
Dual-Signal Feature Spaces Map Protein Subcellular Locations Based on Immunohistochemistry Image and Protein Sequence

Abstract
1. Introduction
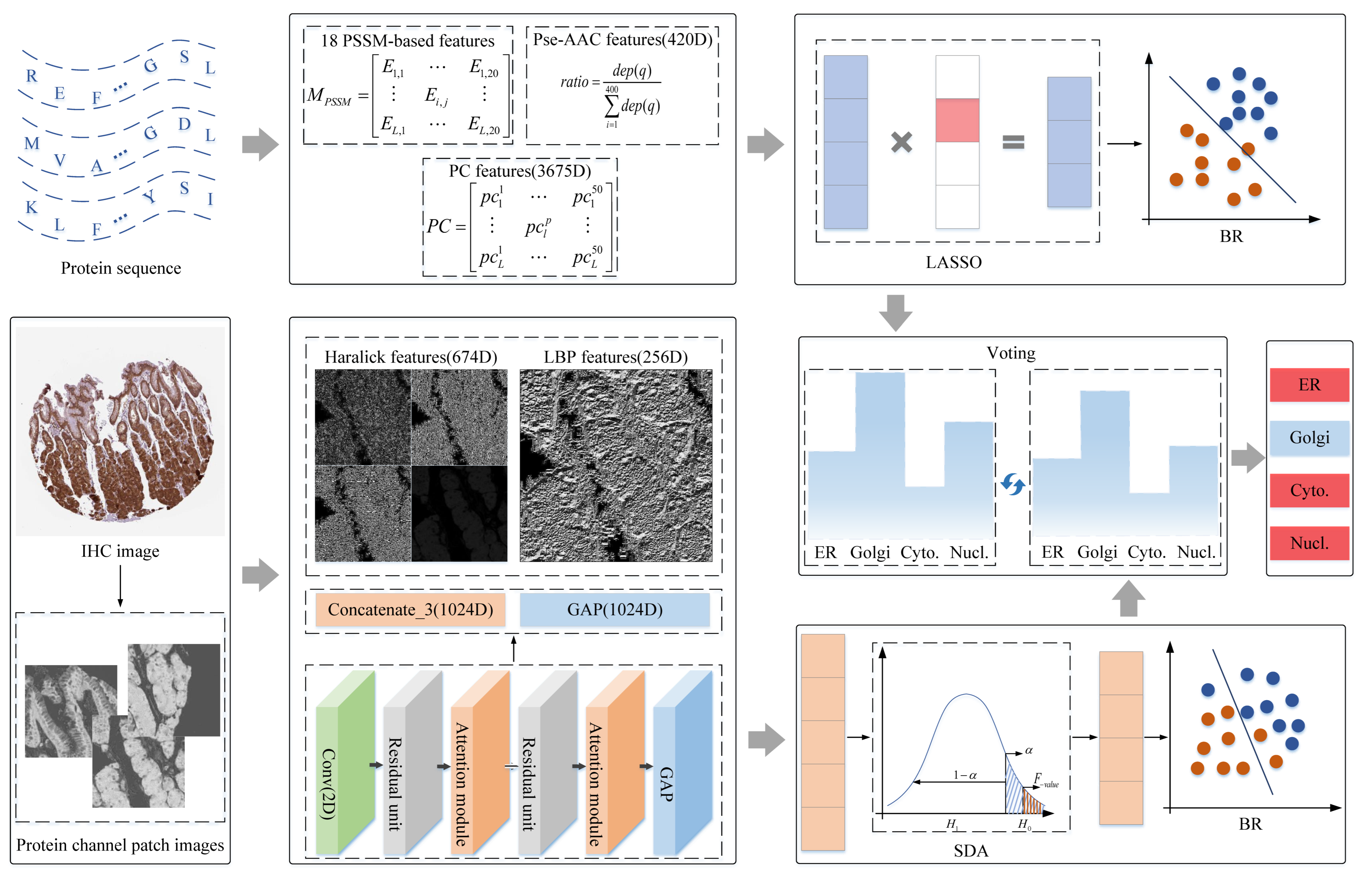
2. Materials and Methods
2.1. The Benchmark Dataset
2.2. Analyzing Subcellular Patterns of Protein from Protein Sequence
2.3. Classifying Protein Subcellular Location Based on Shallow and Abstract Features of IHC Images
2.3.1. Focusing on Protein-Target Regions by Preprocessing
2.3.2. Quantifying IHC Images with Shallow and Abstract Features
2.3.3. Improving Performance of Multiple Classifiers by Centralized Voting Mechanism
3. Results
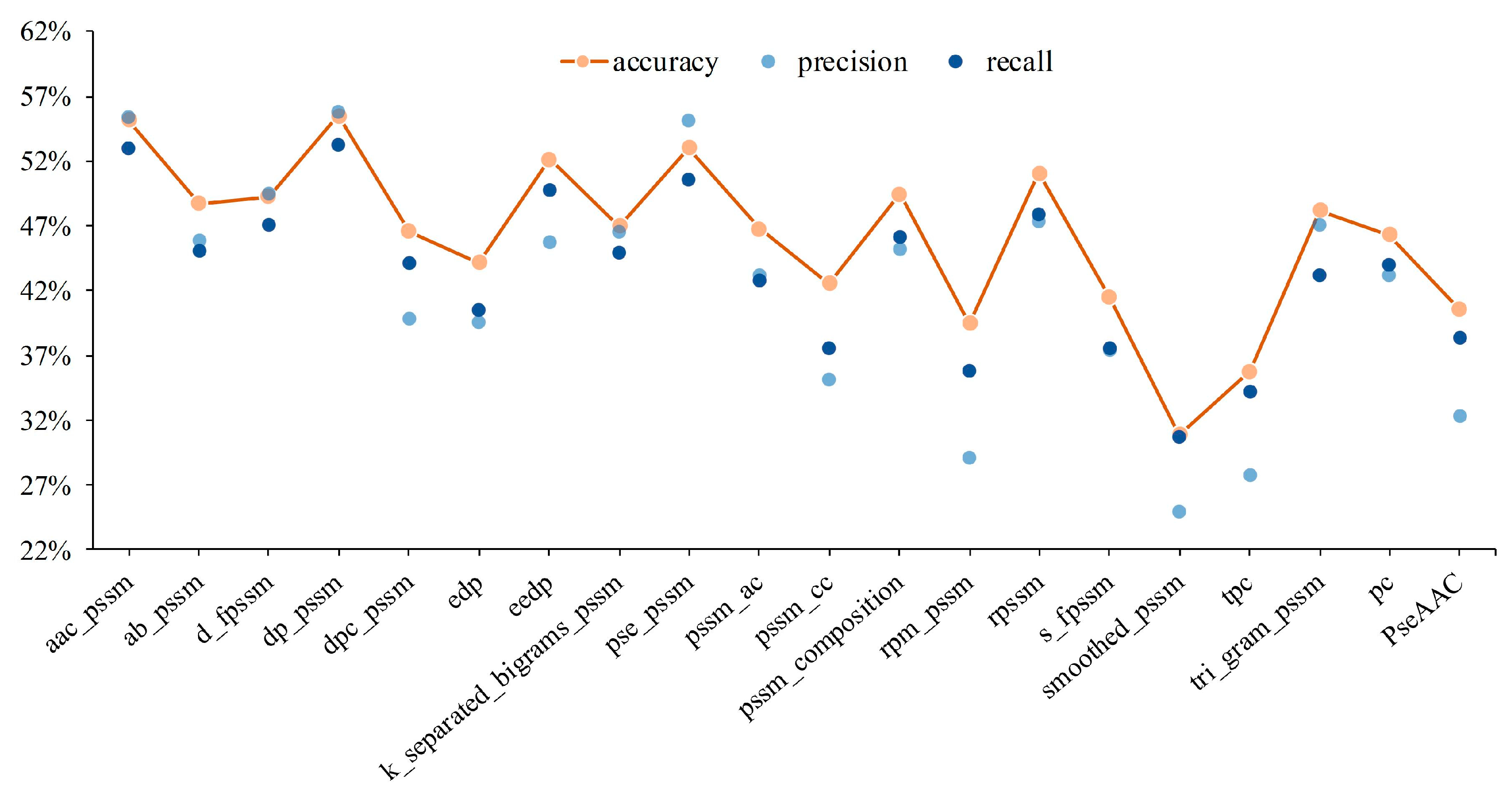
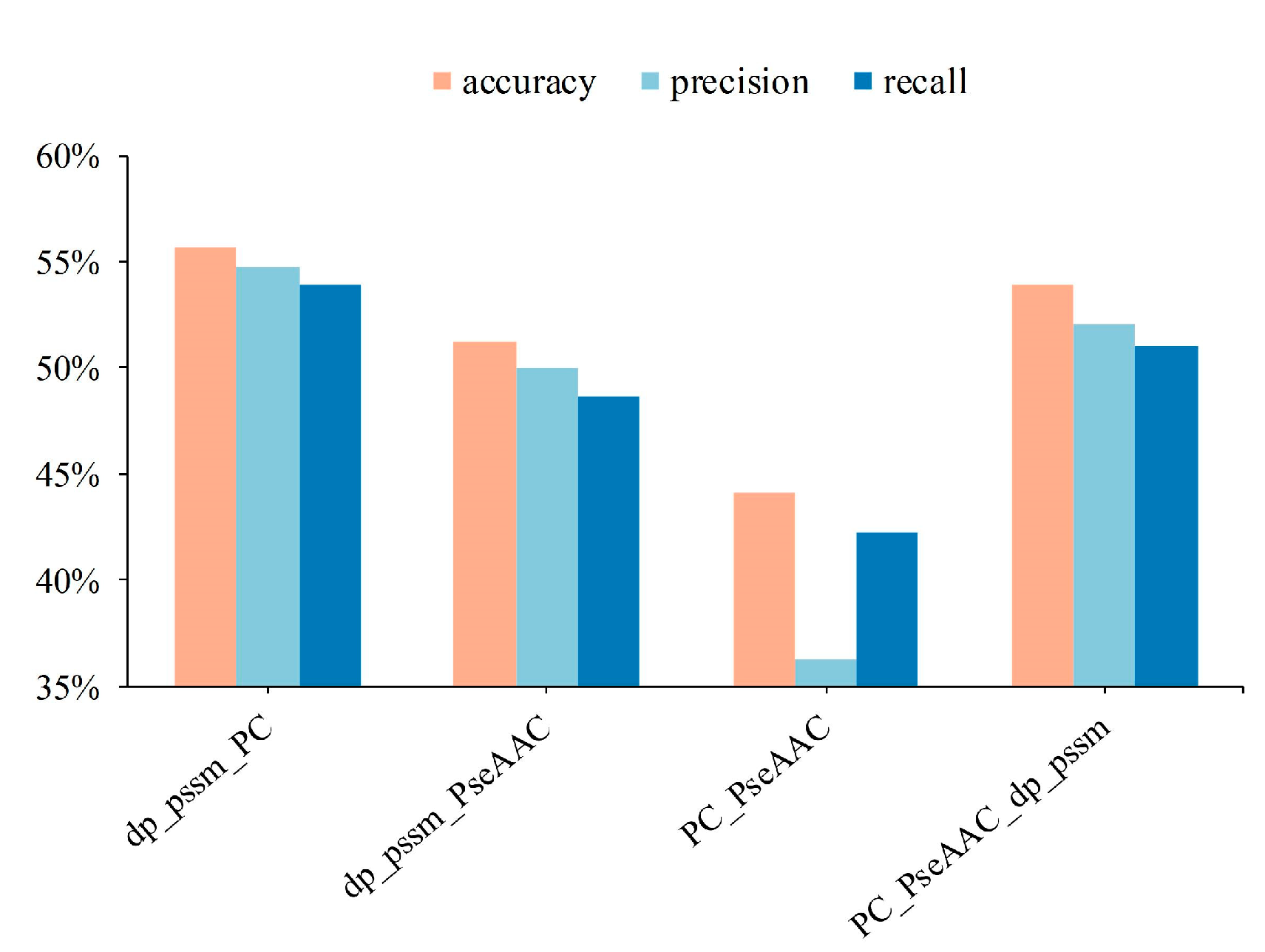
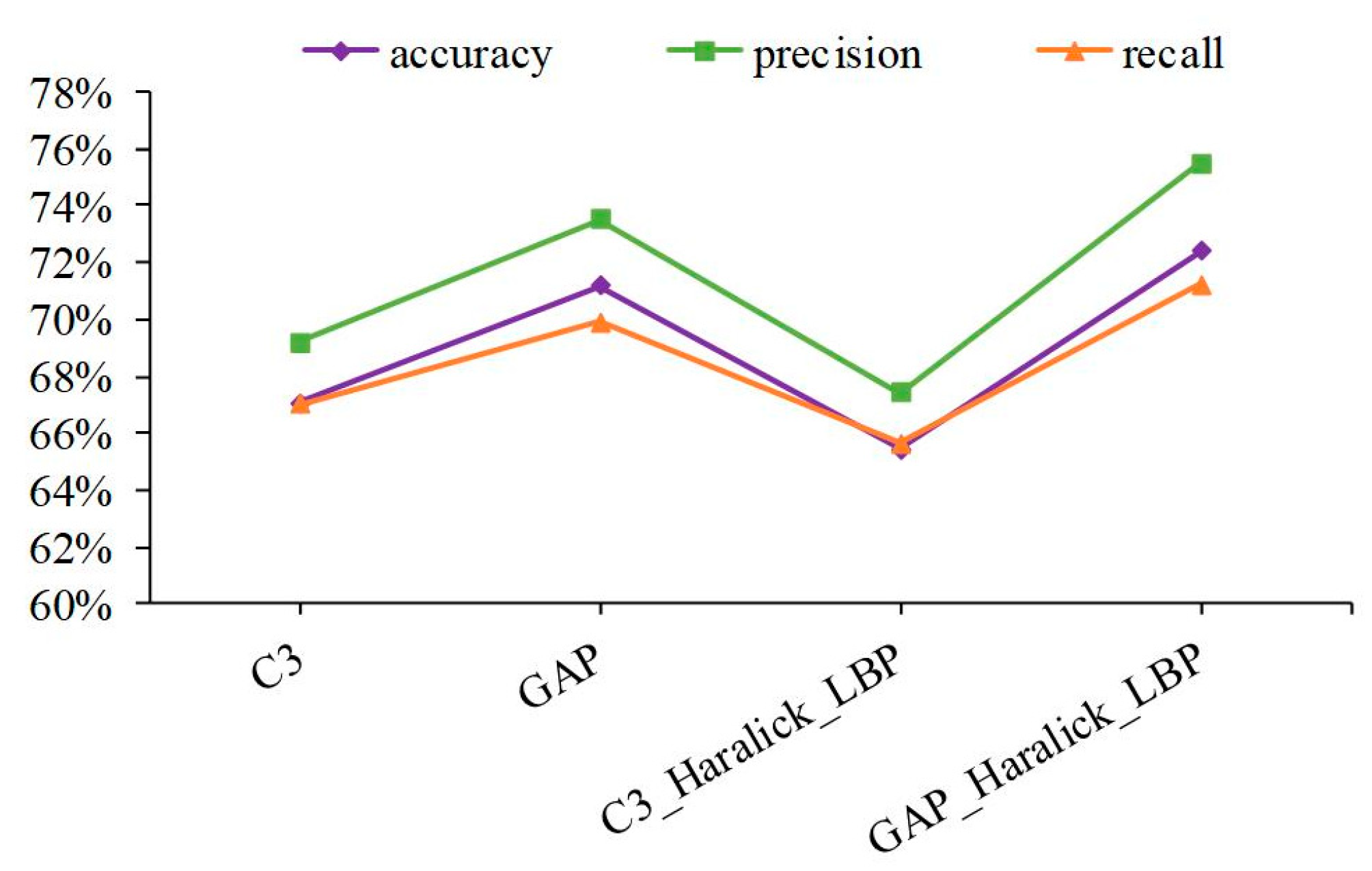
3.1. The Shallow Features of Amino Acid Effectively Act on Protein Subcellular Location
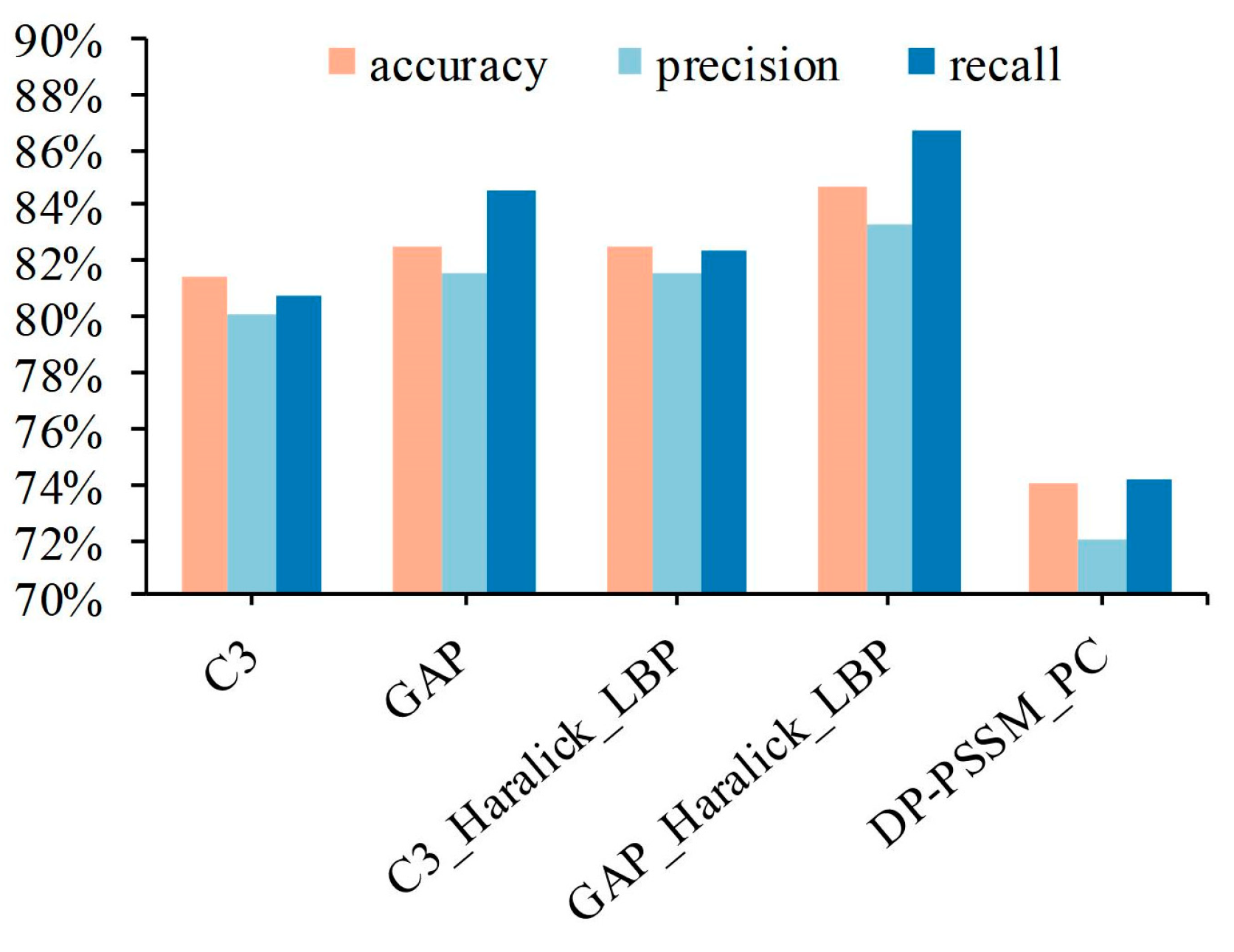
3.2. IHC Images Perform Better in Identifying Protein Subcellular Location Than Protein Sequence
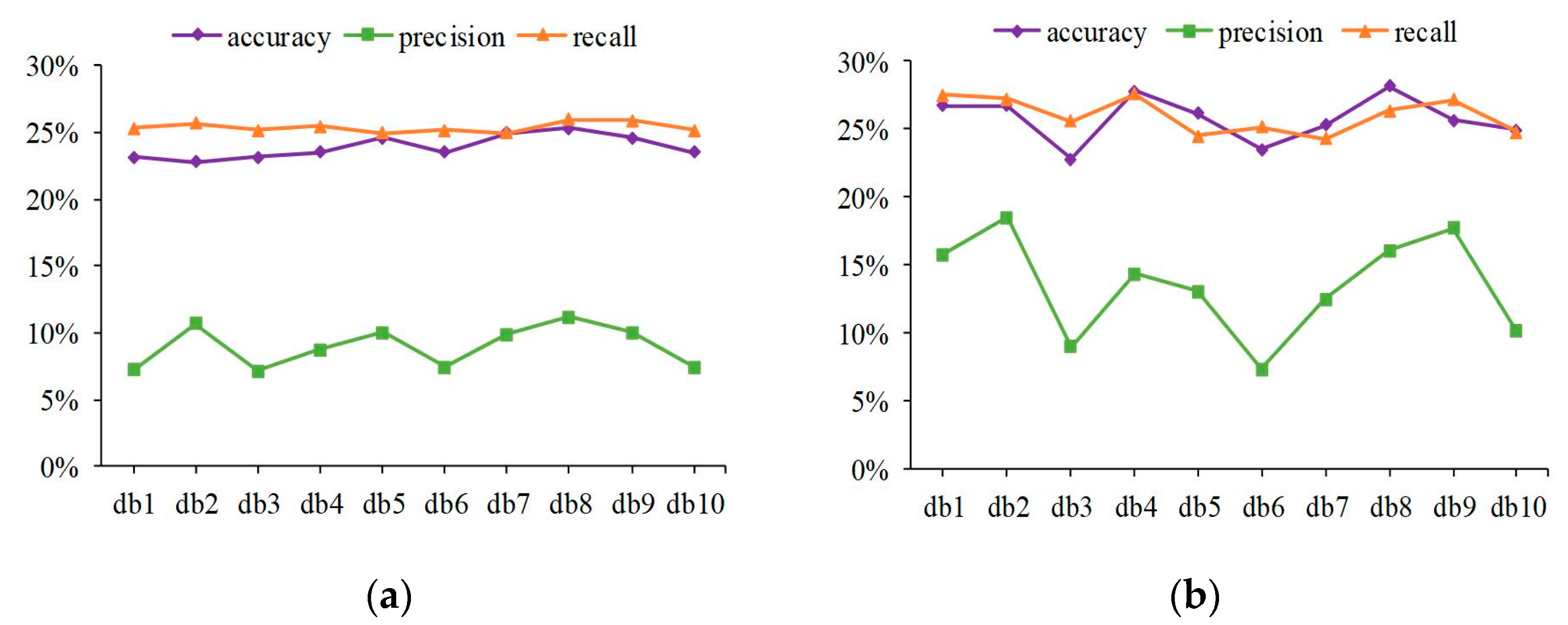
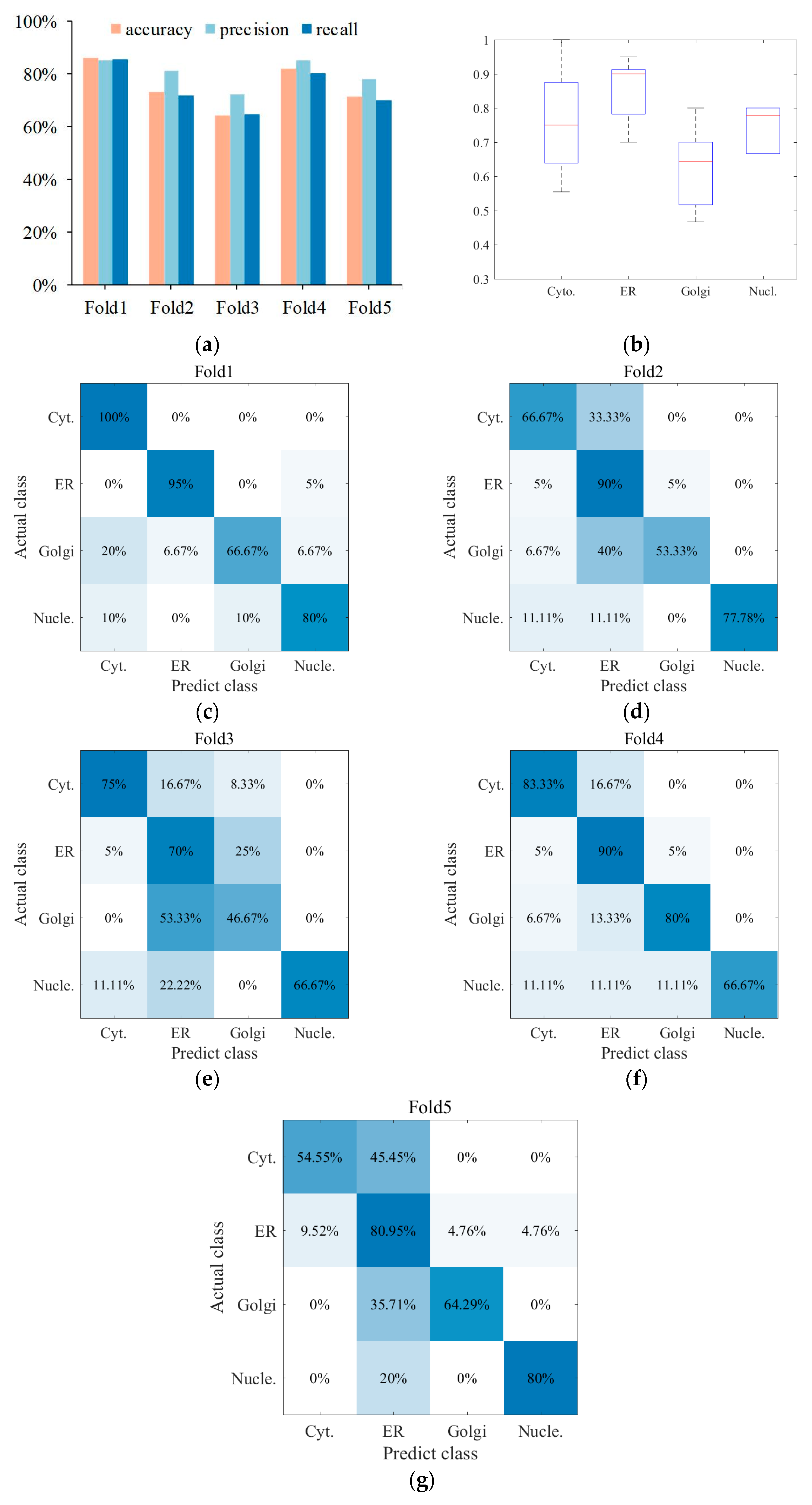
3.3. Superior Performance Obtained with the Centralized Voting Mechanism
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marx, V. Mapping proteins with spatial proteomics. Nat. Methods 2015, 12, 815–819. [Google Scholar] [CrossRef] [PubMed]
- Christopher, J.A.; Geladaki, A.; Dawson, C.S.; Vennard, O.L.; Lilley, K.S. Subcellular Transcriptomics and Proteomics: A Comparative Methods Review. Mol. Cell. Proteom. 2022, 21, 100186. [Google Scholar] [CrossRef] [PubMed]
- Berger, C.M.; Gaume, X.; Bouvet, P. The roles of nucleolin subcellular localization in cancer. Biochimie 2015, 113, 78–85. [Google Scholar] [CrossRef]
