Voltammetry at Hexamethyl-P-Terphenyl Poly(Benzimidazolium) (HMT-PMBI)-Coated Glassy Carbon Electrodes: Charge Transport Properties and Detection of Uric and Ascorbic Acid



Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
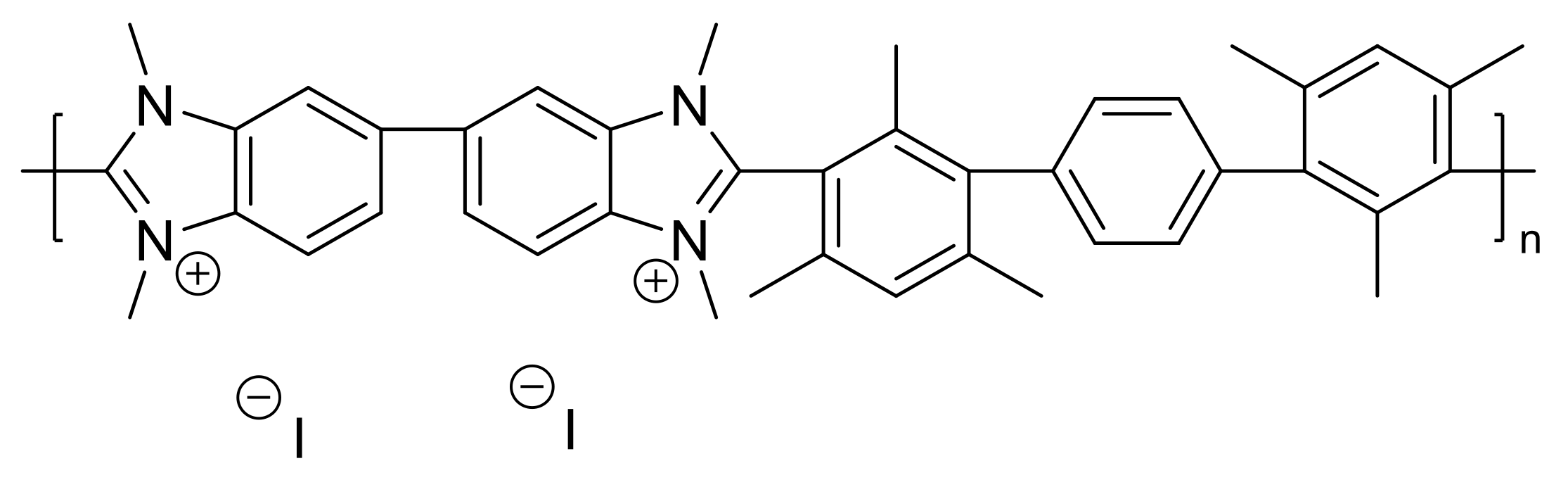
2.2. Synthesis of HMT-PMBI
2.3. Apparatus and Procedures
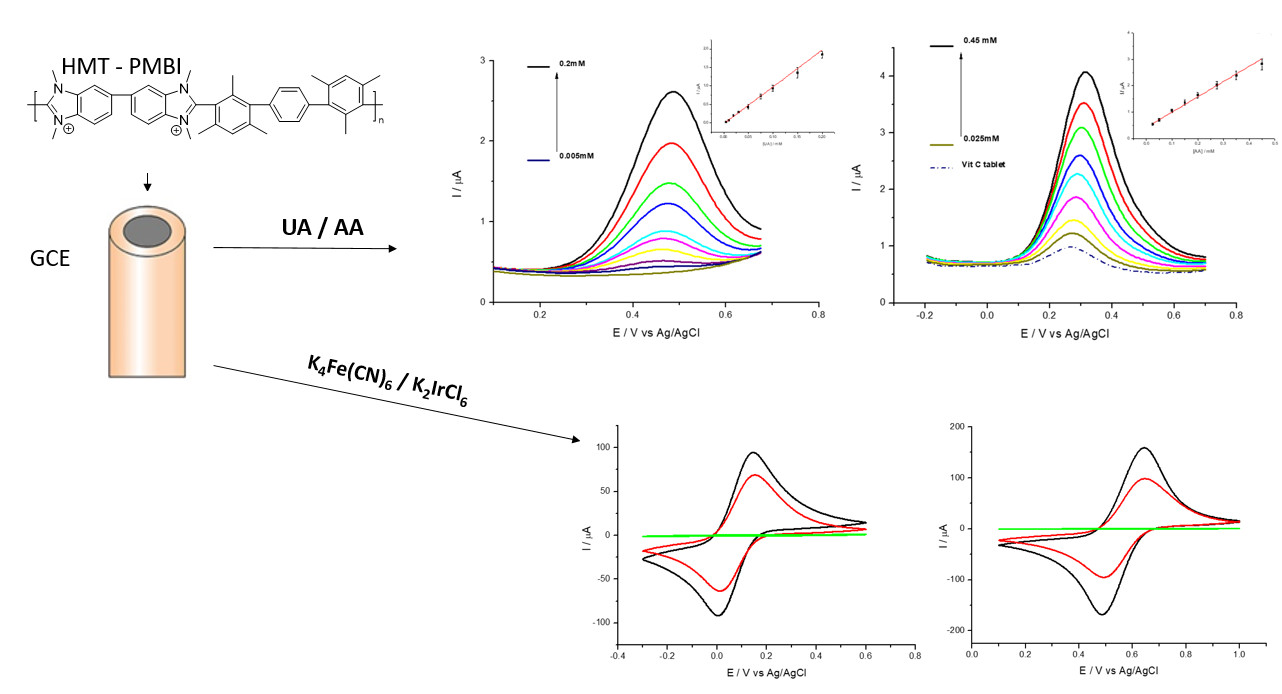
2.4. HMT-PMBI Chemically Modified Electrodes
2.5. Detection of Uric (UA) and Ascorbic (AA) Acid
3. Results and Discussion
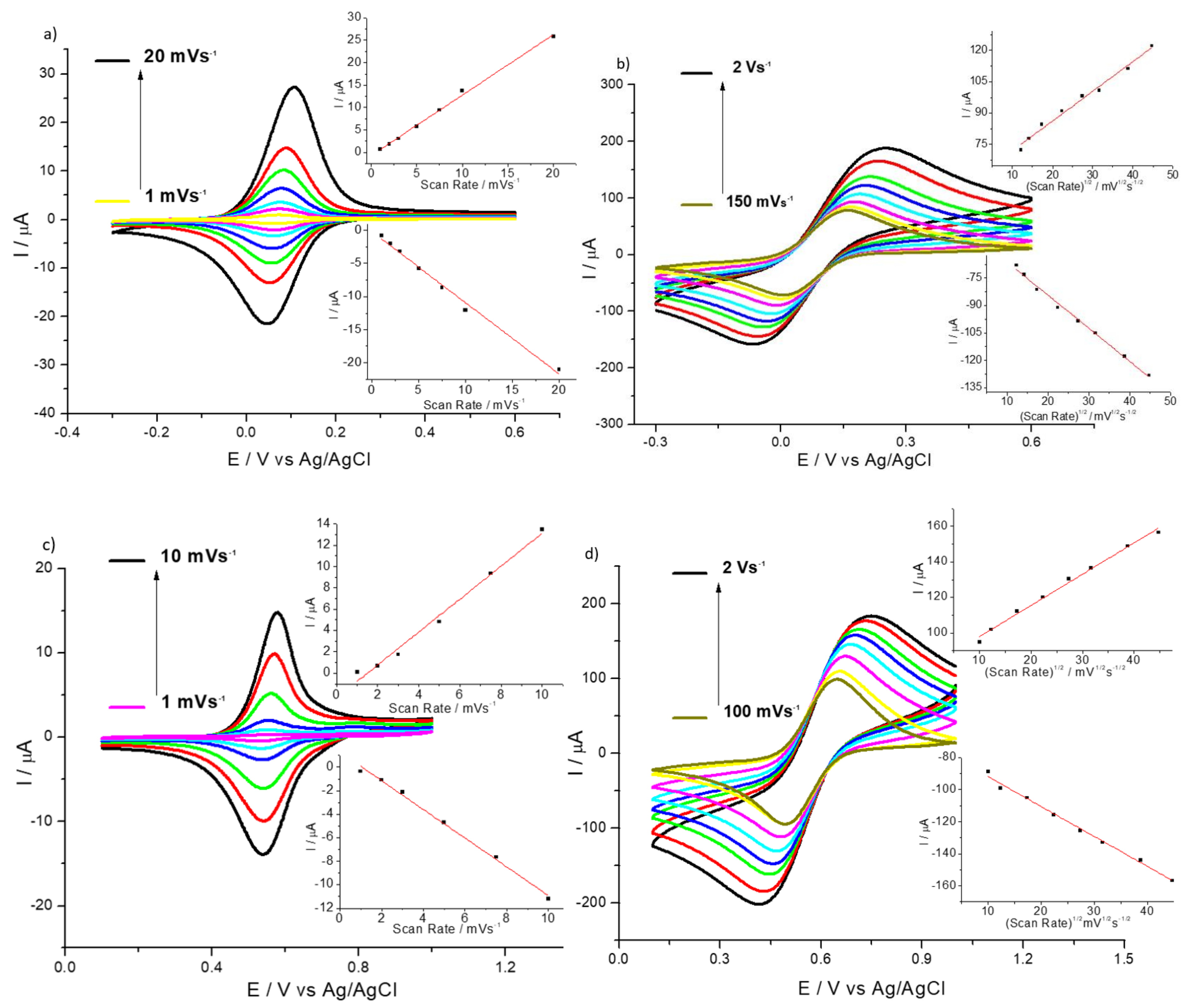
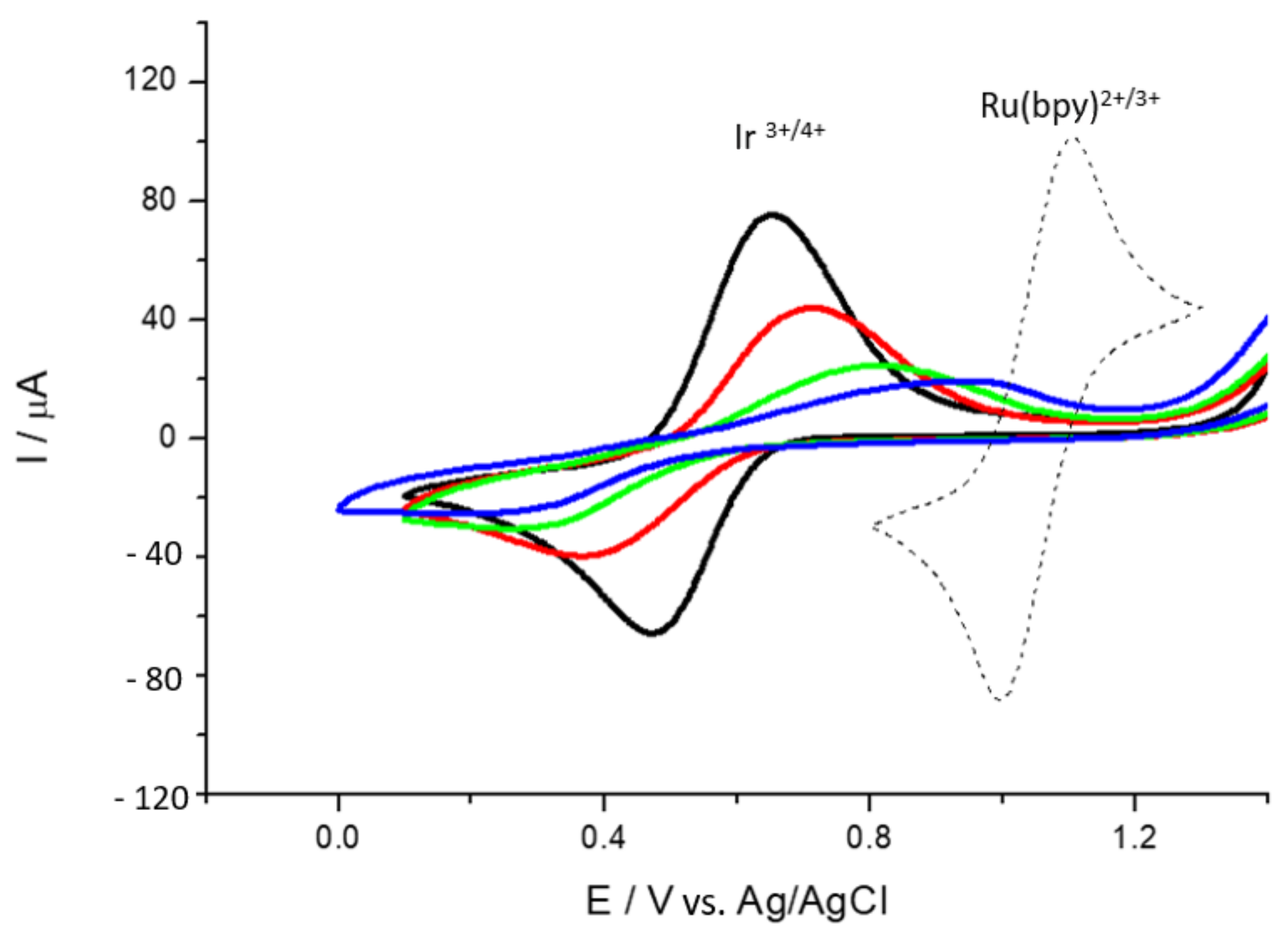
3.1. General Electrochemical Properties
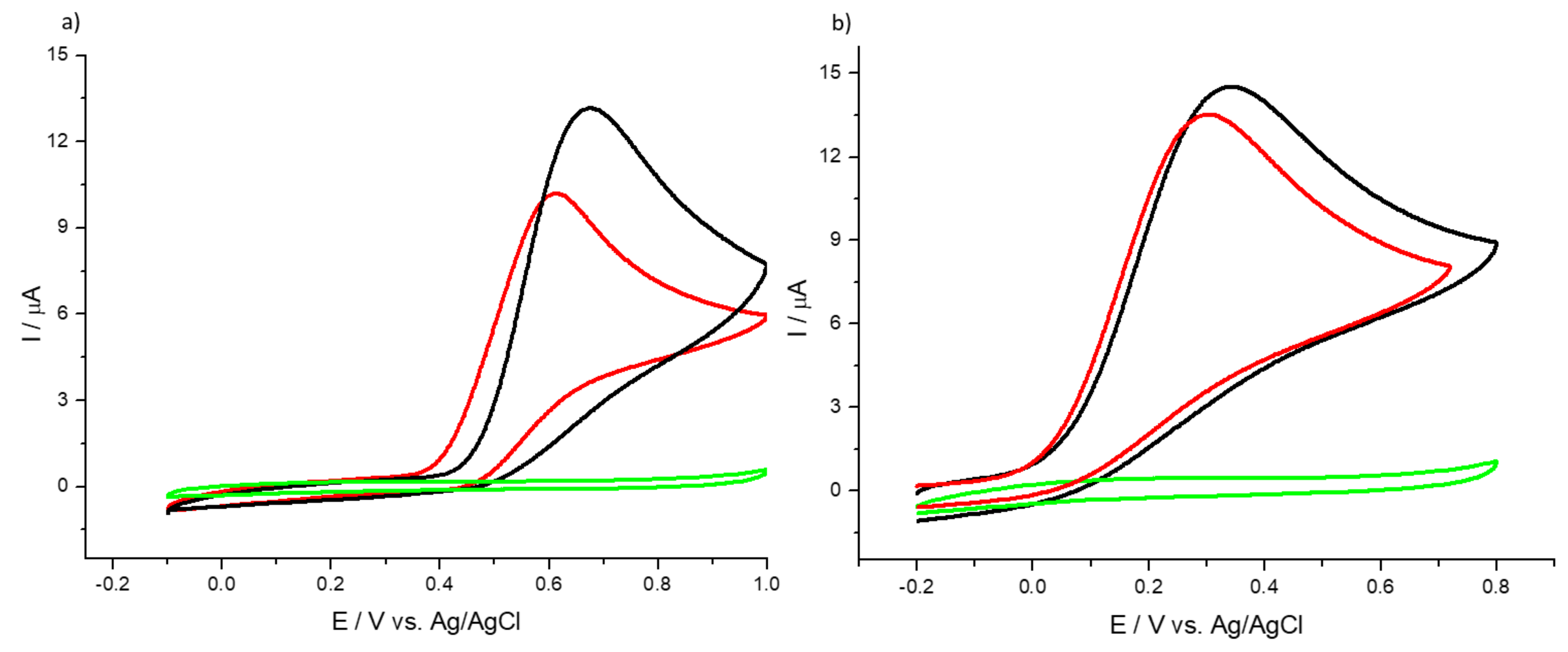
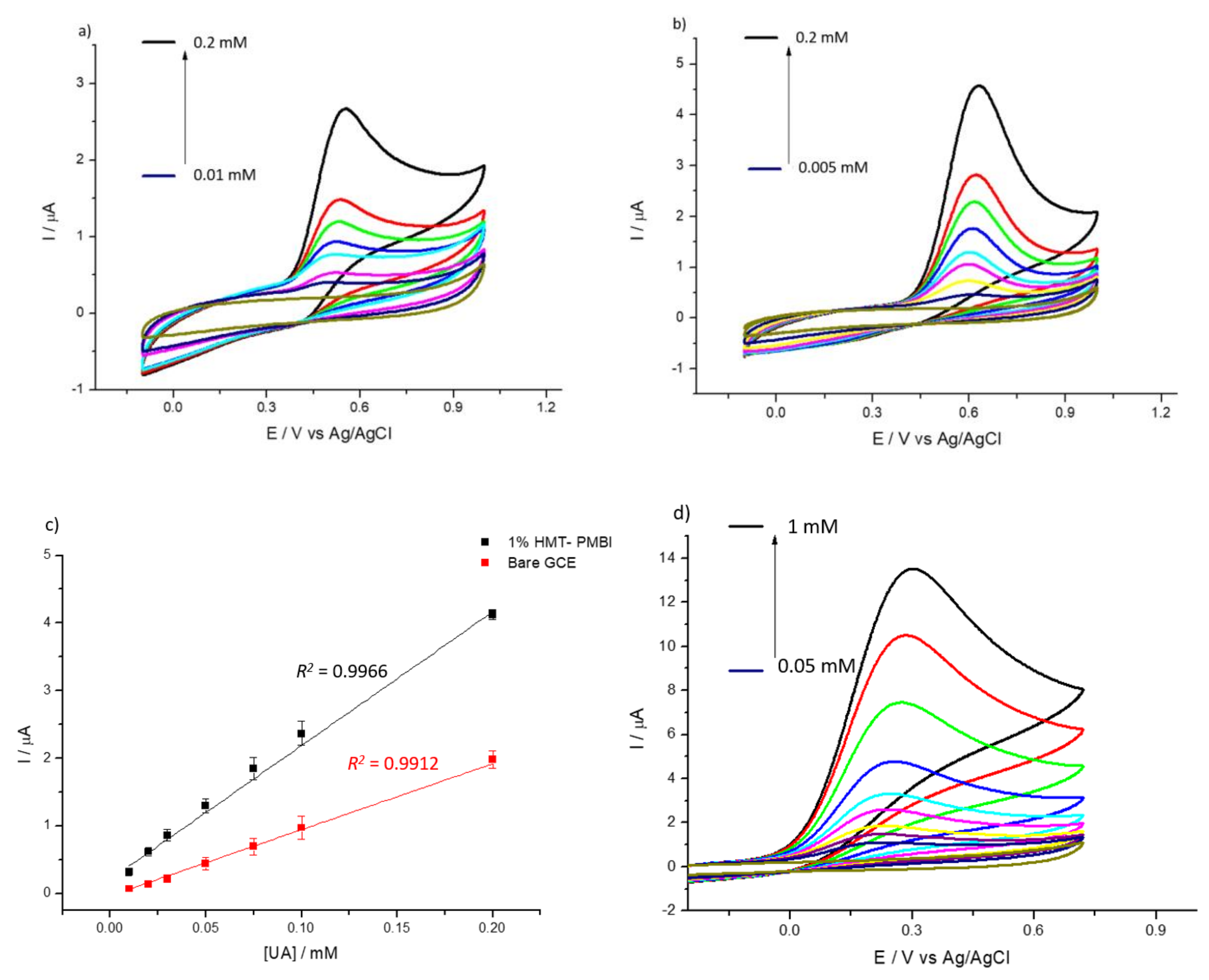
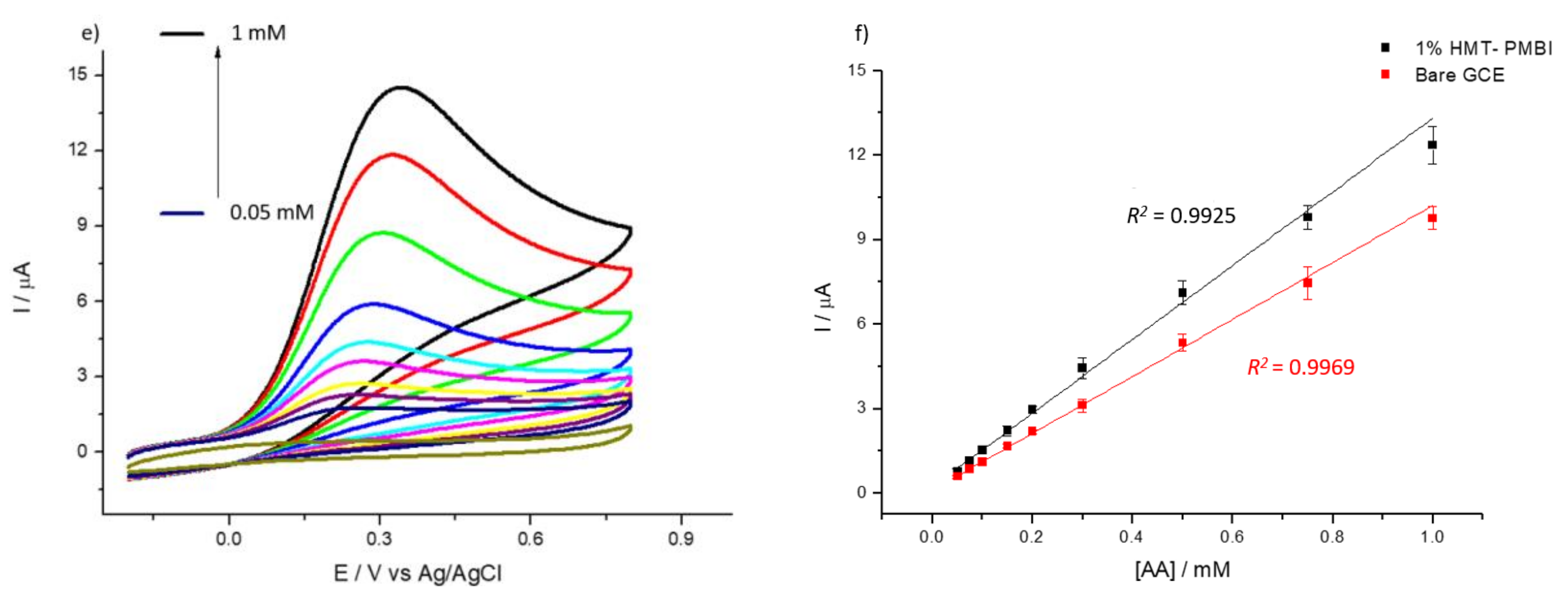
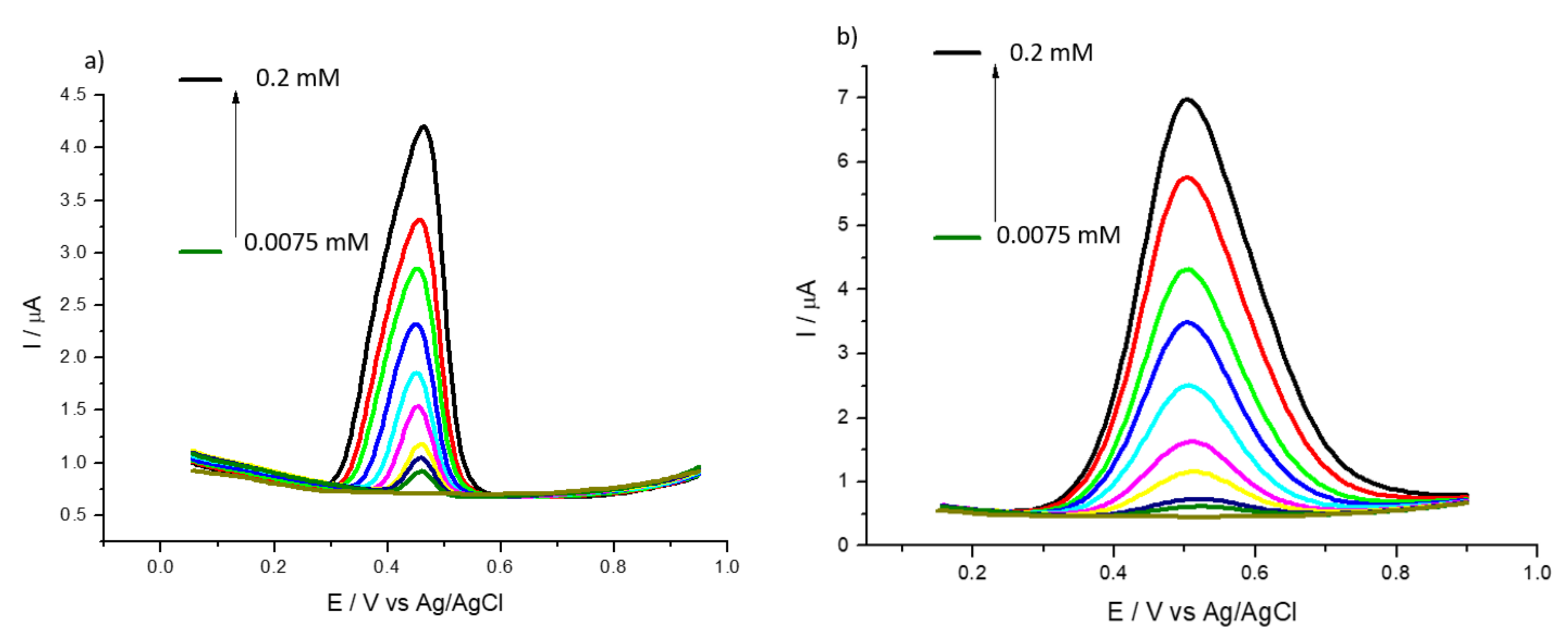
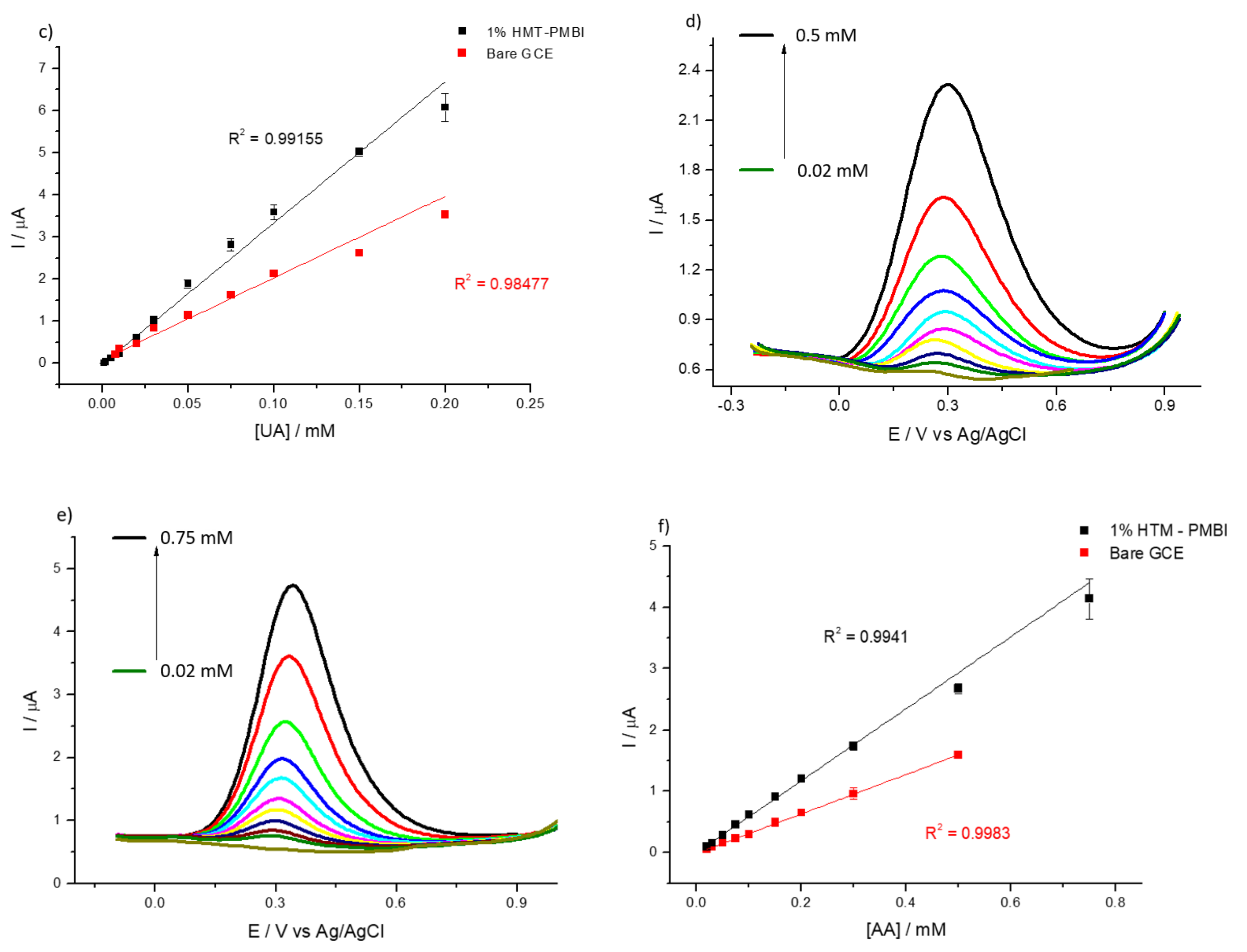
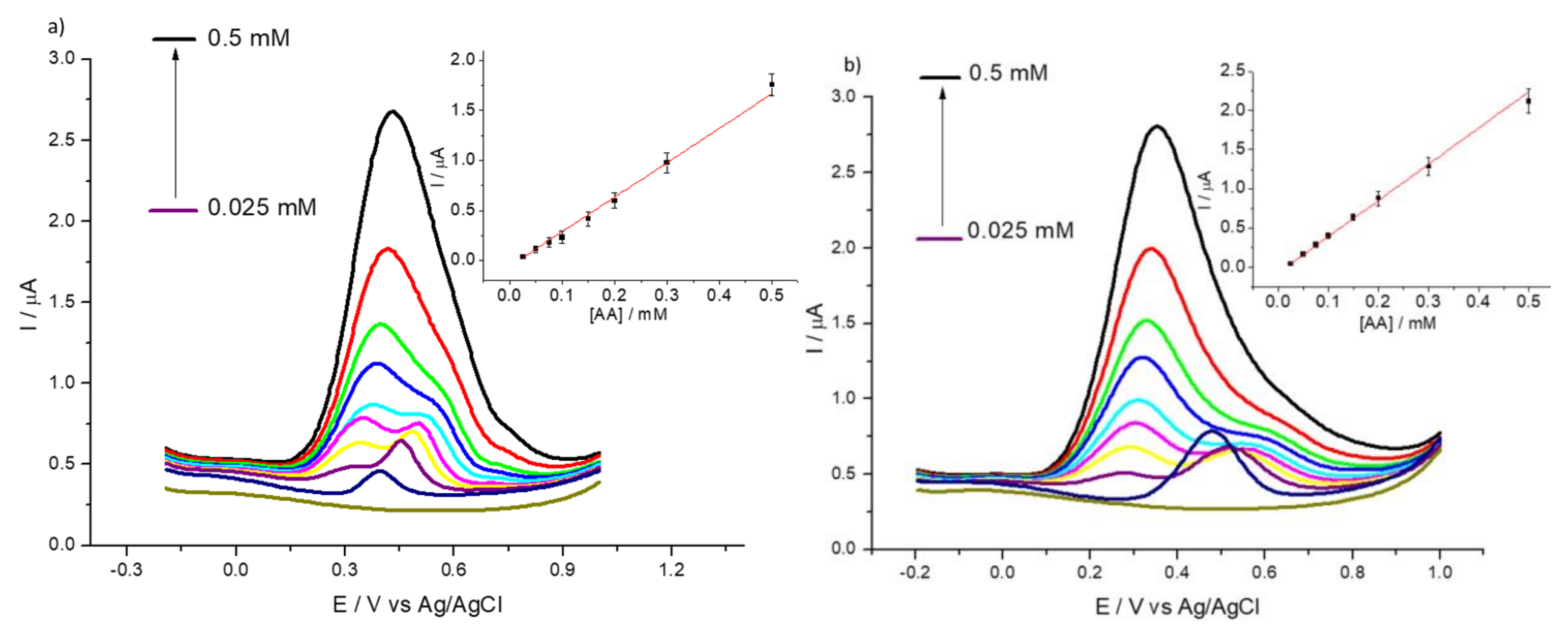
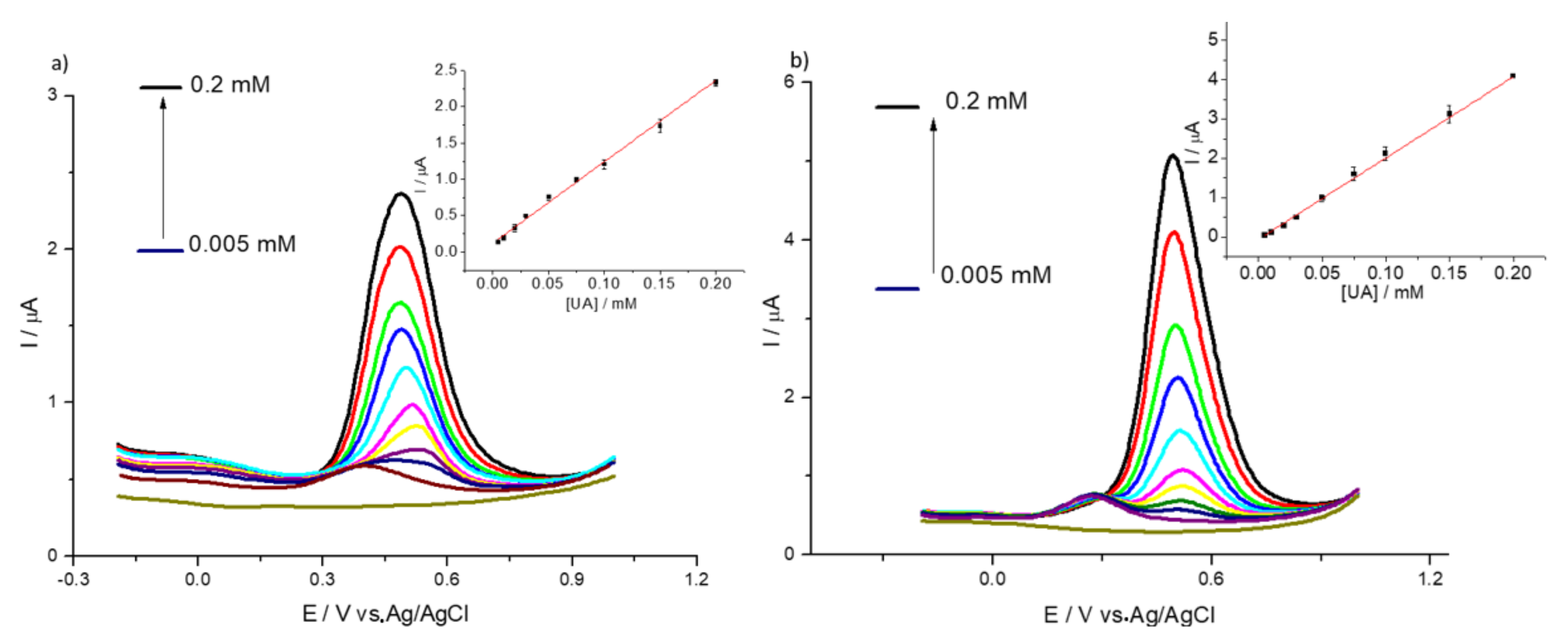
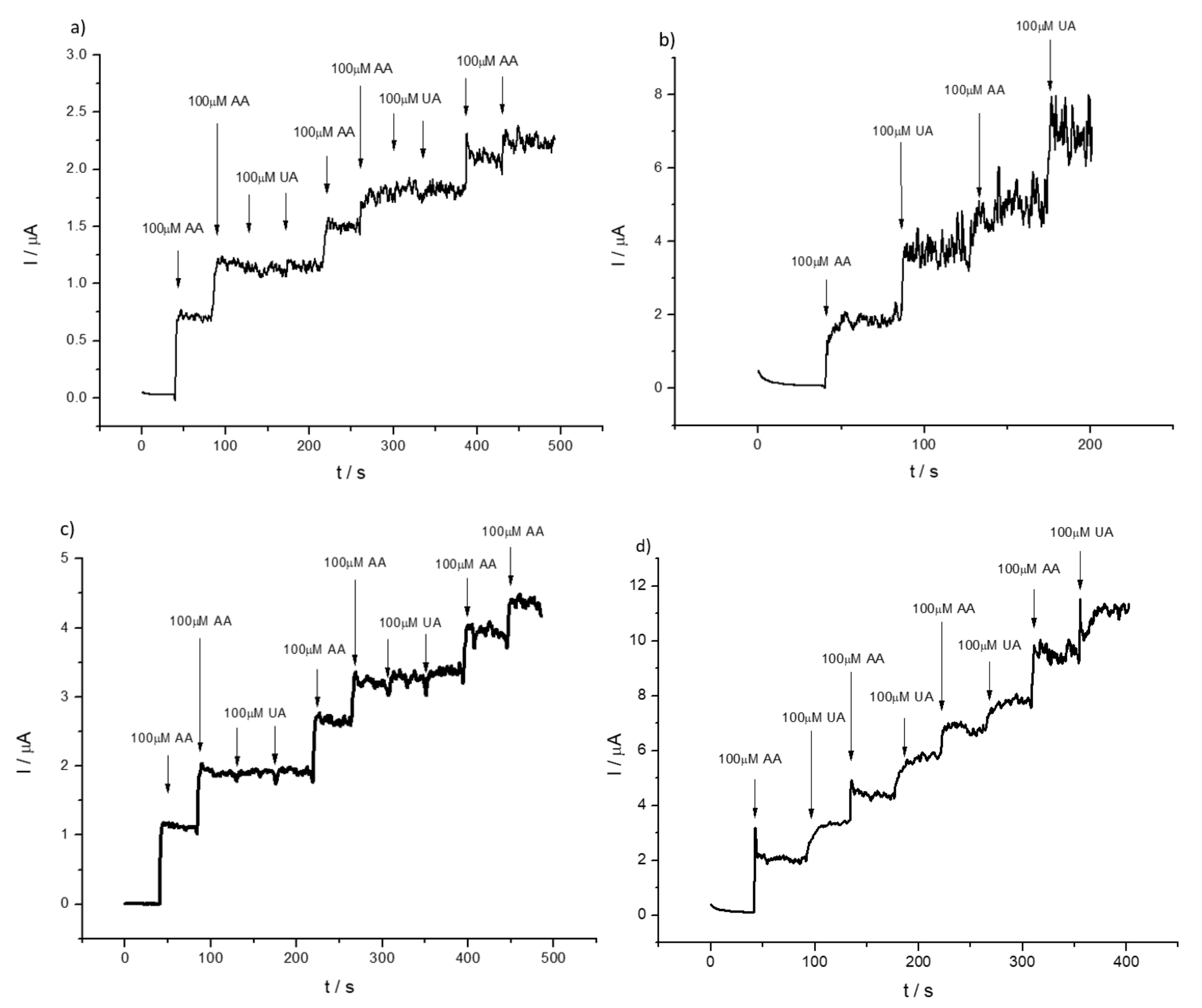
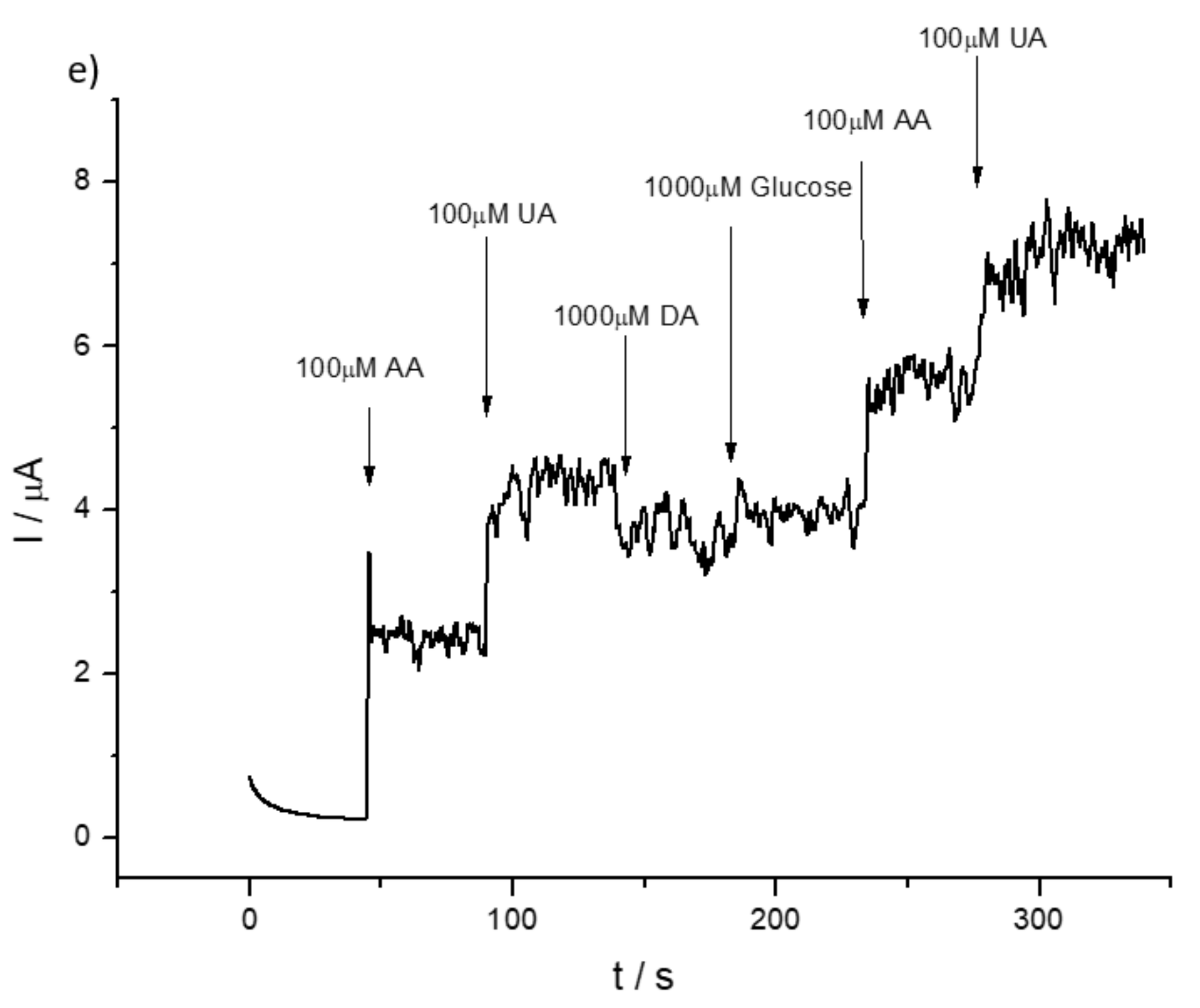
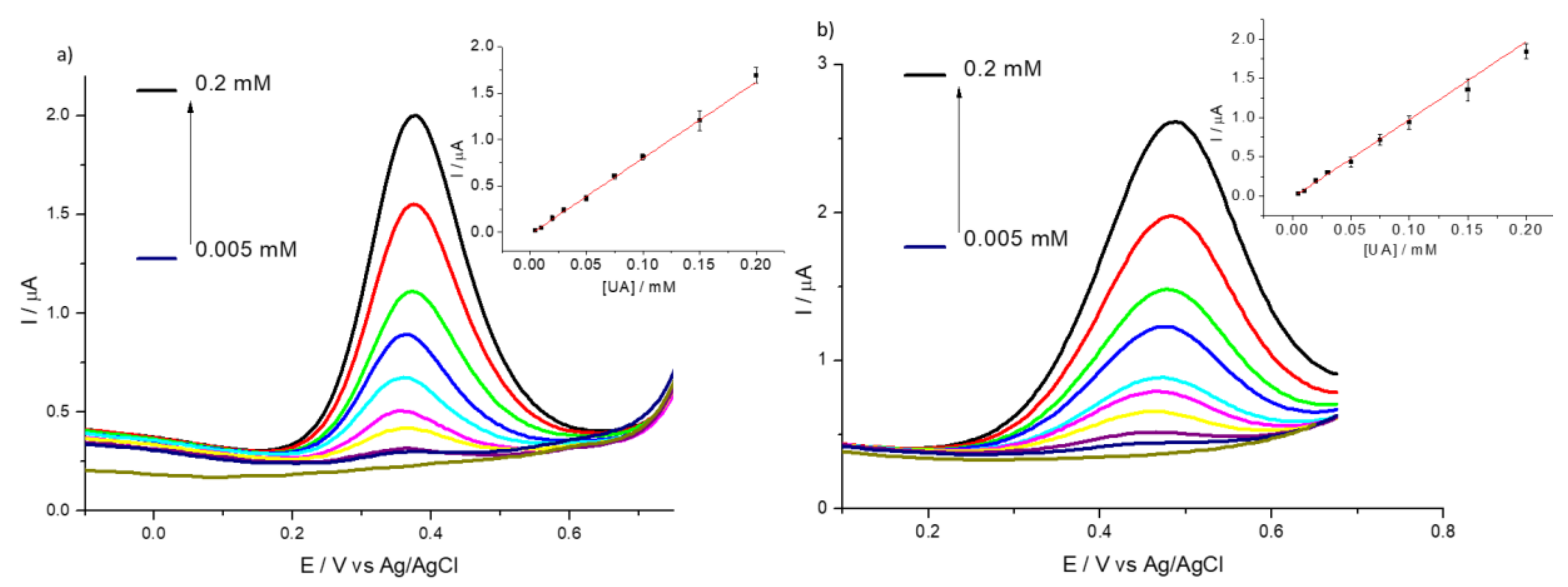
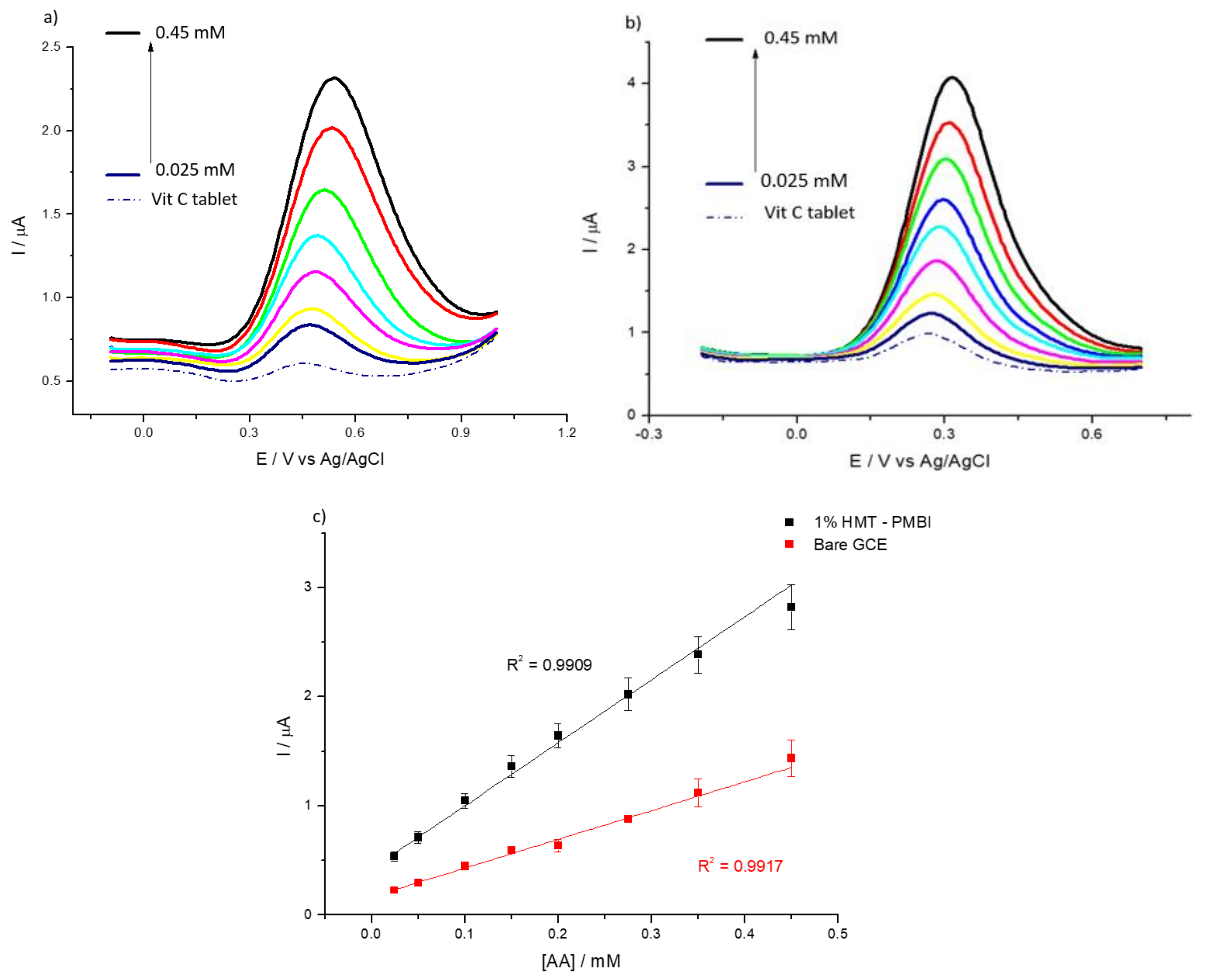
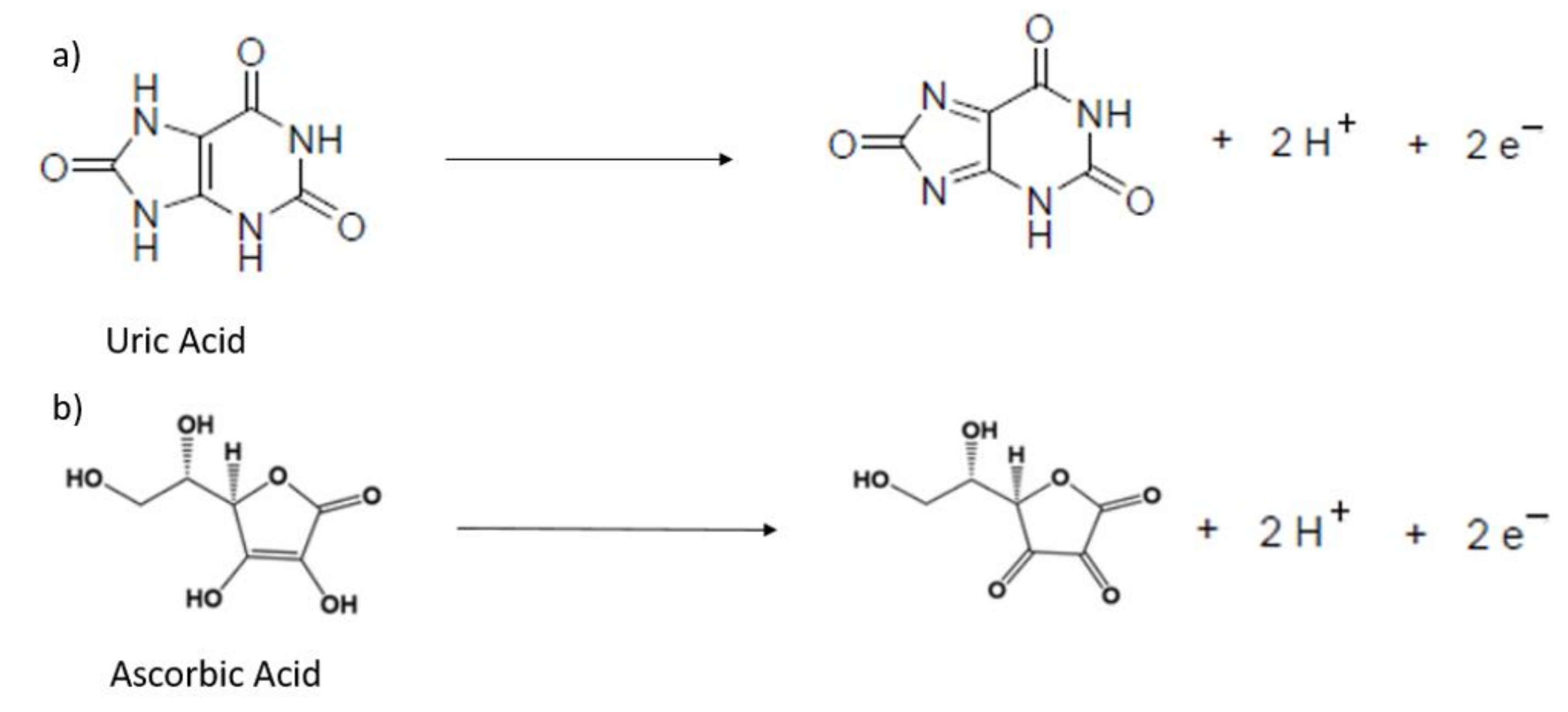
3.2. Detection of Uric and Ascorbic Acids in HMT-PMBI-Coated Electrodes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Forano, C.; Farhat, H.; Mousty, C. Recent trends in electrochemical detection of phosphate in actual waters. Curr. Opin. Electrochem. 2018, 11, 55–61. [Google Scholar] [CrossRef]
