
CK2 in Cancer: Cellular and Biochemical Mechanisms and Potential Therapeutic Target

 , , and
, , and 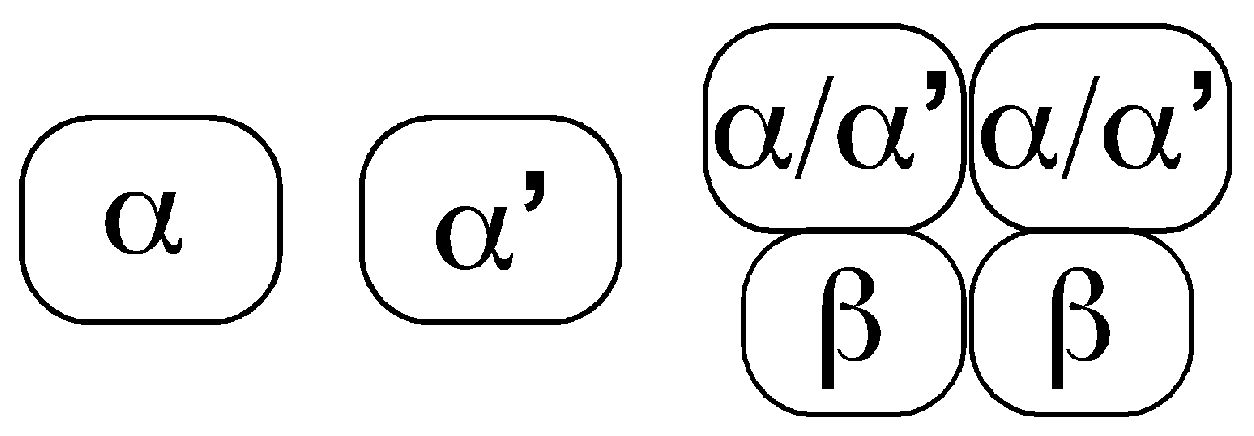{kind=link}
Abstract
:1. Introduction
2. Discussion
2.1. Solid Tumors in Which Over-Expression of CK2 Appears to Contribute to the Cancer Phenotype
2.1.1. Environmentally Induced Cancers
Lung Cancer
Urothelial Cancer
Head and Neck
Mesothelioma
2.1.2. Gastrointestinal Cancers (Often Related to Chronic Inflammation)
Hepatocellular Cancer
Gastric Cancer
Esophageal Cancer
Cholangiocarcinoma
Colorectal Cancer
Pancreatic Cancer
2.1.3. HPV-Related Cancers: Cervical, Head and Neck, Anal and Penile
2.1.4. Other Solid Tumors: Glioblastoma, Melanoma, Ovarian Cancer, Prostate Cancer, Breast Cancer and Renal Cell Carcinoma
Glioblastoma
Melanoma
Ovarian Cancer
Prostate Cancer
Breast Cancer
Renal Cell Carcinoma
2.2. Hematological Malignancies
2.2.1. Leukemia
2.2.2. Non-Hodgkin Lymphoma (NHL)
2.2.3. Myeloma
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Trembley, J.H.; Wu, J.; Unger, G.M.; Kren, B.T.; Ahmed, K. Ck2 Suppression of Apoptosis and Its Implication in Cancer Biology and Therapy; Wiley-Blackwell: Ames, IA, USA, 2013; pp. 219–343. [Google Scholar]
- Seldin, D.C.; Landesman-Bollag, E. The Oncogenic Potential of CK2; Wiley-Blackwell: Ames, IA, USA, 2013. [Google Scholar]
- Ruzzene, M.; Pinna, L.A. Addiction to protein kinase ck2: A common denominator of diverse cancer cells? Biochim. Biophys. Acta 2010, 1804, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Seldin, D.C.; Landesman-Bollag, E.; Farago, M.; Currier, N.; Lou, D.; Dominguez, I. Ck2 as a positive regulator of wnt signalling and tumourigenesis. Mol. Cell. Biochem. 2005, 274, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, I.; Sonenshein, G.E.; Seldin, D.C. Protein kinase ck2 in health and disease: Ck2 and its role in wnt and nf-kappab signaling: Linking development and cancer. Cell. Mol. Life Sci. 2009, 66, 1850–1857. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.X.; Jiang, S.S.; Zhang, X.F.; Zhou, Z.Q.; Pan, Q.Z.; Chen, C.L.; Zhao, J.J.; Tang, Y.; Xia, J.C.; Weng, D.S. Protein kinase ck2α catalytic subunit is overexpressed and serves as an unfavorable prognostic marker in primary hepatocellular carcinoma. Oncotarget 2015, 6, 34800–34817. [Google Scholar] [PubMed]
- Pinna, L.A.; Meggio, F. Protein kinase ck2 (“casein kinase-2”) and its implication in cell division and proliferation. Prog. Cell. Cycle Res. 1997, 3, 77–97. [Google Scholar] [PubMed]
- Ahmed, K.; Davis, A.T.; Wang, H.; Faust, R.A.; Yu, S.; Tawfic, S. Significance of protein kinase ck2 nuclear signaling in neoplasia. J. Cell. Biochem. 2000, 79 (Suppl. 35), 130–135. [Google Scholar] [CrossRef]
- Litchfield, D.W. Protein kinase ck2: Structure, regulation and role in cellular decisions of life and death. Biochem. J. 2003, 369, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, K.A.; Wang, G.; Unger, G.; Slaton, J.; Ahmed, K. Protein kinase ck2—A key suppressor of apoptosis. Adv. Enzyme Regul. 2008, 48, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.; Gerber, D.A.; Cochet, C. Joining the cell survival squad: An emerging role for protein kinase ck2. Trends Cell. Biol. 2002, 12, 226–230. [Google Scholar] [CrossRef]
- Canton, D.A.; Litchfield, D.W. The shape of things to come: An emerging role for protein kinase ck2 in the regulation of cell morphology and the cytoskeleton. Cell. Signal. 2006, 18, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Filhol, O.; Deshiere, A.; Cochet, C. Role of CK2 in the Control of Cell Plasticity in Breast Carcinoma Progression; Wiley-Blackwell: Ames, IA, USA, 2013. [Google Scholar]
- Kramerov, A.A.; Saghizadeh, M.; Caballero, S.; Shaw, L.C.; Li Calzi, S.; Bretner, M.; Montenarh, M.; Pinna, L.A.; Grant, M.B.; Ljubimov, A.V. Inhibition of protein kinase ck2 suppresses angiogenesis and hematopoietic stem cell recruitment to retinal neovascularization sites. Mol. Cell. Biochem. 2008, 316, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Montenarh, M. Protein kinase ck2 and angiogenesis. Adv. Clin. Exp. Med. 2014, 23, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Toselli, P.A.; Russell, L.D.; Seldin, D.C. Globozoospermia in mice lacking the casein kinase ii α’ catalytic subunit. Nat. Genet. 1999, 23, 118–121. [Google Scholar] [PubMed]
- Bibby, A.C.; Litchfield, D.W. The multiple personalities of the regulatory subunit of protein kinase ck2: Ck2 dependent and ck2 independent roles reveal a secret identity for ck2β. Int. J. Biol. Sci. 2005, 1, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Wirkner, U.; Voss, H.; Lichter, P.; Weitz, S.; Ansorge, W.; Pyerin, W. Human casein kinase ii subunit α: Sequence of a processed (pseudo)gene and its localization on chromosome 11. Biochim. Biophys. Acta 1992, 1131, 220–222. [Google Scholar] [CrossRef]
- Ortega, C.E.; Seidner, Y.; Dominguez, I. Mining ck2 in cancer. PLoS ONE 2014, 9, e115609. [Google Scholar] [CrossRef] [PubMed]
- Olsen, B.B.; Issinger, O.G.; Guerra, B. Regulation of DNA-dependent protein kinase by protein kinase ck2 in human glioblastoma cells. Oncogene 2010, 29, 6016–6026. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; McFarland, B.C.; Drygin, D.; Yu, H.; Bellis, S.L.; Kim, H.; Bredel, M.; Benveniste, E.N. Targeting protein kinase CK2 suppresses prosurvival signaling pathways and growth of glioblastoma. Clin. Cancer Res. 2013, 19, 6484–6494. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Vilk, G.; Canton, D.A.; Litchfield, D.W. Phosphorylation regulates the stability of the regulatory ck2β subunit. Oncogene 2002, 21, 3754–3764. [Google Scholar] [CrossRef] [PubMed]
- Tawfic, S.; Yu, S.; Wang, H.; Faust, R.; Davis, A.; Ahmed, K. Protein kinase CK2 signal in neoplasia. Histol. Histopathol. 2001, 16, 573–582. [Google Scholar] [PubMed]
- Macias Alvarez, L.; Revuelta-Cervantes, J.; Dominguez, I. CK2 in embryonic development. In The Wiley-IUBMB Series on Biochemistry and Molecular Biology: Protein Kinase CK2; Wiley: New York, NY, USA, 2013. [Google Scholar]
- Dominguez, I.; Mizuno, J.; Wu, H.; Song, D.H.; Symes, K.; Seldin, D.C. Protein kinase CK2 is required for dorsal axis formation in xenopus embryos. Dev. Biol. 2004, 274, 110–124. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, I.; Mizuno, J.; Wu, H.; Imbrie, G.A.; Symes, K.; Seldin, D.C. A role for CK2α/β in Xenopus early embryonic development. Mol. Cell. Biochem. 2005, 274, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.; Liu, Y.; Xia, R.; Tong, C.; Yue, T.; Jiang, J.; Jia, J. Casein kinase 2 promotes hedgehog signaling by regulating both smoothened and cubitus interruptus. J. Biol. Chem. 2010, 285, 37218–37226. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Qin, H.; Frank, S.J.; Deng, L.; Litchfield, D.W.; Tefferi, A.; Pardanani, A.; Lin, F.T.; Li, J.; Sha, B.; et al. A ck2-dependent mechanism for activation of the jak-stat signaling pathway. Blood 2011, 118, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Di Maira, G.; Salvi, M.; Arrigoni, G.; Marin, O.; Sarno, S.; Brustolon, F.; Pinna, L.A.; Ruzzene, M. Protein kinase CK2 phosphorylates and upregulates akt/pkb. Cell Death Differ. 2005, 12, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Kim, J.J.; Bae, Y.S. Involvement of pi3k-akt-mtor pathway in protein kinase ckii inhibition-mediated senescence in human colon cancer cells. Biochem. Biophys. Res. Commun. 2013, 433, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Torres, J.; Pulido, R. The tumor suppressor pten is phosphorylated by the protein kinase CK2 at its C terminus. Implications for pten stability to proteasome-mediated degradation. J. Biol. Chem. 2001, 276, 993–998. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.J.; Lou, D.Y.; Seldin, D.C.; Lane, W.S.; Neel, B.G. Direct identification of pten phosphorylation sites. FEBS Lett. 2002, 528, 145–153. [Google Scholar] [CrossRef]
- Channavajhala, P.; Seldin, D.C. Functional interaction of protein kinase CK2 and c-myc in lymphomagenesis. Oncogene 2002, 21, 5280–5288. [Google Scholar] [CrossRef] [PubMed]
- Romieu-Mourez, R.; Landesman-Bollag, E.; Seldin, D.C.; Sonenshein, G.E. Protein kinase CK2 promotes aberrant activation of nuclear factor-kappab, transformed phenotype, and survival of breast cancer cells. Cancer Res. 2002, 62, 6770–6778. [Google Scholar] [PubMed]
- Zhang, S.; Long, H.; Yang, Y.L.; Wang, Y.; Hsieh, D.; Li, W.; Au, A.; Stoppler, H.J.; Xu, Z.; Jablons, D.M.; et al. Inhibition of ck2α down-regulates notch1 signalling in lung cancer cells. J. Cell. Mol. Med. 2013, 17, 854–862. [Google Scholar] [CrossRef] [PubMed]
- Heriche, J.K.; Chambaz, E.M. Protein kinase CK2α is a target for the Abl and Bcr-Abl tyrosine kinases. Oncogene 1998, 17, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Diallo, O.T.; Hu, M.; Ehsanian, R.; Yang, X.; Arun, P.; Lu, H.; Korman, V.; Unger, G.; Ahmed, K.; et al. CK2 modulation of Nf-kappaB, TP53, and the malignant phenotype in head and neck cancer by anti-CK2 oligonucleotides in vitro or in vivo via sub-50-nm nanocapsules. Clin. Cancer Res. 2010, 16, 2295–2307. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y.; Han, J.; Kannabiran, V.; Mohan, S.; Cheng, H.; Friedman, J.; Zhang, L.; VanWaes, C.; Chen, Z. Mek inhibitor PD-0325901 overcomes resistance to CK2 inhibitor CX-4945 and exhibits anti-tumor activity in head and neck cancer. Int. J. Biol. Sci. 2015, 11, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Sarno, S.; Reddy, H.; Meggio, F.; Ruzzene, M.; Davies, S.P.; Donella-Deana, A.; Shugar, D.; Pinna, L.A. Selectivity of 4,5,6,7-tetrabromobenzotriazole, an ATP site-directed inhibitor of protein kinase CK2 (‘casein kinase-2’). FEBS Lett. 2001, 496, 44–48. [Google Scholar] [CrossRef]
- Cozza, G.; Mazzorana, M.; Papinutto, E.; Bain, J.; Elliott, M.; di Maira, G.; Gianoncelli, A.; Pagano, M.A.; Sarno, S.; Ruzzene, M.; et al. Quinalizarin as a potent, selective and cell-permeable inhibitor of protein kinase ck2. Biochem. J. 2009, 421, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Hung, M.S.; Xu, Z.; Lin, Y.C.; Mao, J.H.; Yang, C.T.; Chang, P.J.; Jablons, D.M.; You, L. Identification of hematein as a novel inhibitor of protein kinase CK2 from a natural product library. BMC Cancer 2009, 9, 135. [Google Scholar] [CrossRef] [PubMed]
- Pagano, M.A.; Poletto, G.; Di Maira, G.; Cozza, G.; Ruzzene, M.; Sarno, S.; Bain, J.; Elliott, M.; Moro, S.; Zagotto, G.; et al. Tetrabromocinnamic acid (TBCA) and related compounds represent a new class of specific protein kinase CK2 inhibitors. Chembiochem 2007, 8, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Perea, S.E.; Reyes, O.; Baladron, I.; Perera, Y.; Farina, H.; Gil, J.; Rodriguez, A.; Bacardi, D.; Marcelo, J.L.; Cosme, K.; et al. Cigb-300, a novel proapoptotic peptide that impairs the CK2 phosphorylation and exhibits anticancer properties both in vitro and in vivo. Mol. Cell. Biochem. 2008, 316, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui-Jain, A.; Drygin, D.; Streiner, N.; Chua, P.; Pierre, F.; O’Brien, S.E.; Bliesath, J.; Omori, M.; Huser, N.; Ho, C.; et al. CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res. 2010, 70, 10288–10298. [Google Scholar] [CrossRef] [PubMed]
- Zandomeni, R.; Zandomeni, M.C.; Shugar, D.; Weinmann, R. Casein kinase type II is involved in the inhibition by 5,6-dichloro-1-β-D-ribofuranosylbenzimidazole of specific RNA polymerase II transcription. J. Biol. Chem. 1986, 261, 3414–3419. [Google Scholar] [PubMed]
- Hagiwara, M.; Inoue, S.; Tanaka, T.; Nunoki, K.; Ito, M.; Hidaka, H. Differential effects of flavonoids as inhibitors of tyrosine protein kinases and serine/threonine protein kinases. Biochem. Pharmacol. 1988, 37, 2987–2992. [Google Scholar] [PubMed]
- Pagano, M.A.; Meggio, F.; Ruzzene, M.; Andrzejewska, M.; Kazimierczuk, Z.; Pinna, L.A. 2-Dimethylamino-4,5,6,7-tetrabromo-1H-benzimidazole: A novel powerful and selective inhibitor of protein kinase CK2. Biochem. Biophys. Res. Commun. 2004, 321, 1040–1044. [Google Scholar] [CrossRef] [PubMed]
- Yim, H.; Lee, Y.H.; Lee, C.H.; Lee, S.K. Emodin, an anthraquinone derivative isolated from the rhizomes of rheum palmatum, selectively inhibits the activity of casein kinase ii as a competitive inhibitor. Planta Med. 1999, 65, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Gotz, C.; Gratz, A.; Kucklaender, U.; Jose, J. Tf—A novel cell-permeable and selective inhibitor of human protein kinase CK2 induces apoptosis in the prostate cancer cell line LNCaP. Biochim. Biophys. Acta 2012, 1820, 970–977. [Google Scholar] [CrossRef] [PubMed]
- O-charoenrat, P.; Rusch, V.; Talbot, S.G.; Sarkaria, I.; Viale, A.; Socci, N.; Ngai, I.; Rao, P.; Singh, B. Casein kinase ii α subunit and c1-inhibitor are independent predictors of outcome in patients with squamous cell carcinoma of the lung. Clin. Cancer Res. 2004, 10, 5792–5803. [Google Scholar] [CrossRef] [PubMed]
- Hung, M.S.; Lin, Y.C.; Mao, J.H.; Kim, I.J.; Xu, Z.; Yang, C.T.; Jablons, D.M.; You, L. Functional polymorphism of the CK2α intronless gene plays oncogenic roles in lung cancer. PLoS ONE 2010, 5, e11418. [Google Scholar] [CrossRef] [PubMed]
- Daya-Makin, M.; Sanghera, J.S.; Mogentale, T.L.; Lipp, M.; Parchomchuk, J.; Hogg, J.C.; Pelech, S.L. Activation of a tumor-associated protein kinase (p40TAK) and casein kinase 2 in human squamous cell carcinomas and adenocarcinomas of the lung. Cancer Res. 1994, 54, 2262–2268. [Google Scholar] [PubMed]
- Yaylim, I.; Isbir, T. Enhanced casein kinase II (CK II) activity in human lung tumours. Anticancer Res. 2002, 22, 215–218. [Google Scholar] [PubMed]