- Hung, M.-C.; Link, W. Protein localization in disease and therapy. J. Cell Sci. 2011, 124, 3381–3392. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Ding, Y.; Chou, K.-C. Prediction of protein subcellular location using hydrophobic patterns of amino acid sequence. Comput. Biol. Chem. 2006, 30, 367–371. [Google Scholar] [CrossRef]
- Xu, Y.; Shen, H. Review of Research on Biomedical Image Processing Based on Pattern Recognition. J. Electron. Inf. Technol. 2020, 42, 201–213. [Google Scholar] [CrossRef]
- Park, K.-J.; Kanehisa, M. Prediction of protein subcellular locations by support vector machines using compositions of amino acids and amino acid pairs. Bioinformatics 2003, 19, 1656–1663. [Google Scholar] [CrossRef]
- Jeong, J.C.; Lin, X.; Chen, X.-W. On Position-Specific Scoring Matrix for Protein Function Prediction. IEEE/ACM Trans. Comput. Biol. Bioinform. 2010, 8, 308–315. [Google Scholar] [CrossRef]
- Chou, K.-C. Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins Struct. Funct. Bioinform. 2001, 43, 246–255. [Google Scholar] [CrossRef]
- Savojardo, C.; Martelli, P.L.; Fariselli, P.; Casadio, R. TPpred3 detects and discriminates mitochondrial and chloroplastic targeting peptides in eukaryotic proteins. Bioinformatics 2015, 31, 3269–3275. [Google Scholar] [CrossRef]
- Chou, K.C.; Cai, Y.D. Using functional domain composition and support vector machines for prediction of protein subcellular lo cation. J. Biol. Chem. 2002, 277, 45765–45769. [Google Scholar] [CrossRef]
- Scott, M.S.; Thomas, D.Y.; Hallett, M.T. Predicting Subcellular Localization via Protein Motif Co-Occurrence. Genome Res. 2004, 14, 1957–1966. [Google Scholar] [CrossRef][Green Version]
- Chou, K.-C.; Cai, Y.-D. A new hybrid approach to predict subcellular localization of proteins by incorporating gene ontology. Biochem. Biophys. Res. Commun. 2003, 311, 743–747. [Google Scholar] [CrossRef] [PubMed]