- Adhikari, B.; Majumdar, S. Polymers in sensor applications. Prog. Polym. Sci. 2004, 29, 699–766. [Google Scholar] [CrossRef]
- Ronkainen, N.J.; Halsall, H.B.; Heineman, W.R. Electrochemical biosensors. Chem. Soc. Rev. 2010, 39, 1747–1763. [Google Scholar] [CrossRef]
- Zen, J.-M.; Kumar, A.S.; Tsai, D.-M. Recent Updates of Chemically Modified Electrodes in Analytical Chemistry. Electroanalysis 2003, 15, 1073–1087. [Google Scholar] [CrossRef]
- Sajid, M.; Nazal, M.K.; Mansha, M.; Alsharaa, A.; Jillani, S.M.S.; Basheer, C. Chemically modified electrodes for electrochemical detection of dopamine in the presence of uric acid and ascorbic acid: A review. TrAC Trends Anal. Chem. 2016, 76, 15–29. [Google Scholar] [CrossRef]
- Arrigan, D.W.M. Tutorial review. Voltammetric determination of trace metals and organics after accumulation at modified electrodes. Analyst 1994, 119, 1953–1966. [Google Scholar] [CrossRef]
- Ugo, P.; Moretto, L.M. Ion-exchange voltammetry at polymer-coated electrodes: Principles and analytical prospects. Electroanalysis 1995, 7, 1105–1113. [Google Scholar] [CrossRef]