- Ku, M.J.; Park, J.W.; Ryu, B.J.; Son, Y.J.; Kim, S.H.; Lee, S.Y. CK2 inhibitor CX4945 induces sequential inactivation of proteins in the signaling pathways related with cell migration and suppresses metastasis of a549 human lung cancer cells. Bioorg. Med. Chem. Lett. 2013, 23, 5609–5613. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Li, K.; Zhang, S.; Li, Q.; Li, Z.; Zhou, F.; Dong, X.; Liu, L.; Wu, G.; Meng, R. Quinalizarin, a specific CK2 inhibitor, reduces cell viability and suppresses migration and accelerates apoptosis in different human lung cancer cell lines. Indian J. Cancer 2015, 52 (Suppl. 2), 119–124. [Google Scholar]
- Kim, J.; Hwan Kim, S. CK2 inhibitor CX-4945 blocks TGF-β1-induced epithelial-to-mesenchymal transition in A549 human lung adenocarcinoma cells. PLoS ONE 2013, 8, e74342. [Google Scholar]
- Lin, Y.C.; Hung, M.S.; Lin, C.K.; Li, J.M.; Lee, K.D.; Li, Y.C.; Chen, M.F.; Chen, J.K.; Yang, C.T. CK2 inhibitors enhance the radiosensitivity of human non-small cell lung cancer cells through inhibition of stat3 activation. Cancer Biother. Radiopharm. 2011, 26, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Hung, M.S.; Xu, Z.; Chen, Y.; Smith, E.; Mao, J.H.; Hsieh, D.; Lin, Y.C.; Yang, C.T.; Jablons, D.M.; You, L. Hematein, a casein kinase II inhibitor, inhibits lung cancer tumor growth in a murine xenograft model. Int. J. Oncol. 2013, 43, 1517–1522. [Google Scholar] [PubMed]
- Benavent, F.; Capobianco, C.S.; Garona, J.; Cirigliano, S.M.; Perera, Y.; Urtreger, A.J.; Perea, S.E.; Alonso, D.F.; Farina, H.G. CIGB-300, an anti-CK2 peptide, inhibits angiogenesis, tumor cell invasion and metastasis in lung cancer models. Lung Cancer 2016. [Google Scholar] [CrossRef] [PubMed]
- Perera, Y.; Toro, N.D.; Gorovaya, L.; Fernandez, D.E.C.J.; Farina, H.G.; Perea, S.E. Synergistic interactions of the anti-casein kinase 2 CIGB-300 peptide and chemotherapeutic agents in lung and cervical preclinical cancer models. Mol. Clin. Oncol. 2014, 2, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Bliesath, J.; Huser, N.; Omori, M.; Bunag, D.; Proffitt, C.; Streiner, N.; Ho, C.; Siddiqui-Jain, A.; O’Brien, S.E.; Lim, J.K.; et al. Combined inhibition of EGFR and CK2 augments the attenuation of pi3k-akt-mtor signaling and the killing of cancer cells. Cancer Lett. 2012, 322, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Anai, S.; Marco, D.A.; Fujimoto, K.; Konishi, N. Cyclooxygenase 2-dependent and independent activation of Akt through casein kinase 2α contributes to human bladder cancer cell survival. BMC Urol. 2011, 11, 8. [Google Scholar] [CrossRef] [PubMed]
- Jeppesen, D.K.; Nawrocki, A.; Jensen, S.G.; Thorsen, K.; Whitehead, B.; Howard, K.A.; Dyrskjot, L.; Orntoft, T.F.; Larsen, M.R.; Ostenfeld, M.S. Quantitative proteomics of fractionated membrane and lumen exosome proteins from isogenic metastatic and nonmetastatic bladder cancer cells reveal differential expression of EMT factors. Proteomics 2014, 14, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Weidle, U.H.; Birzele, F.; Kollmorgen, G.; Ruger, R. The multiple roles of exosomes in metastasis. Cancer Genom. Proteom. 2017, 14, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Chua, M.M.J.; Dominguez, I. Cancer-type dependent miss-expression of CK2 genes. 2017; in preparation. [Google Scholar]
- Gapany, M.; Faust, R.A.; Tawfic, S.; Davis, A.; Adams, G.L.; Ahmed, K. Association of elevated protein kinase CK2 activity with aggressive behavior of squamous cell carcinoma of the head and neck. Mol. Med. 1995, 1, 659–666. [Google Scholar] [PubMed]
- Faust, R.A.; Gapany, M.; Tristani, P.; Davis, A.; Adams, G.L.; Ahmed, K. Elevated protein kinase CK2 activity in chromatin of head and neck tumors: Association with malignant transformation. Cancer Lett. 1996, 101, 31–35. [Google Scholar] [CrossRef]
- Faust, R.A.; Niehans, G.; Gapany, M.; Hoistad, D.; Knapp, D.; Cherwitz, D.; Davis, A.; Adams, G.L.; Ahmed, K. Subcellular immunolocalization of protein kinase CK2 in normal and carcinoma cells. Int. J. Biochem. Cell. Biol. 1999, 31, 941–949. [Google Scholar] [CrossRef]
- Faust, R.A.; Tawfic, S.; Davis, A.T.; Bubash, L.A.; Ahmed, K. Antisense oligonucleotides against protein kinase CK2-α inhibit growth of squamous cell carcinoma of the head and neck in vitro. Head Neck 2000, 22, 341–346. [Google Scholar] [CrossRef]
- Wang, H.; Davis, A.; Yu, S.; Ahmed, K. Response of cancer cells to molecular interruption of the CK2 signal. Mol. Cell. Biochem. 2001, 227, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.; Duffy, S.; Teknos, T.; Islam, M.; Chen, Z.; Albert, P.S.; Wolf, G.; Van Waes, C. Nuclear factor-kappaB-related serum factors as longitudinal biomarkers of response and survival in advanced oropharyngeal carcinoma. Clin. Cancer Res. 2007, 13, 3182–3190. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Yeh, J.; Van Waes, C. Protein kinase casein kinase 2 mediates inhibitor-kappaB kinase and aberrant nuclear factor-kappaB activation by serum factor(s) in head and neck squamous carcinoma cells. Cancer Res. 2006, 66, 6722–6731. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.W.; Xie, T.X.; Sano, D.; Myers, J.N. IL-6 stabilizes twist and enhances tumor cell motility in head and neck cancer cells through activation of casein kinase 2. PLoS ONE 2011, 6, e19412. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Yang, B.; Shi, S.; Jiang, X. RNA interference (RNAi) mediated stable knockdown of protein casein kinase 2-α (CK2α) inhibits migration and invasion and enhances cisplatin-induced apoptosis in HEp-2 laryngeal carcinoma cells. Acta Histochem. 2014, 116, 1000–1006. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Malhotra, P.S.; Thomas, G.R.; Ondrey, F.G.; Duffey, D.C.; Smith, C.W.; Enamorado, I.; Yeh, N.T.; Kroog, G.S.; Rudy, S.; et al. Expression of proinflammatory and proangiogenic cytokines in patients with head and neck cancer. Clin. Cancer Res. 1999, 5, 1369–1379. [Google Scholar] [PubMed]
- Hosono, S.; Kajiyama, H.; Terauchi, M.; Shibata, K.; Ino, K.; Nawa, A.; Kikkawa, F. Expression of Twist increases the risk for recurrence and for poor survival in epithelial ovarian carcinoma patients. Br. J. Cancer 2007, 96, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S.A.; Taylor, J.M.; Terrell, J.E.; Islam, M.; Li, Y.; Fowler, K.E.; Wolf, G.T.; Teknos, T.N. Interleukin-6 predicts recurrence and survival among head and neck cancer patients. Cancer 2008, 113, 750–757. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Yan, C.; Quan, X.X.; Yang, X.; Zhang, J.; Bian, Y.; Chen, Z.; Van Waes, C. CK2 phosphorylates and inhibits TAp73 tumor suppressor function to promote expression of cancer stem cell genes and phenotype in head and neck cancer. Neoplasia 2014, 16, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Unger, G.M.; Kren, B.T.; Korman, V.L.; Kimbrough, T.G.; Vogel, R.I.; Ondrey, F.G.; Trembley, J.H.; Ahmed, K. Mechanism and efficacy of sub-50-nm tenfibgen nanocapsules for cancer cell-directed delivery of anti-CK2 RNAi to primary and metastatic squamous cell carcinoma. Mol. Cancer Ther. 2014, 13, 2018–2029. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yang, Y.L.; Wang, Y.; You, B.; Dai, Y.; Chan, G.; Hsieh, D.; Kim, I.J.; Fang, L.T.; Au, A.; et al. CK2α, over-expressed in human malignant pleural mesothelioma, regulates the Hedgehog signaling pathway in mesothelioma cells. J. Exp. Clin. Cancer Res. 2014, 33, 93. [Google Scholar] [PubMed]
