
On the Prediction of In Vitro Arginine Glycation of Short Peptides Using Artificial Neural Networks

 ,
,  ,
, 
Abstract
:1. Introduction
2. Methodology
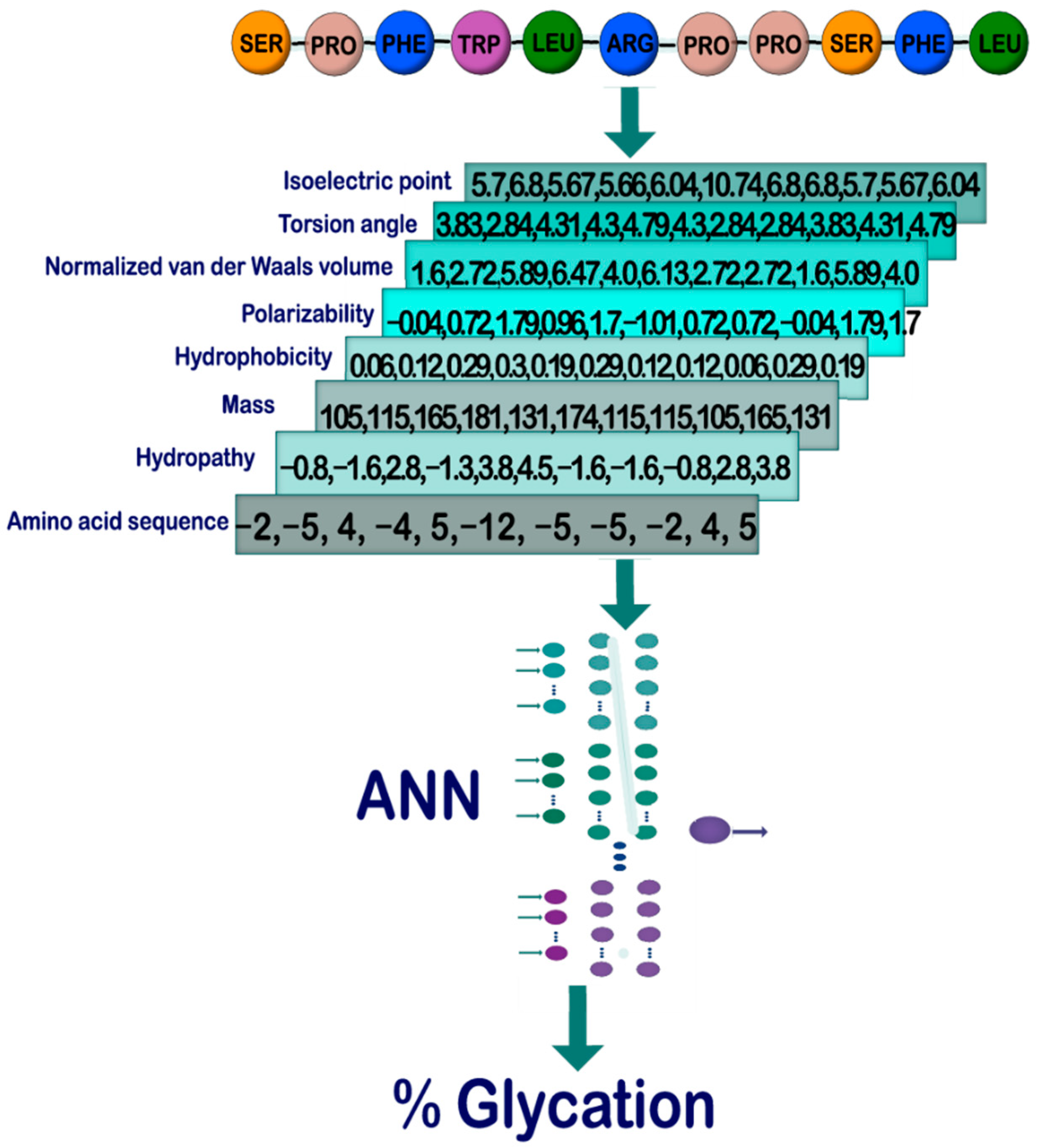
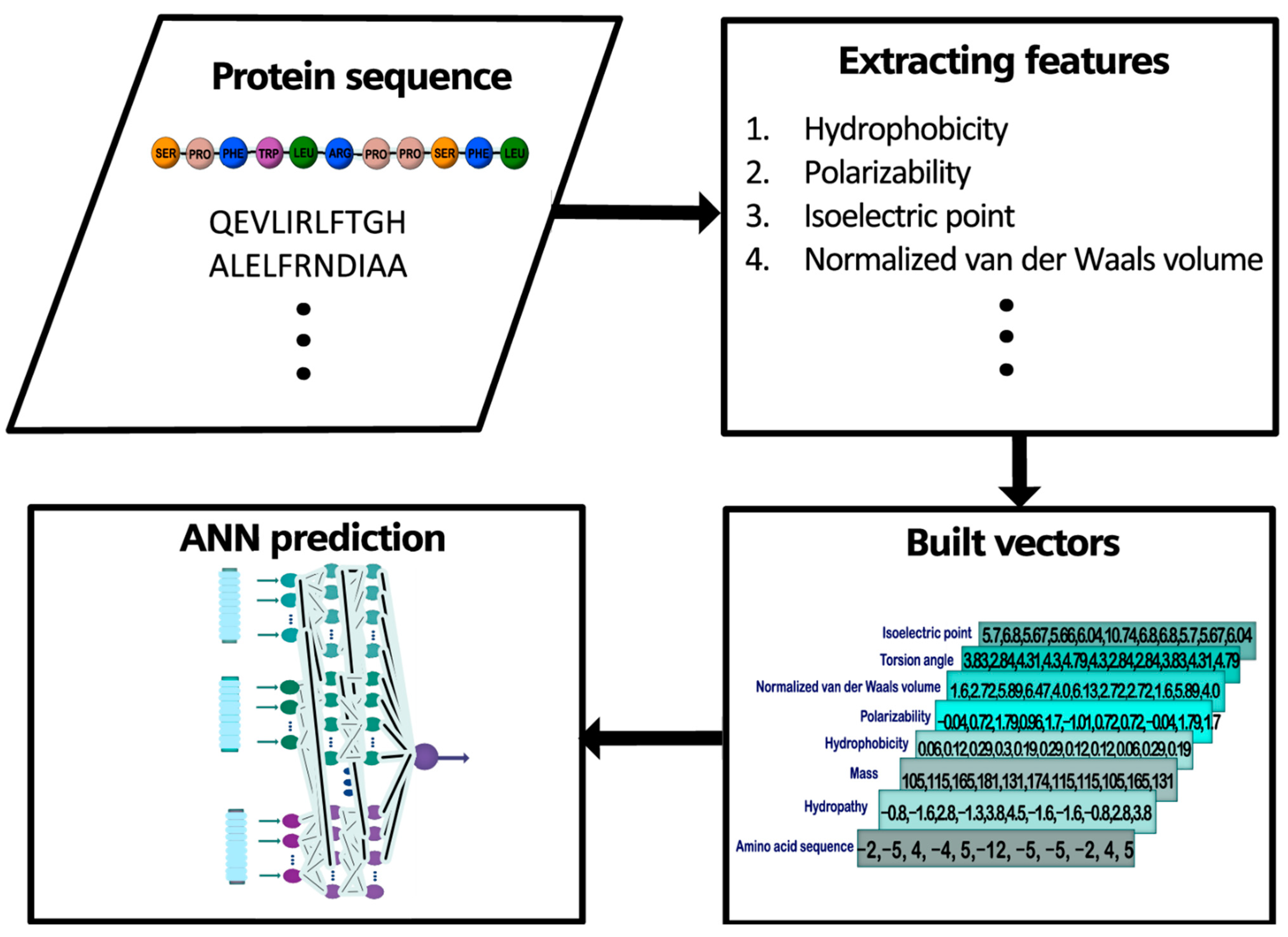
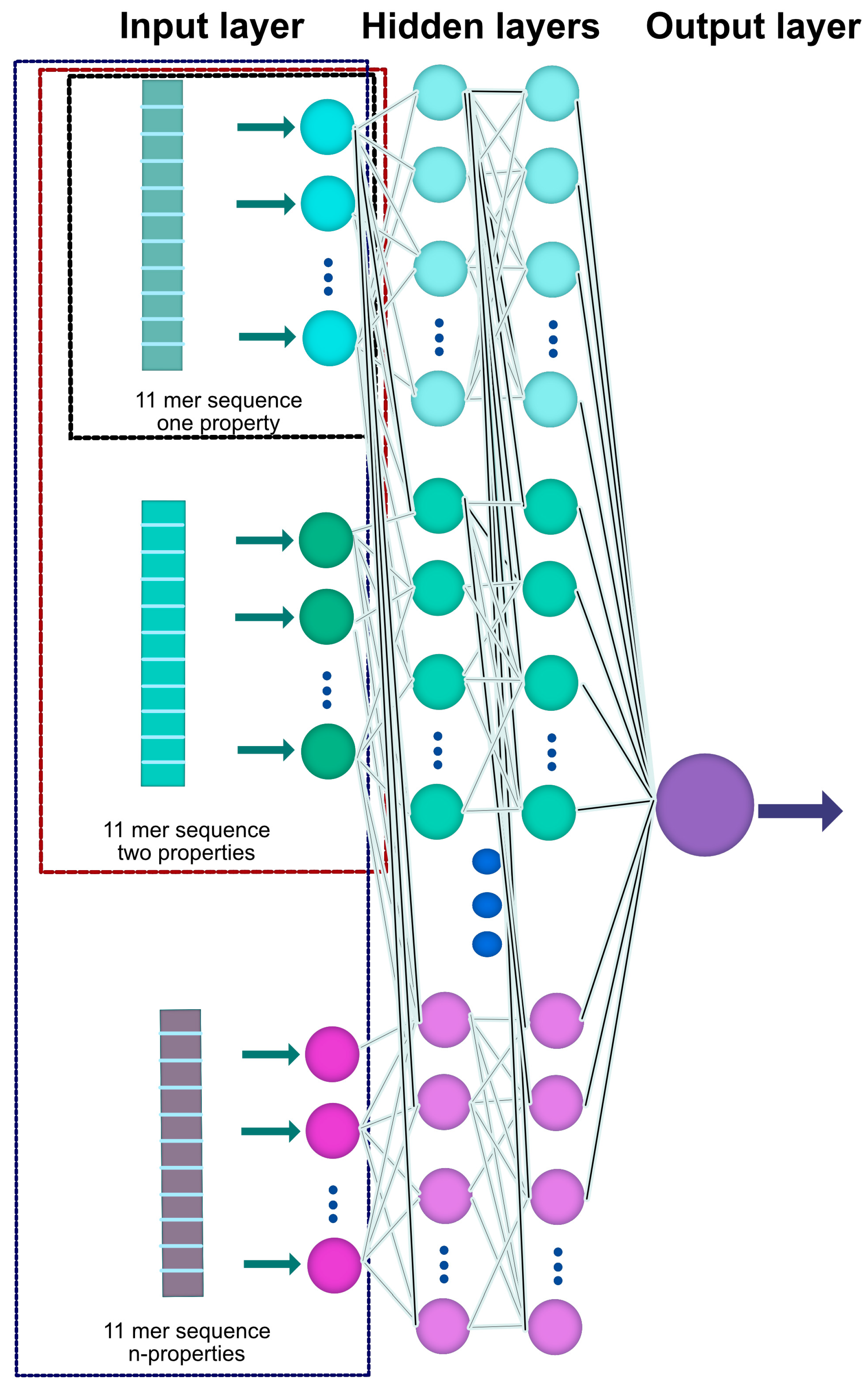
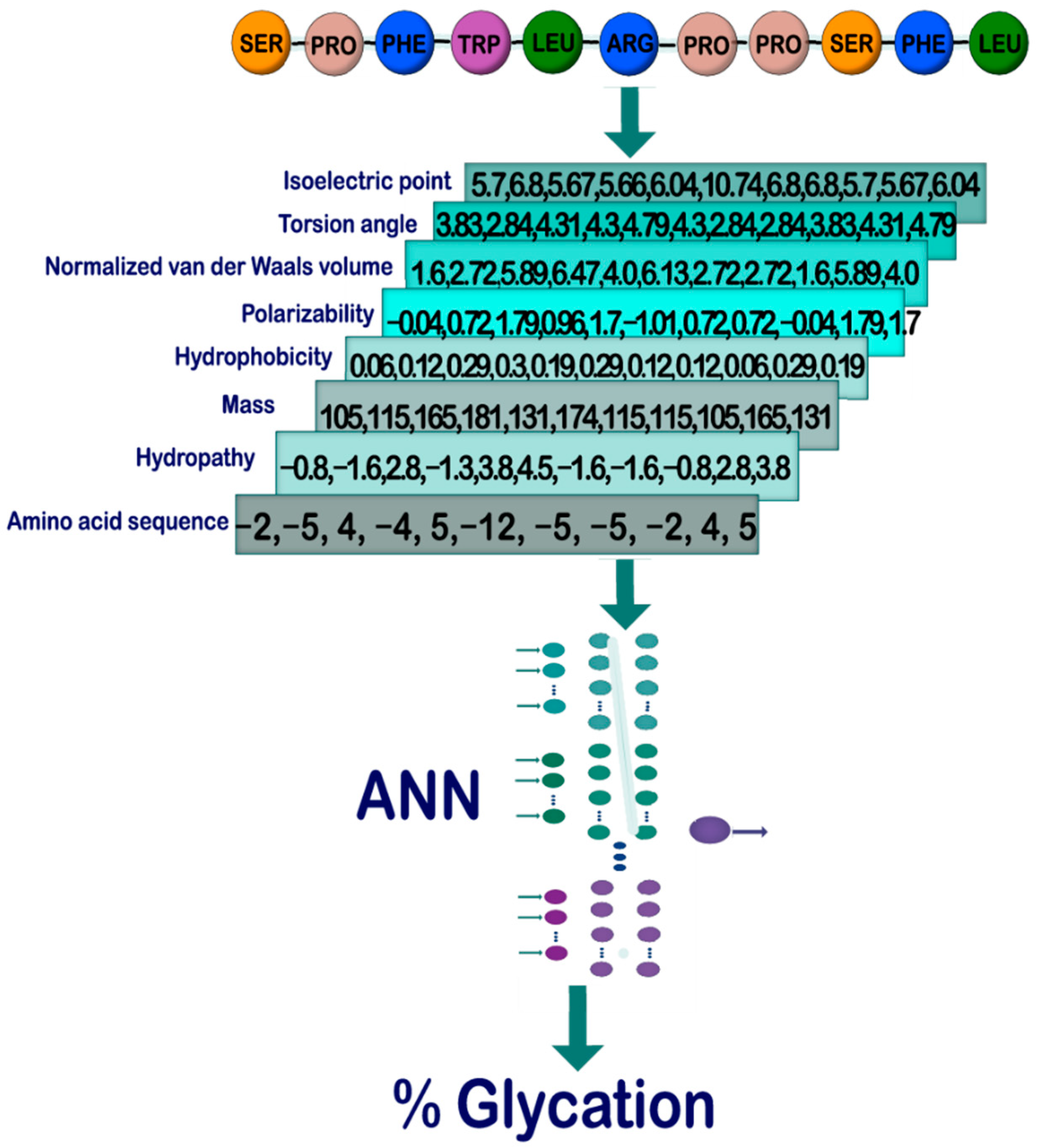
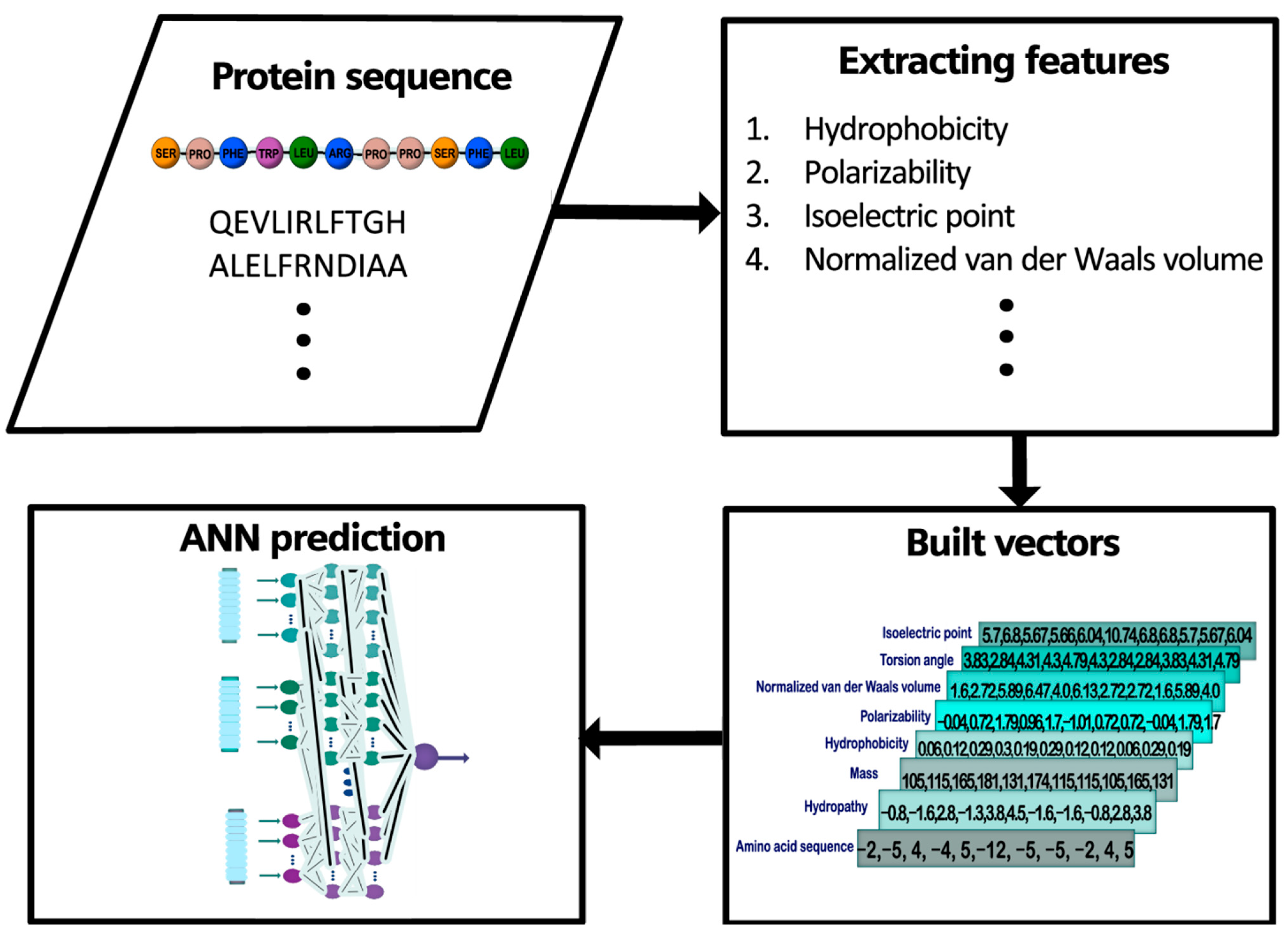
2.1. General Outline
2.2. Database Construction and Study Cases
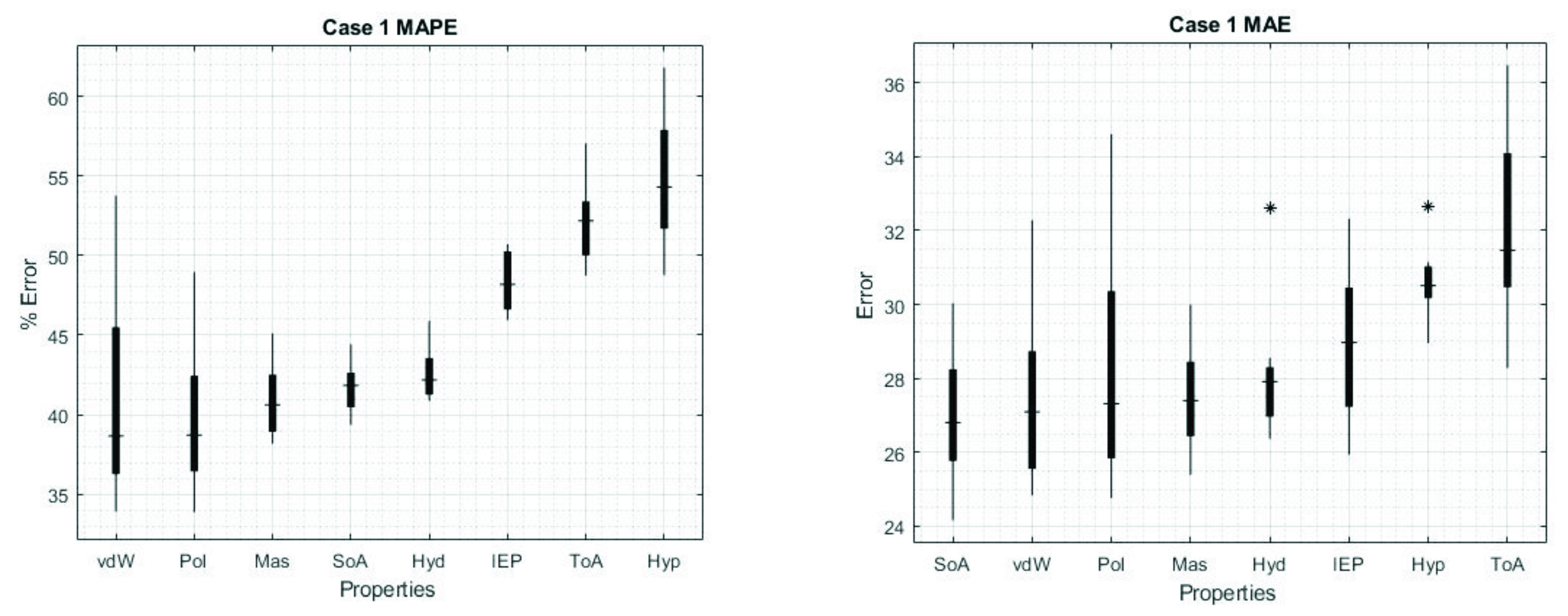
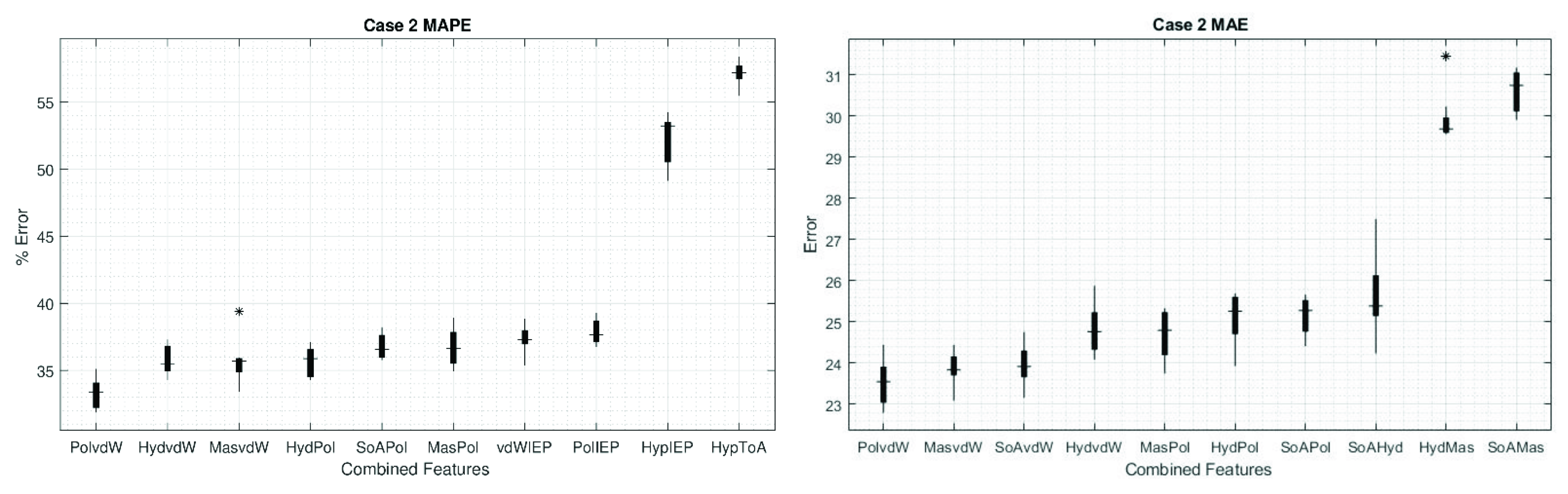
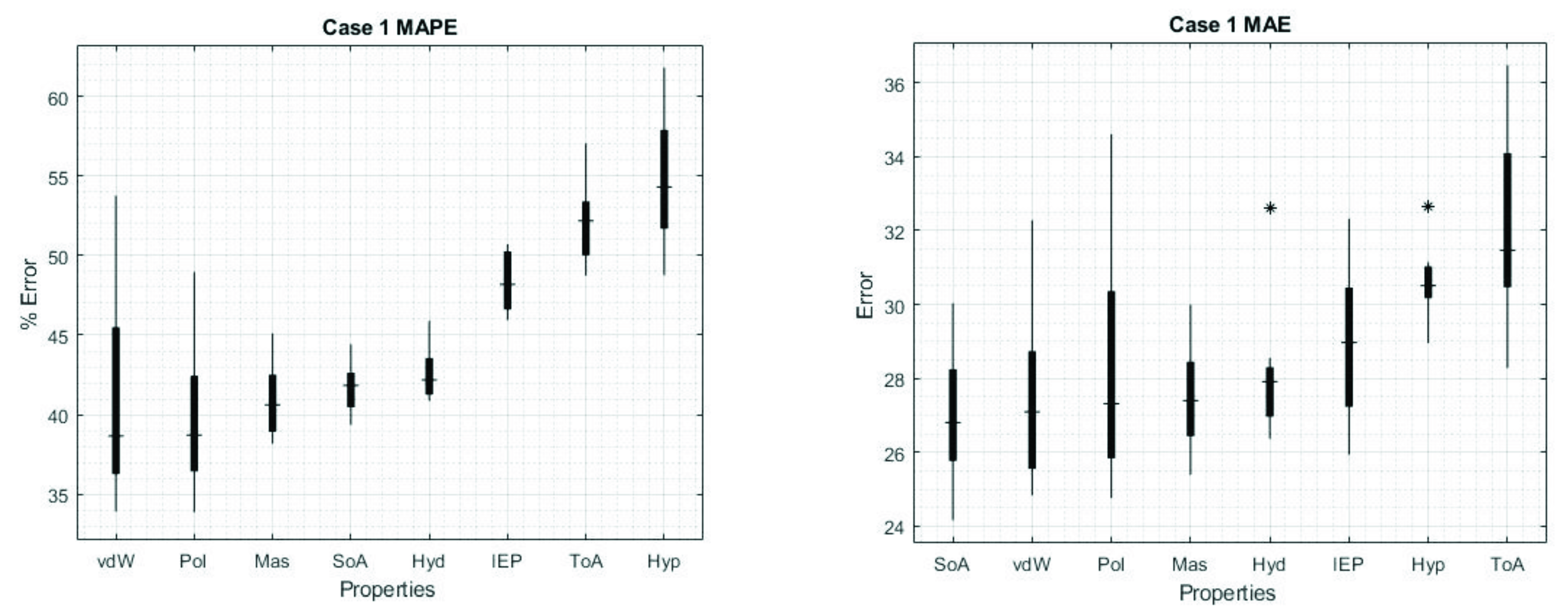
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sigal, A.; Milo, R.; Cohen, A.; Geva-Zatorsky, N.; Klein, Y.; Liron, Y.; Rosenfeld, N.; Danon, T.; Perzov, N.; Alon, U. Variability and Memory of Protein Levels in Human Cells. Nature 2006, 444, 643–646. [Google Scholar] [CrossRef]
- Ponomarenko, E.A.; Poverennaya, E.V.; Ilgisonis, E.V.; Pyatnitskiy, M.A.; Kopylov, A.T.; Zgoda, V.G.; Lisitsa, A.V.; Archakov, A.I. The Size of the Human Proteome: The Width and Depth. Int. J. Anal. Chem. 2016, 2016, 7436849. [Google Scholar] [CrossRef]
- Ho, J.M.L.; Miller, C.A.; Smith, K.A.; Mattia, J.R.; Bennett, M.R. Improved Pyrrolysine Biosynthesis through Phage Assisted Non-Continuous Directed Evolution of the Complete Pathway. Nat. Commun. 2021, 12, 3914. [Google Scholar] [CrossRef]
- Müller, M.M. Post-Translational Modifications of Protein Backbones: Unique Functions, Mechanisms, and Challenges. Biochemistry 2018, 57, 177–185. [Google Scholar] [CrossRef]