- Smitha, B.; Sridhar, S.; Khan, A. Solid polymer electrolyte membranes for fuel cell applications—A review. J. Membr. Sci. 2005, 259, 10–26. [Google Scholar] [CrossRef]
- Couture, G.; Alaaeddine, A.; Boschet, F.; Ameduri, B. Polymeric materials as anion-exchange membranes for alkaline fuel cells. Prog. Polym. Sci. 2011, 36, 1521–1557. [Google Scholar] [CrossRef]
- Kusoglu, A.; Weber, A.Z. New Insights into Perfluorinated Sulfonic-Acid lonomers. Chem. Rev. 2017, 117, 987–1104. [Google Scholar] [CrossRef]
- Pan, Z.; An, L.; Zhao, T.; Tang, Z. Advances and challenges in alkaline anion exchange membrane fuel cells. Prog. Energy Combust. Sci. 2018, 66, 141–175. [Google Scholar] [CrossRef]
- Geise, G.M.; Lee, H.-S.; Miller, D.J.; Freeman, B.D.; McGrath, J.E.; Paul, D.R. Water purification by membranes: The role of polymer science. J. Polym. Sci. Part B Polym. Phys. 2010, 48, 1685–1718. [Google Scholar] [CrossRef]
- Nagarale, R.; Gohil, G.; Shahi, V.K. Recent developments on ion-exchange membranes and electro-membrane processes. Adv. Colloid Interface Sci. 2006, 119, 97–130. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.; Meier-Haack, J. Preparation of ion-exchange materials and membranes. Desalination 2014, 342, 156–174. [Google Scholar] [CrossRef]