- Shi, Y.; Moura, U.; Opitz, I.; Soltermann, A.; Rehrauer, H.; Thies, S.; Weder, W.; Stahel, R.A.; Felley-Bosco, E. Role of hedgehog signaling in malignant pleural mesothelioma. Clin. Cancer Res. 2012, 18, 4646–4656. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; He, J.; Zhang, F.; Li, H.; Yue, D.; Wang, C.; Jablons, D.M.; He, B.; Lui, N. SMO expression level correlates with overall survival in patients with malignant pleural mesothelioma. J. Exp. Clin. Cancer Res. 2013, 32, 7. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Chang, Y.G.; Bae, H.J.; Eun, J.W.; Shen, Q.; Park, S.J.; Shin, W.C.; Lee, E.K.; Park, S.; Ahn, Y.M.; et al. Oncogenic potential of CK2α and its regulatory role in EGF-induced HDAC2 expression in human liver cancer. FEBS J. 2014, 281, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Sui, C.; Meng, F.; Tian, X.; Fu, L.; Li, Y.; Qi, X.; Cui, H.; Liu, Y.; Jiang, Y. Stable knockdown of protein kinase CK2-α (CK2α) inhibits migration and invasion and induces inactivation of hedgehog signaling pathway in hepatocellular carcinoma Hep G2 cells. Acta Histochem. 2014, 116, 1501–1508. [Google Scholar] [CrossRef] [PubMed]
- Sass, G.; Klinger, N.; Sirma, H.; Hashemolhosseini, S.; Hellerbrand, C.; Neureiter, D.; Wege, H.; Ocker, M.; Tiegs, G. Inhibition of experimental HCC growth in mice by use of the kinase inhibitor DMAT. Int. J. Oncol. 2011, 39, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Potvin, B.; Huang, T.; Hilgard, P.; Spray, D.C.; Suadicani, S.O.; Wolkoff, A.W.; Stanley, P.; Stockert, R.J. A novel casein kinase 2 α-subunit regulates membrane protein traffic in the human hepatoma cell line HuH-7. J. Biol. Chem. 2001, 276, 2075–2082. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.R.; Kim, K.; Lee, K.H.; Kim, S.J.; Kim, J. Inhibition of casein kinase 2 enhances the death ligand- and natural kiler cell-induced hepatocellular carcinoma cell death. Clin. Exp. Immunol. 2008, 152, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.S.; Park, S.H.; Kim, K.M.; Kwon, K.S.; Kim, C.Y.; Lee, H.K.; Park, B.H.; Park, H.S.; Lee, H.; Moon, W.S.; et al. CK2α phosphorylates DBC1 and is involved in the progression of gastric carcinoma and predicts poor survival of gastric carcinoma patients. J. Int. Cancer 2015, 136, 797–809. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.Y.; Fang, C.L.; Chen, Y.; Li, C.F.; Chen, S.H.; Kuo, C.Y.; Tai, C.; Uen, Y.H. Overexpression of nuclear protein kinase CK2 Beta subunit and prognosis in human gastric carcinoma. Ann. Surg. Oncol. 2010, 17, 1695–1702. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Lee do, Y.; Yu da, Y.; Kim, S.; Lee, Y.C. Helicobacter pylori induces cell migration and invasion through casein kinase 2 in gastric epithelial cells. Helicobacter 2014, 19, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.H.; Lin, K.Y.; Chang, C.C.; Fang, C.L.; Lin, C.P. Aloe-emodin-induced apoptosis in human gastric carcinoma cells. Food Chem. Toxicol. 2007, 45, 2296–2303. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Chen, Q.; Wang, Q.; Sun, Y.; Wang, S.; Li, A.; Xu, S.; Roe, O.D.; Wang, M.; Zhang, R.; et al. JWA reverses cisplatin resistance via the CK2-XRCC1 pathway in human gastric cancer cells. Cell Death Dis. 2014, 5, e1551. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.Y.; Choi, H.K.; Choi, K.C.; Park, S.Y.; Ota, I.; Yook, J.I.; Lee, Y.H.; Kim, K.; Yoon, H.G. Nuclear hormone receptor corepressor promotes esophageal cancer cell invasion by transcriptional repression of interferon-gamma-inducible protein 10 in a casein kinase 2-dependent manner. Mol. Biol. Cell. 2012, 23, 2943–2954. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Lee, Y.; Kim, W.; Ko, H.; Choi, H.; Kim, K. Caspase-2 primes cancer cells for TRAIL-mediated apoptosis by processing procaspase-8. EMBO J. 2005, 24, 3532–3542. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.; Kim, S.; Jin, C.H.; Lee, E.; Ham, S.; Yook, J.I.; Kim, K. Protein kinase casein kinase 2-mediated upregulation of N-cadherin confers anoikis resistance on esophageal carcinoma cells. Mol. Cancer Res. 2012, 10, 1032–1038. [Google Scholar] [CrossRef] [PubMed]
- Juan, H.C.; Tsai, H.T.; Chang, P.H.; Huang, C.Y.; Hu, C.P.; Wong, F.H. Insulin-like growth factor 1 mediates 5-fluorouracil chemoresistance in esophageal carcinoma cells through increasing survivin stability. Apoptosis 2011, 16, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Xu, J.; Ding, G.; Cao, L. Overexpressions of CK2β and XIAP are associated with poor prognosis of patients with cholangiocarcinoma. Pathol. Oncol. Res. 2014, 20, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Kotawong, K.; Thitapakorn, V.; Roytrakul, S.; Phaonakrop, N.; Viyanant, V.; Na-Bangchang, K. Plasma peptidome as a source of biomarkers for diagnosis of cholangiocarcinoma. Asian Pac. J. Cancer Prev. 2016, 17, 1163–1168. [Google Scholar] [CrossRef] [PubMed]
- Izeradjene, K.; Douglas, L.; Delaney, A.; Houghton, J.A. Influence of casein kinase II in tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in human rhabdomyosarcoma cells. Clin. Cancer Res. 2004, 10, 6650–6660. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Luo, H.; Zeng, Q.; Dong, Z.; Wu, D.; Liu, L. Protein kinase CK2α is overexpressed in colorectal cancer and modulates cell proliferation and invasion via regulating EMT-related genes. J. Transl. Med. 2011, 9, 97. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.Y.; Tai, C.; Hsu, J.C.; Li, C.F.; Fang, C.L.; Lai, H.C.; Hseu, Y.C.; Lin, Y.F.; Uen, Y.H. Overexpression of nuclear protein kinase CK2 α catalytic subunit (CK2α) as a poor prognosticator in human colorectal cancer. PLoS ONE 2011, 6, e17193. [Google Scholar]
- Duncan, J.S.; Litchfield, D.W. Too much of a good thing: The role of protein kinase CK2 in tumorigenesis and prospects for therapeutic inhibition of CK2. Biochim. Biophys. Acta 2008, 1784, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Niechi, I.; Silva, E.; Cabello, P.; Huerta, H.; Carrasco, V.; Villar, P.; Cataldo, L.R.; Marcelain, K.; Armisen, R.; Varas-Godoy, M.; et al. Colon cancer cell invasion is promoted by protein kinase CK2 through increase of endothelin-converting enzyme-1c protein stability. Oncotarget 2015, 6, 42749–42760. [Google Scholar] [PubMed]
- Lambert, L.A.; Whyteside, A.R.; Turner, A.J.; Usmani, B.A. Isoforms of endothelin-converting enzyme-1 (ECE-1) have opposing effects on prostate cancer cell invasion. Br. J. Cancer 2008, 99, 1114–1120. [Google Scholar] [CrossRef] [PubMed]
