
To Assist Oncologists: An Efficient Machine Learning-Based Approach for Anti-Cancer Peptides Classification

 ,
,  , ,
, ,  and
and
Abstract
1. Introduction
- Prior peptide classification models were developed using a single feature descriptors method without any modification that captured meaningless information against each peptide sequence.
- To improve the accuracy of peptide classification, most of the studies followed a fusion strategy to collect a diverse and massive number of features, resulting in homogeneous patterns and high dimension descriptors that affect the model performance.
- Due to the lack of an effective vaccine, the increase in drug-resistant, and the fatal nature of cancer, we present a novel intelligent framework for efficiently distinguishing anticancer peptides from unstructured peptides sequences. The proposed paradigm is beneficial to the development of vaccines against cancer peptides.
- The variety and numbers of peptides in databanks are rapidly increasing due to the advancement of sequence technology. We deeply investigated the literature and concluded that most of the researchers use numerous flavors of encoding techniques, which exhibit poor performance when extracting contextual information from peptide sequences, resulting in non-representative algorithms. In this paper, a novel approach is presented that engages a diverse collection of features to obtain using statistical methods. The proposed model extracts contextual features to accurately categorize the nature of peptides.
- The PseAAC method demonstrates an incredible performance in various protein sequence classification that comprises three physicochemical properties including hydrophilicity, hydrophobicity, and charge of basic amino acids however, sometimes it gives poor results when the sequence of peptides is short in length. To improve the prediction strength, we added some new physicochemical properties including flexibility, irreplaceability, solvent accessible surface area, polarity, polarizability, and rigidity.
- We performed numerous possible combinations of features against an ensemble classifier to evaluate the strength of individual components. The proposed model shows convincing results and provides new state-of-the-art (SOTA) accuracy over testing peptide sequences.
2. Related Work
3. Materials and Methods
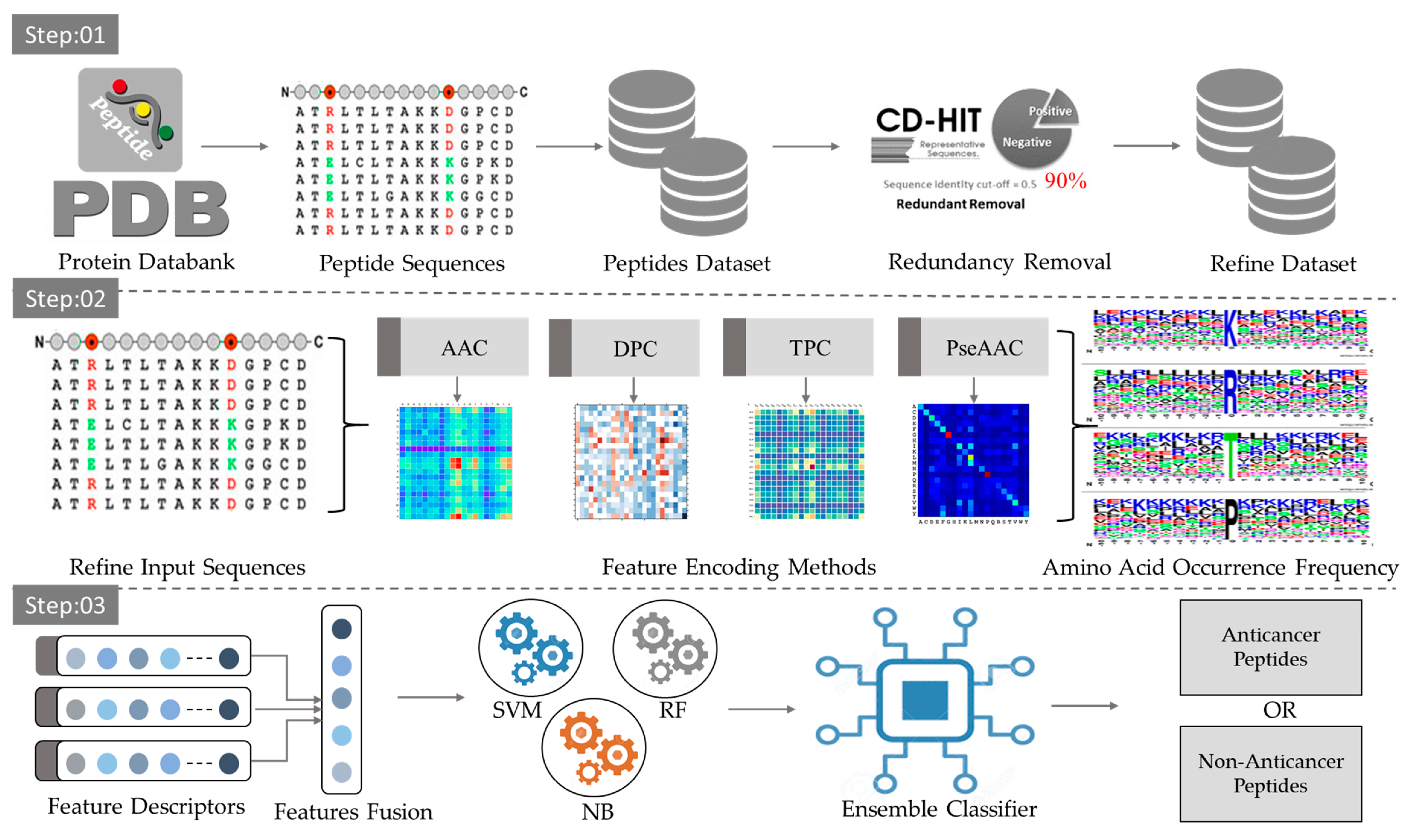
3.1. Dataset
3.2. Preprocessing
3.3. Peptide Encoding Methods
3.3.1. Amino Acid Composition (AAC)
3.3.2. Dipeptide Composition (DPC)
3.3.3. Tripeptide Composition (TPC)
3.3.4. Improved Pseudo Amino Acid Composition (IPseAAC)
3.4. Optimal Feature Selection Technique
- (a)
- The average value of each attribute can be calculated as:
- (b)
- The gap between the average values of X and Xi:
- (c)
- The covariance matrix can be calculated as:where B = {}in(Q*P)
- (d)
- The eigenvalue of Cm is computed as:where the largest eigenvalues ‘∂1’should be less than the highest of second ‘∂2’ and so on.