- Qi, L.; Thomas, E.L.; White, S.H.; Smith, S.K.; Lee, C.A.; Wilson, L.R.; Sombers, L.A. Unmasking the Effects of L-DOPA on Rapid Dopamine Signaling with an Improved Approach for Nafion Coating Carbon-Fiber Microelectrodes. Anal. Chem. 2016, 88, 8129–8136. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, P.; Dankoski, E.C.; Petrovic, J.; Keithley, R.B.; Wightman, R.M. Voltammetric Detection of 5-Hydroxytryptamine Release in the Rat Brain. Anal. Chem. 2009, 81, 9462–9471. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.-C.; Chen, P.-Y. Electrochemical Detection of 2-Naphthol at a Glassy Carbon Electrode Modified with Tosflex Film. Electroanalysis 2007, 19, 1315–1321. [Google Scholar] [CrossRef]
- Ugo, P.; Bertoncello, P.; Vezzà, F. Langmuir–Blodgett films of different ionomeric polymers deposited on electrode surfaces. Electrochim. Acta 2004, 49, 3785–3793. [Google Scholar] [CrossRef][Green Version]
- Ugo, P.; Moretto, L.; De Boni, A.; Scopece, P.; Mazzocchin, G. Iron(II) and iron(III) determination by potentiometry and ion-exchange voltammetry at ionomer-coated electrodes. Anal. Chim. Acta 2002, 474, 147–160. [Google Scholar] [CrossRef]
- Yang, H. Anodic stripping voltammetric determination of bismuth(III) using a Tosflex-coated mercury film electrode. Talanta 1999, 50, 977–984. [Google Scholar] [CrossRef]
- Ugo, P.; Moretto, L.M.; Mazzocchin, G.A. Voltammetric determination of trace mercury in chloride media at glassy carbon electrodes modified with polycationic ionomers. Anal. Chim. Acta 1995, 305, 74–82. [Google Scholar] [CrossRef]
- Jones, T.R.; Hernandez-Aldave, S.; Kaspar, R.B.; Letterio, M.P.; Yan, Y.; Bertoncello, P. Tris(2,4,6-trimethoxyphenyl)polysulfone-methylene quaternary phosphonium chloride (TPQPCl) ionomer chemically modified electrodes: An electroanalytical study towards sensing applications. Electrochim. Acta 2019, 311, 160–169. [Google Scholar] [CrossRef]
- Hernandez-Aldave, S.; Kaspar, R.B.; Letterio, M.P.; Tarat, A.; Yan, Y.; Bertoncello, P. Quaternary phosphonium-based (TPQPCl)-ionomer/graphite nanoplatelets composite chemically modified electrodes: A novel platform for sensing applications. J. Mater. Chem. C 2018, 6, 13293–13304. [Google Scholar] [CrossRef]
- Shigehara, K.; Oyama, N.; Anson, F.C. Electrochemical responses of electrodes coated with redox polymers. Evidence for control of charge-transfer rates across polymeric layers by electron exchange between incorporated redox sites. J. Am. Chem. Soc. 1981, 103, 2552–2558. [Google Scholar] [CrossRef]
- Buttry, D.A.; Anson, F.C. Electron hopping vs molecular-diffusion as charge-transfer mechanisms in redox polymer-films. J. Electroanal. Chem. 1981, 130, 333–338. [Google Scholar] [CrossRef]
- Buttry, D.A.; Anson, F.C. Effects of electron exchange and single-file diffusion on charge propagation in nafion films containing redox couples. J. Am. Chem. Soc. 1983, 105, 685–689. [Google Scholar] [CrossRef]
- Buttry, D.A.; Saveant, J.M.; Anson, F.C. Enhancement of charge-transport rates by redox cross-reactions between reactants incorporated in Nafion coatings. J. Phys. Chem. 1984, 88, 3086–3091. [Google Scholar] [CrossRef]
- Amatore, C.; Savéant, J.; Tessier, D. Charge transfer at partially blocked surfaces A model for the case of microscopic active and inactive sites. J. Electroanal. Chem. 1983, 147, 39–51. [Google Scholar] [CrossRef]
- Martin, C.R.; Rubinstein, I.; Bard, A.J. Polymer-films on electrodes.9. electron and mass-transfer in nafion films containing tris(2,2′-bipyridine)ruthenium(2+). J. Am. Chem. Soc. 1982, 104, 4817–4824. [Google Scholar] [CrossRef]
- Rubinstein, I.; Bard, A.J. Polymer-films on electrodes.4. nafion-coated electrodes and electrogenerated chemi-luminescence of surface-attached RU(BPY)32+. J. Am. Chem. Soc 1980, 102, 6641–6642. [Google Scholar] [CrossRef]
- Rubinstein, I.; Bard, A.J. Polymer-films on electrodes. 5. electrochemistry and chemi-luminescence at nafion-coated electrodes. J. Am. Chem. Soc. 1981, 103, 5007–5013. [Google Scholar] [CrossRef]
- White, H.S.; Leddy, J.; Bard, A.J. Polymer films on electrodes. Investigation of charge-transport mechanisms in Nafion polymer modified electrodes. J. Am. Chem. Soc. 1982, 104, 4811–4817. [Google Scholar] [CrossRef]
- Williams, S.R.; Long, T.E. Recent advances in the synthesis and structure–property relationships of ammonium ionenes. Prog. Polym. Sci. 2009, 34, 762–782. [Google Scholar] [CrossRef]
- Bara, J.E.; O’Harra, K.E. Recent Advances in the Design of Ionenes: Toward Convergence with High-Performance Polymers. Macromol. Chem. Phys. 2019, 220, 17. [Google Scholar] [CrossRef]
- Pham, T.H.; Olsson, J.S.; Jannasch, P. N-Spirocyclic Quaternary Ammonium lonenes for Anion-Exchange Membranes. J. Am. Chem. Soc. 2017, 139, 2888–2891. [Google Scholar] [CrossRef]
- Wright, A.G.; Holdcroft, S. Hydroxide-Stable Ionenes. ACS Macro Lett. 2014, 3, 444–447. [Google Scholar] [CrossRef]
- Lee, K.M.; Wycisk, R.; Litt, M.; Pintauro, P.N. Alkaline fuel cell membranes from xylylene block ionenes. J. Membr. Sci. 2011, 383, 254–261. [Google Scholar] [CrossRef]
- Glantzounis, G.; Tsimoyiannis, E.; Kappas, A.; Galaris, D. Uric Acid and Oxidative Stress. Curr. Pharm. Des. 2005, 11, 4145–4151. [Google Scholar] [CrossRef]
- Maesaka, J.K.; Fishbane, S. Regulation of renal urate excretion: A critical review. Am. J. Kidney Dis. 1998, 32, 917–933. [Google Scholar] [CrossRef]
- Jin, M. Uric acid, hyperuricemia and vascular diseases. Front. Biosci. 2012, 17, 656–699. [Google Scholar] [CrossRef]
- Maiuolo, J.; Oppedisano, F.; Gratteri, S.; Muscoli, C.; Mollace, V. Regulation of uric acid metabolism and excretion. Int. J. Cardiol. 2016, 213, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Omar, M.N.; Salleh, A.B.; Lim, H.N.; Tajudin, A.A.; Ngee, L.H. Electrochemical detection of uric acid via uricase-immobilized graphene oxide. Anal. Biochem. 2016, 509, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-H.; Shih, Y.-C.; Wu, C.-Y.; Yuan, C.-J.; Yang, Y.-S.; Li, Y.-K.; Wu, T.-K. Detection of serum uric acid using the optical polymeric enzyme biochip system. Biosens. Bioelectron. 2004, 19, 1627–1633. [Google Scholar] [CrossRef] [PubMed]
- Raj, C.R.; Ohsaka, T. Voltammetric detection of uric acid in the presence of ascorbic acid at a gold electrode modified with a self-assembled monolayer of heteroaromatic thiol. J. Electroanal. Chem. 2003, 540, 69–77. [Google Scholar]
- Johnson, R.J.; Kang, D.-H.; