A Reliable BioFET Immunosensor for Detection of p53 Tumour Suppressor in Physiological-Like Environment

 ,
, 
 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
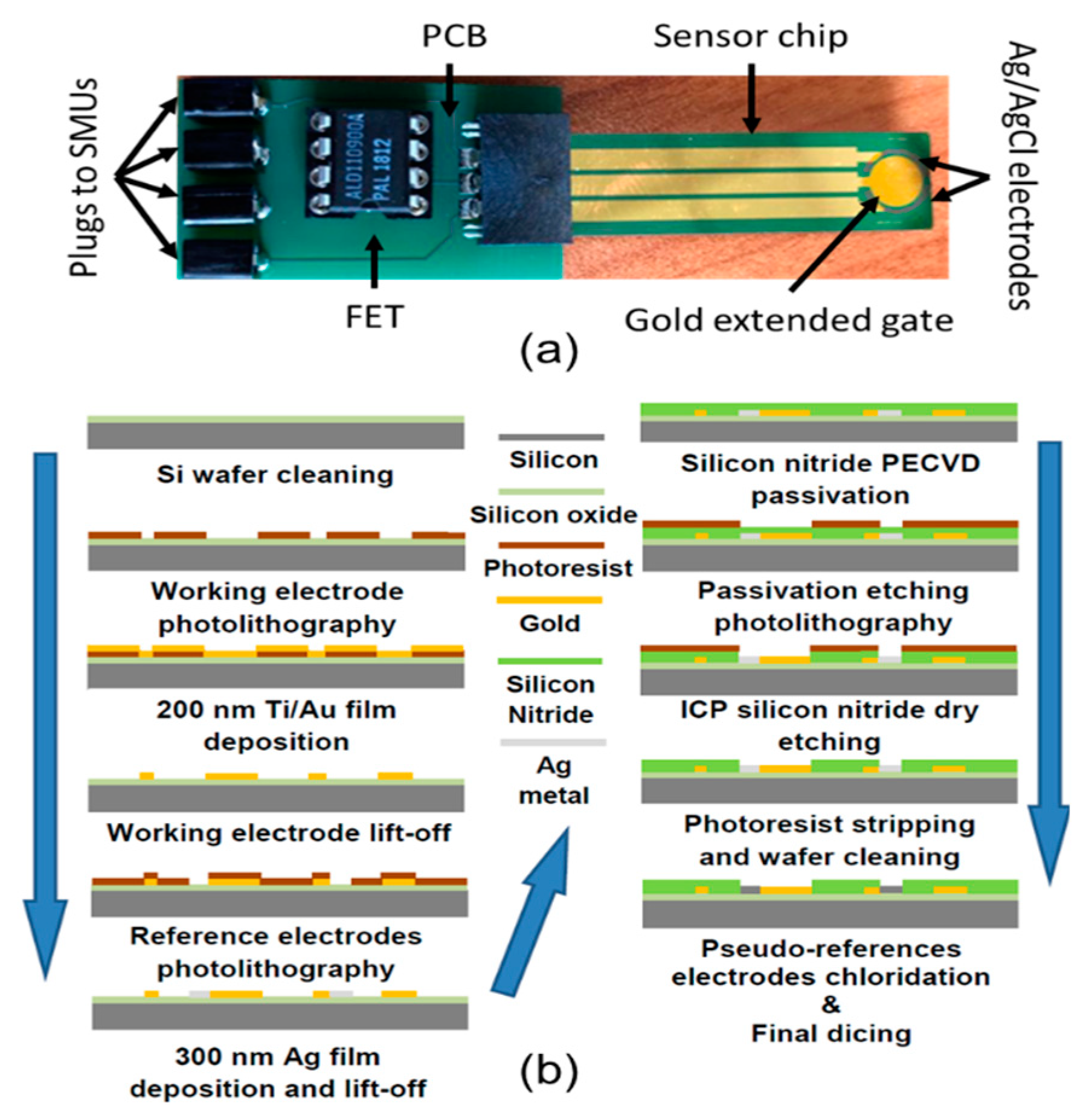
2.1. Experimental Setup
2.2. Sensing Chip Microfabrication
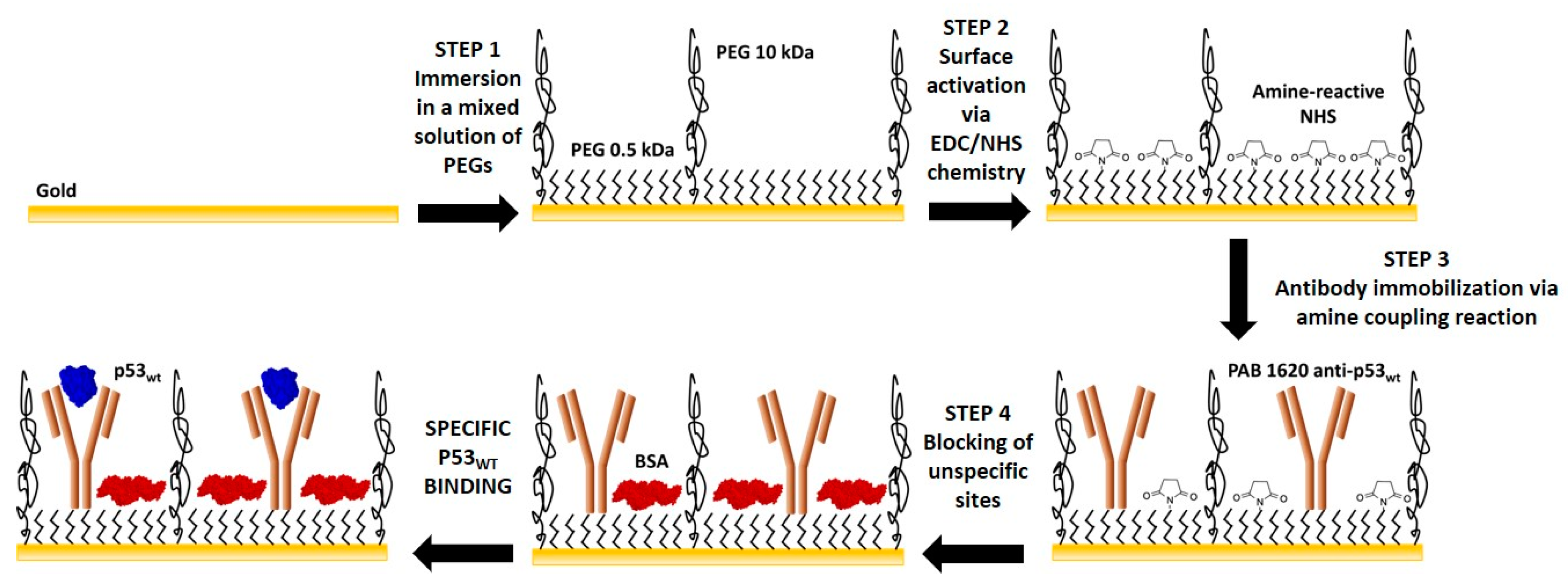
2.3. Extended-Gate Electrode Surface Biofunctionalization
2.4. Biosensing Experiments
3. Results and Discussion
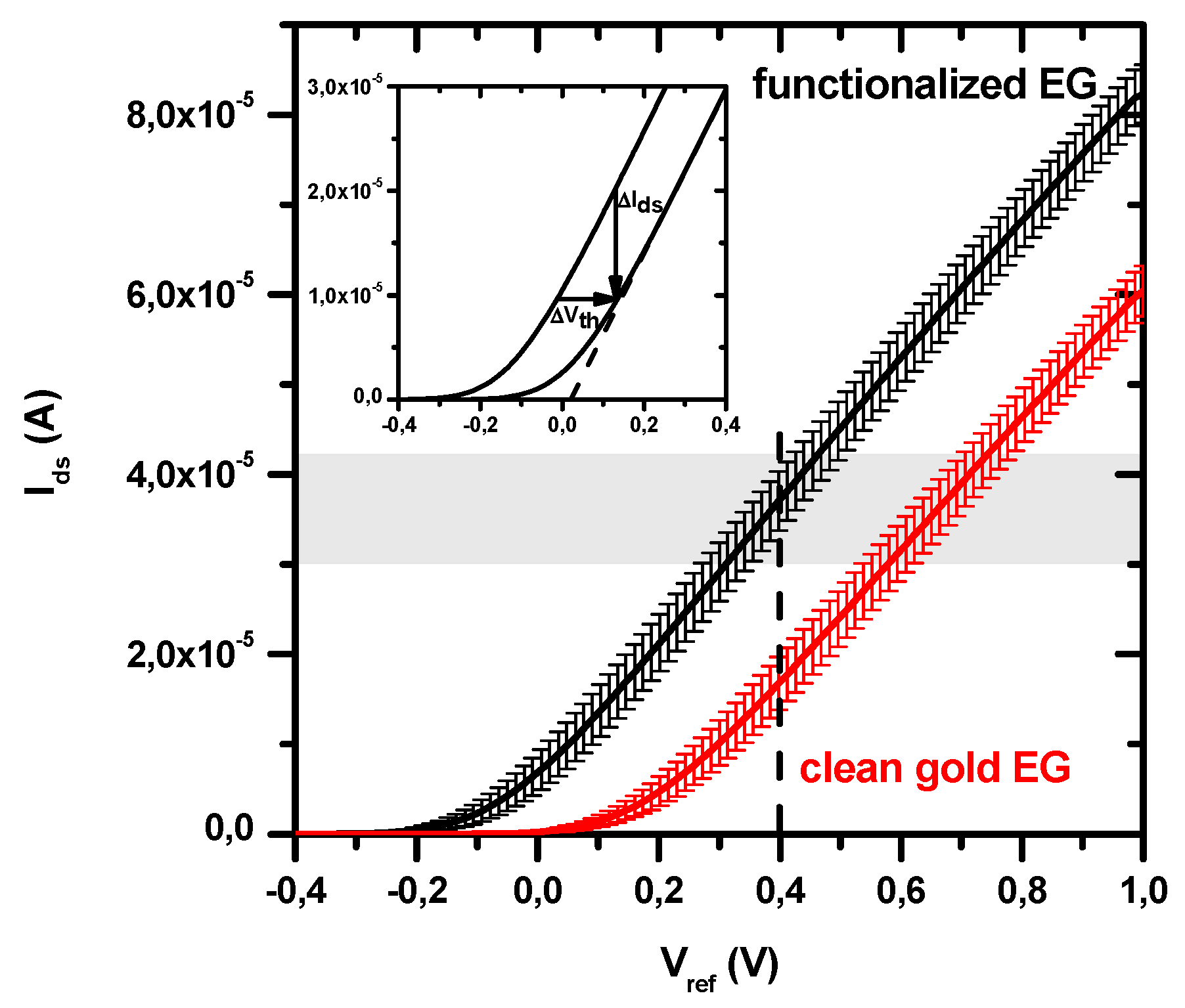
3.1. BioFET Transfer Characteristics
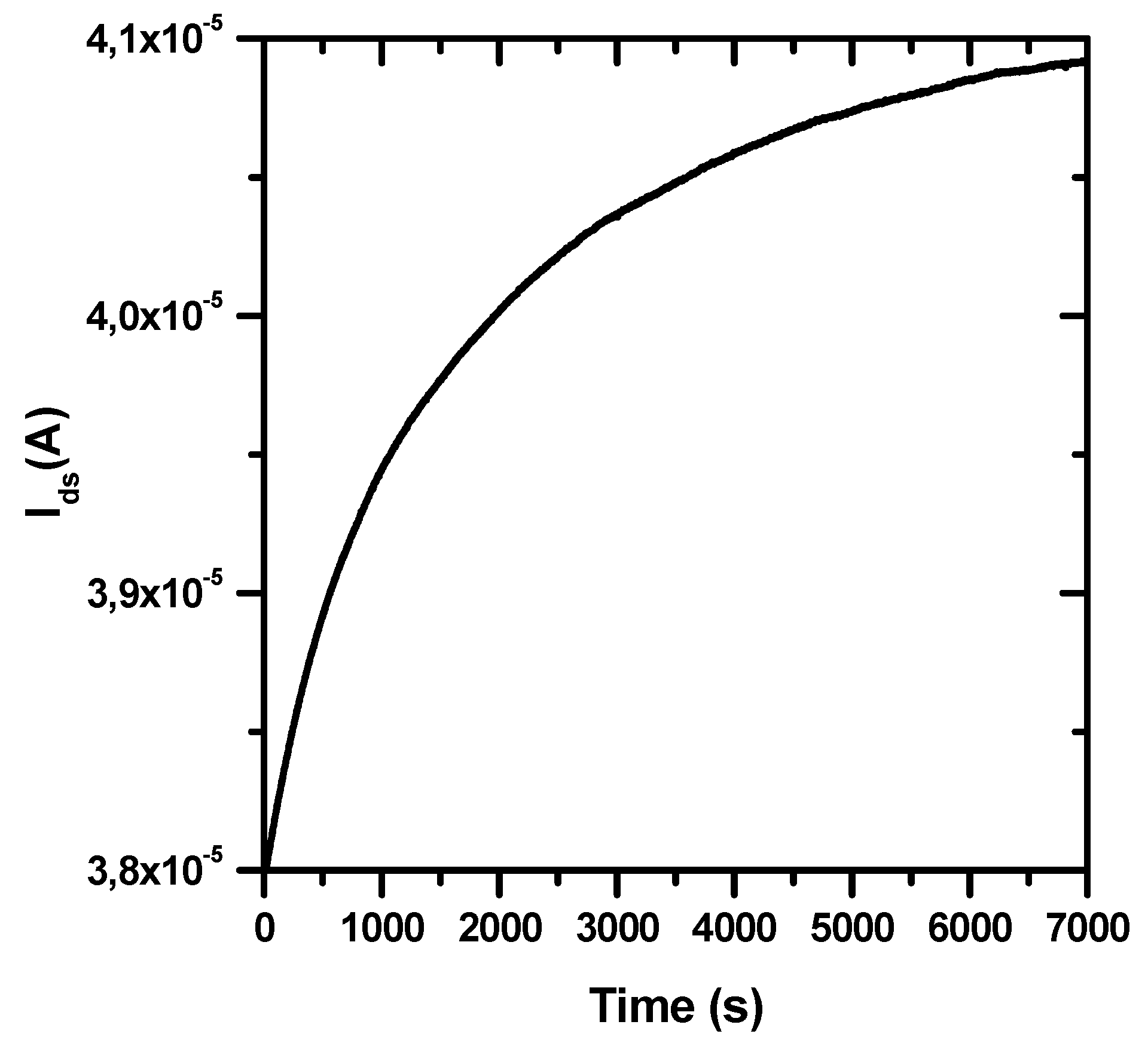
3.2. Stability over Time
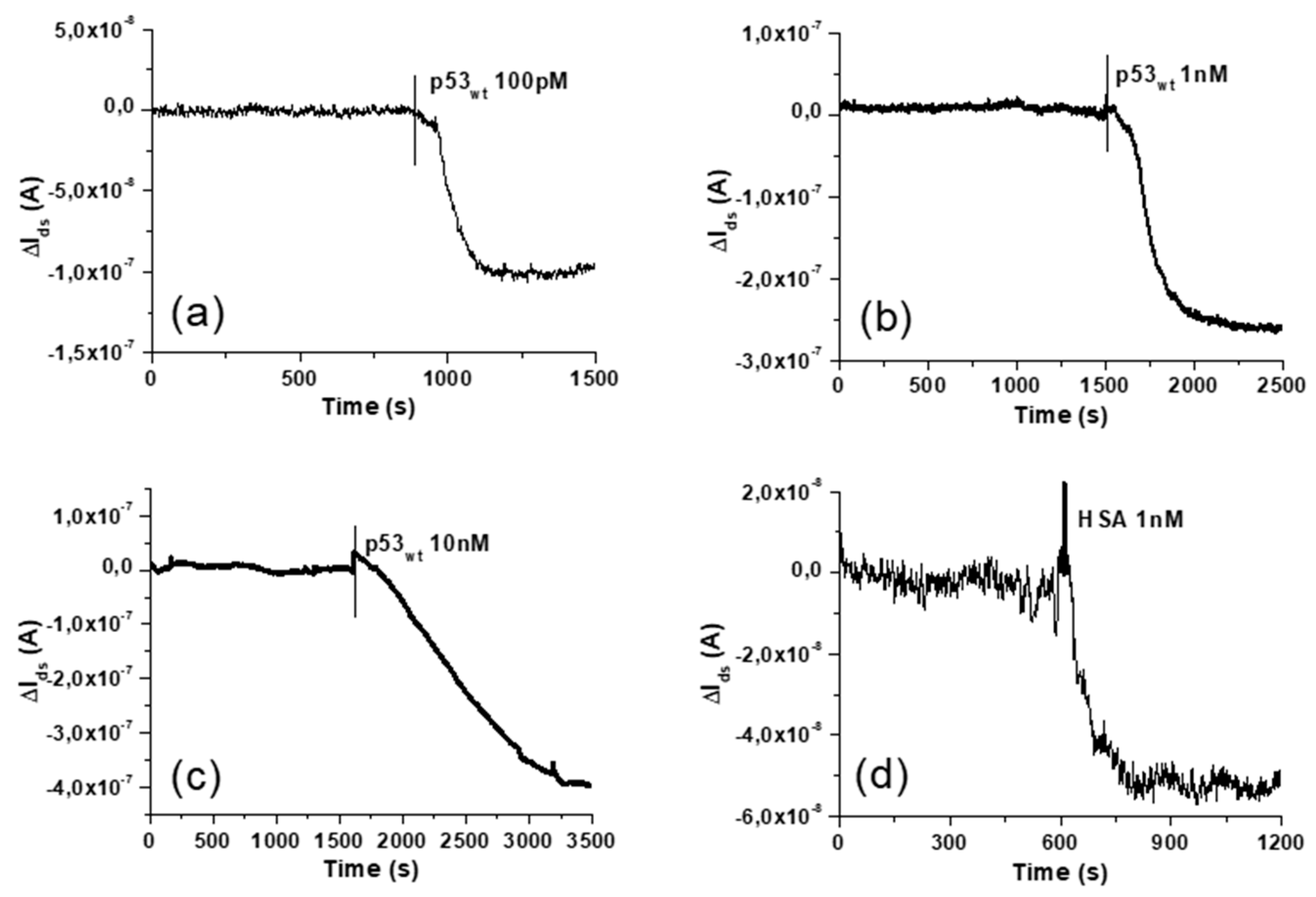
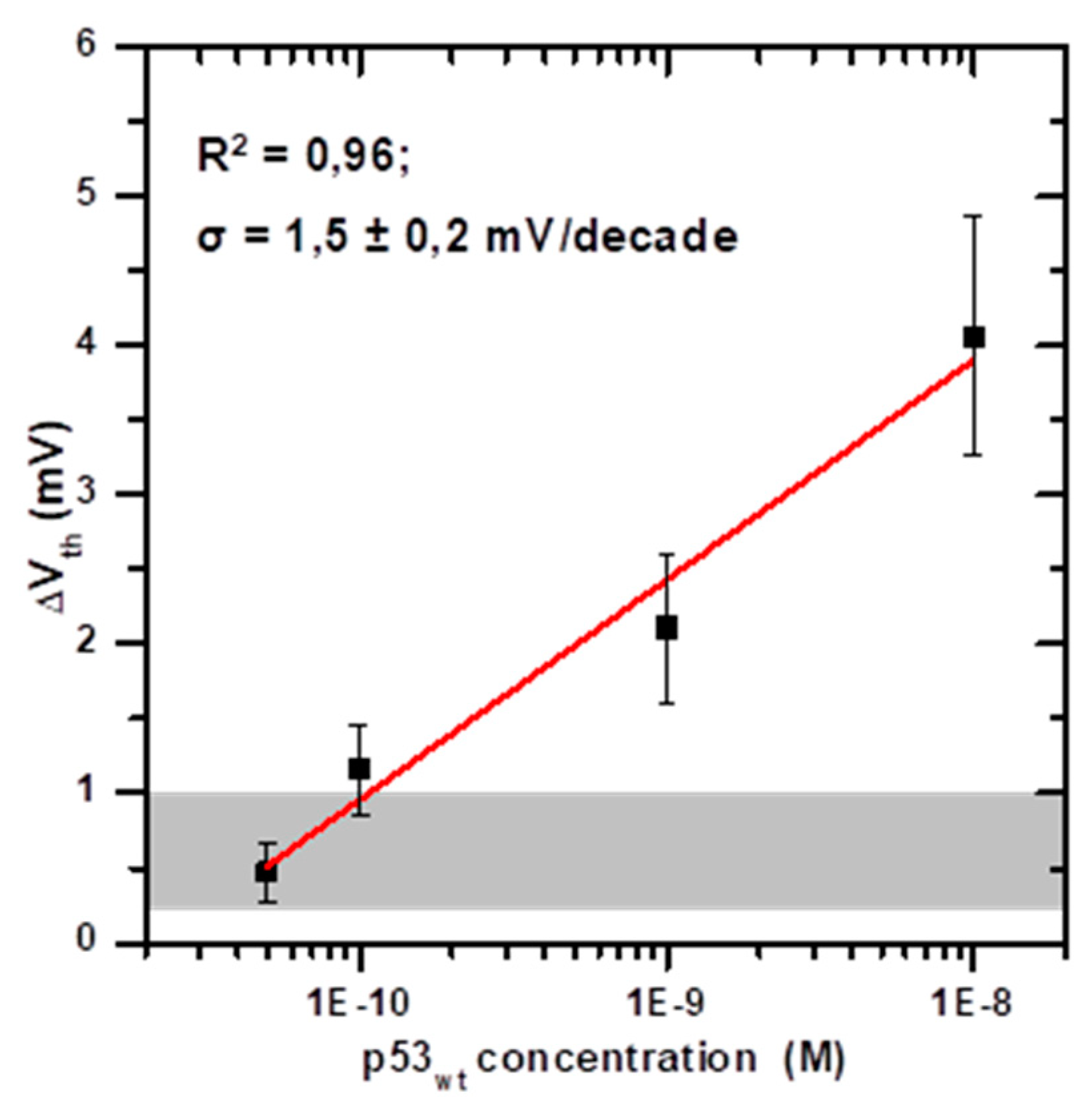
3.3. Response to p53wt: Extended-Gate Surface Potential Changes and Detection Limit
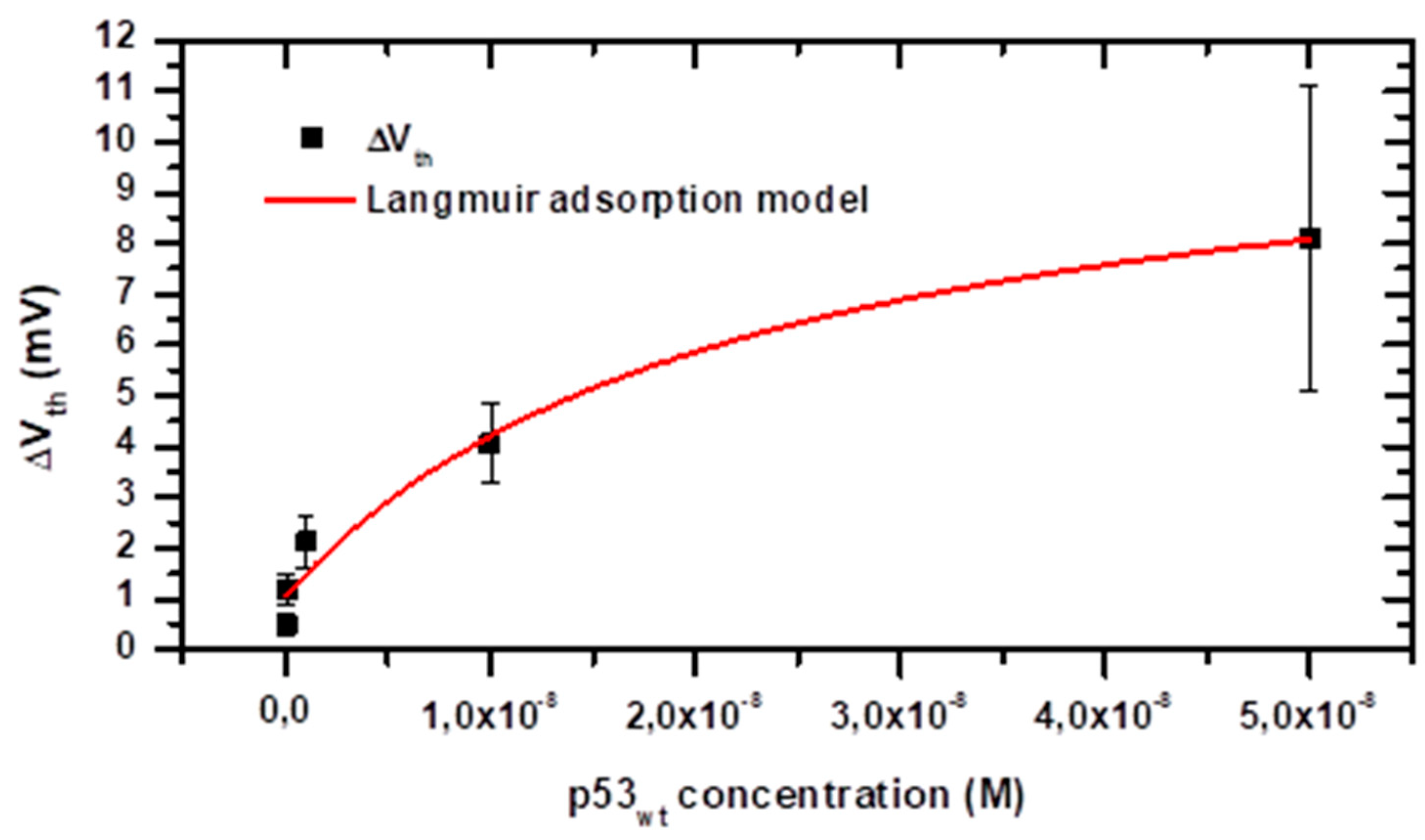
3.4. Energetics of the Adsorption Process
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lane, D.; Levine, A. p53 Research: The Past Thirty Years and the Next Thirty Years. Cold Spring Harb. Perspect. Biol. 2010, 2, a00893. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Fersht, A.R. Structural Biology of the Tumor Suppressor p53. Annu. Rev. Biochem. 2008, 77, 557–582. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; He, C.; Zhang, Z.; Wang, J. Sensitive chemiluminescence detection of wild-type p53 protein captured by surface-confined consensus DNA duplexes. Biosens. Bioelectron. 2013, 47, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhu, X.; Wu, M.; Xia, N.; Wang, J.; Zhou, F. Simultaneous and label-free determination of wild-type and mutant p53 at a single surface plasmon resonance chip preimmobilized with consensus DNA and monoclonal antibody. Anal. Chem. 2009, 81, 8441–8446. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 Mutations in Human Cancers: Origins, Consequences, and Clinical Use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef] [PubMed]
- Hemminki, K.; Partanen, R.; Koskinen, H.; Smith, S.; Carney, W.; Brandt-Rauf, P.W. The Molecular Epidemiology of Oncoproteins: Serum p53 Protein in Patients With Asbestosis. Chest 1996, 109, 22S–26S. [Google Scholar] [CrossRef] [PubMed]
- Lutz, W.; Nowakowska-Swirta, E. Gene p53 mutations, protein p53, and anti-p53 antibodies as biomarkers of cancer process. Int. J. Occup. Med. Environ. Health 2002, 15, 209–218. [Google Scholar]
- Afsharan, H.; Navaeipour, F.; Khalilzadeh, B.; Tajalli, H.; Mollabashi, M.; Ahar, M.J.; Rashidi, M.R. Highly sensitive electrochemiluminescence detection of p53 protein using functionalized Ru-silica nanoporous@gold nanocomposite. Biosens. Bioelectron. 2016, 80, 146–153. [Google Scholar] [CrossRef]
- Assah, E.; Goh, W.; Zheng, X.T.; Lim, T.X.; Li, J.; Lane, D.; Ghadessy, F.; Tan, Y.N. Rapid colorimetric detection of p53 protein function using DNA-gold nanoconjugates with applications for drug discovery and cancer diagnostics. Colloids Surf. B Biointerfaces 2018, 169, 214–221. [Google Scholar] [CrossRef]
- Jia, C.P.; Zhong, X.Q.; Hua, B.; Liu, M.Y.; Jing, F.X.; Lou, X.H.; Yao, S.H.; Xiang, J.Q.; Jin, Q.H.; Zhao, J.L. Nano-ELISA for highly sensitive protein detection. Biosens. Bioelectron. 2009, 24, 2836–2841. [Google Scholar] [CrossRef]