- (e)
- Evaluate the eigenvector as:
3.5. Classification Algorithms
3.5.1. Support Vector Machine (SVM)
3.5.2. K-Nearest Neighbor (KNN)
3.5.3. Random Forest (RF)
3.5.4. Ensemble Classifier Mechanism
3.6. Model Evaluation
3.6.1. System Configuration and Data Setting
3.6.2. Evaluation Metrics
4. Experimental Results
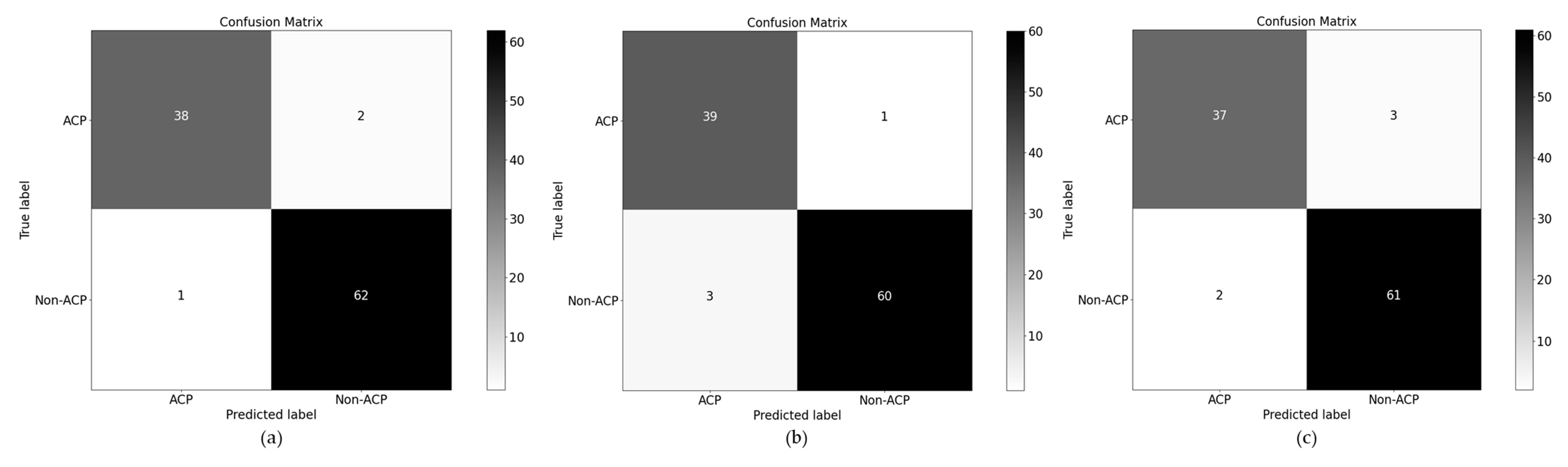
4.1. Ablation Study over PSD1
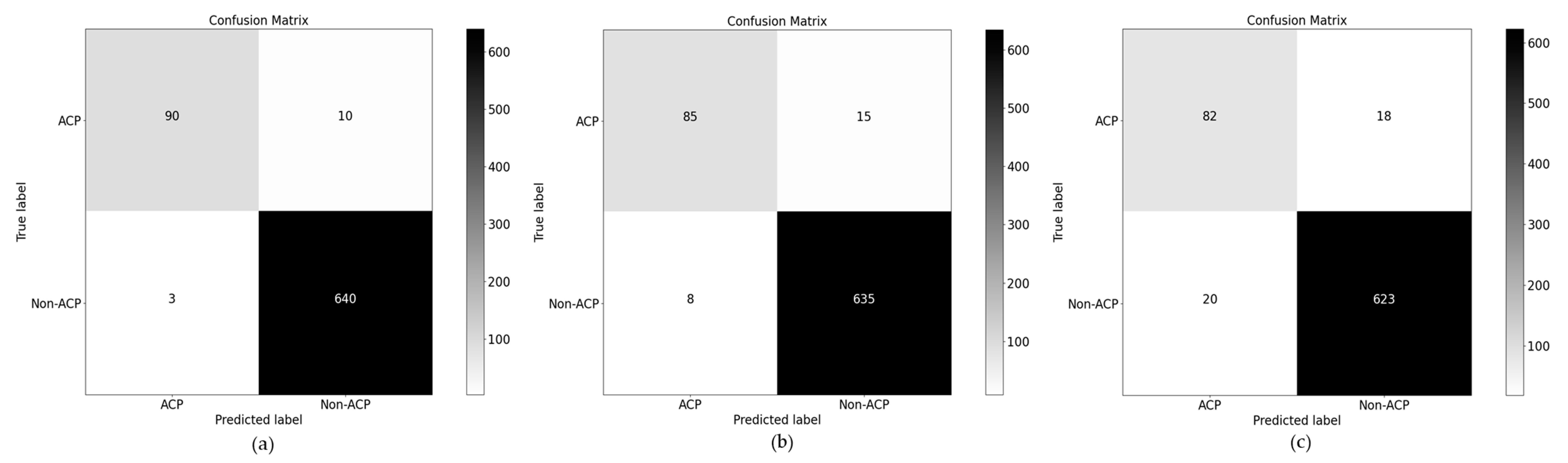
4.2. Ablation Study over PSD2
4.3. Results Assessment with SOTA Methods over PSD1
4.4. Results Assessment with SOTA Methods over PSD2
5. Conclusions and Future Research Direction
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Nomenclature
| AAC | Amino acid composition | SVM | Support vector machine |
| ATC | Atomic composition | PCA | Principal component analysis |
| DPC | Dipeptide composition | ACPs | Anti-cancer peptides |
| MCC | Matthews correlation coefficient | NACPs | Non Anti-cancer peptides |
| ML | Machine learning | TPC | Tripeptide composition |
| RF | Random Forest | NB | Naïve Bayes |
| PseAAC | Pseudo amino acid composition | KNN | K-nearest neighbor |
| IPseAAC | Improved pseudo amino acid composition | LR | Logistic regressions |
| CD-HIT | Cluster database at high identity with tolerance | GRU | Gaited recurrent unit |
| LSTM | Long short-term memory | SOTA | State-of-the-art |
References
- Ferlay, J.; Shin, H.-R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer 2010, 127, 2893–2917. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA A Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Kanavos, P. The rising burden of cancer in the developing world. Ann. Oncol. 2006, 17, viii15–viii23. [Google Scholar] [CrossRef] [PubMed]
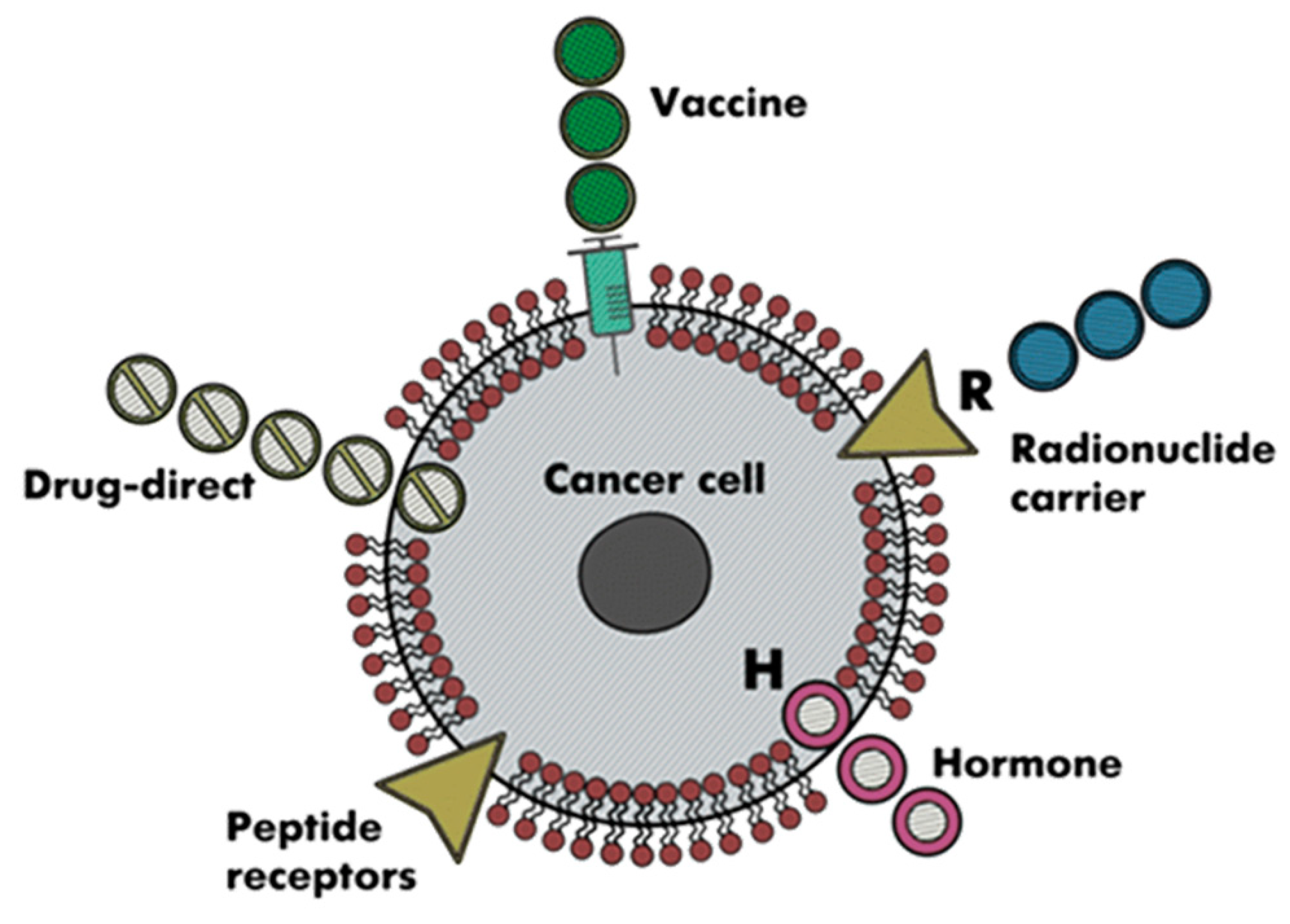
- Thundimadathil, J. Cancer Treatment Using Peptides: Current Therapies and Future Prospects. J. Amino Acids 2012, 2012, 967347. [Google Scholar] [CrossRef] [PubMed]
- Harris, F.; Dennison, S.R.; Singh, J.; Phoenix, D.A. On the selectivity and efficacy of defense peptides with respect to cancer cells. Med. Res. Rev. 2011, 33, 190–234. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, I.; Fernando, J.; Mainez, J.; Sancho, P. TGF-beta Signaling in Cancer Treatment. Curr. Pharm. Des. 2014, 20, 2934–2947. [Google Scholar] [CrossRef]
- Karbalaeemohammad, S.; Naderi-Manesh, H. Two novel anticancer peptides from Aurein1. 2. Int. J. Pept. Res. Ther. 2011, 17, 159–164. [Google Scholar] [CrossRef]
- Khan, F.; Akbar, S.; Basit, A.; Khan, I.; Akhlaq, H. Identification of anticancer peptides using optimal feature space of Chou’s split amino acid composition and support vector machine. In Proceedings of the 2017 4th International Conference on Biomedical and Bioinformatics Engineering, Seoul, Korea, 12–14 November 2017. [Google Scholar]