- Smollich, M.; Gotte, M.; Kersting, C.; Fischgrabe, J.; Kiesel, L.; Wulfing, P. Selective ETAR antagonist atrasentan inhibits hypoxia-induced breast cancer cell invasion. Breast Cancer Res. Treat. 2008, 108, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Smollich, M.; Gotte, M.; Yip, G.W.; Yong, E.S.; Kersting, C.; Fischgrabe, J.; Radke, I.; Kiesel, L.; Wulfing, P. On the role of endothelin-converting enzyme-1 (ECE-1) and neprilysin in human breast cancer. Breast Cancer Res. Treat. 2007, 106, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Rayhman, O.; Klipper, E.; Muller, L.; Davidson, B.; Reich, R.; Meidan, R. Small interfering RNA molecules targeting endothelin-converting enzyme-1 inhibit endothelin-1 synthesis and the invasive phenotype of ovarian carcinoma cells. Cancer Res. 2008, 68, 9265–9273. [Google Scholar] [CrossRef] [PubMed]
- Izeradjene, K.; Douglas, L.; Delaney, A.; Houghton, J.A. Casein kinase II (CK2) enhances death-inducing signaling complex (DISC) activity in TRAIL-induced apoptosis in human colon carcinoma cell lines. Oncogene 2005, 24, 2050–2058. [Google Scholar] [CrossRef] [PubMed]
- Hamacher, R.; Saur, D.; Fritsch, R.; Reichert, M.; Schmid, R.M.; Schneider, G. Casein kinase II inhibition induces apoptosis in pancreatic cancer cells. Oncol. Rep. 2007, 18, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Guerra, B.; Hochscherf, J.; Jensen, N.B.; Issinger, O.G. Identification of a novel potent, selective and cell permeable inhibitor of protein kinase CK2 from the NIH/NCI diversity set library. Mol. Cell. Biochem. 2015, 406, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Giroux, V.; Iovanna, J.L.; Garcia, S.; Dagorn, J.C. Combined inhibition of PAK7, MAP3K7 and CK2α kinases inhibits the growth of MiaPaCa2 pancreatic cancer cell xenografts. Cancer Gene Ther. 2009, 16, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Kreutzer, J.N.; Ruzzene, M.; Guerra, B. Enhancing chemosensitivity to gemcitabine via RNA interference targeting the catalytic subunits of protein kinase CK2 in human pancreatic cancer cells. BMC Cancer 2010, 10, 440. [Google Scholar] [CrossRef] [PubMed]
- Tugizov, S.; Berline, J.; Herrera, R.; Penaranda, M.E.; Nakagawa, M.; Palefsky, J. Inhibition of human papillomavirus type 16 E7 phosphorylation by the s100 MRP-8/14 protein complex. J. Virol. 2005, 79, 1099–1112. [Google Scholar] [CrossRef] [PubMed]
- Massimi, P.; Banks, L. Differential phosphorylation of the HPV-16 E7 oncoprotein during the cell cycle. Virology 2000, 276, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Cao, X.C.; Xiao, Q.; Quan, M.F. Apigenin inhibits HeLa sphere-forming cells through inactivation of casein kinase 2α. Mol. Med. Rep. 2015, 11, 665–669. [Google Scholar] [PubMed]
- Solares, A.M.; Santana, A.; Baladron, I.; Valenzuela, C.; Gonzalez, C.A.; Diaz, A.; Castillo, D.; Ramos, T.; Gomez, R.; Alonso, D.F.; et al. Safety and preliminary efficacy data of a novel casein kinase 2 (CK2) peptide inhibitor administered intralesionally at four dose levels in patients with cervical malignancies. BMC Cancer 2009, 9, 146. [Google Scholar] [CrossRef] [PubMed]
- Sarduy, M.R.; Garcia, I.; Coca, M.A.; Perera, A.; Torres, L.A.; Valenzuela, C.M.; Baladron, I.; Solares, M.; Reyes, V.; Hernandez, I.; et al. Optimizing CIGB-300 intralesional delivery in locally advanced cervical cancer. Br. J. Cancer 2015, 112, 1636–1643. [Google Scholar] [CrossRef] [PubMed]
- Dixit, D.; Sharma, V.; Ghosh, S.; Mehta, V.S.; Sen, E. Inhibition of Casein kinase-2 induces p53-dependent cell cycle arrest and sensitizes glioblastoma cells to tumor necrosis factor (TNFα)-induced apoptosis through SIRT1 inhibition. Cell. Death Dis. 2012, 3, e271. [Google Scholar] [CrossRef] [PubMed]
- Nitta, R.T.; Gholamin, S.; Feroze, A.H.; Agarwal, M.; Cheshier, S.H.; Mitra, S.S.; Li, G. Casein kinase 2α regulates glioblastoma brain tumor-initiating cell growth through the β-catenin pathway. Oncogene 2015, 34, 3688–3699. [Google Scholar] [CrossRef] [PubMed]
- Mandal, T.; Bhowmik, A.; Chatterjee, A.; Chatterjee, U.; Chatterjee, S.; Ghosh, M.K. Reduced phosphorylation of Stat3 at Ser-727 mediated by casein kinase 2 - Protein phosphatase 2A enhances Stat3 Tyr-705 induced tumorigenic potential of glioma cells. Cell. Signal. 2014, 26, 1725–1734. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Font, L.; Alcaraz, E.; Plana, M.; Candiota, A.P.; Itarte, E.; Arus, C. Protein kinase CK2 content in GL261 mouse glioblastoma. Pathol. Oncol. Res. 2016, 22, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Kaminska, B.; Ellert-Miklaszewska, A.; Oberbek, A.; Wisniewski, P.; Kaza, B.; Makowska, M.; Bretner, M.; Kazimierczuk, Z. Efficacy and mechanism of anti-tumor action of new potential CK2 inhibitors toward glioblastoma cells. Int. J. Oncol. 2009, 35, 1091–1100. [Google Scholar] [CrossRef] [PubMed]
- Prudent, R.; Moucadel, V.; Nguyen, C.H.; Barette, C.; Schmidt, F.; Florent, J.C.; Lafanechere, L.; Sautel, C.F.; Duchemin-Pelletier, E.; Spreux, E.; et al. Antitumor activity of pyridocarbazole and benzopyridoindole derivatives that inhibit protein kinase CK2. Cancer Res. 2010, 70, 9865–9874. [Google Scholar] [CrossRef] [PubMed]
- Moucadel, V.; Prudent, R.; Sautel, C.F.; Teillet, F.; Barette, C.; Lafanechere, L.; Receveur-Brechot, V.; Cochet, C. Antitumoral activity of allosteric inhibitors of protein kinase CK2. Oncotarget 2011, 2, 997–1010. [Google Scholar] [CrossRef] [PubMed]
- Olsen, B.B.; Svenstrup, T.H.; Guerra, B. Downregulation of protein kinase CK2 induces autophagic cell death through modulation of the mTOR and MAPK signaling pathways in human glioblastoma cells. Int. J. Oncol. 2012, 41, 1967–1976. [Google Scholar] [PubMed]
- Joughin, B.A.; Naegle, K.M.; Huang, P.H.; Yaffe, M.B.; Lauffenburger, D.A.; White, F.M. An integrated comparative phosphoproteomic and bioinformatic approach reveals a novel class of MPM-2 motifs upregulated in EGFRvIII-expressing glioblastoma cells. Mol. Biosyst. 2009, 5, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.T.; Patil, R.; Galstyan, A.; Gangalum, P.R.; Cavenee, W.K.; Furnari, F.B.; Ljubimov, V.A.; Chesnokova, A.; Kramerov, A.A.; Ding, H.; et al. Simultaneous blockade of interacting CK2 and EGFR pathways by tumor-targeting nanobioconjugates increases therapeutic efficacy against glioblastoma multiforme. J. Control. Release 2016, 244, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Ritt, D.A.; Morrison, D.K.; Der, C.J.; Cox, A.D. Protein kinase CK2α maintains extracellular signal-regulated kinase (ERK) activity in a CK2α kinase-independent manner to promote resistance to inhibitors of RAF and MEK but not ERK in BRAF mutant melanoma. J. Biol. Chem. 2016, 291, 17804–17815. [Google Scholar] [CrossRef] [PubMed]
- Mitev, V.; Miteva, L.; Botev, I.; Houdebine, L.M. Enhanced Casein kinase II activity in metastatic melanoma. J. Dermatol. Sci. 1994, 8, 45–49. [Google Scholar] [CrossRef]
- Cheng, X.; Merz, K.H.; Vatter, S.; Christ, J.; Wolfl, S.; Eisenbrand, G. 7,7′-diazaindirubin—A small molecule inhibitor of Casein kinase 2 in vitro and in cells. Bioorg. Med. Chem. 2014, 22, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Parker, R.; Clifton-Bligh, R.; Molloy, M.P. Phosphoproteomics of mapk inhibition in braf-mutated cells and a role for the lethal synergism of dual braf and ck2 inhibition. Mol Cancer Ther 2014, 13, 1894–1906. [Google Scholar] [CrossRef] [PubMed]
- Pathak, H.B.; Zhou, Y.; Sethi, G.; Hirst, J.; Schilder, R.J.; Golemis, E.A.; Godwin, A.K. A synthetic lethality screen using a focused siRNA library to identify sensitizers to dasatinib therapy for the treatment of epithelial ovarian cancer. PLoS ONE 2015, 10, e0144126. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.S.; Kim, S.O.; Leung, P.C.; Auersperg, N.; Pelech, S.L. Profiling of protein kinases in the neoplastic transformation of human ovarian surface epithelium. Gynecol. Oncol. 2001, 82, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Tang, A.Q.; Cao, X.C.; Tian, L.; He, L.; Liu, F. Apigenin inhibits the self-renewal capacity of human ovarian cancer SKOV3derived sphere-forming cells. Mol. Med. Rep. 2015, 11, 2221–2226. [Google Scholar] [PubMed]
- Siddiqui-Jain, A.; Bliesath, J.; Macalino, D.; Omori, M.; Huser, N.; Streiner, N.; Ho, C.B.; Anderes, K.; Proffitt, C.; O’Brien, S.E.; et al. CK2 inhibitor CX-4945 suppresses DNA repair response triggered by DNA-targeted anticancer drugs and augments efficacy: Mechanistic rationale for drug combination therapy. Mol. Cancer Ther. 2012, 11, 994–1005. [Google Scholar] [CrossRef] [PubMed]
- Kulbe, H.; Iorio, F.; Chakravarty, P.; Milagre, C.S.; Moore, R.; Thompson, R.G.; Everitt, G.; Canosa, M.; Montoya, A.; Drygin, D.; et al. Integrated transcriptomic and proteomic analysis identifies protein kinase CK2 as a key signaling node in an inflammatory cytokine network in ovarian cancer cells. Oncotarget 2016, 7, 15648–15661. [Google Scholar] [CrossRef] [PubMed]
- Qaiser, F.; Trembley, J.H.; Sadiq, S.; Muhammad, I.; Younis, R.; Hashmi, S.N.; Murtaza, B.; Rector, T.S.; Naveed, A.K.; Ahmed, K. Examination of CK2α and NF-kappaB p65 expression in human benign prostatic hyperplasia and prostate cancer tissues. Mol. Cell. Biochem. 2016, 420, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Laramas, M.; Pasquier, D.; Filhol, O.; Ringeisen, F.; Descotes, J.L.; Cochet, C. Nuclear localization of protein kinase CK2 catalytic subunit (CK2α) is associated with poor prognostic factors in human prostate cancer. Eur. J. Cancer 2007, 43, 928–934. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.; Kren, B.T.; Abedin, M.J.; Vogel, R.I.; Shaughnessy, D.P.; Nacusi, L.; Korman, V.L.; Li, Y.; Dehm, S.M.; Zimmerman, C.L.; et al. CK2 targeted RNAi therapeutic delivered via malignant cell-directed tenfibgen nanocapsule: Dose and molecular mechanisms of response in xenograft prostate tumors. Oncotarget 2016, 7, 61789–61805. [Google Scholar] [CrossRef] [PubMed]
- Yenice, S.; Davis, A.T.; Goueli, S.A.; Akdas, A.; Limas, C.; Ahmed, K. Nuclear casein kinase 2 (CK-2) activity in human normal, benign hyperplastic, and cancerous prostate. Prostate 1994, 24, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.Y.; Lim, B.J.; Choi, H.K.; Hong, S.W.; Jang, H.S.; Kim, C.; Chun, K.H.; Choi, K.C.; Yoon, H.G. CK2-NcoR signaling cascade promotes prostate tumorigenesis. Oncotarget 2013, 4, 972–983. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.C.; Hessenauer, A.; Gotz, C.; Montenarh, M. DMAT, an inhibitor of protein kinase CK2 induces reactive oxygen species and DNA double strand breaks. Oncol. Rep. 2009, 21, 1593–1597. [Google Scholar] [PubMed]
- Trembley, J.H.; Unger, G.M.; Gomez, O.C.; Abedin, J.; Korman, V.L.; Vogel, R.I.; Niehans, G.; Kren, B.T.; Ahmed, K. Tenfibgen-DMAT nanocapsule delivers CK2 inhibitor DMAT to prostate cancer xenograft tumors causing inhibition of cell proliferation. Mol. Cell. Pharmacol. 2014, 6, 15–25. [Google Scholar] [PubMed]
- Trembley, J.H.; Unger, G.M.; Korman, V.L.; Tobolt, D.K.; Kazimierczuk, Z.; Pinna, L.A.; Kren, B.T.; Ahmed, K. Nanoencapsulated anti-CK2 small molecule drug or siRNA specifically targets malignant cancer but not benign cells. Cancer Lett. 2012, 315, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Unger, G.; Ahmad, K.A.; Slaton, J.W.; Ahmed, K. Downregulation of CK2 induces apoptosis in cancer cells—A potential approach to cancer therapy. Mol. Cell. Biochem. 2005, 274, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Yao, K.; Youn, H.; Gao, X.; Huang, B.; Zhou, F.; Li, B.; Han, H. Casein kinase 2 inhibition attenuates androgen receptor function and cell proliferation in prostate cancer cells. Prostate 2012, 72, 1423–1430. [Google Scholar] [CrossRef] [PubMed]
- Hessenauer, A.; Schneider, C.C.; Gotz, C.; Montenarh, M. CK2 inhibition induces apoptosis via the ER stress response. Cell. Signal. 2011, 23, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Pierre, F.; Chua, P.C.; O’Brien, S.E.; Siddiqui-Jain, A.; Bourbon, P.; Haddach, M.; Michaux, J.; Nagasawa, J.; Schwaebe, M.K.; Stefan, E.; et al. Pre-clinical characterization of CX-4945, a potent and selective small molecule inhibitor of CK2 for the treatment of cancer. Mol. Cell. Biochem. 2011, 356, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Qaiser, F.; Trembley, J.H.; Kren, B.T.; Wu, J.J.; Naveed, A.K.; Ahmed, K. Protein kinase CK2 inhibition induces cell death via early impact on mitochondrial function. J. Cell. Biochem. 2014, 115, 2103–2115. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Ahmad, K.A.; Harris, N.H.; Ahmed, K. Impact of protein kinase CK2 on inhibitor of apoptosis proteins in prostate cancer cells. Mol. Cell. Biochem. 2008, 316, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Orzechowska, E.; Kozlowska, E.; Staron, K.; Trzcinska-Danielewicz, J. Time schedule-dependent effect of the CK2 inhibitor TBB on PC-3 human prostate cancer cell viability. Oncol. Rep. 2012, 27, 281–285. [Google Scholar] [PubMed]
- Wang, G.; Ahmad, K.A.; Ahmed, K. Role of protein kinase CK2 in the regulation of tumor necrosis factor-related apoptosis inducing ligand-induced apoptosis in prostate cancer cells. Cancer Res. 2006, 66, 2242–2249. [Google Scholar] [CrossRef] [PubMed]
- Gray, G.K.; McFarland, B.C.; Rowse, A.L.; Gibson, S.A.; Benveniste, E.N. Therapeutic CK2 inhibition attenuates diverse prosurvival signaling cascades and decreases cell viability in human breast cancer cells. Oncotarget 2014, 5, 6484–6496. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.S.; Park, S.H.; Jamiyandorj, U.; Kim, K.M.; Noh, S.J.; Kim, J.R.; Park, H.J.; Kwon, K.S.; Jung, S.H.; Park, H.S.; et al. CK2α/CSNK2A1 phosphorylates SIRT6 and is involved in the progression of breast carcinoma and predicts shorter survival of diagnosed patients. Am. J. Pathol. 2016, 186, 3297–3315. [Google Scholar] [CrossRef] [PubMed]