- Jiang, T.; Song, Y.; Du, D.; Liu, X.; Lin, Y. Detection of p53 Protein Based on Mesoporous Pt–Pd Nanoparticles with Enhanced Peroxidase-like Catalysis. ACS Sens. 2016, 1, 717–724. [Google Scholar] [CrossRef]
- Bizzarri, A.R.; Moscetti, I.; Cannistraro, S. Surface enhanced Raman spectroscopy based immunosensor for ultrasensitive and selective detection of wild type p53 and mutant p53R175H. Anal. Chim. Acta 2018, 1029, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Giannetto, M.; Bianchi, M.V.; Mattarozzi, M.; Careri, M. Competitive amperometric immunosensor for determination of p53 protein in urine with carbon nanotubes/gold nanoparticles screen-printed electrodes: A potential rapid and noninvasive screening tool for early diagnosis of urinary tract carcinoma. Anal. Chim. Acta 2017, 991, 133–141. [Google Scholar] [CrossRef]
- Wang, X.; Wang, X.; Wang, X.; Chen, F.; Zhu, K.; Xu, Q.; Tang, M. Novel electrochemical biosensor based on functional composite nanofibers for sensitive detection of p53 tumor suppressor gene. Anal. Chim. Acta 2013, 765, 63–69. [Google Scholar] [CrossRef]
- Han, S.H.; Kim, S.K.; Park, K.; Yi, S.Y.; Park, H.J.; Lyu, H.K.; Kim, M.; Chung, B.H. Detection of mutant p53 using field-effect transistor biosensor. Anal. Chim. Acta 2010, 665, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Turner, A.P.F. Biosensors: Sense and sensibility. Chem. Soc. Rev. 2013, 42, 3184–3196. [Google Scholar] [CrossRef] [PubMed]
- Lowe, B.M.; Sun, K.; Zeimpekis, I.; Skylaris, C.K.; Green, N.G. Field-effect sensors-from pH sensing to biosensing: Sensitivity enhancement using streptavidin-biotin as a model system. Analyst 2017, 142, 4173–4200. [Google Scholar] [CrossRef]
- Bergveld, P. The development and application of FET-based biosensors. Biosensors 1986, 2, 15–33. [Google Scholar] [CrossRef]
- Kaisti, M. Detection principles of biological and chemical FET sensors. Biosens. Bioelectron. 2017, 98, 437–448. [Google Scholar] [CrossRef]
- Vacic, A.; Criscione, J.M.; Rajan, N.K.; Stern, E.; Fahmy, T.M.; Reed, M.A. Determination of molecular configuration by debye length modulation. J. Am. Chem. Soc. 2011, 133, 13886–13889. [Google Scholar] [CrossRef]
- Stern, E.; Wagner, R.; Sigworth, F.J.; Breaker, R.; Fahmy, T.M.; Reed, M.A. Importance of the debye screening length on nanowire field effect transistor sensors. Nano Lett. 2007, 7, 3405–3409. [Google Scholar] [CrossRef] [PubMed]
- Palazzo, G.; De Tullio, D.; Magliulo, M.; Mallardi, A.; Intranuovo, F.; Mulla, M.Y.; Favia, P.; Vikholm-Lundin, I.; Torsi, L. Detection beyond Debye’s length with an electrolyte-gated organic field-effect transistor. Adv. Mater. 2015, 27, 911–916. [Google Scholar] [CrossRef]
- Nakatsuka, N.; Yang, K.A.; Abendroth, J.M.; Cheung, K.M.; Xu, X.; Yang, H.; Zhao, C.; Zhu, B.; Rim, Y.S.; Yang, Y.; et al. Aptamer-field-effect transistors overcome Debye length limitations for small-molecule sensing. Science 2018, 362, 319–324. [Google Scholar] [CrossRef]