- Virnig, B.A.; Baxter, N.N.; Habermann, E.B.; Feldman, R.D.; Bradley, C.J. A Matter Of Race: Early-Versus Late-Stage Cancer Diagnosis. Health Aff. 2009, 28, 160–168. [Google Scholar] [CrossRef]
- Hazelton, W.D.; Luebeck, E.G. Biomarker-based early cancer detection: Is it achievable? Sci. Transl. Med. 2011, 3, 109fs9. [Google Scholar] [CrossRef]
- Omenn, G.S. Strategies for Genomic and Proteomic Profiling of Cancers. Stat. Biosci. 2014, 8, 1–7. [Google Scholar] [CrossRef]
- Mahassni, S.H.; Al-Reemi, R.M. Apoptosis and necrosis of human breast cancer cells by an aqueous extract of garden cress (Lepidium sativum) seeds. Saudi J. Biol. Sci. 2013, 20, 131–139. [Google Scholar] [CrossRef]
- Gerber, B.; Freund, M.; Reimer, T. Recurrent breast cancer: Treatment strategies for maintaining and prolonging good quality of life. Dtsch. Arztebl. Int. 2010, 107, 85. [Google Scholar]
- Marqus, S.; Pirogova, E.; Piva, T.J. Evaluation of the use of therapeutic peptides for cancer treatment. J. Biomed. Sci. 2017, 24, 21. [Google Scholar] [CrossRef]
- McGregor, D.P. Discovering and improving novel peptide therapeutics. Curr. Opin. Pharmacol. 2008, 8, 616–619. [Google Scholar] [CrossRef]
- Schulte, I.; Tammen, H.; Selle, H.; Schulz-Knappe, P. Peptides in body fluids and tissues as markers of disease. Expert Rev. Mol. Diagn. 2005, 5, 145–157. [Google Scholar] [CrossRef]
- Diamandis, E.P. Peptidomics for Cancer Diagnosis: Present and Future. J. Proteome Res. 2006, 5, 2079–2082. [Google Scholar] [CrossRef]
- Schaduangrat, N.; Nantasenamat, C.; Prachayasittikul, V.; Shoombuatong, W. ACPred: A Computational Tool for the Prediction and Analysis of Anticancer Peptides. Molecules 2019, 24, 1973. [Google Scholar] [CrossRef]
- Chou, K.-C.; Cai, Y.-D. Prediction and classification of protein subcellular location-sequence-order effect and pseudo amino acid composition. J. Cell. Biochem. 2003, 90, 1250–1260. [Google Scholar] [CrossRef]
- Chou, K.-C. Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins Struct. Funct. Bioinform. 2001, 43, 246–255. [Google Scholar] [CrossRef]
- Dehzangi, A.; Heffernan, R.; Sharma, A.; Lyons, J.; Paliwal, K.; Sattar, A. Gram-positive and Gram-negative protein subcellular localization by incorporating evolutionary-based descriptors into Chou׳s general PseAAC. J. Theor. Biol. 2015, 364, 284–294. [Google Scholar] [CrossRef]
- Meher, P.K.; Sahu, T.K.; Saini, V.; Rao, A.R. Predicting antimicrobial peptides with improved accuracy by incorporating the compositional, physico-chemical and structural features into Chou’s general PseAAC. Sci. Rep. 2017, 7, srep42362. [Google Scholar] [CrossRef]
- Chen, W.; Ding, H.; Feng, P.; Lin, H.; Chou, K.-C. iACP: A sequence-based tool for identifying anticancer peptides. Oncotarget 2016, 7, 16895–16909. [Google Scholar] [CrossRef]
- Manavalan, B.; Basith, S.; Shin, T.H.; Choi, S.; Kim, M.O.; Lee, G. MLACP: Machine-learning-based prediction of anticancer peptides. Oncotarget 2017, 8, 77121–77136. [Google Scholar] [CrossRef]
- Xu, L.; Liang, G.; Wang, L.; Liao, C. A Novel Hybrid Sequence-Based Model for Identifying Anticancer Peptides. Genes 2018, 9, 158. [Google Scholar] [CrossRef] [PubMed]
- Boopathi, V.; Subramaniyam, S.; Malik, A.; Lee, G.; Manavalan, B.; Yang, D.-C. mACPpred: A Support Vector Machine-Based Meta-Predictor for Identification of Anticancer Peptides. Int. J. Mol. Sci. 2019, 20, 1964. [Google Scholar] [CrossRef]
- Li, Q.; Zhou, W.; Wang, D.; Wang, S.; Li, Q. Prediction of anticancer peptides using a low-dimensional feature model. Front. Bioeng. Biotechnol. 2020, 8, 892. [Google Scholar] [CrossRef]
- Akbar, S.; Hayat, M.; Tahir, M.; Chong, K.T. cACP-2LFS: Classification of Anticancer Peptides Using Sequential Discriminative Model of KSAAP and Two-Level Feature Selection Approach. IEEE Access 2020, 8, 131939–131948. [Google Scholar] [CrossRef]
- Agrawal, P.; Bhagat, D.; Mahalwal, M.; Sharma, N.; Raghava, G.P.S. AntiCP 2.0: An updated model for predicting anticancer peptides. Brief. Bioinform. 2021, 22, bbaa153. [Google Scholar] [CrossRef]
- Tyagi, A.; Kapoor, P.; Kumar, R.; Chaudhary, K.; Gautam, A.; Raghava, G.P.S. In Silico Models for Designing and Discovering Novel Anticancer Peptides. Sci. Rep. 2013, 3, srep02984. [Google Scholar] [CrossRef] [PubMed]
- Li, F.-M.; Wang, X.-Q. Identifying anticancer peptides by using improved hybrid compositions. Sci. Rep. 2016, 6, srep33910. [Google Scholar] [CrossRef]
- Akbar, S.; Hayat, M.; Iqbal, M.; Jan, M.A. iACP-GAEnsC: Evolutionary genetic algorithm based ensemble classification of anticancer peptides by utilizing hybrid feature space. Artif. Intell. Med. 2017, 79, 62–70. [Google Scholar] [CrossRef]
- Kabir, M.; Arif, M.; Ahmad, S.; Ali, Z.; Swati, Z.N.K.; Yu, D.-J. Intelligent computational method for discrimination of anticancer peptides by incorporating sequential and evolutionary profiles information. Chemom. Intell. Lab. Syst. 2018, 182, 158–165. [Google Scholar] [CrossRef]
- Vijayakumar, S.; Lakshmi, P. ACPP: A web server for prediction and design of anti-cancer peptides. Int. J. Pept. Res. Ther. 2015, 21, 99–106. [Google Scholar] [CrossRef]
- Hajisharifi, Z.; Piryaiee, M.; Beigi, M.M.; Behbahani, M.; Mohabatkar, H. Predicting anticancer peptides with Chou′s pseudo amino acid composition and investigating their mutagenicity via Ames test. J. Theor. Biol. 2014, 341, 34–40. [Google Scholar] [CrossRef]
- Novkovic, M.; Simunić, J.; Bojović, V.; Tossi, A.; Juretić, D. DADP: The database of anuran defense peptides. Bioinformatics 2012, 28, 1406–1407. [Google Scholar] [CrossRef]
- Wang, G.; Li, X.; Wang, Z. APD2: The updated antimicrobial peptide database and its application in peptide design. Nucleic Acids Res. 2008, 37, D933–D937. [Google Scholar] [CrossRef]
- Liu, B.; Wu, H.; Chou, K.-C. Pse-in-One 2.0: An Improved Package of Web Servers for Generating Various Modes of Pseudo Components of DNA, RNA, and Protein Sequences. Nat. Sci. 2017, 9, 67–91. [Google Scholar] [CrossRef]
- Akbar, S.; Rehman, A.U.; Hayat, M.; Sohail, M. cACP: Classifying anticancer peptides using discriminative intelligent model via Chou’s 5-step rules and general pseudo components. Chemom. Intell. Lab. Syst. 2019, 196, 103912. [Google Scholar] [CrossRef]
- Zhang, C.-T.; Chou, K.-C. An optimization approach to predicting protein structural class from amino acid composition. Protein Sci. 1992, 1, 401–408. [Google Scholar] [CrossRef]
- Hu, L.; Huang, T.; Shi, X.; Lu, W.-C.; Cai, Y.-D.; Chou, K.-C. Predicting Functions of Proteins in Mouse Based on Weighted Protein-Protein Interaction Network and Protein Hybrid Properties. PLoS ONE 2011, 6, e14556. [Google Scholar] [CrossRef]
- Cai, Y.-D.; Feng, K.-Y.; Lu, W.-C.; Chou, K.-C. Using LogitBoost classifier to predict protein structural classes. J. Theor. Biol. 2006, 238, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.-B.; Chou, K.-C. PseAAC: A flexible web server for generating various kinds of protein pseudo amino acid composition. Anal. Biochem. 2008, 373, 386–388. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Wang, X.; Xu, C.; Gao, Y. PseAAC-Builder: A cross-platform stand-alone program for generating various special Chou’s pseudo-amino acid compositions. Anal. Biochem. 2012, 425, 117–119. [Google Scholar] [CrossRef]
- Cao, D.-S.; Xu, Q.-S.; Liang, Y.-Z. propy: A tool to generate various modes of Chou’s PseAAC. Bioinformatics 2013, 29, 960–962. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Gu, S.; Jiao, Y. PseAAC-General: Fast building various modes of general form of Chou’s pseudo-amino acid composition for large-scale protein datasets. Int. J. Mol. Sci. 2014, 15, 3495–3506. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.-C. Pseudo Amino Acid Composition and its Applications in Bioinformatics, Proteomics and System Biology. Curr. Proteom. 2009, 6, 262–274. [Google Scholar] [CrossRef]
- Chou, K.-C. Some remarks on protein attribute prediction and pseudo amino acid composition. J. Theor. Biol. 2011, 273, 236–247. [Google Scholar] [CrossRef]
- Tahir, M.; Tayara, H.; Chong, K.T. iRNA-PseKNC (2methyl): Identify RNA 2’-O-methylation sites by convolution neural network and Chou’s pseudo components. J. Theor. Biol. 2019, 465, 1–6. [Google Scholar] [CrossRef]
- Liu, B.; Liu, F.; Wang, X.; Kuo-Chen, C.; Fang, L.; Chou, K.-C. Pse-in-One: A web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucleic Acids Res. 2015, 43, W65–W71. [Google Scholar] [CrossRef]
- Hayat, M.; Khan, A. Predicting membrane protein types by fusing composite protein sequence features into pseudo amino acid composition. J. Theor. Biol. 2011, 271, 10–17. [Google Scholar] [CrossRef]
- Khan, Z.U.; Hayat, M.; Khan, M.A. Discrimination of acidic and alkaline enzyme using Chou’s pseudo amino acid composition in conjunction with probabilistic neural network model. J. Theor. Biol. 2015, 365, 197–203. [Google Scholar] [CrossRef]
- Khan, S.U.; Baik, R. MPPIF-Net: Identification of Plasmodium Falciparum Parasite Mitochondrial Proteins Using Deep Features with Multilayer Bi-directional LSTM. Processes 2020, 8, 725. [Google Scholar] [CrossRef]
- Khan, S.U.; Hussain, T.; Ullah, A.; Baik, S.W. Deep-ReID: Deep features and autoencoder assisted image patching strategy for person re-identification in smart cities surveillance. Multimed. Tools Appl. 2021, 1–22. [Google Scholar] [CrossRef]
- Khan, S.U.; Haq, I.U.; Khan, N.; Muhammad, K.; Hijji, M.; Baik, S.W. Learning to rank: An intelligent system for person reidentification. Int. J. Intell. Syst. 2022. [Google Scholar] [CrossRef]
- Khan, N.; Haq, I.U.; Ullah, F.U.M.; Khan, S.U.; Lee, M.Y. CL-Net: ConvLSTM-Based Hybrid Architecture for Batteries’ State of Health and Power Consumption Forecasting. Mathematics 2021, 9, 3326. [Google Scholar] [CrossRef]
- Khan, N.; Ullah, F.U.M.; Haq, I.U.; Khan, S.U.; Lee, M.Y.; Baik, S.W. AB-Net: A Novel Deep Learning Assisted Framework for Renewable Energy Generation Forecasting. Mathematics 2021, 9, 2456. [Google Scholar] [CrossRef]
- Haq, I.; Ullah, A.; Khan, S.; Khan, N.; Lee, M.; Rho, S.; Baik, S. Sequential Learning-Based Energy Consumption Prediction Model for Residential and Commercial Sectors. Mathematics 2021, 9, 605. [Google Scholar] [CrossRef]
- Ullah, F.; Khan, N.; Hussain, T.; Lee, M.; Baik, S. Diving Deep into Short-Term Electricity Load Forecasting: Comparative Analysis and a Novel Framework. Mathematics 2021, 9, 611. [Google Scholar] [CrossRef]
- Khan, N.; Haq, I.U.; Khan, S.U.; Rho, S.; Lee, M.Y.; Baik, S.W. DB-Net: A novel dilated CNN based multi-step forecasting model for power consumption in integrated local energy systems. Int. J. Electr. Power Energy Syst. 2021, 133, 107023. [Google Scholar] [CrossRef]
- Khan, S.U.; Haq, I.U.; Khan, Z.A.; Khan, N.; Lee, M.Y.; Baik, S.W. Atrous Convolutions and Residual GRU Based Architecture for Matching Power Demand with Supply. Sensors 2021, 21, 7191. [Google Scholar] [CrossRef]
- Hajisharifi, Z.; Mohabatkar, H. In silico prediction of anticancer peptides by TRAINER tool. Mol. Biol. Res. Commun. 2013, 2, 39–45. [Google Scholar] [CrossRef]
- Ge, L.; Liu, J.; Zhang, Y.; Dehmer, M. Identifying anticancer peptides by using a generalized chaos game representation. J. Math. Biol. 2018, 78, 441–463. [Google Scholar] [CrossRef]
- Ahmed, S.; Muhammod, R.; Khan, Z.H.; Adilina, S.; Sharma, A.; Shatabda, S.; Dehzangi, A. ACP-MHCNN: An accurate multi-headed deep-convolutional neural network to predict anticancer peptides. Sci. Rep. 2021, 11, 23676. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| References | Features | Evaluation | Classifier |
