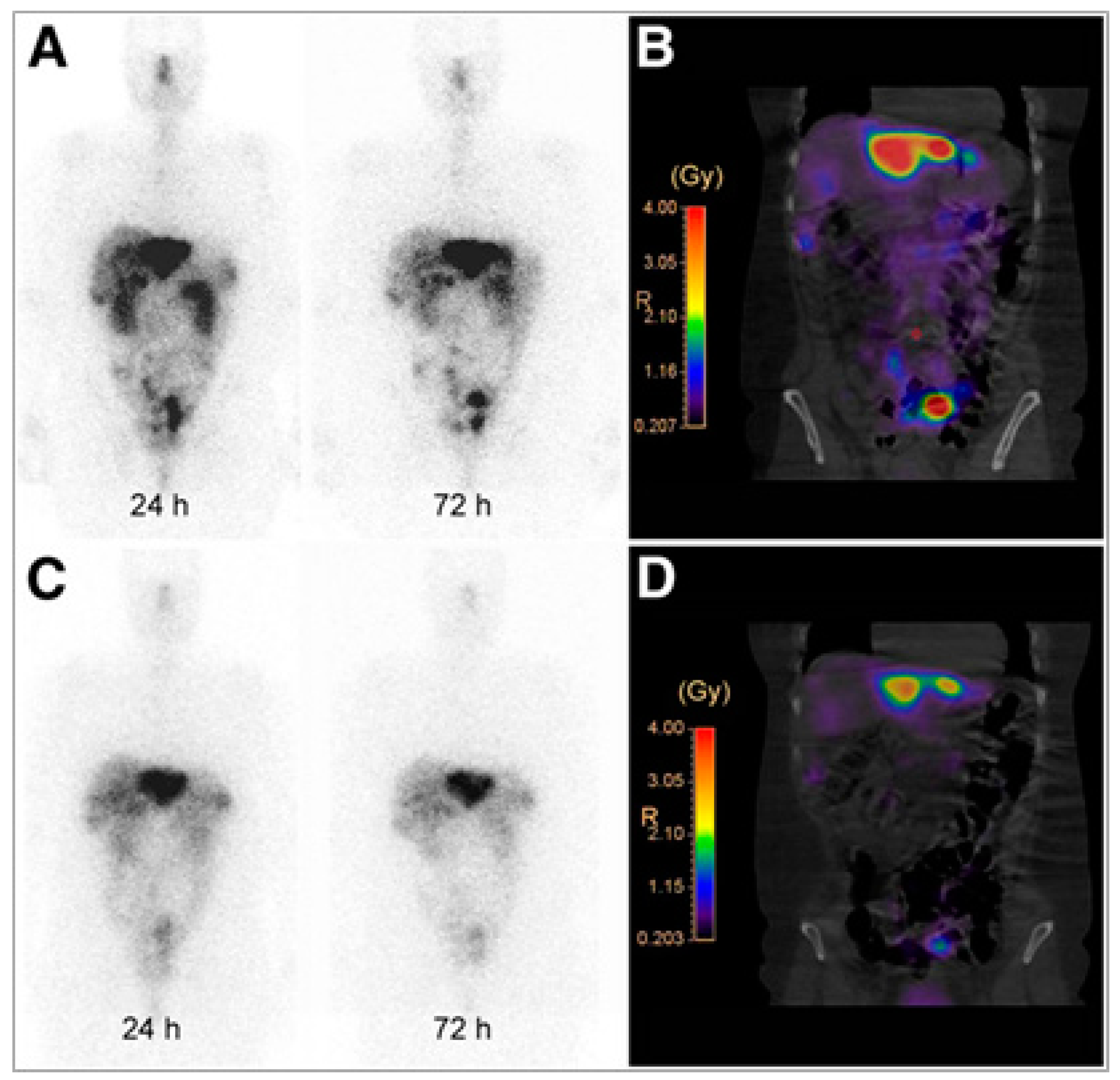
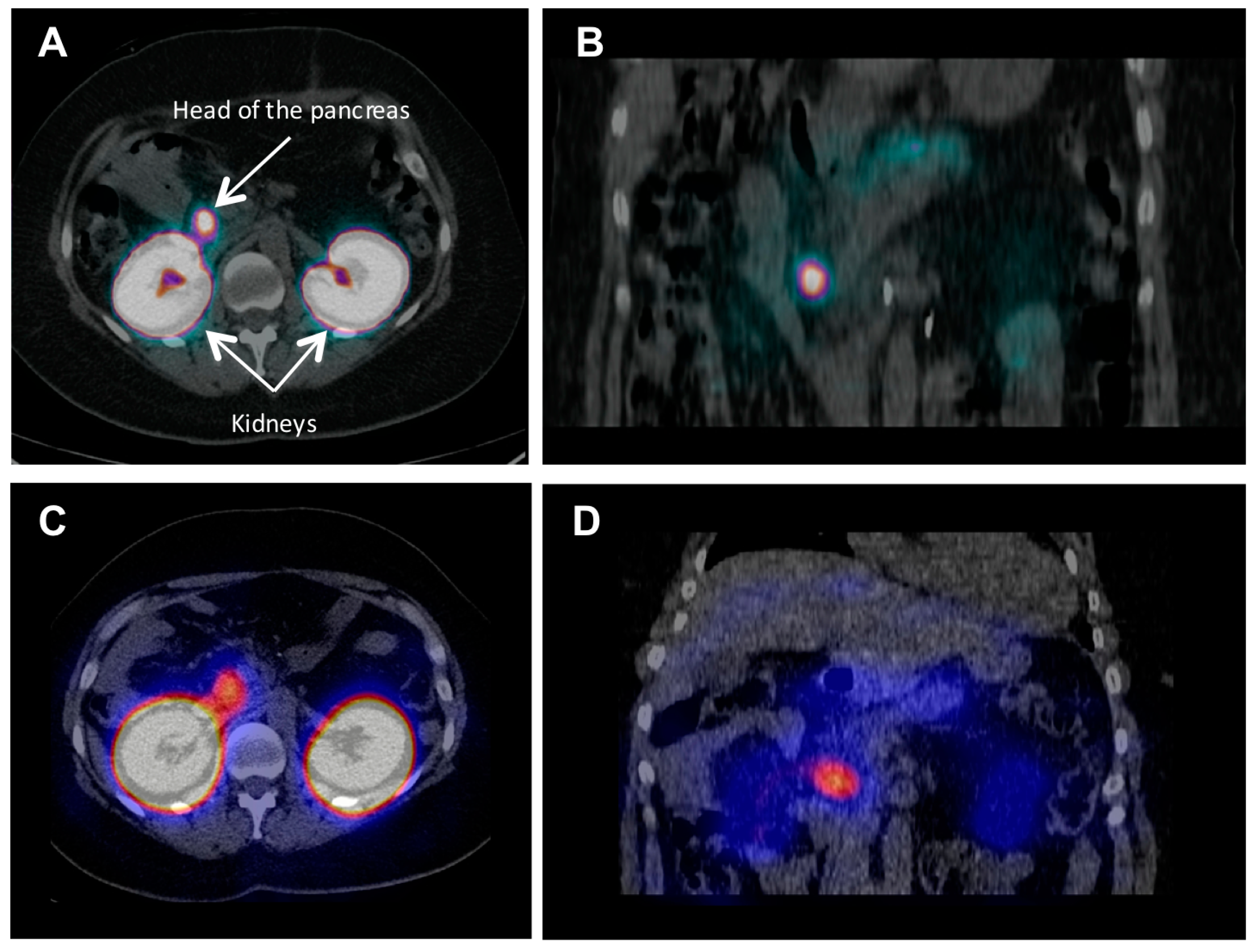

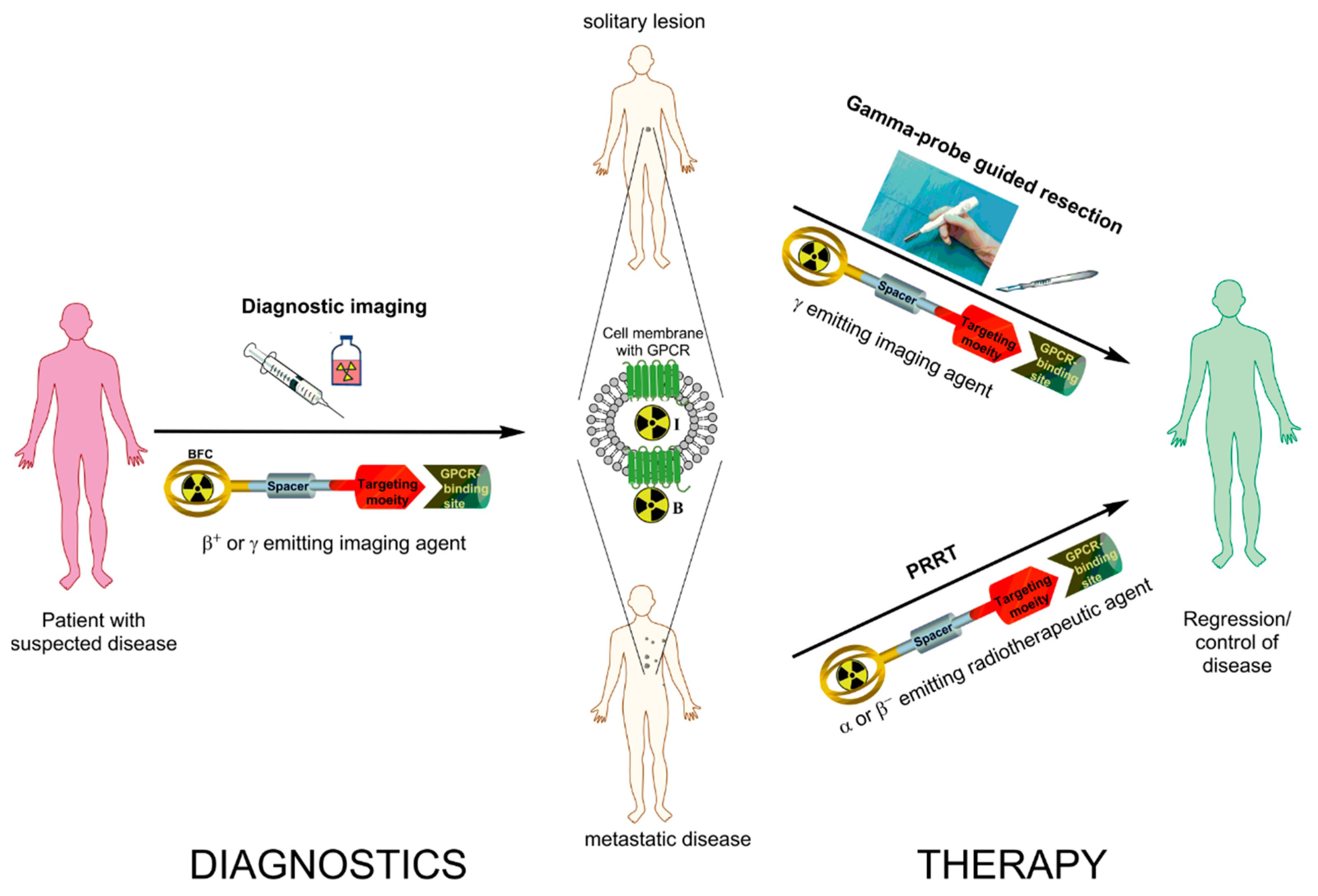{kind=link}
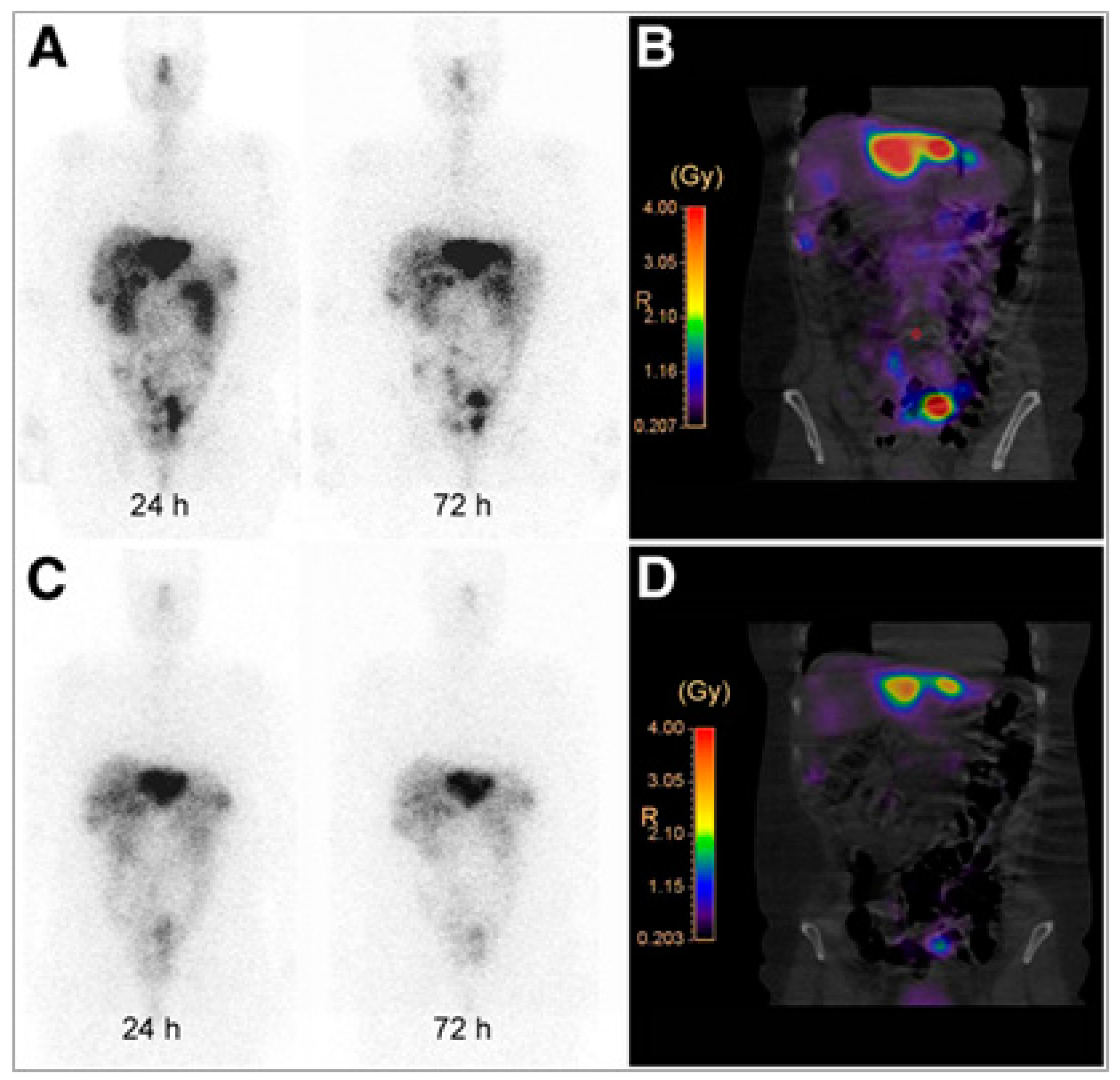{kind=link}
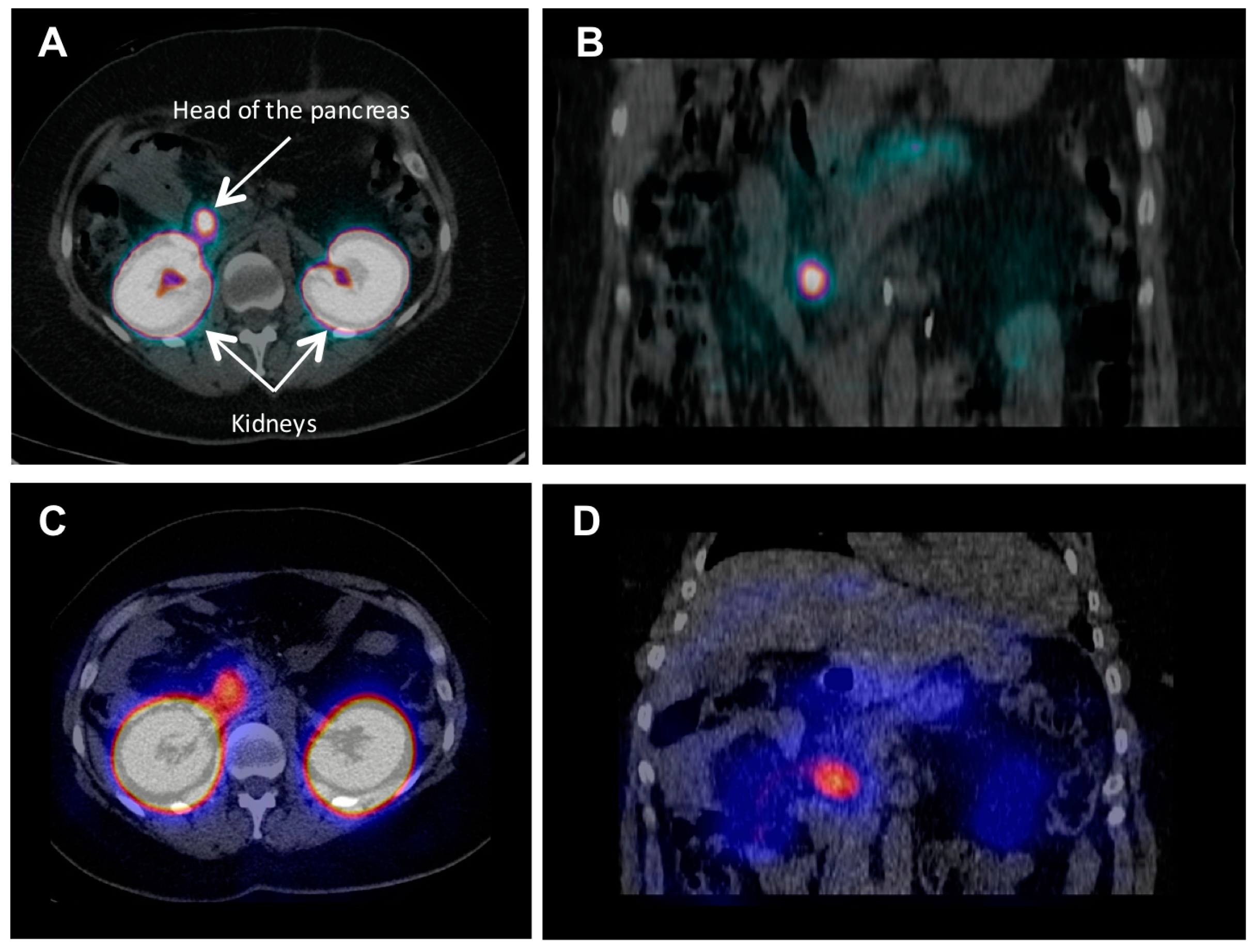{kind=link}
| Code | Chemical Structure |
|---|---|
| Somatostatin receptor agonists | |
| OC | d-Phe-cyclo(Cys-Phe-d-Trp-Lys-Thr-Cys)Thr(ol) |
| TOC | d-Phe-cyclo(Cys-Tyr-d-Trp-Lys-Thr-Cys)Thr(ol) |
| TATE | d-Phe-cyclo(Cys-Tyr-d-Trp-Lys-Thr-Cys)Thr |
| NOC | d-Phe-cyclo(Cys-1-Nal-d-Trp-Lys-Thr-Cys)Thr(ol) |