- Kren, B.T.; Unger, G.M.; Abedin, M.J.; Vogel, R.I.; Henzler, C.M.; Ahmed, K.; Trembley, J.H. Preclinical evaluation of cyclin dependent kinase 11 and casein kinase 2 survival kinases as RNA interference targets for triple negative breast cancer therapy. Breast Cancer Res. 2015, 17, 19. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.D.; Nguyen, T.; Carriere, P.P.; Tilghman, S.L.; Williams, C. Protein kinase CK2 expression predicts relapse survival in ERα dependent breast cancer, and modulates erα expression in vitro. Int. J. Environ. Res. Public Health 2015, 13. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Wang, X.; Chen, G.Y.; Dalerba, P.; Gurney, A.; Hoey, T.; Sherlock, G.; Lewicki, J.; Shedden, K.; Clarke, M.F. The prognostic role of a gene signature from tumorigenic breast-cancer cells. N. Engl. J. Med. 2007, 356, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Landesman-Bollag, E.; Romieu-Mourez, R.; Song, D.H.; Sonenshein, G.E.; Cardiff, R.D.; Seldin, D.C. Protein kinase CK2 in mammary gland tumorigenesis. Oncogene 2001, 20, 3247–3257. [Google Scholar] [CrossRef] [PubMed]
- Feliciano, A.; Castellvi, J.; Artero-Castro, A.; Leal, J.A.; Romagosa, C.; Hernandez-Losa, J.; Peg, V.; Fabra, A.; Vidal, F.; Kondoh, H.; et al. miR-125b acts as a tumor suppressor in breast tumorigenesis via its novel direct targets ENPEP, CK2-α, CCNJ, and MEGF9. PLoS ONE 2013, 8, e76247. [Google Scholar] [CrossRef] [PubMed]
- Munstermann, U.; Fritz, G.; Seitz, G.; Lu, Y.P.; Schneider, H.R.; Issinger, O.G. Casein kinase II is elevated in solid human tumours and rapidly proliferating non-neoplastic tissue. Eur. J. Biochem. 1990, 189, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Das, N.; Datta, N.; Chatterjee, U.; Ghosh, M.K. Estrogen receptor α transcriptionally activates casein kinase 2 α: A pivotal regulator of promyelocytic leukaemia protein (PML) and AKT in oncogenesis. Cell Signal. 2016, 28, 675–687. [Google Scholar] [CrossRef] [PubMed]
- Currier, N.; Solomon, S.E.; Demicco, E.G.; Chang, D.L.; Farago, M.; Ying, H.; Dominguez, I.; Sonenshein, G.E.; Cardiff, R.D.; Xiao, Z.X.; et al. Oncogenic signaling pathways activated in DMBA-induced mouse mammary tumors. Toxicol. Pathol. 2005, 33, 726–737. [Google Scholar] [CrossRef] [PubMed]
- Ford, H.L.; Landesman-Bollag, E.; Dacwag, C.S.; Stukenberg, P.T.; Pardee, A.B.; Seldin, D.C. Cell cycle-regulated phosphorylation of the human SIX1 homeodomain protein. J. Biol. Chem. 2000, 275, 22245–22254. [Google Scholar] [CrossRef] [PubMed]
- Tapia, J.C.; Torres, V.A.; Rodriguez, D.A.; Leyton, L.; Quest, A.F. Casein kinase 2 (CK2) increases survivin expression via enhanced β-catenin-T cell factor/lymphoid enhancer binding factor-dependent transcription. Proc. Natl. Acad. Sci. USA 2006, 103, 15079–15084. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Kang, B.S.; Bae, Y.S. Premature senescence in human breast cancer and colon cancer cells by tamoxifen-mediated reactive oxygen species generation. Life Sci. 2014, 97, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.F.; Guo, S.; Demicco, E.G.; Romieu-Mourez, R.; Landesman-Bollag, E.; Seldin, D.C.; Sonenshein, G.E. Inducible IkappaB kinase/IkappaB kinase epsilon expression is induced by CK2 and promotes aberrant nuclear factor-kappaB activation in breast cancer cells. Cancer Res. 2005, 65, 11375–11383. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Chen, L.; Hu, Z.; Li, H.; Li, J.; Wei, C.; Huang, Y.; Song, H.; Fang, L. Alterations of microRNAs are associated with impaired growth of MCF-7 breast cancer cells induced by inhibition of casein kinase 2. Int. J. Clin. Exp. Pathol. 2014, 7, 4008–4015. [Google Scholar] [PubMed]
- Hagan, C.R.; Regan, T.M.; Dressing, G.E.; Lange, C.A. CK2-dependent phosphorylation of progesterone receptors (PR) on Ser81 regulates PR-B isoform-specific target gene expression in breast cancer cells. Mol. Cell. Biol. 2011, 31, 2439–2452. [Google Scholar] [CrossRef] [PubMed]
- McManaway, M.E.; Eckberg, W.R.; Anderson, W.A. Characterization and hormonal regulation of casein kinase II activity in heterotransplanted human breast tumors in nude mice. Exp. Clin. Endocrinol. 1987, 90, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Drygin, D.; Ho, C.B.; Omori, M.; Bliesath, J.; Proffitt, C.; Rice, R.; Siddiqui-Jain, A.; O’Brien, S.; Padgett, C.; Lim, J.K.; et al. Protein kinase CK2 modulates IL-6 expression in inflammatory breast cancer. Biochem. Biophys. Res. Commun. 2011, 415, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Yde, C.W.; Frogne, T.; Lykkesfeldt, A.E.; Fichtner, I.; Issinger, O.G.; Stenvang, J. Induction of cell death in antiestrogen resistant human breast cancer cells by the protein kinase CK2 inhibitor DMAT. Cancer Lett. 2007, 256, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, Y.; Takano, H.; Fojo, T. Cellular adaptation to drug exposure: Evolution of the drug-resistant phenotype. Cancer Res. 1997, 57, 5086–5092. [Google Scholar] [PubMed]
- Rabjerg, M.; Guerra, B.; Olivan-Viguera, A.; Nedergaard Mikkelsen, M.L.; Kohler, R.; Issinger, O.G.; Marcussen, N. Nuclear localization of the CK2α-subunit correlates with poor prognosis in clear cell renal cell carcinoma. Oncotarget 2016, 8, 1613–1627. [Google Scholar]
- Rabjerg, M.; Bjerregaard, H.; Halekoh, U.; Jensen, B.L.; Walter, S.; Marcussen, N. Molecular characterization of clear cell renal cell carcinoma identifies CSNK2A1, SPP1 and DEFB1 as promising novel prognostic markers. APMIS 2016, 124, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Caroline Roelants, S.G.; Duchemin-Pelletier, E.; McLeer-Florin, A.; Tisseyre, C.; Aubert, C.; Champelovier, P.; Boutonnat, J.; Descotes, J.L.; Rambeaud, J.-J.; Arnoux, V.; et al. Dysregulated expression of protein kinase ck2 in renal cancer. In Protein Kinase CK2 Cellular Function in Normal and Disease States; Khalil, O.-G.I., Ryszard Szyszka, A., Eds.; Springer: Cham, Switzerland, 2015; Volume 12, pp. 241–257. [Google Scholar]
- Stalter, G.; Siemer, S.; Becht, E.; Ziegler, M.; Remberger, K.; Issinger, O.G. Asymmetric expression of protein kinase CK2 subunits in human kidney tumors. Biochem. Biophys. Res. Commun. 1994, 202, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Ampofo, E.; Kietzmann, T.; Zimmer, A.; Jakupovic, M.; Montenarh, M.; Gotz, C. Phosphorylation of the von Hippel-Lindau protein (VHL) by protein kinase CK2 reduces its protein stability and affects p53 and HIF-1α mediated transcription. Int. J. Biochem. Cell. Biol. 2010, 42, 1729–1735. [Google Scholar] [CrossRef] [PubMed]
- German, P.; Bai, S.; Liu, X.D.; Sun, M.; Zhou, L.; Kalra, S.; Zhang, X.; Minelli, R.; Scott, K.L.; Mills, G.B.; et al. Phosphorylation-dependent cleavage regulates von Hippel Lindau proteostasis and function. Oncogene 2016, 35, 4973–4980. [Google Scholar] [CrossRef] [PubMed]
- Lolkema, M.P.; Gervais, M.L.; Snijckers, C.M.; Hill, R.P.; Giles, R.H.; Voest, E.E.; Ohh, M. Tumor suppression by the von Hippel-Lindau protein requires phosphorylation of the acidic domain. J. Biol. Chem. 2005, 280, 22205–22211. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.M.; Soares, M.V.; Ribeiro, P.; Caldas, J.; Povoa, V.; Martins, L.R.; Melao, A.; Serra-Caetano, A.; de Sousa, A.B.; Lacerda, J.F.; et al. Adult B-cell acute lymphoblastic leukemia cells display decreased PTEN activity and constitutive hyperactivation of PI3K/Akt pathway despite high PTEN protein levels. Haematologica 2014, 99, 1062–1068. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Gowda, C.; Pan, X.; Ding, Y.; Tong, Y.; Tan, B.H.; Wang, H.; Muthusami, S.; Ge, Z.; Sachdev, M.; et al. Targeting casein kinase II restores Ikaros tumor suppressor activity and demonstrates therapeutic efficacy in high-risk leukemia. Blood 2015, 126, 1813–1822. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.; Yunes, J.A.; Cardoso, B.A.; Martins, L.R.; Jotta, P.Y.; Abecasis, M.; Nowill, A.E.; Leslie, N.R.; Cardoso, A.A.; Barata, J.T. PTEN posttranslational inactivation and hyperactivation of the PI3K/Akt pathway sustain primary t cell leukemia viability. J. Clin. Investig. 2008, 118, 3762–3774. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Eom, J.I.; Cheong, J.-W.; Choi, A.J.; Lee, J.K.; Yang, W.I.; Min, Y.H. Protein kinase CK2α as an unfavorable prognostic marker and novel therapeutic target in acute myeloid leukemia. Clin. Cancer Res. 2007, 13, 1019–1028. [Google Scholar] [CrossRef] [PubMed]
- Quotti Tubi, L.; Gurrieri, C.; Brancalion, A.; Bonaldi, L.; Bertorelle, R.; Manni, S.; Piazza, F. Inhibition of protein kinase CK2 with the clinical-grade small ATP-competitive compound CX-4945 or by RNA interference unveils its role in acute myeloid leukemia cell survival, p53-dependent apoptosis and daunorubicin-induced cytotoxicity. J. Hematol. Oncol. 2013, 6, 78. [Google Scholar] [CrossRef] [PubMed]
- Martins, L.R.; Lucio, P.; Silva, M.C.; Anderes, K.L.; Gameiro, P.; Silva, M.G.; Barata, J.T. Targeting CK2 overexpression and hyperactivation as a novel therapeutic tool in chronic lymphocytic leukemia. Blood 2010, 116, 2724–2731. [Google Scholar] [CrossRef] [PubMed]
- Gowda, C.; Sachdev, M.; Muthisami, S.; Kapadia, M.; Petrovic-Dovat, L.; Hartman, M.; Ding, Y.; Song, C.; Payne, J.L.; Tan, B.H.; et al. Casein kinase II (CK2) as a therapeutic target for hematological malignancies. Curr. Pharm. Des. 2016, in press. [Google Scholar] [CrossRef]
- Buontempo, F.; Orsini, E.; Martins, L.R.; Antunes, I.; Lonetti, A.; Chiarini, F.; Tabellini, G.; Evangelisti, C.; Evangelisti, C.; Melchionda, F.; et al. Cytotoxic activity of the casein kinase 2 inhibitor CX-4945 against T-cell acute lymphoblastic leukemia: Targeting the unfolded protein response signaling. Leukemia 2014, 28, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Martins, L.R.; Lucio, P.; Melao, A.; Antunes, I.; Cardoso, B.A.; Stansfield, R.; Bertilaccio, M.T.; Ghia, P.; Drygin, D.; Silva, M.G.; et al. Activity of the clinical-stage CK2-specific inhibitor CX-4945 against chronic lymphocytic leukemia. Leukemia 2014, 28, 179–182. [Google Scholar] [CrossRef] [PubMed]
- Seldin, D.C. New models of lymphoma in transgenic mice. Curr. Opin. Immunol. 1995, 7, 665–673. [Google Scholar] [CrossRef]
- Seldin, D.C.; Leder, P. Casein kinase II α transgene-induced murine lymphoma: Relation to theileriosis in cattle. Science 1995, 267, 894–897. [Google Scholar] [CrossRef] [PubMed]
- Pizzi, M.; Piazza, F.; Agostinelli, C.; Fuligni, F.; Benvenuti, P.; Mandato, E.; Casellato, A.; Rugge, M.; Semenzato, G.; Pileri, S.A. Protein kinase CK2 is widely expressed in follicular, burkitt and diffuse large b-cell lymphomas and propels malignant B-cell growth. Oncotarget 2015, 6, 6544–6552. [Google Scholar] [CrossRef] [PubMed]
- Manni, S.; Brancalion, A.; Mandato, E.; Tubi, L.Q.; Colpo, A.; Pizzi, M.; Cappellesso, R.; Zaffino, F.; Di Maggio, S.A.; Cabrelle, A.; et al. Protein kinase CK2 inhibition down modulates the NF-kappaB and STAT3 survival pathways, enhances the cellular proteotoxic stress and synergistically boosts the cytotoxic effect of bortezomib on multiple myeloma and mantle cell lymphoma cells. PLoS ONE 2013, 8, e75280. [Google Scholar] [CrossRef] [PubMed]
- Piazza, F.A.; Ruzzene, M.; Gurrieri, C.; Montini, B.; Bonanni, L.; Chioetto, G.; Di Maira, G.; Barbon, F.; Cabrelle, A.; Zambello, R.; et al. Multiple myeloma cell survival relies on high activity of protein kinase CK2. Blood 2006, 108, 1698–1707. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Ma, J.; Zhu, H.Y.; Zhang, X.H.; Du, Z.Y.; Xu, Y.J.; Yu, X.D. Apigenin inhibits proliferation and induces apoptosis in human multiple myeloma cells through targeting the trinity of CK2, Cdc37 and Hsp90. Mol. Cancer 2011, 10, 104. [Google Scholar] [CrossRef] [PubMed]
- Piazza, F.; Manni, S.; Semenzato, G. Novel players in multiple myeloma pathogenesis: Role of protein kinases CK2 and GSK3. Leukemia Res. 2013, 37, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Manni, S.; Brancalion, A.; Tubi, L.Q.; Colpo, A.; Pavan, L.; Cabrelle, A.; Ave, E.; Zaffino, F.; Di Maira, G.; Ruzzene, M.; et al. Protein kinase CK2 protects multiple myeloma cells from ER stress-induced apoptosis and from the cytotoxic effect of HSP90 inhibition through regulation of the unfolded protein response. Clin. Cancer Res. 2012, 18, 1888–1900. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G.; Venerando, A.; Sarno, S.; Pinna, L.A. The selectivity of CK2 inhibitor quinalizarin: A reevaluation. Biomed. Res. Int. 2015, 2015, 734127. [Google Scholar] [CrossRef] [PubMed]
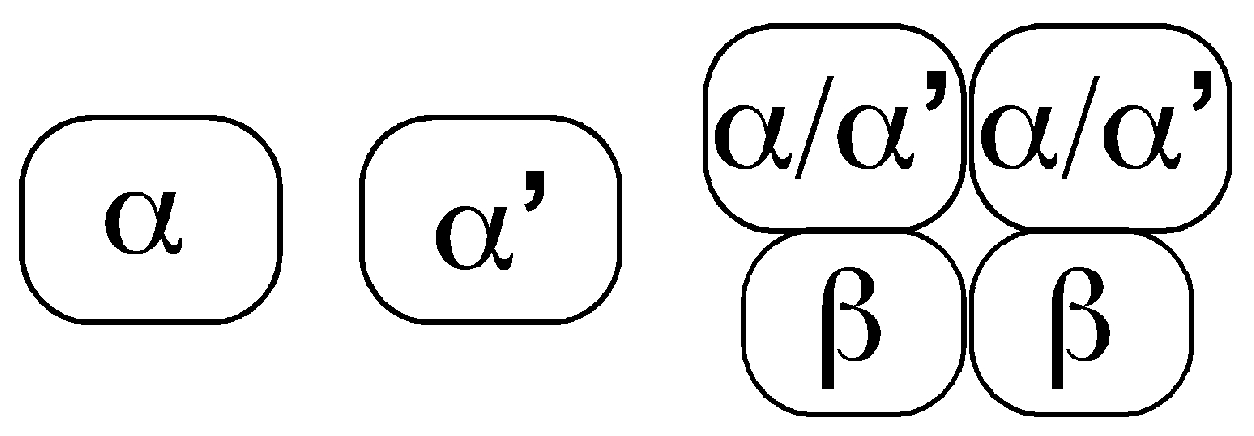
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chua, M.M.J.; Ortega, C.E.; Sheikh, A.; Lee, M.; Abdul-Rassoul, H.; Hartshorn, K.L.; Dominguez, I. CK2 in Cancer: Cellular and Biochemical Mechanisms and Potential Therapeutic Target. Pharmaceuticals 2017, 10, 18. https://doi.org/10.3390/ph10010018
Chua MMJ, Ortega CE, Sheikh A, Lee M, Abdul-Rassoul H, Hartshorn KL, Dominguez I. CK2 in Cancer: Cellular and Biochemical Mechanisms and Potential Therapeutic Target. Pharmaceuticals. 2017; 10(1):18. https://doi.org/10.3390/ph10010018
Chicago/Turabian StyleChua, Melissa M.J., Charina E. Ortega, Ayesha Sheikh, Migi Lee, Hussein Abdul-Rassoul, Kevan L. Hartshorn, and Isabel Dominguez. 2017. "CK2 in Cancer: Cellular and Biochemical Mechanisms and Potential Therapeutic Target" Pharmaceuticals 10, no. 1: 18. https://doi.org/10.3390/ph10010018
APA StyleChua, M. M. J., Ortega, C. E., Sheikh, A., Lee, M., Abdul-Rassoul, H., Hartshorn, K. L., & Dominguez, I. (2017). CK2 in Cancer: Cellular and Biochemical Mechanisms and Potential Therapeutic Target. Pharmaceuticals, 10(1), 18. https://doi.org/10.3390/ph10010018