
Convolutional Neural Network Applied to SARS-CoV-2 Sequence Classification

 ,
,  , , , ,
, , , ,  and
and 
(This article belongs to the Section Intelligent Sensors)
Abstract
:1. Introduction
- Develop and validate a new genomic viral classification based on the k-mers’ image representation and a CNN.
- Develop an alignment-free method to classify SARS-CoV-2 sequences as viruses from different realms, families, genera, and subgenera.
- Develop a deep learning algorithm that can efficiently classify the complete cDNA sequences of the virus.
2. Materials and Methods
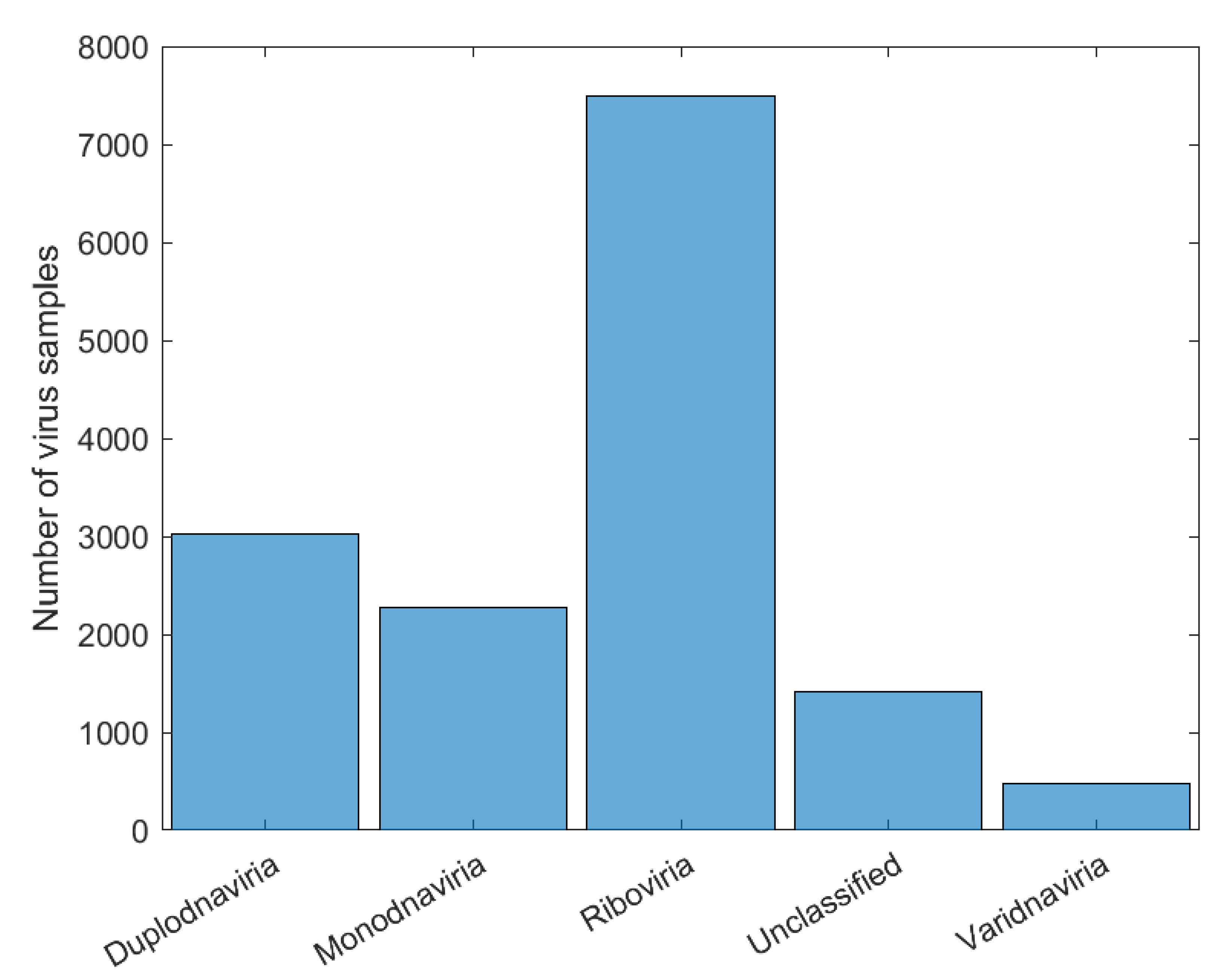
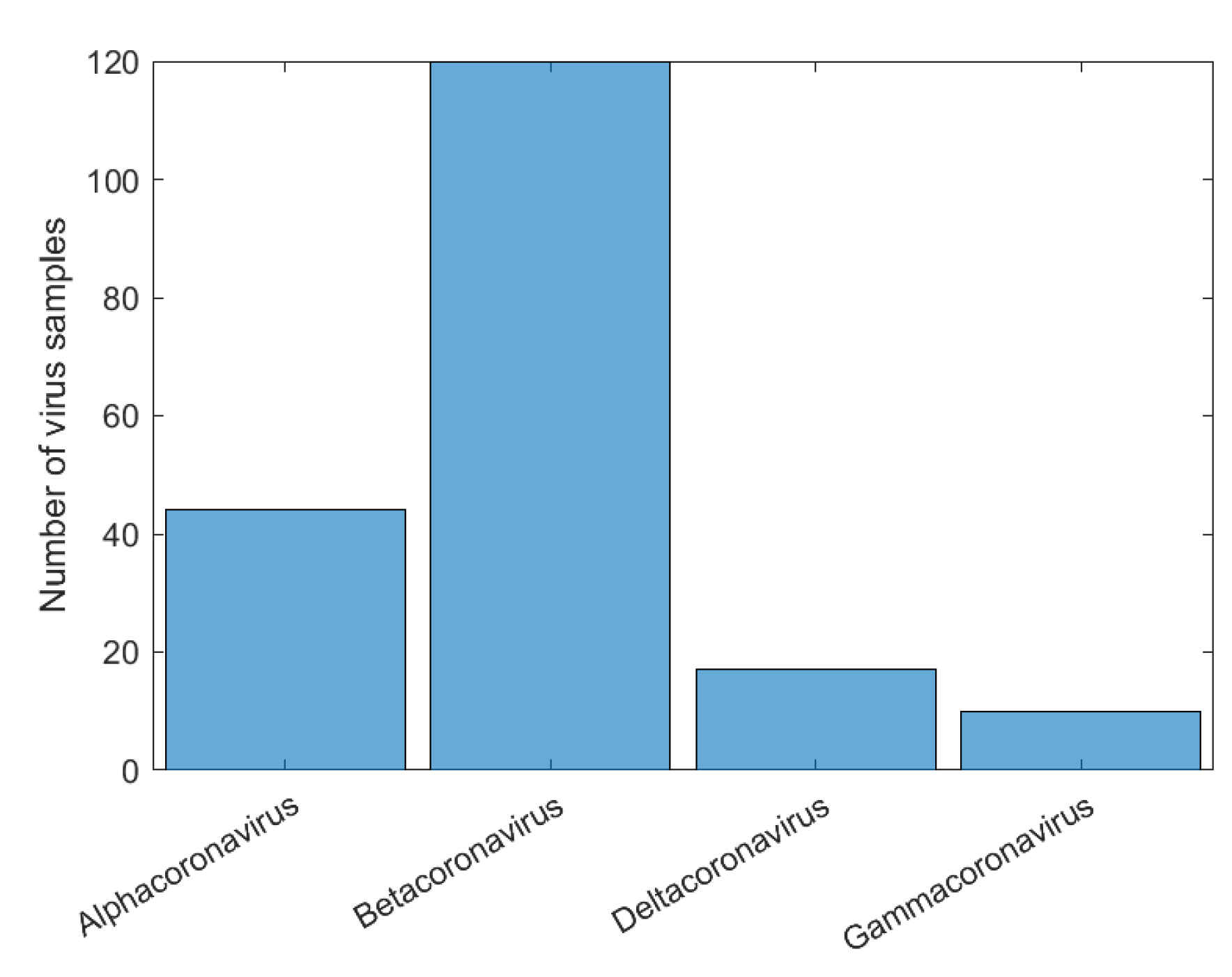
2.1. Data
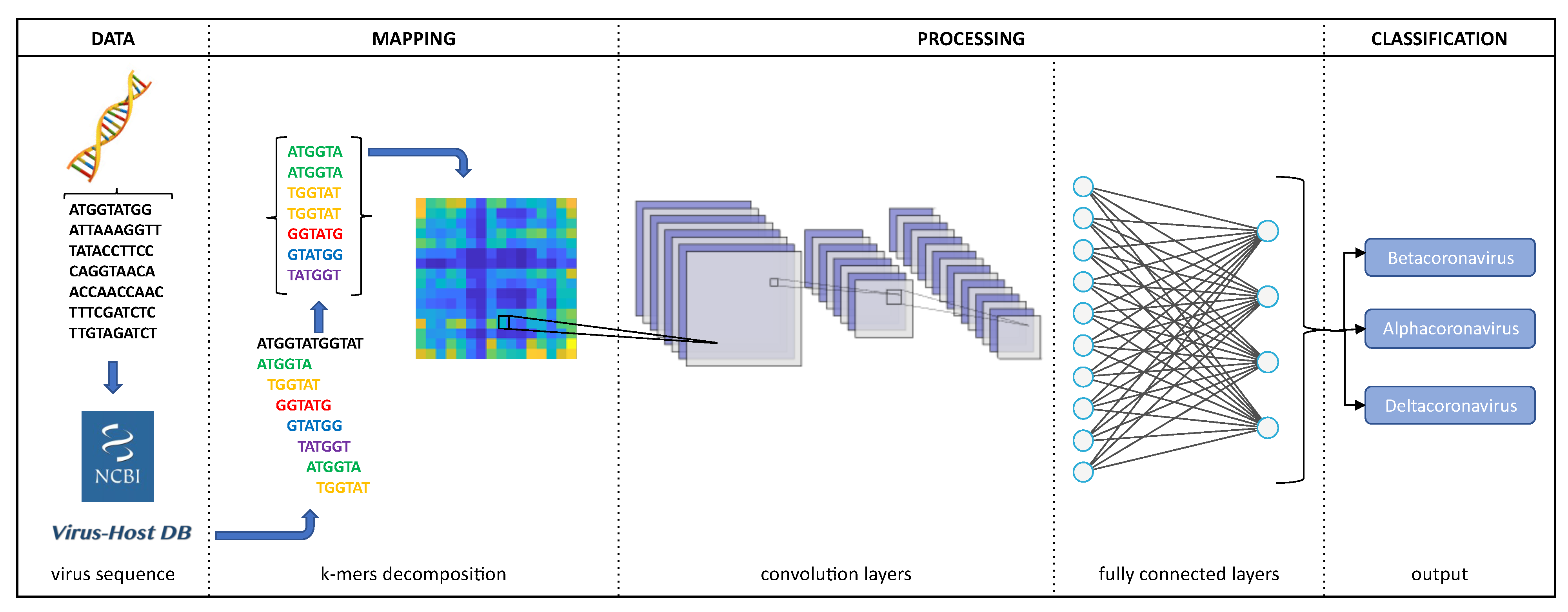
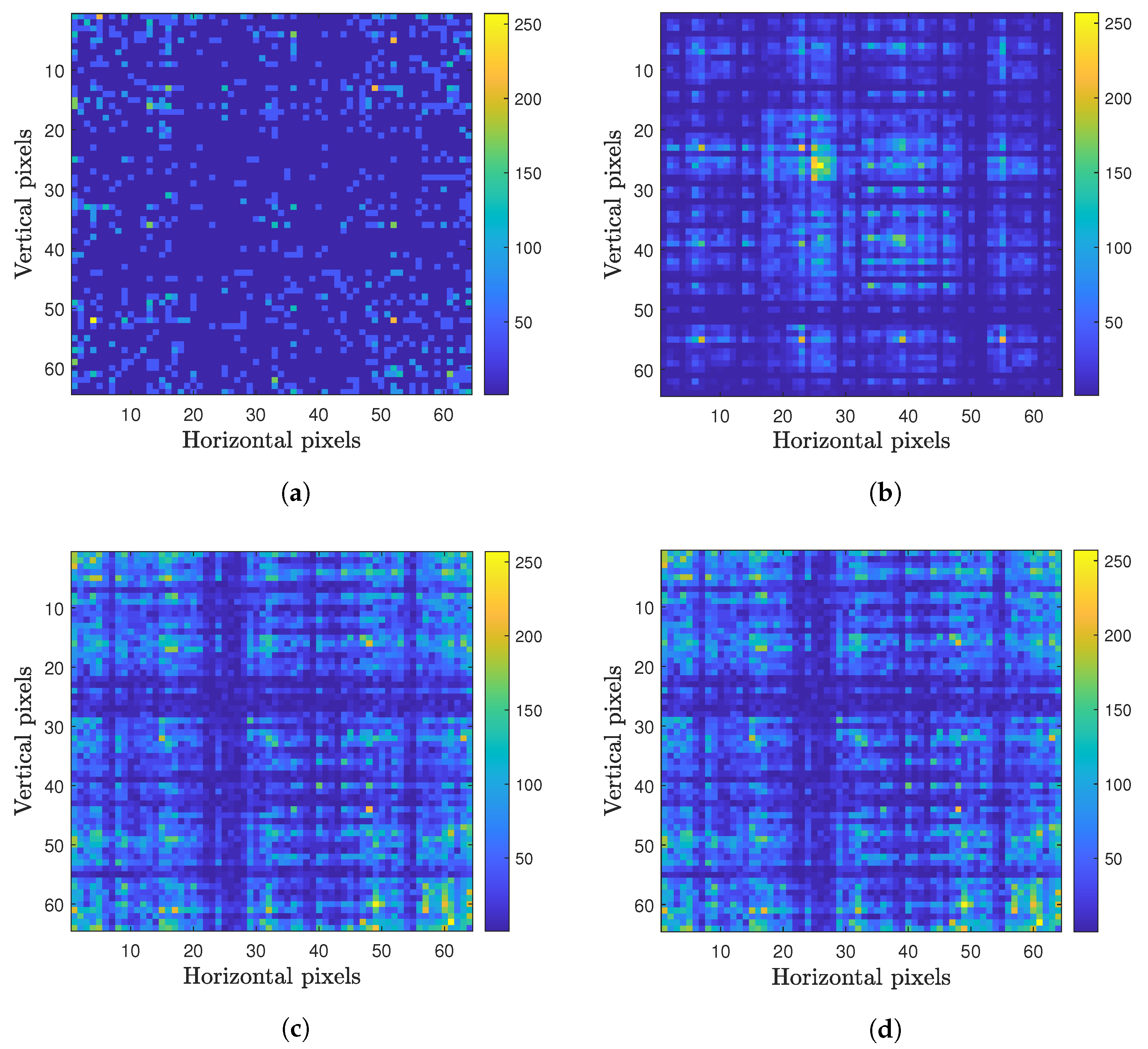
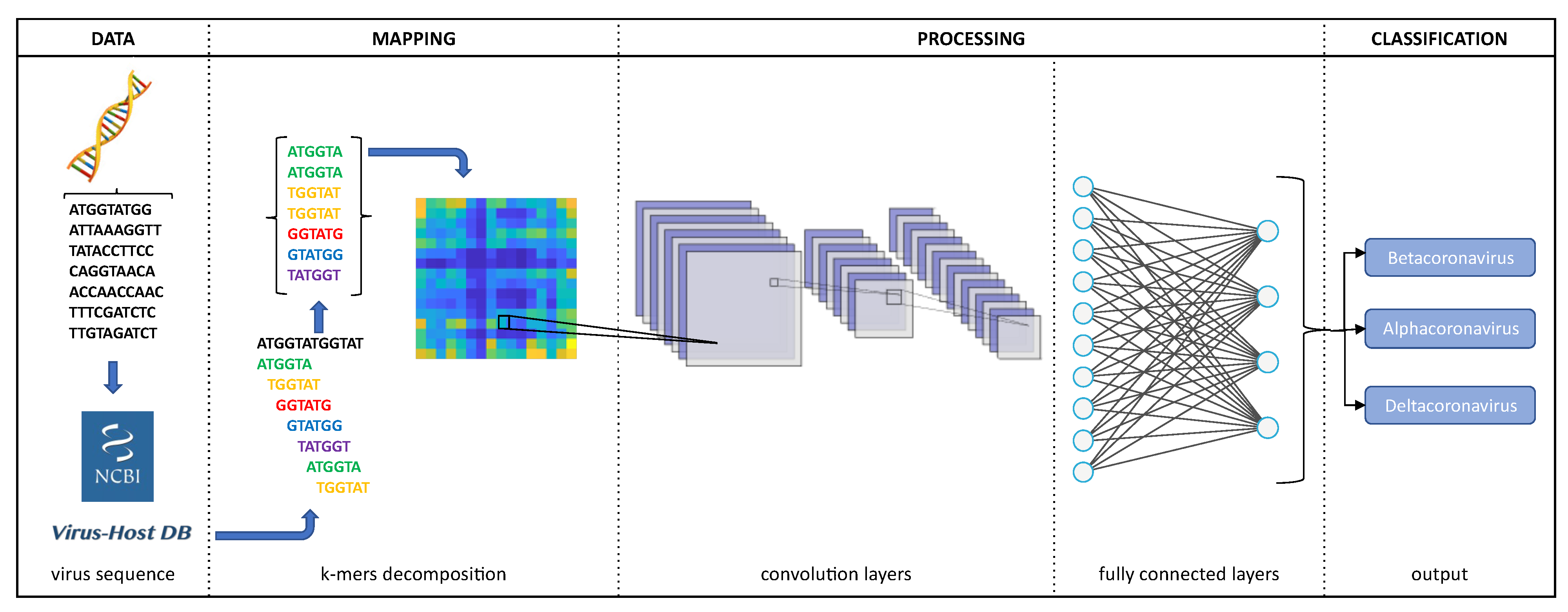
2.2. Image Representation
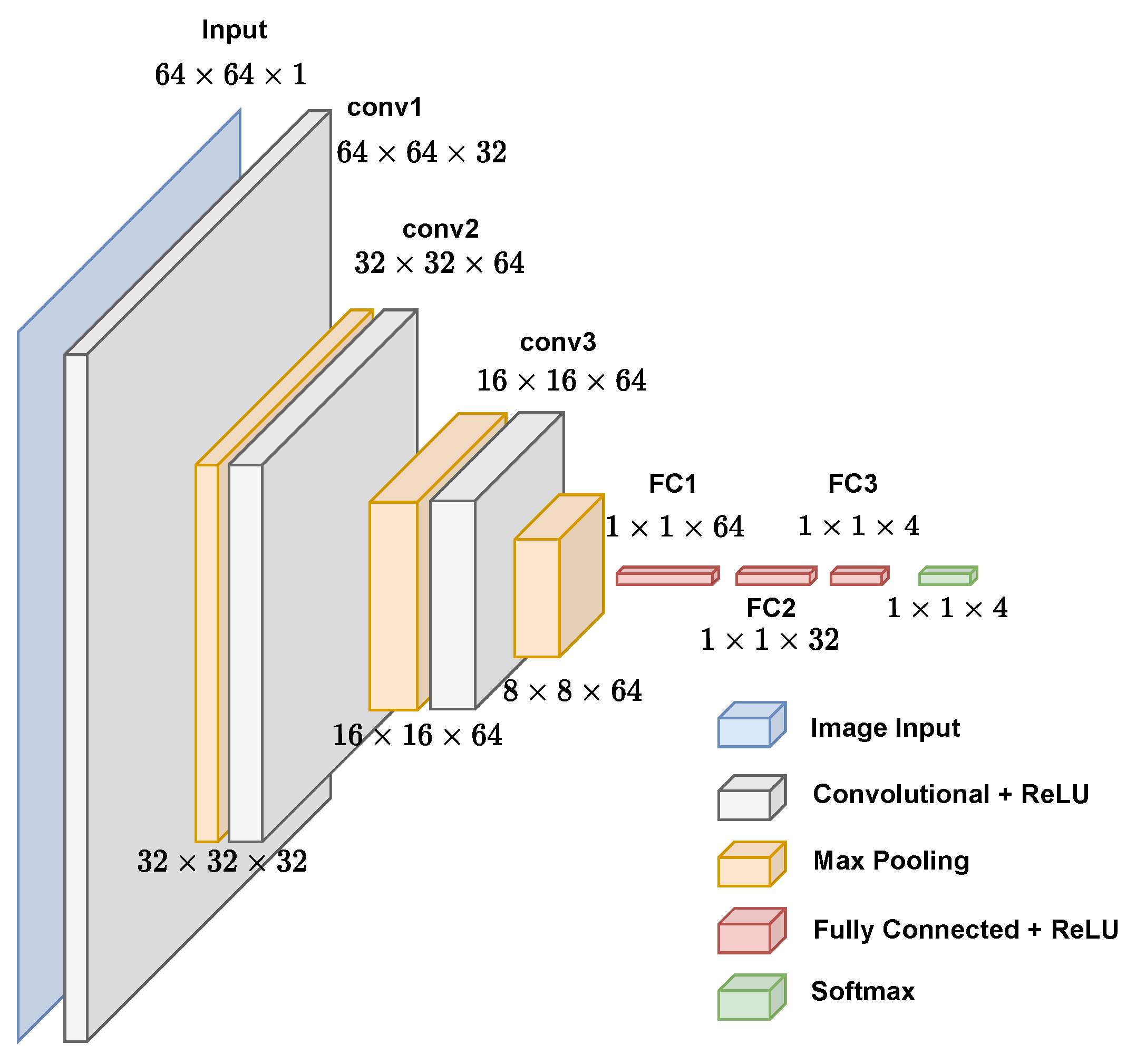
2.3. Processing
3. Results and Discussion
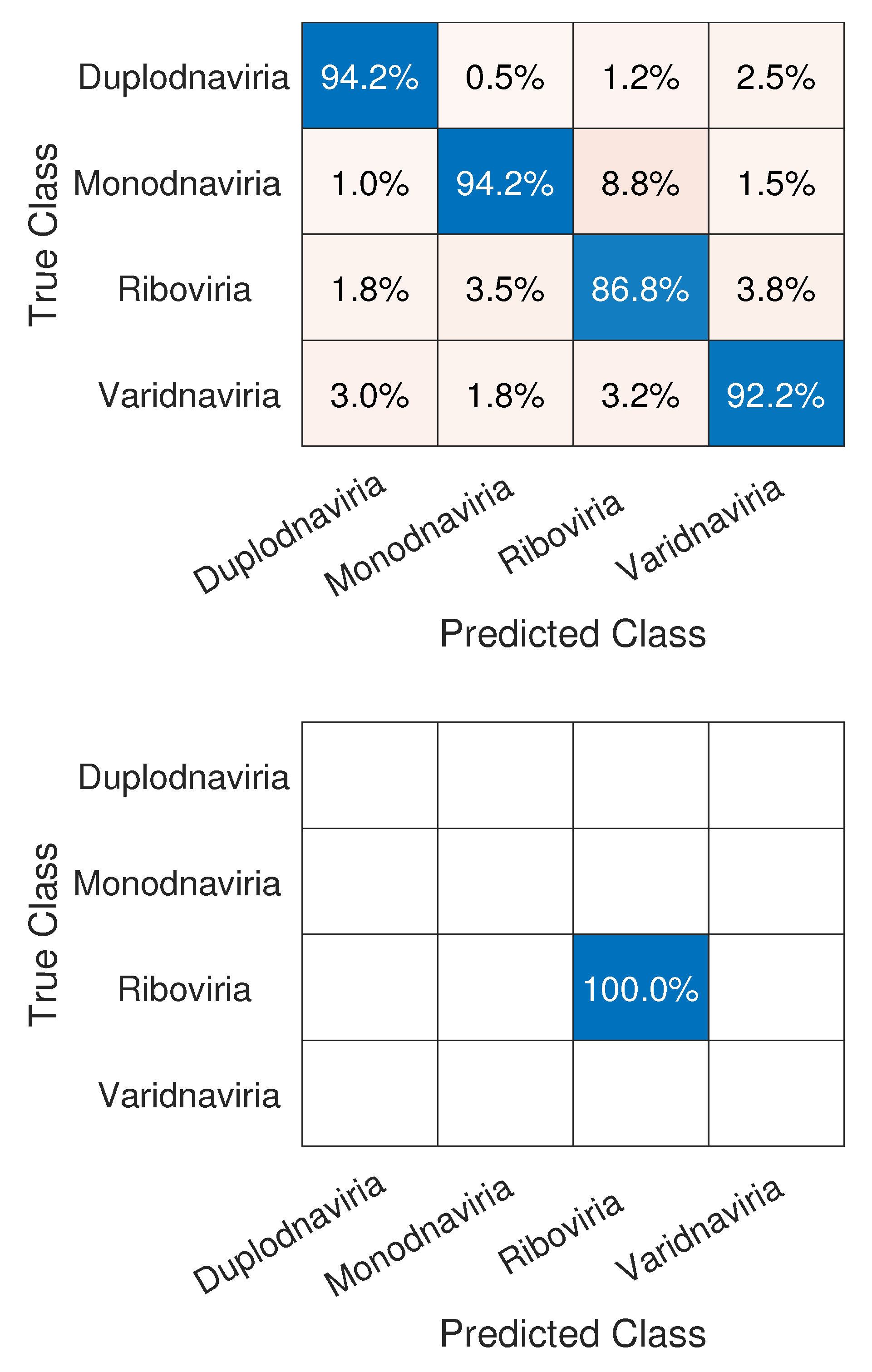
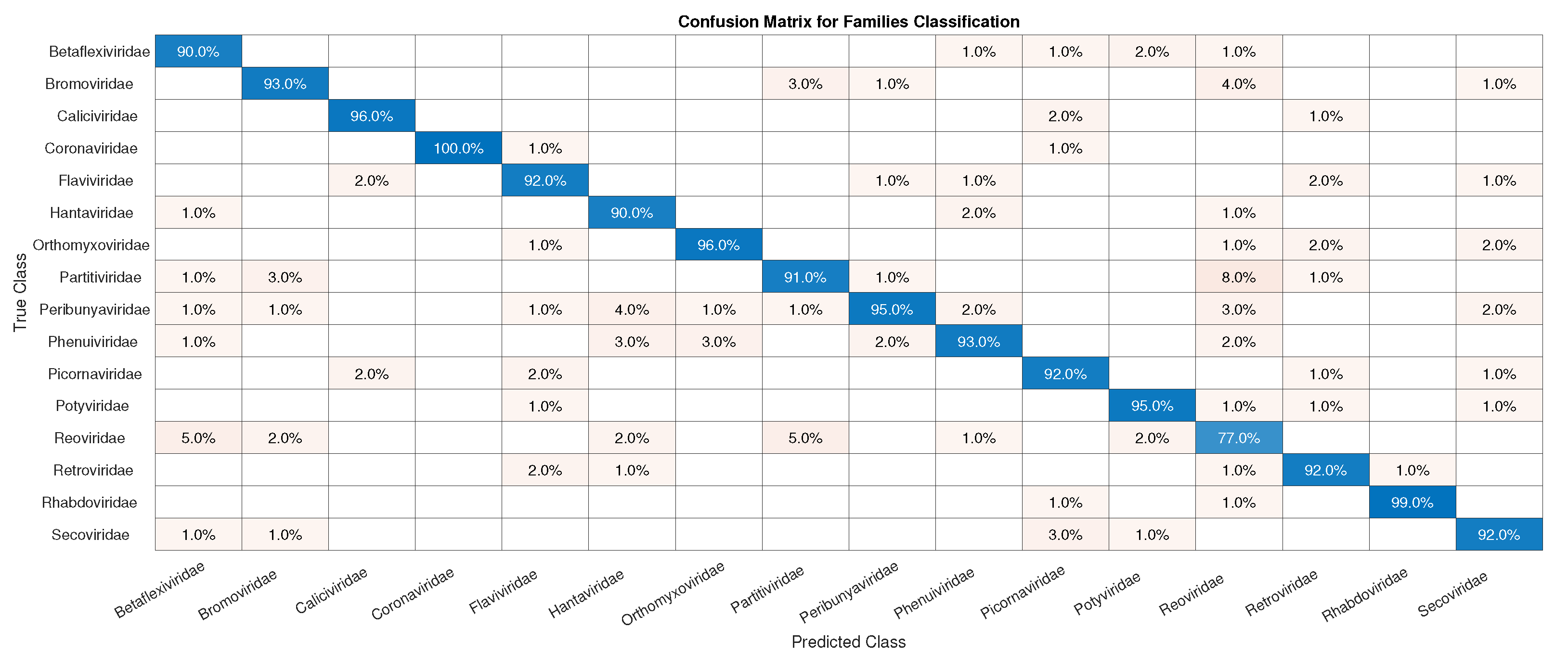
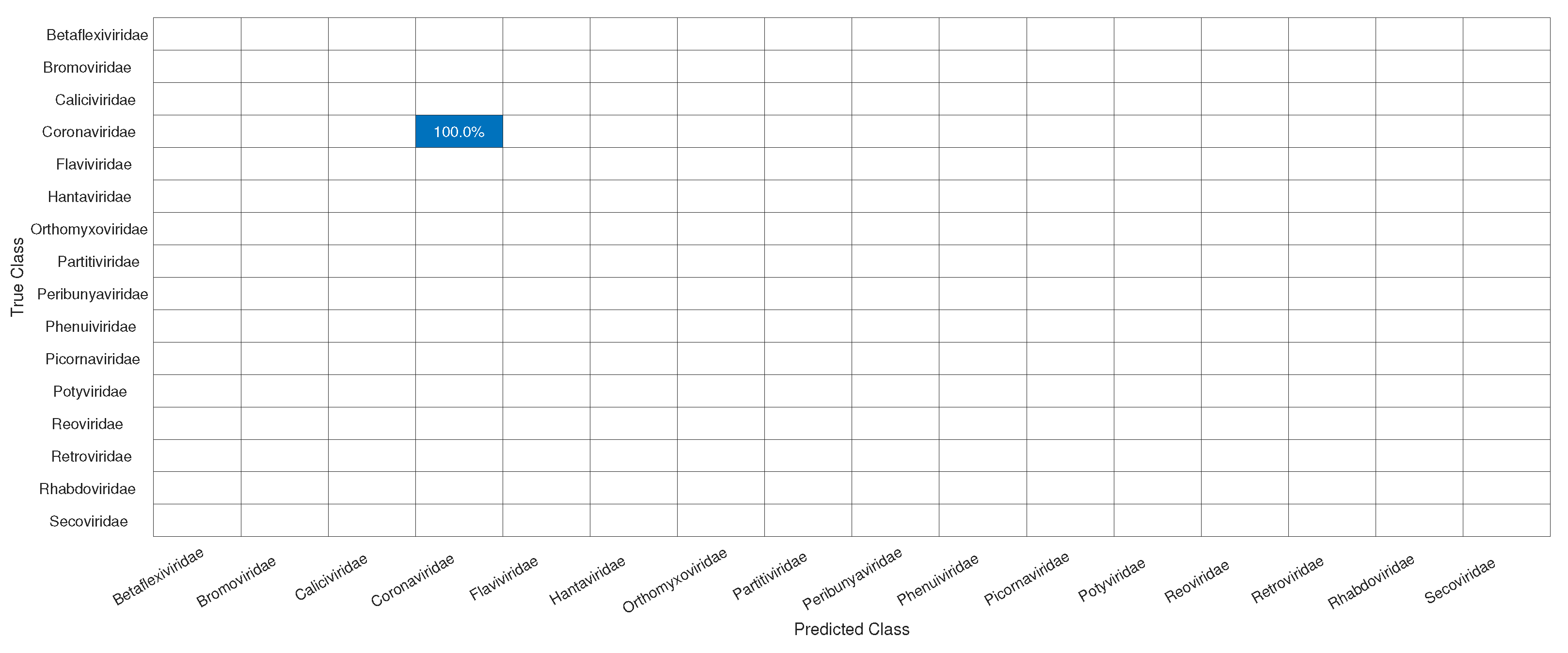
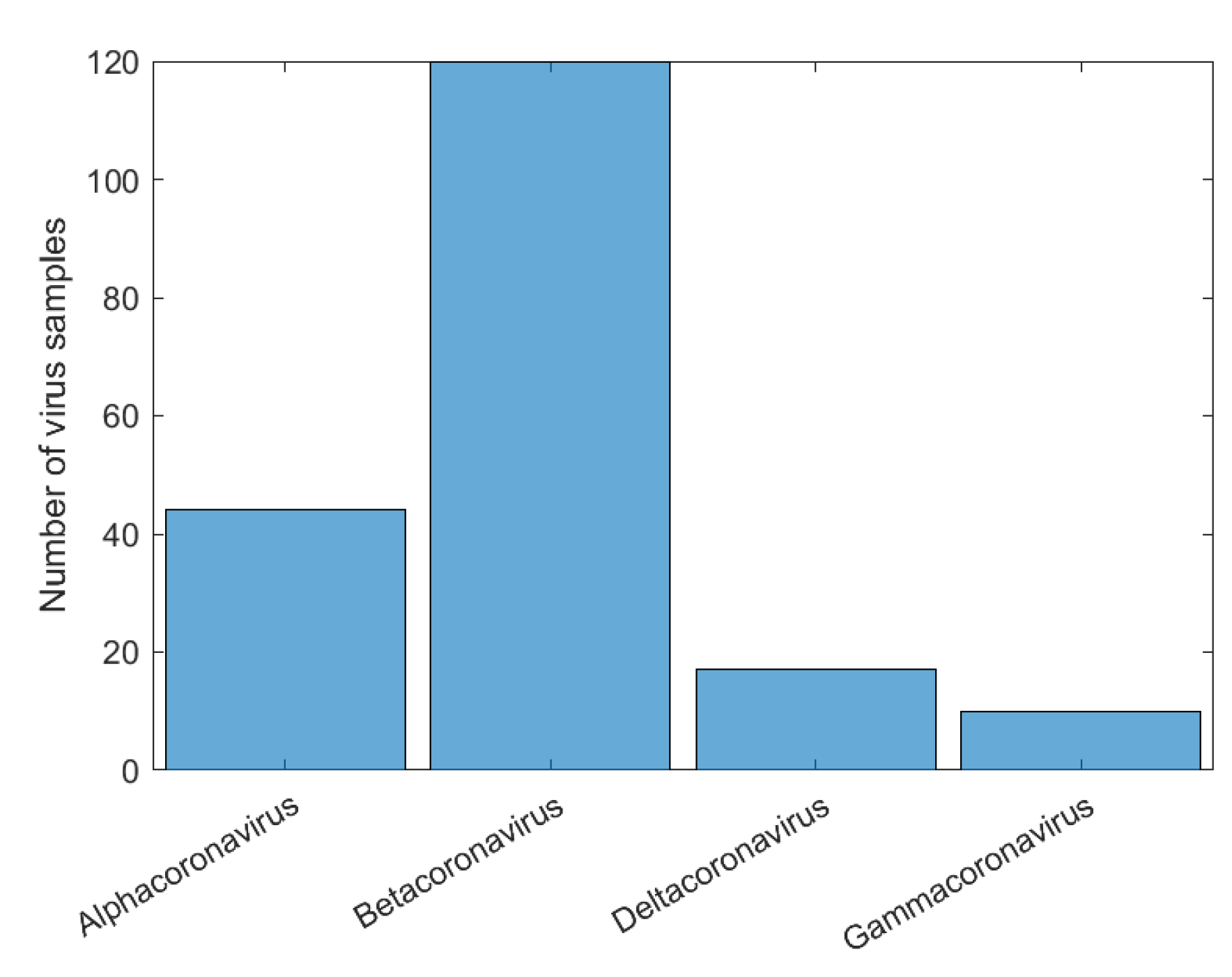
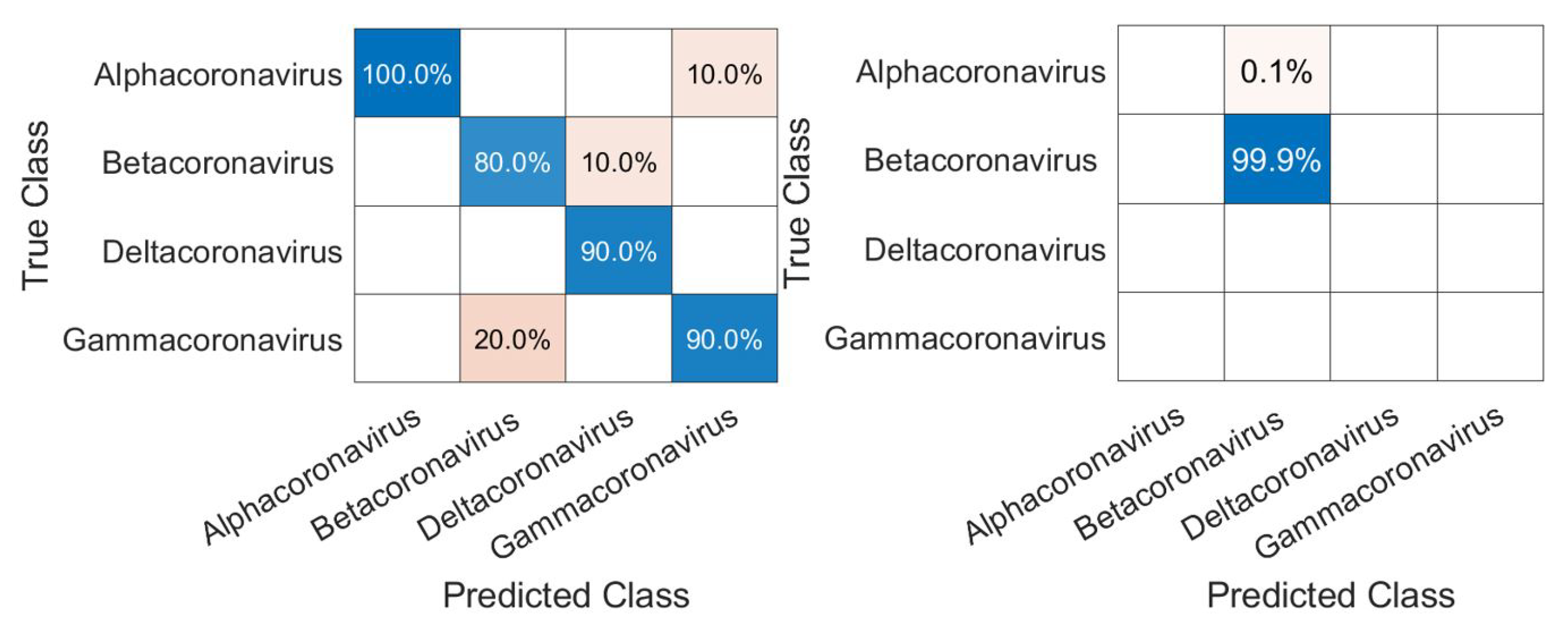
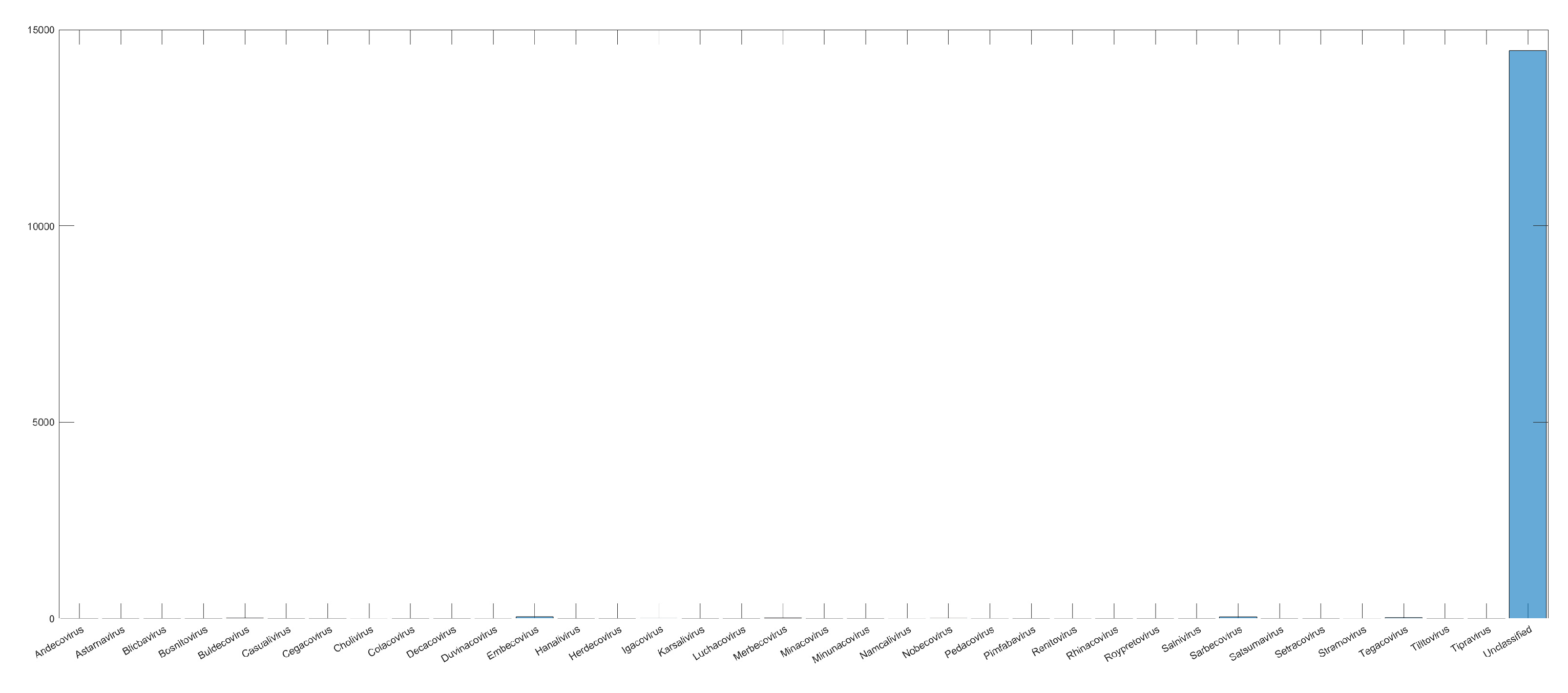
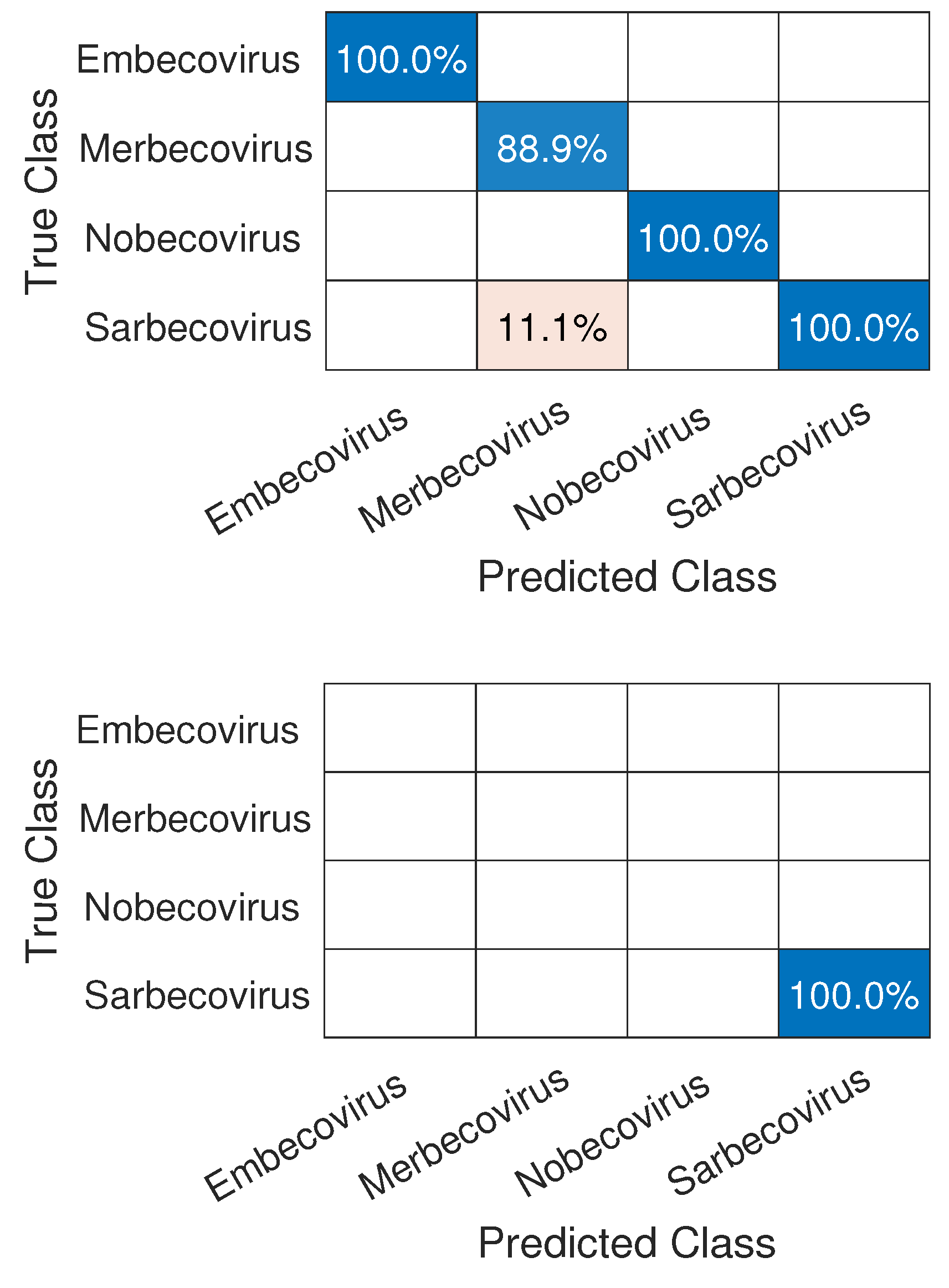
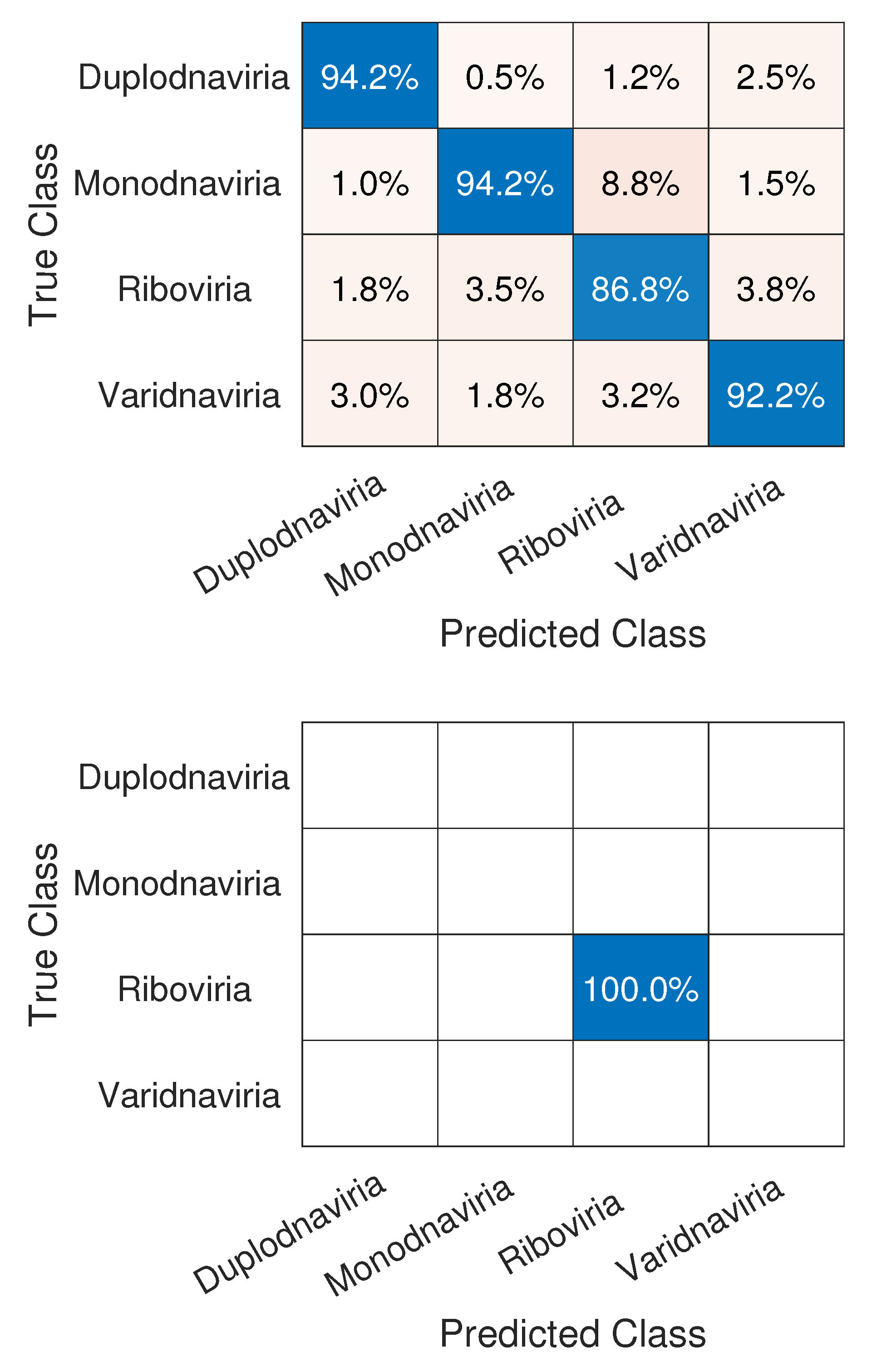
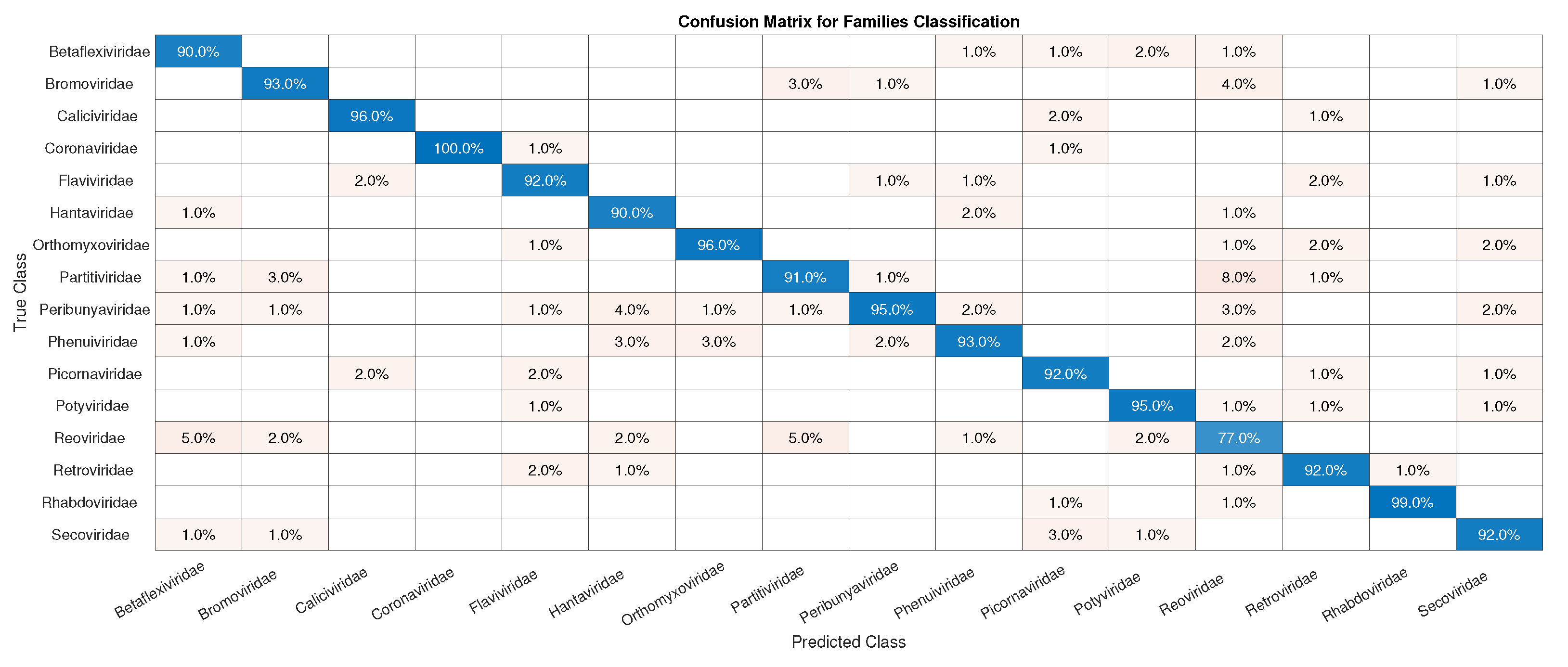
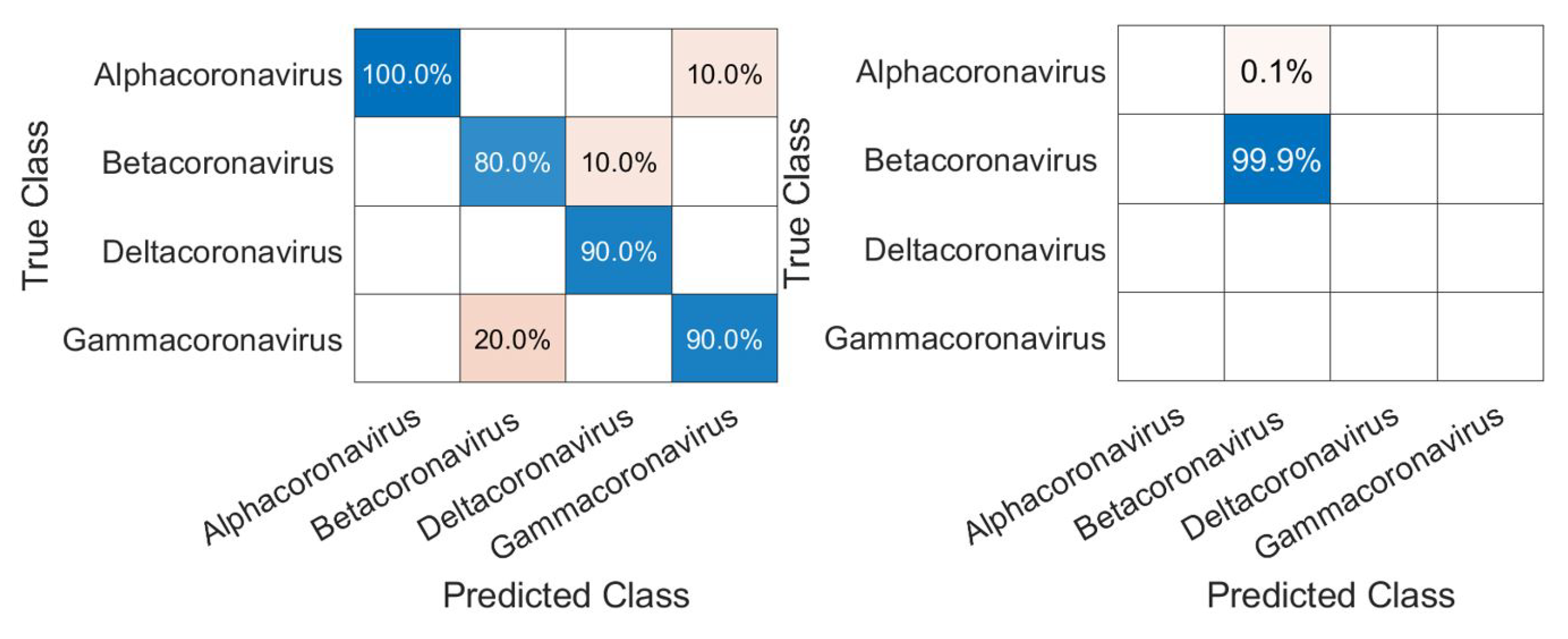
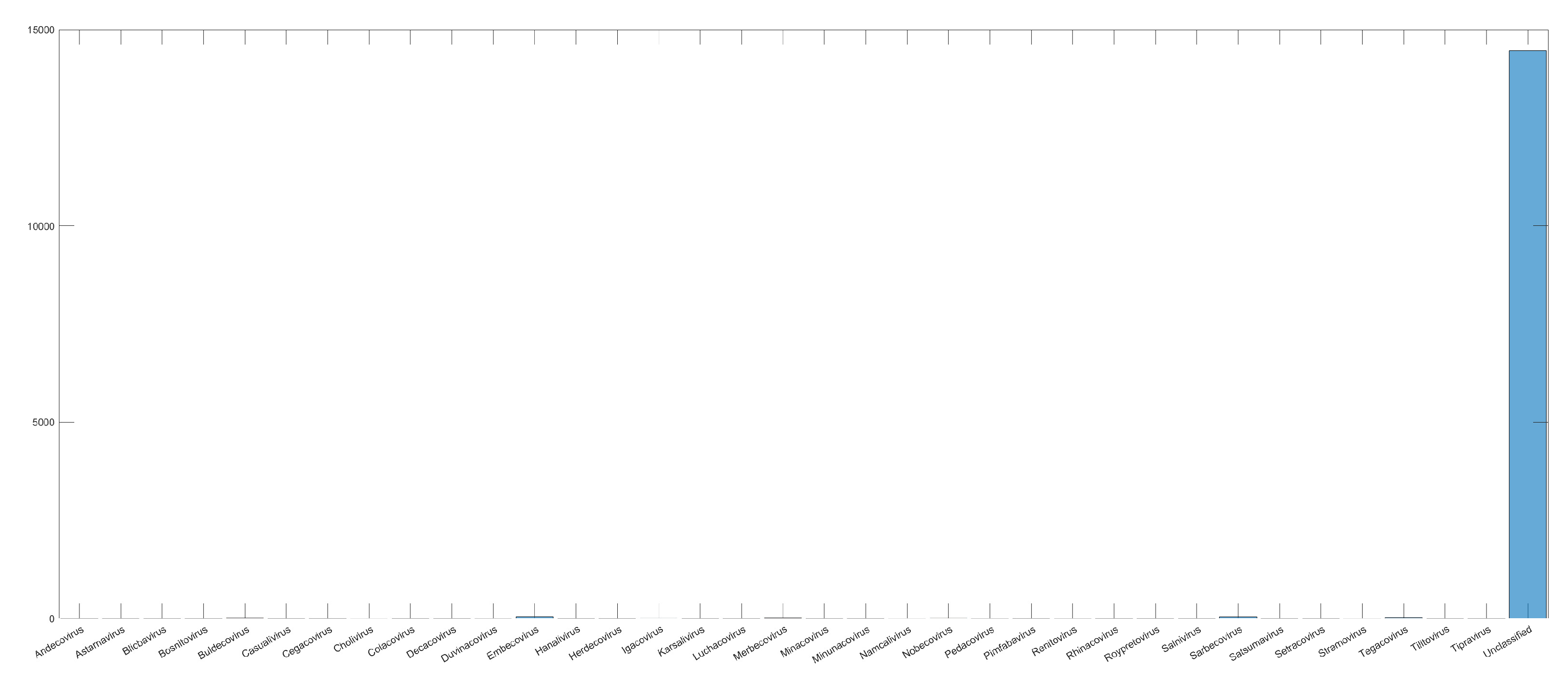
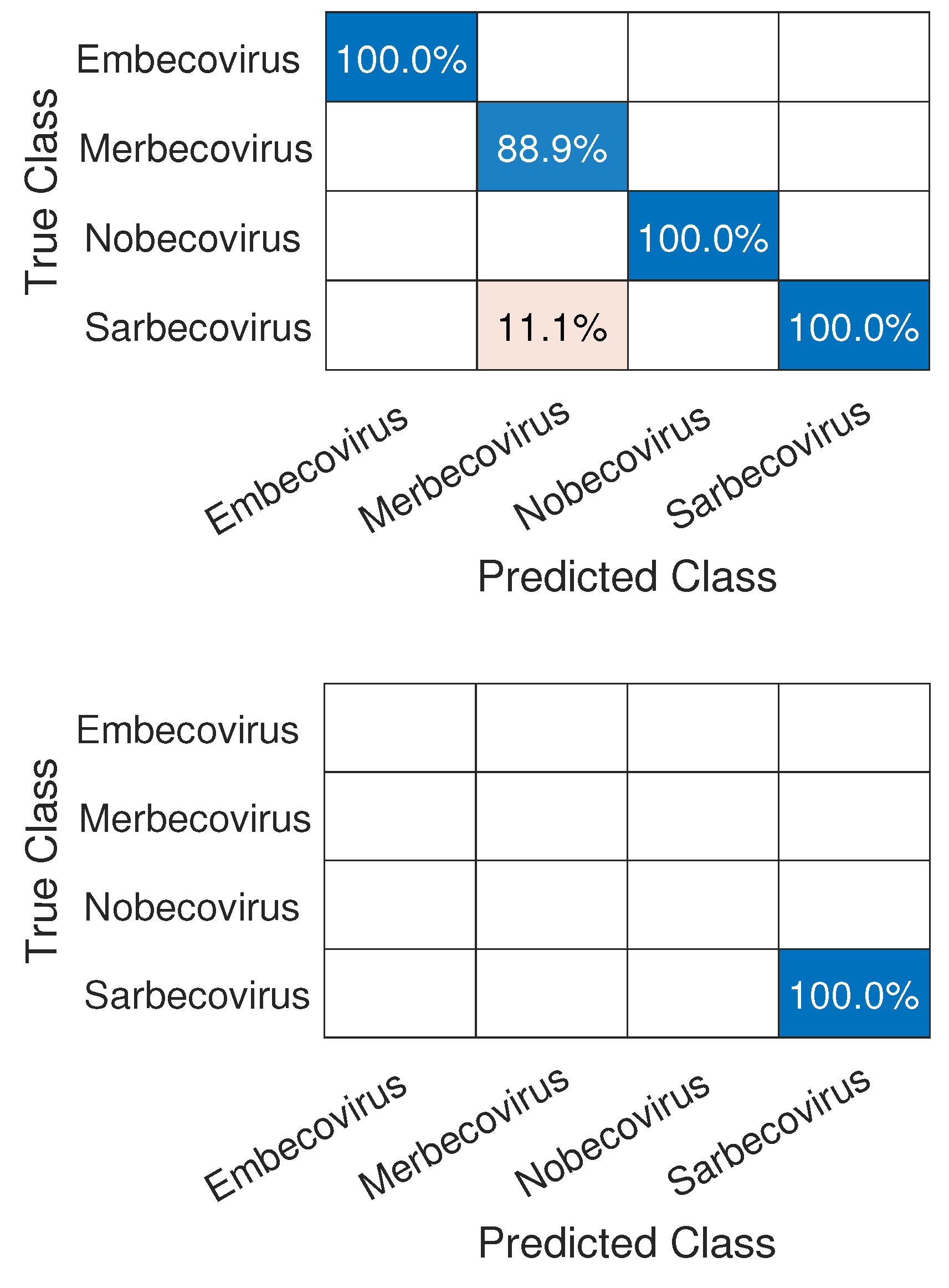
3.1. CNN Architecture, Confusion Matrices, and Histograms
3.2. Related Works Comparison
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Woo, P.C.Y.; Huang, Y.; Lau, S.K.P.; Yuen, K.Y. Coronavirus Genomics and Bioinformatics Analysis. Viruses 2010, 2, 1804–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, J.; Li, F.; Shi, Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [Green Version]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARSCoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef]
- Jungreis, I.; Nelson, C.W.; Ardern, Z.; Finkel, Y.; Krogan, N.J.; Sato, K.; Ziebuhr, J.; Stern-Ginossar, N.; Pavesi, A.; Firth, A.E.; et al. Conflicting and ambiguous names of overlapping ORFs in the SARS-CoV-2 genome: A homology-based resolution. Virology 2021, 558, 145–151. [Google Scholar] [CrossRef]
- Guo, Y.R.; Cao, Q.D.; Hong, Z.S.; Tan, Y.Y.; Chen, S.D.; Jin, H.J.; Tan, K.S.; Wang, D.Y.; Yan, Y. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak—An update on the status. Mil. Med. Res. 2020, 7, 11. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Wu, Q.; Zhang, Z. Probable pangolin origin of SARS-CoV-2 associated with the COVID-19 outbreak. Curr. Biol. 2020, 6, 1346–1351. [Google Scholar] [CrossRef]
- Randhawa, G.S.; Soltysiak, M.P.M.; Roz, H.E.; de Souza, C.P.E.; Hill, K.A.; Kari, L. Machine learning using intrinsic genomic signatures for rapid classification of novel pathogens: COVID-19 case study. PLoS ONE 2020, 15, e0232391. [Google Scholar] [CrossRef] [Green Version]
- A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 1970, 48, 443–453. [CrossRef]
- Smith, T.F.; Waterman, M.S. Identification of common molecular subsequences. J. Mol. Biol. 1981, 147, 195–197. [Google Scholar] [CrossRef]
- Pearson, W.R.; Lipman, D.J. Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci. USA 1988, 85, 2444–2448. [Google Scholar] [CrossRef] [Green Version]
- Improved sensitivity of nucleic acid database searches using application-specific scoring matrices. Methods 1991, 3, 66–70. [CrossRef]
- Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [CrossRef]
- Pinello, L.; Lo Bosco, G.; Yuan, G.C. Applications of alignment-free methods in epigenomics. Briefings Bioinform. 2013, 15, 419–430. [Google Scholar] [CrossRef] [Green Version]
- Vinga, S.; Almeida, J. Alignment-free sequence comparison—A review. Bioinformatics 2003, 19, 513–523. [Google Scholar] [CrossRef]
- Zielezinski, A.; Vinga, S.; Almeida, J.; Karlowski, W.M. Alignment-free sequence comparison: Benefits, applications, and tools. Genome Biol. 2017, 18, 186. [Google Scholar] [CrossRef] [Green Version]
- Morgenstern, B. Sequence Comparison without Alignment: The SpaM approaches. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Zielezinski, A.; Girgis, H.Z.; Bernard, G.; Leimeister, C.A.; Tang, K.; Dencker, T.; Lau, A.K.; Röhling, S.; Choi, J.J.; Waterman, M.S.; et al. Benchmarking of alignment-free sequence comparison methods. Genome Biol. 2019, 20, 144. [Google Scholar] [CrossRef] [Green Version]
- Barbosa, R.d.M.; Fernandes, M.A. Chaos game representation dataset of SARS-CoV-2 genome. Data Brief 2020, 30, 105618. [Google Scholar] [CrossRef]