- Savojardo, C.; Martelli, P.L.; Fariselli, P.; Profiti, G.; Casadio, R. BUSCA: An integrative web server to predict subcellular localization of proteins. Nucleic Acids Res. 2018, 46, W459–W466. [Google Scholar] [CrossRef]
- Shen, H.B.; Chou, K.C. A top-down approach to enhance the power of predicting human protein subcellular localization: Hum-mPLoc 2.0. Anal. Biochem. 2009, 394, 269–274. [Google Scholar] [CrossRef]
- Guo, X.; Liu, F.; Ju, Y.; Wang, Z.; Wang, C. Human Protein Subcellular Localization with Integrated Source and Multi-label Ensemble Classifier. Sci. Rep. 2016, 6, 28087. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Yang, Y.; Shen, H.-B. Hum-mPLoc 3.0: Prediction enhancement of human protein subcellular localization through modeling the hidden correlations of gene ontology and functional domain features. Bioinformatics 2017, 33, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.-H.; Lv, Y.-P.; Li, L.; Xu, H.-M.; Ji, B.-B.; Chen, J.; Li, C.; Liao, B.; Nan, X.-Y. Protein sequence information extraction and subcellular localization prediction with gapped k-Mer method. BMC Bioinform. 2019, 20, 719. [Google Scholar] [CrossRef] [PubMed]
- Armenteros, J.J.A.; Sønderby, C.K.; Sønderby, S.K.; Nielsen, H.; Winther, O. DeepLoc: Prediction of protein subcellular localization using deep learning. Bioinformatics 2017, 33, 3387–3395. [Google Scholar] [CrossRef]
- Liao, Z.; Pan, G.; Sun, C.; Tang, J. Predicting subcellular location of protein with evolution information and sequence-based deep learning. BMC Bioinform. 2021, 22, 515. [Google Scholar] [CrossRef]
- Cong, H.; Liu, H.; Chen, Y.; Cao, Y. Self-evoluting framework of deep convolutional neural network for multilocus protein subcellular localization. Med. Biol. Eng. Comput. 2020, 58, 3017–3038. [Google Scholar] [CrossRef] [PubMed]