- Shen, M.Y.; Li, B.R.; Li, Y.K. Silicon nanowire field-effect-transistor based biosensors: From sensitive to ultra-sensitive. Biosens. Bioelectron. 2014, 60, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Sorgenfrei, S.; Chiu, C.-Y.; Johnston, M.L.; Nuckolls, C.; Shepard, K.L. Debye Screening in Single-Molecule Carbon Nanotube Field-Effect Sensors. Nano Lett. 2011, 11, 3739–3743. [Google Scholar] [CrossRef] [PubMed]
- Gao, N.; Zhou, W.; Jiang, X.; Hong, G.; Fu, T.-M.; Lieber, C.M. General strategy for biodetection in high ionic strength solutions using transistor-based nanoelectronic sensors. Nano Lett. 2015, 15, 2143–2148. [Google Scholar] [CrossRef]
- Gao, N.; Gao, T.; Yang, X.; Dai, X.; Zhou, W.; Zhang, A.; Lieber, C.M. Specific detection of biomolecules in physiological solutions using graphene transistor biosensors. Proc. Natl. Acad. Sci. USA 2016, 113, 14633–14638. [Google Scholar] [CrossRef]
- Gutiérrez-Sanz, Ó.; Andoy, N.M.; Filipiak, M.S.; Haustein, N.; Tarasov, A. Direct, Label-Free, and Rapid Transistor-Based Immunodetection in Whole Serum. ACS Sens. 2017, 2, 1278–1286. [Google Scholar] [CrossRef] [PubMed]
- Haustein, N.; Gutiérrez-Sanz, Ó.; Tarasov, A. Analytical Model to Describe the Effect of Polyethylene Glycol on Ionic Screening of Analyte Charges in Transistor-Based Immunosensing. ACS Sens. 2019, 4, 874–882. [Google Scholar] [CrossRef] [PubMed]
- Kaisti, M.; Kerko, A.; Aarikka, E.; Saviranta, P.; Boeva, Z.; Soukka, T.; Lehmusvuori, A. Real-Time wash-free detection of unlabeled PNA-DNA hybridization using discrete FET sensor. Sci. Rep. 2017, 7, 1–8. [Google Scholar] [CrossRef]
- Pullano, S.A.; Critello, C.D.; Mahbub, I.; Tasneem, N.T.; Shamsir, S.; Islam, S.K.; Greco, M.; Fiorillo, A.S. EGFET-based sensors for bioanalytical applications: A review. Sensors 2018, 18, 4042. [Google Scholar] [CrossRef] [PubMed]