- Jeffrey, H. Chaos game representation of gene structure. Nucleic Acids Res. 1990, 18, 2163–2170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Löchel, H.F.; Eger, D.; Sperlea, T.; Heider, D. Deep learning on chaos game representation for proteins. Bioinformatics 2020, 36, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, R.d.M.; Fernandes, M.A. Data stream dataset of SARS-CoV-2 genome. Data Brief 2020, 31, 105829. [Google Scholar] [CrossRef]
- Randhawa, G.S.; Hill, K.A.; Kari, L. ML-DSP: Machine Learning with Digital Signal Processing for ultrafast, accurate, and scalable genome classification at all taxonomic levels. BMC Genom. 2019, 20, 267. [Google Scholar] [CrossRef] [Green Version]
- Libbrecht, M.W.; Noble, W.S. Machine learning applications in genetics and genomics. Nat. Rev. Genet. 2015, 16, 321–332. [Google Scholar] [CrossRef] [Green Version]
- Liu, B. BioSeq-Analysis: A platform for DNA, RNA and protein sequence analysis based on machine learning approaches. Briefings Bioinform. 2019, 20, 1280–1294. [Google Scholar] [CrossRef] [Green Version]
- Fiannaca, A.; La Paglia, L.; La Rosa, M.; Bosco, L.; Renda, G.; Rizzo, R.; Gaglio, S.; Urso, A. Deep learning models for bacteria taxonomic classification of metagenomic data. BMC Bioinform. 2018, 19, 198. [Google Scholar] [CrossRef]
- Randhawa, G.S.; Soltysiak, M.P.; Roz, H.E.; de Souza, C.P.; Hill, K.A.; Kari, L. Machine learning analysis of genomic signatures provides evidence of associations between Wuhan 2019-nCoV and bat betacoronaviruses. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Remita, M.A.; Halioui, A.; Diouara, A.A.M.; Daigle, B.; Kiani, G.; Diallo, A.B. A machine learning approach for viral genome classification. BMC Bioinform. 2017, 18, 208. [Google Scholar] [CrossRef] [Green Version]
- Mock, F.; Viehweger, A.; Barth, E.; Marz, M. Viral host prediction with Deep Learning. bioRxiv 2019. [Google Scholar] [CrossRef]
- Zhu, H.; Guo, Q.; Li, M.; Wang, C.; Fang, Z.; Wang, P.; Tan, J.; Wu, S.; Xiao, Y. Host and infectivity prediction of Wuhan 2019 novel coronavirus using deep learning algorithm. BioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Desai, H.P.; Parameshwaran, A.P.; Sunderraman, R.; Weeks, M. Comparative Study Using Neural Networks for 16S Ribosomal Gene Classification. J. Comput. Biol. 2020, 27, 248–258. [Google Scholar] [CrossRef] [Green Version]
- Zou, J.; Huss, M.; Abid, A.; Mohammadi, P.; Torkamani, A.; Telenti, A. A primer on deep learning in genomics. Nat. Genet. 2019, 51, 12–18. [Google Scholar] [CrossRef]
- Min, S.; Lee, B.; Yoon, S. Deep learning in bioinformatics. Briefings Bioinform. 2017, 18, 851–869. [Google Scholar] [CrossRef] [Green Version]
- Eraslan, G.; Avsec, Ž.; Gagneur, J.; Theis, F.J. Deep learning: New computational modelling techniques for genomics. Nat. Rev. Genet. 2019, 20, 389–403. [Google Scholar] [CrossRef]
- Rizzo, R.; Fiannaca, A.; La Rosa, M.; Urso, A. A Deep Learning Approach to DNA Sequence Classification. In Proceedings of the Computational Intelligence Methods for Bioinformatics and Biostatistics, Naples, Italy, 10–12 September 2015; Angelini, C., Rancoita, P.M., Rovetta, S., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 129–140. [Google Scholar]
- Nguyen, N.G.; Tran, V.A.; Ngo, D.L.; Phan, D.; Lumbanraja, F.R.; Faisal, M.R.; Abapihi, B.; Kubo, M.; Satou, K. DNA sequence classification by convolutional neural network. J. Biomed. Sci. Eng. 2016, 9, 280. [Google Scholar] [CrossRef] [Green Version]
- Tampuu, A.; Bzhalava, Z.; Dillner, J.; Vicente, R. ViraMiner: Deep learning on raw DNA sequences for identifying viral genomes in human samples. PLoS ONE 2019, 14, e0222271. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Song, K.; Deng, C.; Ahlgren, N.A.; Fuhrman, J.A.; Li, Y.; Xie, X.; Poplin, R.; Sun, F. Identifying viruses from metagenomic data using deep learning. Quant. Biol. 2020, 8, 64–77. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Rincon, A.; Tonda, A.; Mendoza-Maldonado, L.; Claassen, E.; Garssen, J.; Kraneveld, A.D. Accurate Identification of SARS-CoV-2 from Viral Genome Sequences using Deep Learning. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Shang, J.; Sun, Y. CHEER: HierarCHical taxonomic classification for viral mEtagEnomic data via deep leaRning. Methods 2021, 189, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, M.G.F.; Câmara, G.B.M.; Barbosa, R.d.M.; Fernandes, M.A.C. Deep learning based on stacked sparse autoencoder applied to viral genome classification of SARS-CoV-2 virus. bioRxiv 2021. [Google Scholar] [CrossRef]
- Fernandes, M.A.C. k-mers 1D and 2D representation dataset of SARS-CoV-2 nucleotide sequences. Mendeley Data 2020. [Google Scholar] [CrossRef]
- Mahmud, M.; Kaiser, M.S.; Hussain, A.; Vassanelli, S. Applications of Deep Learning and Reinforcement Learning to Biological Data. IEEE Trans. Neural Networks Learn. Syst. 2018, 29, 2063–2079. [Google Scholar] [CrossRef] [Green Version]
- Acheson, N.H. Fundamentals of Molecular Virology; Wiley: Hoboken, NJ, USA, 2007. [Google Scholar]
- Fabijańska, A.; Grabowski, S. Viral genome deep classifier. IEEE Access 2019, 7, 81297–81307. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Datasets | Content | Number of Classes |
|---|---|---|
| Dataset1 | 1553 samples | SARS-CoV-2 species |
| Dataset2 | 14,684 samples | 4 realms 95 families 1160 genera 36 subgenera |
| Mini-Batch Size | 1st Convolutional Filter | 2nd Convolutional Filter | 3rd Convolutional Filter | Stride Size | K-Fold Size | CNN Accuracy (%) | Standard Deviation |
|---|---|---|---|---|---|---|---|
| 128 | 1 | 10 | |||||
| 128 | 2 | 10 | |||||
| 64 | 2 | 10 | |||||
| 128 | 2 | 10 | |||||
| 128 | 2 | 5 |
| Experiment | Validation Accuracy (Mean ± Standard Deviation) | Validation Error (Mean ± Standard Deviation) |
|---|---|---|
| Exp1 | ||
| Exp2 | ||
| Exp3 | ||
| Exp4 |
| Reference | Maximum Sequence Size (bp) | Dataset Size (Numbers of Sequences) | Number of Convolutional Layers | Accuracy | AUROC |
|---|---|---|---|---|---|
| Fabijańska and | 5 | – | – | ||
| Grabowski (2019) [47] | |||||
| Ren et al. (2020) [40] | 3000 | 1 | – | ||
| Tampuue et al. (2019) [39] | 300 | – | 2 | – | |
| Lopez-Rincon et al. (2020) [41] | 553 | 3 | – | ||
| Shang and Sun (2020) [42] | 250 | 4 | 79– | - | |
| This work | 3 | – |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Câmara, G.B.M.; Coutinho, M.G.F.; Silva, L.M.D.d.; Gadelha, W.V.d.N.; Torquato, M.F.; Barbosa, R.d.M.; Fernandes, M.A.C. Convolutional Neural Network Applied to SARS-CoV-2 Sequence Classification. Sensors 2022, 22, 5730. https://doi.org/10.3390/s22155730
Câmara GBM, Coutinho MGF, Silva LMDd, Gadelha WVdN, Torquato MF, Barbosa RdM, Fernandes MAC. Convolutional Neural Network Applied to SARS-CoV-2 Sequence Classification. Sensors. 2022; 22(15):5730. https://doi.org/10.3390/s22155730
Chicago/Turabian StyleCâmara, Gabriel B. M., Maria G. F. Coutinho, Lucileide M. D. da Silva, Walter V. do N. Gadelha, Matheus F. Torquato, Raquel de M. Barbosa, and Marcelo A. C. Fernandes. 2022. "Convolutional Neural Network Applied to SARS-CoV-2 Sequence Classification" Sensors 22, no. 15: 5730. https://doi.org/10.3390/s22155730
APA StyleCâmara, G. B. M., Coutinho, M. G. F., Silva, L. M. D. d., Gadelha, W. V. d. N., Torquato, M. F., Barbosa, R. d. M., & Fernandes, M. A. C. (2022). Convolutional Neural Network Applied to SARS-CoV-2 Sequence Classification. Sensors, 22(15), 5730. https://doi.org/10.3390/s22155730