- Cong, H.; Liu, H.; Cao, Y.; Chen, Y.; Liang, C. Multiple Protein Subcellular Locations Prediction Based on Deep Convolutional Neural Networks with Self-Attention Mechanism. Interdiscip. Sci. Comput. Life Sci. 2022, 14, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Morrison, A.O.; Gardner, J.M. Microscopic Image Photography Techniques of the Past, Present, and Future. Arch. Pathol. Lab. Med. 2015, 139, 1558–1564. [Google Scholar] [CrossRef] [PubMed]
- Newberg, J.; Murphy, R.F. A Framework for the Automated Analysis of Subcellular Patterns in Human Protein Atlas Images. J. Proteome Res. 2008, 7, 2300–2308. [Google Scholar] [CrossRef]
- Jackson, C.; Murphy, R.F.; Kovacevic, J. Intelligent Acquisition and Learning of Fluorescence Microscope Data Models. IEEE Trans. Image Process. 2009, 18, 2071–2084. [Google Scholar] [CrossRef]
- Liu, G.-H.; Zhang, B.-W.; Qian, G.; Wang, B.; Mao, B.; Bichindaritz, I. Bioimage-Based Prediction of Protein Subcellular Location in Human Tissue with Ensemble Features and Deep Networks. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019, 17, 1966–1980. [Google Scholar] [CrossRef] [PubMed]
- Tahir, M.; Khan, A.; Majid, A. Protein subcellular localization of fluorescence imagery using spatial and transform domain features. Bioinformatics 2012, 28, 91–97. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xu, Y.-Y.; Yang, F.; Zhang, Y.; Shen, H.-B. An image-based multi-label human protein subcellular localization predictor (iLocator) reveals protein mislocalizations in cancer tissues. Bioinformatics 2013, 29, 2032–2040. [Google Scholar] [CrossRef]
- Yang, F.; Xu, Y.-Y.; Wang, S.-T.; Shen, H.-B. Image-based classification of protein subcellular location patterns in human reproductive tissue by ensemble learning global and local features. Neurocomputing 2014, 131, 113–123. [Google Scholar] [CrossRef]
- Nanni, L.; Brahnam, S.; Ghidoni, S.; Lumini, A. Bioimage classification with handcrafted and learned features. IEEE/ACM Trans. Comput. Biol. Bioinform. 2018, 16, 874–885. [Google Scholar] [CrossRef]
- Yang, F.; Liu, Y.; Wang, Y.; Yin, Z.; Yang, Z. MIC_Locator: A novel image-based protein subcellular location multi-label prediction model based on multi-scale monogenic signal representation and intensity encoding strategy. BMC Bioinform. 2019, 20, 522. [Google Scholar] [CrossRef] [PubMed]
- Xue, Z.-Z.; Wu, Y.; Gao, Q.-Z.; Zhao, L.; Xu, Y.-Y. Automated classification of protein subcellular localization in immunohistochemistry images to reveal biomarkers in colon cancer. BMC Bioinform. 2020, 21, 398. [Google Scholar] [CrossRef] [PubMed]
- Ullah, M.; Han, K.; Hadi, F.; Xu, J.; Song, J.; Yu, D.-J. PScL-HDeep: Image-based prediction of protein subcellular location in human tissue using ensemble learning of handcrafted and deep learned features with two-layer feature selection. Brief. Bioinform. 2021, 22, bbab278. [Google Scholar] [CrossRef]
- Pärnamaa, T.; Parts, L. Accurate Classification of Protein Subcellular Localization from High-Throughput Microscopy Images Using Deep Learning. G3 Genes Genomes Genet. 2017, 7, 1385–1392. [Google Scholar] [CrossRef] [PubMed]
- Long, W.; Yang, Y.; Shen, H.-B. ImPLoc: A multi-instance deep learning model for the prediction of protein subcellular localization based on immunohistochemistry images. Bioinformatics 2020, 36, 2244–2250. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.-X.; Yang, Y.; Xu, Y.; Shen, H. Incorporating label correlations into deep neural networks to classify protein subcellular location patterns in immunohistochemistry images. Proteins Struct. Funct. Bioinform. 2022, 90, 493–503. [Google Scholar] [CrossRef]