- Guan, W.; Duan, X.; Reed, M.A. Highly specific and sensitive non-enzymatic determination of uric acid in serum and urine by extended gate field effect transistor sensors. Biosens. Bioelectron. 2014, 51, 225–231. [Google Scholar] [CrossRef]
- Johnson, B.N.; Mutharasan, R. Regeneration of gold surfaces covered by adsorbed thiols and proteins using liquid-phase hydrogen peroxide-mediated UV-photooxidation. J. Phys. Chem. C 2013, 117, 1335–1341. [Google Scholar] [CrossRef]
- Chen, S.; Nyholm, L.; Jokilaakso, N.; Karlstrm, A.E.; Linnros, J.; Smith, U.; Zhang, S.L. Current instability for silicon nanowire field-effect sensors operating in electrolyte with platinum gate electrodes. Electrochem. Solid-State Lett. 2011, 14. [Google Scholar] [CrossRef]
- Ortiz-Conde, A.; García-Sánchez, F.J.; Muci, J.; Terán Barrios, A.; Liou, J.J.; Ho, C.S. Revisiting MOSFET threshold voltage extraction methods. Microelectron. Reliab. 2013, 53, 90–104. [Google Scholar] [CrossRef]
- Waleed Shinwari, M.; Jamal Deen, M.; Landheer, D. Study of the electrolyte-insulator-semiconductor field-effect transistor (EISFET) with applications in biosensor design. Microelectron. Reliab. 2007, 47, 2025–2057. [Google Scholar] [CrossRef]
- Uematsu, Y.; Kajisa, T.; Sakata, T. Fundamental Characteristics of a Glucose Transistor with a Chemically Functional Interface. ChemElectroChem 2017, 4, 2225–2231. [Google Scholar] [CrossRef]