| Somatostatin receptor antagonists | |
| BASS | p-NO2-Phe-cyclo(d-Cys-Tyr-d-Trp-Lys-Thr-Cys)d-Tyr-NH2 |
| LM3 | p-Cl-Phe-cyclo(d-Cys-Tyr-d-Aph(Cbm)-Lys-Thr-Cys)d-Tyr-NH2 |
| JR10 | p-NO2-Phe-cyclo(d-Cys-Tyr-d-Aph(Cbm)-Lys-Thr-Cys)d-Tyr-NH2 |
| JR11 | p-Cl-Phe-cyclo(d-Cys-Aph(Hor)-d-Aph(Cbm)-Lys-Thr-Cys]-d-Tyr-NH2 |
| Code | Chemical Structure | Reference |
|---|---|---|
| CCK8 analogs | ||
| CCK8 | d-Asp-Tyr-Met-Gly-Trp-Met-Asp-Phe-NH2 | |
| sCCK8 | d-Asp-Tyr(OSO3H)-Met-Gly-Trp-Met-Asp-Phe-NH2 | |
| CCK8(Nle) | d-Asp-Tyr-Nle-Gly-Trp-Nle-Asp-Phe-NH2 | P: [108,109]; C: [101,102,110] |
| Minigastrin analogs | ||
| MG | Leu1-Glu2-Glu3-Glu4-Glu5-Glu6-Ala7-Tyr8-Gly9-Trp10-Met11-Asp12-Phe13-NH2 | P, C: [105] |
| MG0 | d-Glu1-Glu2-Glu3-Glu4-Glu5-Glu6-Ala7-Tyr8-Gly9-Trp10-Met11-Asp12-Phe13-NH2 | P: [106]; C: [99,107]; CP |
| MG11 | d-Glu-Ala-Tyr-Gly-Trp-Met-Asp-Phe-NH2 | P: [109,111]; C: [98,110]; CP |
| Demogastrin 2 (N4-conjugate) | N4-Gly-d-Glu-(Glu)5-Ala-Tyr-Gly-Trp-Met-Asp-Phe-NH2 | P: [112]; C: [103] |
| H2-Met, APH070 | His-His-Glu-Ala-Tyr-Gly-Trp-Met-Asp-Phe-NH2 | P: [113]; CP |
| Cyclo-MG1 (DOTA-conjugate) | DOTA-DGlu-(Ala-Tyr)-d-Lys-Trp-Met-Asp-Phe-NH2(cycloDGlu-DLys) | P: [114]; CP |
| MGD5 (divalent; DOTA-conjugate) | DOTA-Gly-Ser-Cys-(Glu-Ala-Tyr-Gly-Trp-Nle-Asp-Phe-NH2)2 | P: [115]; CP |
| PP-F10 (DOTA-conjugate) | DOTA-(d-Gln)6-Ala-Tyr-Gly-Trp-Met-Asp-Phe-NH2 | P, C: [116]; CP |
| PP-F11 (DOTA-conjugate) | DOTA-(d-Glu)6-Ala-Tyr-Gly-Trp-Met-Asp-Phe-NH2 | P: [117,118]; CP |
| C: in clinical trial | ||
| PP-F11-N (DOTA-conjugate) | DOTA-(d-Glu)6-Ala-Tyr-Gly-Trp-Nle-Asp-Phe-NH2 | P: [119] |
| C: in clinical trial | ||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fani, M.; Peitl, P.K.; Velikyan, I. Current Status of Radiopharmaceuticals for the Theranostics of Neuroendocrine Neoplasms. Pharmaceuticals 2017, 10, 30. https://doi.org/10.3390/ph10010030
Fani M, Peitl PK, Velikyan I. Current Status of Radiopharmaceuticals for the Theranostics of Neuroendocrine Neoplasms. Pharmaceuticals. 2017; 10(1):30. https://doi.org/10.3390/ph10010030
Chicago/Turabian StyleFani, Melpomeni, Petra Kolenc Peitl, and Irina Velikyan. 2017. "Current Status of Radiopharmaceuticals for the Theranostics of Neuroendocrine Neoplasms" Pharmaceuticals 10, no. 1: 30. https://doi.org/10.3390/ph10010030
APA StyleFani, M., Peitl, P. K., & Velikyan, I. (2017). Current Status of Radiopharmaceuticals for the Theranostics of Neuroendocrine Neoplasms. Pharmaceuticals, 10(1), 30. https://doi.org/10.3390/ph10010030