- Xue, M.-Q.; Zhu, X.-L.; Wang, G.; Xu, Y.-Y. DULoc: Quantitatively unmixing protein subcellular location patterns in immunofluorescence images based on deep learning features. Bioinformatics 2022, 38, 827–833. [Google Scholar] [CrossRef]
- Wang, F.; Wei, L. Multi-scale deep learning for the imbalanced multi-label protein subcellular localization prediction based on immunohistochemistry images. Bioinformatics 2022, 38, 2602–2611. [Google Scholar] [CrossRef]
- Ouyang, W.; Winsnes, C.F.; Hjelmare, M.; Cesnik, A.J.; Åkesson, L.; Xu, H.; Sullivan, D.P.; Dai, S.; Lan, J.; Jinmo, P.; et al. Analysis of the Human Protein Atlas Image Classification competition. Nat. Methods 2019, 16, 1254–1261. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, F.; Ma, Q.; Wang, Z.; Wang, S.; Zou, K. Human protein subcellular localization prediction based on error correcting output coding strategy while combining immunohistochemistry image and amino acid sequence. In Proceedings of the 4th International Conference on Advanced Electronic Materials, Computers and Software Engineering (AEMCSE), Changsha, China, 26–28 March 2021; IEEE: Piscataway, NJ, USA, 2021; pp. 876–881. [Google Scholar]
- Wang, G.; Xue, M.-Q.; Shen, H.-B.; Xu, Y.-Y. Learning protein subcellular localization multi-view patterns from heterogeneous data of imaging, sequence and networks. Briefings Bioinform. 2022, 23, bbab539. [Google Scholar] [CrossRef]
- Nakai, K.; Wei, L. Recent Advances in the Prediction of Subcellular Localization of Proteins and Related Topics. Front. Bioinform. 2022, 2, 910531. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yang, B.; Revote, J.; Leier, A.; Marquez-Lago, T.T.; Webb, G.; Song, J.; Chou, K.-C.; Lithgow, T. POSSUM: A bioinformatics toolkit for generating numerical sequence feature descriptors based on PSSM profiles. Bioinformatics 2017, 33, 2756–2758. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.-B.; Chou, K.-C. PseAAC: A flexible web server for generating various kinds of protein pseudo amino acid composition. Anal. Biochem. 2008, 373, 386–388. [Google Scholar] [CrossRef]