- Singh, N.K.; Thungon, P.D.; Estrela, P.; Goswami, P. Development of an aptamer-based field effect transistor biosensor for quantitative detection of Plasmodium falciparum glutamate dehydrogenase in serum samples. Biosens. Bioelectron. 2019, 123, 30–35. [Google Scholar] [CrossRef]
- Cheng, S.; Hotani, K.; Hideshima, S.; Kuroiwa, S.; Nakanishi, T.; Hashimoto, M.; Mori, Y.; Osaka, T. Field effect transistor biosensor using antigen binding fragment for detecting tumor marker in human serum. Materials 2014, 7, 2490–2500. [Google Scholar] [CrossRef]
- Minamiki, T.; Sasaki, Y.; Tokito, S.; Minami, T. Label-free direct electrical detection of a histidine-rich protein with sub-femtomolar sensitivity using an organic field-effect transistor. ChemistryOpen 2017, 6, 472–475. [Google Scholar] [CrossRef]
- Duan, X.; Li, Y.; Rajan, N.K.; Routenberg, D.A.; Modis, Y.; Reed, M.A. Quantification of the affinities and kinetics of protein interactions using silicon nanowire biosensors. Nat. Nanotechnol. 2012, 7, 401–407. [Google Scholar] [CrossRef]
- Xu, S.; Zhang, C.; Jiang, S.; Hu, G.; Li, X.; Zou, Y.; Liu, H.; Li, J.; Li, Z.; Wang, X.; et al. Graphene foam field-effect transistor for ultra-sensitive label-free detection of ATP. Sens. Actuators B Chem. 2019, 284, 125–133. [Google Scholar] [CrossRef]
- Kase, H.; Negishi, R.; Arifuku, M.; Kiyoyanagi, N.; Kobayashi, Y. Biosensor response from target molecules with inhomogeneous charge localization. J. Appl. Phys. 2018, 124. [Google Scholar] [CrossRef]
- Tarasov, A.; Tsai, M.Y.; Flynn, E.M.; Joiner, C.A.; Taylor, R.C.; Vogel, E.M. Gold-coated graphene field-effect transistors for quantitative analysis of protein-antibody interactions. 2D Mater. 2015, 2, 44008. [Google Scholar] [CrossRef]
- Ravera, M.W.; Cárcamo, J.; Brissette, R.; Alam-Moghé, A.; Dedova, O.; Cheng, W.; Hsiao, K.C.; Klebanov, D.; Shen, H.; Tang, P.; et al. Identification of an allosteric binding site on the transcription factor p53 using a phage-displayed peptide library. Oncogene 1998, 16, 1993–1999. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baldacchini, C.; Montanarella, A.F.; Francioso, L.; Signore, M.A.; Cannistraro, S.; Bizzarri, A.R. A Reliable BioFET Immunosensor for Detection of p53 Tumour Suppressor in Physiological-Like Environment. Sensors 2020, 20, 6364. https://doi.org/10.3390/s20216364
Baldacchini C, Montanarella AF, Francioso L, Signore MA, Cannistraro S, Bizzarri AR. A Reliable BioFET Immunosensor for Detection of p53 Tumour Suppressor in Physiological-Like Environment. Sensors. 2020; 20(21):6364. https://doi.org/10.3390/s20216364
Chicago/Turabian StyleBaldacchini, Chiara, Antonino Francesco Montanarella, Luca Francioso, Maria Assunta Signore, Salvatore Cannistraro, and Anna Rita Bizzarri. 2020. "A Reliable BioFET Immunosensor for Detection of p53 Tumour Suppressor in Physiological-Like Environment" Sensors 20, no. 21: 6364. https://doi.org/10.3390/s20216364
APA StyleBaldacchini, C., Montanarella, A. F., Francioso, L., Signore, M. A., Cannistraro, S., & Bizzarri, A. R. (2020). A Reliable BioFET Immunosensor for Detection of p53 Tumour Suppressor in Physiological-Like Environment. Sensors, 20(21), 6364. https://doi.org/10.3390/s20216364