- Gavin, J.W.; Jon, S.T.; Toone, E.J. Natural Product Glycosyltransferases: Properties and Applications. Adv. Enzymol. Relat. Areas Mol. Biol. 2009, 76, 55–119. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, Z.; Jia, J.; Du, T.; Zhang, N.; Tang, Y.; Fang, Y.; Fang, D. Overview of Histone Modification. In Advances in Experimental Medicine and Biology; Springer Nature: Berlin/Heidelberg, Germany, 2021; pp. 1–16. [Google Scholar] [CrossRef]
- Rabbani, N.; Thornalley, P.J. Dicarbonyl Stress in Cell and Tissue Dysfunction Contributing to Ageing and Disease. Biochem. Biophys. Res. Commun. 2015, 458, 221–226. [Google Scholar] [CrossRef]
- Ahmed, N.; Babaei-Jadidi, R.; Howell, S.K.; Beisswenger, P.J.; Thornalley, P.J. Degradation Products of Proteins Damaged by Glycation, Oxidation and Nitration in Clinical Type 1 Diabetes. Diabetologia 2005, 48, 1590–1603. [Google Scholar] [CrossRef]
- Oya, T.; Hattori, N.; Mizuno, Y.; Miyata, S.; Maeda, S.; Osawa, T.; Uchida, K. Methylglyoxal Modification of Protein. Chemical and Immunochemical Characterization of Methylglyoxal-Arginine Adducts. J. Biol. Chem. 1999, 274, 18492–18502. [Google Scholar] [CrossRef]
- Rabbani, N.; Thornalley, P.J. Protein Glycation—Biomarkers of Metabolic Dysfunction and Early-Stage Decline in Health in the Era of Precision Medicine. Redox Biol. 2021, 42, 101920. [Google Scholar] [CrossRef]
- Mercado-Uribe, H.; Andrade-Medina, M.; Espinoza-Rodríguez, J.H.; Carrillo-Tripp, M.; Scheckhuber, C.Q. Analyzing Structural Alterations of Mitochondrial Intermembrane Space Superoxide Scavengers Cytochrome-c and SOD1 after Methylglyoxal Treatment. PLoS ONE 2020, 15, e0232408. [Google Scholar] [CrossRef]
- Phillips, S.A.; Thornalley, P.J. The Formation of Methylglyoxal from Triose Phosphates. Investigation Using a Specific Assay for Methylglyoxal. Eur. J. Biochem. 1993, 212, 101–105. [Google Scholar] [CrossRef]
- Rabbani, N.; Thornalley, P.J. Measurement of Methylglyoxal by Stable Isotopic Dilution Analysis LC-MS/MS with Corroborative Prediction in Physiological Samples. Nat. Protoc. 2014, 9, 1969–1979. [Google Scholar] [CrossRef]
- Thornalley, P.J. Glyoxalase I--Structure, Function and a Critical Role in the Enzymatic Defence against Glycation. Biochem. Soc. Trans. 2003, 31 Pt 6, 1343–1348. [Google Scholar] [CrossRef]
- Mannervik, B. Molecular Enzymology of the Glyoxalase System. Drug Metabol. Drug Interact. 2008, 23, 13–27. [Google Scholar] [CrossRef]
- Kumar Pasupulati, A.; Chitra, P.S.; Reddy, G.B. Advanced Glycation End Products Mediated Cellular and Molecular Events in the Pathology of Diabetic Nephropathy. Biomol. Concepts 2016, 7, 293–309. [Google Scholar] [CrossRef]
- Schalkwijk, C.G.; Stehouwer, C.D.A. Methylglyoxal, a Highly Reactive Dicarbonyl Compound, in Diabetes, Its Vascular Complications, and Other Age-Related Diseases. Physiol. Rev. 2020, 100, 407–461. [Google Scholar] [CrossRef]
- Morcos, M.; Du, X.; Pfisterer, F.; Hutter, H.; Sayed, A.A.R.; Thornalley, P.; Ahmed, N.; Baynes, J.; Thorpe, S.; Kukudov, G.; et al. Glyoxalase-1 Prevents Mitochondrial Protein Modification and Enhances Lifespan in Caenorhabditis elegans. Aging Cell 2008, 7, 260–269. [Google Scholar] [CrossRef]
- Scheckhuber, C.Q.; Mack, S.J.; Strobel, I.; Ricciardi, F.; Gispert, S.; Osiewacz, H.D. Modulation of the Glyoxalase System in the Aging Model Podospora Anserina: Effects on Growth and Lifespan. Aging 2010, 2, 969–980. [Google Scholar] [CrossRef]
- Fan, X.; Monnier, V.M. Protein Posttranslational Modification (PTM) by Glycation: Role in Lens Aging and Age-Related Cataractogenesis. Exp. Eye Res. 2021, 210, 108705. [Google Scholar] [CrossRef]
- Scheckhuber, C.Q. Studying the Mechanisms and Targets of Glycation and Advanced Glycation End-Products in Simple Eukaryotic Model Systems. Int. J. Biol. Macromol. 2019, 127, 85–94. [Google Scholar] [CrossRef]
- Sjoblom, N.M.; Kelsey, M.M.G.; Scheck, R.A. A Systematic Study of Selective Protein Glycation. Angew. Chem. Int. Ed. 2018, 57, 16077–16082. [Google Scholar] [CrossRef]
- Johansen, M.B.; Kiemer, L.; Brunak, S. Analysis and Prediction of Mammalian Protein Glycation. Glycobiology 2006, 16, 844–853. [Google Scholar] [CrossRef]
- Rabuñal, J.R.; Dorado, J. Artificial Neural Networks in Real-Life Applications. In Artificial Neural Networks in Real-Life Applications; IGI Global: Hershey, PA, USA, 2006; pp. 1–375. [Google Scholar] [CrossRef]
- Ju, Z.; Sun, J.; Li, Y.; Wang, L. Predicting Lysine Glycation Sites Using Bi-Profile Bayes Feature Extraction. Comput. Biol. Chem. 2017, 71, 98–103. [Google Scholar] [CrossRef]
- Liu, Y.; Gu, W.; Zhang, W.; Wang, J. Predict and Analyze Protein Glycation Sites with the MRMR and IFS Methods. Biomed. Res. Int. 2015, 2015, 561547. [Google Scholar] [CrossRef]
- Yu, J.; Shi, S.; Zhang, F.; Chen, G.; Cao, M. PredGly: Predicting Lysine Glycation Sites for Homo Sapiens Based on XGboost Feature Optimization. Bioinformatics 2019, 35, 2749–2756. [Google Scholar] [CrossRef]
- Xu, Y.; Li, L.; Ding, J.; Wu, L.Y.; Mai, G.; Zhou, F. Gly-PseAAC: Identifying Protein Lysine Glycation through Sequences. Gene 2017, 602, 1–7. [Google Scholar] [CrossRef]
- Zhao, X.; Zhao, X.; Bao, L.; Zhang, Y.; Dai, J.; Yin, M. Glypre: In Silico Prediction of Protein Glycation Sites by Fusing Multiple Features and Support Vector Machine. Molecules 2017, 22, 1891. [Google Scholar] [CrossRef]
- Islam, M.M.; Saha, S.; Rahman, M.M.; Shatabda, S.; Farid, D.M.; Dehzangi, A. IProtGly-SS: Identifying Protein Glycation Sites Using Sequence and Structure Based Features. Proteins Struct. Funct. Bioinform. 2018, 86, 777–789. [Google Scholar] [CrossRef]
- Reddy, H.M.; Sharma, A.; Dehzangi, A.; Shigemizu, D.; Chandra, A.A.; Tsunoda, T. GlyStruct: Glycation Prediction Using Structural Properties of Amino Acid Residues. BMC Bioinform. 2019, 19, 547. [Google Scholar] [CrossRef]
- Xu, H.; Zhou, J.; Lin, S.; Deng, W.; Zhang, Y.; Xue, Y. PLMD: An Updated Data Resource of Protein Lysine Modifications. J. Genet. Genom. 2017, 44, 243–250. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, Y.; Gao, T.; Pan, Z.; Cheng, H.; Yang, Q.; Cheng, Z.; Guo, A.; Ren, J.; Xue, Y. CPLM: A Database of Protein Lysine Modifications. Nucleic Acids Res. 2014, 42, D531–D536. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Cao, J.; Gao, X.; Zhou, Y.; Wen, L.; Yang, X.; Yao, X.; Ren, J.; Xue, Y. CPLA 1.0: An Integrated Database of Protein Lysine Acetylation. Nucleic Acids Res. 2011, 39 (Suppl. 1), D1029–D1034. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Xue, M.; Thornalley, P.J. Dicarbonyls and Glyoxalase in Disease Mechanisms and Clinical Therapeutics. Glycoconj. J. 2016, 33, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, K.; Koike, S.; Suzuki, T.; Ogasawara, Y. Carbonylation of Skin Collagen Induced by Reaction with Methylglyoxal. Biochem. Biophys. Res. Commun. 2021, 562, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Toyoshima, M.; Hisano, Y.; Balan, S.; Iwayama, Y.; Aono, H.; Futamura, Y.; Osada, H.; Owada, Y.; Yoshikawa, T. Glyoxalase I Disruption and External Carbonyl Stress Impair Mitochondrial Function in Human Induced Pluripotent Stem Cells and Derived Neurons. Transl. Psychiatry 2021, 11, 275. [Google Scholar] [CrossRef]
- Bora, S.; Adole, P.S.; Motupalli, N.; Pandit, V.R.; Vinod, K.V. Association between Carbonyl Stress Markers and the Risk of Acute Coronary Syndrome in Patients with Type 2 Diabetes Mellitus–A Pilot Study. Diabetes Metab. Syndr. Clin. Res. Rev. 2020, 14, 1751–1755. [Google Scholar] [CrossRef]
- Al-Naser, M.; Elshafei, M.; Al-Sarkhi, A. Artificial Neural Network Application for Multiphase Flow Patterns Detection: A New Approach. J. Pet. Sci. Eng. 2016, 145, 548–564. [Google Scholar] [CrossRef]
- Rosa, E.S.; Salgado, R.M.; Ohishi, T.; Mastelari, N. Performance Comparison of Artificial Neural Networks and Expert Systems Applied to Flow Pattern Identification in Vertical Ascendant Gas-Liquid Flows. Int. J. Multiph. Flow 2010, 36, 738–754. [Google Scholar] [CrossRef]
- Kingma, D.P.; Ba, J. Adam: A Method for Stochastic Optimization. In Proceedings of the 3rd International Conference on Learning Representations, San Diego, CA, USA, 7–9 May 2015. [Google Scholar]
- Eckle, K.; Schmidt-Hieber, J. A Comparison of Deep Networks with ReLU Activation Function and Linear Spline-Type Methods. Neural Netw. 2019, 110, 232–242. [Google Scholar] [CrossRef]
- Rumelhart, D.E.; Hinton, G.E.; Williams, R.J. Learning Representations by Back-Propagating Errors. Nature 1986, 323, 533–536. [Google Scholar] [CrossRef]
- Pan, C.; Tabatabaei, S.K.; Tabatabaei Yazdi, S.M.H.; Hernandez, A.G.; Schroeder, C.M.; Milenkovic, O. Rewritable Two-Dimensional DNA-Based Data Storage with Machine Learning Reconstruction. Nat. Commun. 2022, 13, 2984. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Wang, Z.; Wang, H.; Pang, Y.; Lee, T.-Y. Characterization and Identification of Lysine Crotonylation Sites Based on Machine Learning Method on Both Plant and Mammalian. Sci. Rep. 2020, 10, 20447. [Google Scholar] [CrossRef] [PubMed]
- Scheckhuber, C.Q. Arg354 in the Catalytic Centre of Bovine Liver Catalase Is Protected from Methylglyoxal-Mediated Glycation. BMC Res. Notes 2015, 8, 830. [Google Scholar] [CrossRef] [PubMed]
- Markus, G.; Tritsch, G.L.; Parthasarathy, R. A model for hydropathy-based peptide interactions. Arch. Biochem. Biophys. 1989, 272, 433–439. [Google Scholar] [CrossRef]
- Chiavari, G.; Galletti, G.C. Pyrolysis—gas chromatography/mass spectrometry of amino acids. J. Anal. Appl. Pyrolysis 1992, 24, 123–137. [Google Scholar] [CrossRef]
- Fauchère, J.L.; Charton, M.; Kier, L.B.; Verloop, A.; Pliska, V. Amino acid side chain parameters for correlation studies in biology and pharmacology. Int. J. Pept. Protein Res. 1988, 32, 269–278. [Google Scholar] [CrossRef]
- Lefranc, M.-P. Amino Acids. 2001. Available online: https://www.imgt.org/IMGTeducation/Aide-memoire/_UK/aminoacids/ (accessed on 7 June 2021).
- Bachem. Peptide Calculator. 2017. Available online: https://www.bachem.com/knowledge-center/peptide-calculator/ (accessed on 7 June 2021).
- Jha, K.; Saha, S.; Tanveer, M. Prediction of protein-protein interactions using stacked auto-encoder. Trans. Emerging Tel Technol. 2021, e4256. [Google Scholar] [CrossRef]
- Rosenblatt, F. Principles of Neurodynamics: Perceptrons and the Theory of Brain Mechanisms; Spartan Books: Washington, DC, USA, 1962. [Google Scholar]
- Rojas, R. Neural Networks: A Systematic Introduction; Springer: Berlin/Heidelberg, Germany, 1996. [Google Scholar]
- Bishop, C. Machine Learning for Pattern Recognition; Springer: Berlin/Heidelberg, Germany, 2006. [Google Scholar]
- Goodfellow, I.; Bengio, Y.; Courville, A. A Deep Learning; The MIT Press: Cambridge, MA, USA, 2016. [Google Scholar]
- Ketkar, N. Deep Learning with Python: A Hands-on Introduction; Apress, NYC: New York, NY, USA, 2017. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cases | Features | ANN Layer Architecture |
|---|---|---|
| Case 1A | Sequence of amino acid | 11 × 4 × 3 × 2 × 1 |
| Case 1B | Hydropathy | 11 × 4 × 3 × 2 × 1 |
| Case 1C | Mass | 11 × 4 × 3 × 2 × 1 |
| Case 1D | Polarizability | 11 × 4 × 3 × 2 × 1 |
| Case 1E | Hydrophobicity | 11 × 4 × 3 × 2 × 1 |
| Case 1F | Normalized van der Waals volume | 11 × 4 × 3 × 2 × 1 |
| Case 1G | Torsion angle | 11 × 4 × 3 × 2 × 1 |
| Case 1H | Isoelectric point | 11 × 4 × 3 × 2 × 1 |
| Case 2 | Polarization + normalized van der Waals volume | 22 × 10 × 1 |
| Case 3 | Sequence of amino acid + hydropathy + normalized van der Waals volume | 33 × 40 × 1 |
| MAPE | ||||||||
|---|---|---|---|---|---|---|---|---|
| vdW | Mas | Pol | SoA | Hyd | IEP | ToA | Hyp | |
| vdW | 39.75/27.49 | 35.71 | 33.3 | 37.61 | 35.77 | 37.34 | 40.62 | 39.45 |
| Mas | 24.83 | 40.93/27.5 | 36.74 | 46.28 | 40 | 39.05 | 46.47 | 39.36 |
| Pol | 23.53 | 25.5 | 41.06/28.3 | 36.79 | 35.66 | 37.88 | 41.51 | 41.73 |
| SoA | 25.63 | 29.93 | 25.74 | 41.71/26.98 | 40 | 46.59 | 44.96 | 44.74 |
| Hyd | 25.69 | 29.87 | 25.66 | 26.36 | 42.6/28.17 | 45.4 | 46.58 | 46.22 |
| IEP | 23.96 | 26.11 | 23.87 | 26.81 | 27.02 | 48.34/28.95 | 46.15 | 52.23 |
| ToA | 24.69 | 28.85 | 25.15 | 28.42 | 28.83 | 26.08 | 52.12/32.11 | 57.13 |
| Hyp | 26.21 | 26.77 | 26.51 | 27.48 | 27.8 | 27.57 | 30.61 | 54.79/30.63 |
| MAE |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Que-Salinas, U.; Martinez-Peon, D.; Reyes-Figueroa, A.D.; Ibarra, I.; Scheckhuber, C.Q. On the Prediction of In Vitro Arginine Glycation of Short Peptides Using Artificial Neural Networks. Sensors 2022, 22, 5237. https://doi.org/10.3390/s22145237
Que-Salinas U, Martinez-Peon D, Reyes-Figueroa AD, Ibarra I, Scheckhuber CQ. On the Prediction of In Vitro Arginine Glycation of Short Peptides Using Artificial Neural Networks. Sensors. 2022; 22(14):5237. https://doi.org/10.3390/s22145237
Chicago/Turabian StyleQue-Salinas, Ulices, Dulce Martinez-Peon, Angel D. Reyes-Figueroa, Ivonne Ibarra, and Christian Quintus Scheckhuber. 2022. "On the Prediction of In Vitro Arginine Glycation of Short Peptides Using Artificial Neural Networks" Sensors 22, no. 14: 5237. https://doi.org/10.3390/s22145237
APA StyleQue-Salinas, U., Martinez-Peon, D., Reyes-Figueroa, A. D., Ibarra, I., & Scheckhuber, C. Q. (2022). On the Prediction of In Vitro Arginine Glycation of Short Peptides Using Artificial Neural Networks. Sensors, 22(14), 5237. https://doi.org/10.3390/s22145237