- Chou, K.-C. Using amphiphilic pseudo amino acid composition to predict enzyme subfamily classes. Bioinformatics 2005, 21, 10–19. [Google Scholar] [CrossRef]
- Chou, K.-C.; Cai, Y.-D. Prediction of Membrane Protein Types by Incorporating Amphipathic Effects. J. Chem. Inf. Model. 2005, 45, 407–413. [Google Scholar] [CrossRef]
- Shen, H.-B.; Chou, K.-C. Ensemble classifier for protein fold pattern recognition. Bioinformatics 2006, 22, 1717–1722. [Google Scholar] [CrossRef]
- Chou, K.-C. Prediction of Protein Subcellular Locations by Incorporating Quasi-Sequence-Order Effect. Biochem. Biophys. Res. Commun. 2000, 278, 477–483. [Google Scholar] [CrossRef]
- Zou, H.; Zhan, C. Using Multi-Level Correlation Information to Identify Amyloidogenic Peptides. ChemistrySelect 2022, 7, e202104578. [Google Scholar] [CrossRef]
- Tibshirani, R. Regression shrinkage and selection via the lasso. J. R. Stat. Soc. Ser. B Methodol. 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Boutell, M.R.; Luo, J.; Shen, X.; Brown, C.M. Learning multi-label scene classification. Pattern Recognit. 2004, 37, 1757–1771. [Google Scholar] [CrossRef]
- Nanni, L.; Brahnam, S.; Lumini, A. Novel features for automated cell phenotype image classification. In Advances in Computational Biology; Springer: New York, NY, USA, 2010; pp. 207–213. [Google Scholar]
- He, K.; Zhang, X.; Ren, S.; Sun, J. Deep residual learning for image recognition. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Las Vegas, NV, USA, 27–30 June 2016; pp. 770–778. [Google Scholar]