|---|---|---|---|
| [23] | PseACC, g-gap dipeptide | Accuracy, Sensitivity, Specificity, MCC | SVM |
| [24] | AAC, DPC, ATC, and PCP | ------ | SVM, RFT |
| [25] | g-gap dipeptide | Accuracy, Sensitivity, Specificity, MCC, F1-score | SVM |
| [26] | Composition-based, physicochemical properties and profiles | Accuracy, Sensitivity, Specificity, MCC, AUC, p-value | SVM, LR, KNN, RF |
| [27] | AAC, Conjoint triad, PAAC, GAAC | Accuracy, Sensitivity, Specificity, MCC, F1-score | SVM, RFT, LibD3C |
| [28] | K-space amino acid pair, Composite physiochemical properties, auto covariance, | Accuracy, Sensitivity, Specificity, MCC | SVM, RFT, FKNN |
| [29] | AAC, DPC, Terminus composition, binary profile | ------ | Tree based |
| [30] | Binary profile, DPC | Accuracy, Sensitivity, Specificity, MCC, AUC | SVM |
| [31] | ReduceAAC, AAC, average chemical shift | Sensitivity, Specificity, MCC, QA | SVM |
| [32] | PAAC, RAAC, g-gap dipeptide | Accuracy, Sensitivity, Specificity, MCC | SVM, KNN, PNN, RF, GRNN |
| [33] | Pseudo position specific scoring matrix, Composite protein sequence, Split-AAC | Accuracy, Sensitivity, Specificity, MCC, G-mean, F-measure, Precision, Recall | SVM, KNN, PNN |
| [34] | Protein relatedness measure | Sensitivity, Specificity, MCC, AUC, Overall accuracy | SVM, AdaBoost |
| [35] | PAAC, Local alignment kernel | Accuracy, Sensitivity, Specificity, MCC | SVM |
| Datasets | ACP | NACPs | Total Data | Training Data | Testing Data |
|---|---|---|---|---|---|
| PSD1 (Benchmark) [23] | 138 | 206 | 344 | 241 | 103 |
| PSD2 (Main dataset) [39] | 225 | 2250 | 2475 | 1732 | 743 |
| Method | PSD1 | ||||
|---|---|---|---|---|---|
| Accuracy | Sensitivity | Specificity | MCC | F1-Score | |
| AAC | 86.41 | 88.24 | 85.51 | 0.71 | 81.08 |
| DPC | 88.35 | 91.18 | 96.86 | 0.75 | 83.78 |
| TPC | 85.44 | 87.88 | 84.29 | 0.69 | 79.45 |
| IPseAAC | 88.35 | 83.33 | 91.80 | 0.75 | 85.37 |
| AAC+DPC | 92.23 | 92.11 | 92.31 | 0.83 | 89.74 |
| AAC+TPC | 90.29 | 84.09 | 94.92 | 0.80 | 88.10 |
| AAC+IPseAAC | 91.26 | 84.44 | 96.55 | 0.82 | 89.41 |
| DPC+TPC | 89.32 | 89.19 | 89.39 | 0.77 | 85.71 |
| DPC+IPseAAC | 93.20 | 88.37 | 96.67 | 0.86 | 91.57 |
| TPC+IPseAAC | 91.29 | 89.74 | 92.19 | 0.82 | 88.61 |
| Proposed Model (λ = 1) | 97.09 | 97.44 | 96.88 | 0.94 | 96.20 |
| Proposed Model (λ = 2) | 96.12 | 92.86 | 98.36 | 0.82 | 95.12 |
| Proposed Model (λ = 3) | 95.15 | 94.87 | 95.31 | 0.90 | 93.67 |
| Method | PSD2 | ||||
|---|---|---|---|---|---|
| Accuracy | Sensitivity | Specificity | MCC | F1-Score | |
| AAC | 88.96 | 58.82 | 93.76 | 0.53 | 59.41 |
| DPC | 91.25 | 68.42 | 94.60 | 0.61 | 66.67 |
| TPC | 86.27 | 48.94 | 91.68 | 0.39 | 47.42 |
| IPseAAC | 90.98 | 66.67 | 94.72 | 0.61 | 66.33 |
| AAC+DPC | 93.00 | 75.53 | 95.53 | 0.69 | 73.20 |
| AAC+TPC | 94.05 | 79.79 | 96.12 | 0.73 | 77.32 |
| AAC+ IPseAAC | 92.87 | 72.82 | 96.09 | 0.69 | 73.89 |
| DPC+TPC | 95.29 | 84.21 | 96.91 | 0.79 | 82.05 |
| DPC+ IPseAAC | 92.73 | 71.30 | 96.38 | 0.69 | 74.04 |
| TPC+ IPseAAC | 91.79 | 80.00 | 92.92 | 0.60 | 63.03 |
| Proposed Model (λ = 1) | 98.25 | 96.77 | 98.46 | 0.92 | 93.26 |
| Proposed Model (λ = 2) | 96.90 | 91.40 | 97.69 | 0.86 | 88.08 |
| Proposed Model (λ = 3) | 94.89 | 80.39 | 97.19 | 0.78 | 81.19 |
| Model/Year | Accuracy | Sensitivity | Specificity | MCC | F1-Score |
|---|---|---|---|---|---|
| SPAP [62] 2013 | 87.00 | 92.00 | 86.00 | 0.74 | - |
| LAK [35] 2014 | 92.68 | 89.70 | 85.18 | 0.78 | - |
| iACP [23] 2016 | 95.06 | 89.86 | 98.54 | 0.89 | - |
| IAP [31] 2016 | 93.61 | 89.86 | 96.12 | 0.86 | - |
| iACP-GAEnsC [32] 2017 | 96.45 | 95.36 | 97.57 | 0.91 | - |
| SAP [25] 2018 | 91.86 | 86.23 | 95.63 | 0.83 | 89.47 |
| LDFM [27] 2020 | 92.73 | 87.70 | 96.10 | 0.84 | 92.70 |
| Proposed Model (λ = 1) | 97.09 | 97.44 | 96.88 | 0.93 | 96.20 |
| Proposed Model (λ = 2) | 96.12 | 92.86 | 98.36 | 0.91 | 95.12 |
| Proposed Model (λ = 3) | 95.15 | 94.87 | 95.31 | 0.89 | 93.67 |
| Model/Year | Accuracy | Sensitivity | Specificity | MCC | F1-Score |
|---|---|---|---|---|---|
| NT5CT5 [30] 2013 | 92.65 | 74.67 | 94.44 | 0.61 | - |
| GCGR [63] 2018 | 96.36 | 69.33 | 99.07 | 0.76 | - |
| cACP [39] 2019 | 96.91 | 77.32 | 98.12 | 0.79 | - |
| ACP-MHCNN [64] 2021 | 91.0 | 97.6 | 84.2 | 0.82 | - |
| Proposed (λ = 1) | 98.25 | 96.77 | 98.46 | 0.92 | 93.26 |
| Proposed (λ = 2) | 96.90 | 91.40 | 97.69 | 0.86 | 88.08 |
| Proposed (λ = 3) | 94.89 | 80.39 | 97.19 | 0.78 | 81.19 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alsanea, M.; Dukyil, A.S.; Afnan; Riaz, B.; Alebeisat, F.; Islam, M.; Habib, S. To Assist Oncologists: An Efficient Machine Learning-Based Approach for Anti-Cancer Peptides Classification. Sensors 2022, 22, 4005. https://doi.org/10.3390/s22114005
Alsanea M, Dukyil AS, Afnan, Riaz B, Alebeisat F, Islam M, Habib S. To Assist Oncologists: An Efficient Machine Learning-Based Approach for Anti-Cancer Peptides Classification. Sensors. 2022; 22(11):4005. https://doi.org/10.3390/s22114005
Chicago/Turabian StyleAlsanea, Majed, Abdulsalam S. Dukyil, Afnan, Bushra Riaz, Farhan Alebeisat, Muhammad Islam, and Shabana Habib. 2022. "To Assist Oncologists: An Efficient Machine Learning-Based Approach for Anti-Cancer Peptides Classification" Sensors 22, no. 11: 4005. https://doi.org/10.3390/s22114005
APA StyleAlsanea, M., Dukyil, A. S., Afnan, Riaz, B., Alebeisat, F., Islam, M., & Habib, S. (2022). To Assist Oncologists: An Efficient Machine Learning-Based Approach for Anti-Cancer Peptides Classification. Sensors, 22(11), 4005. https://doi.org/10.3390/s22114005