- Zhuang, P.; Wang, Y.; Qiao, Y. Learning attentive pairwise interaction for fine-grained classification. In Proceedings of the AAAI Conference on Artificial Intelligence, New York, NY, USA, 7–12 February 2020; Volume 34, pp. 13130–13137. [Google Scholar]
- Juan EY, T.; Li, W.J.; Jhang, J.H.; Chiu, C.H. Predicting protein subcellular localizations for gram-negative bacteria using DP-PSSM and support vector machines. In Proceedings of the 2009 International Conference on Complex, Intelligent and Software Intensive Systems, Fukuoka, Japan, 16–19 March 2009; IEEE: Piscataway, NJ, USA, 2009; pp. 836–841. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subcellular Location | Sequence | Image |
|---|---|---|
| ER | 101 | 101 |
| Golgi | 74 | 74 |
| Cytosol | 59 | 59 |
| Nucleoplasm | 47 | 47 |
| Total | 281 | 281 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zou, K.; Wang, S.; Wang, Z.; Zou, H.; Yang, F. Dual-Signal Feature Spaces Map Protein Subcellular Locations Based on Immunohistochemistry Image and Protein Sequence. Sensors 2023, 23, 9014. https://doi.org/10.3390/s23229014
Zou K, Wang S, Wang Z, Zou H, Yang F. Dual-Signal Feature Spaces Map Protein Subcellular Locations Based on Immunohistochemistry Image and Protein Sequence. Sensors. 2023; 23(22):9014. https://doi.org/10.3390/s23229014
Chicago/Turabian StyleZou, Kai, Simeng Wang, Ziqian Wang, Hongliang Zou, and Fan Yang. 2023. "Dual-Signal Feature Spaces Map Protein Subcellular Locations Based on Immunohistochemistry Image and Protein Sequence" Sensors 23, no. 22: 9014. https://doi.org/10.3390/s23229014
APA StyleZou, K., Wang, S., Wang, Z., Zou, H., & Yang, F. (2023). Dual-Signal Feature Spaces Map Protein Subcellular Locations Based on Immunohistochemistry Image and Protein Sequence. Sensors, 23(22), 9014. https://doi.org/10.3390/s23229014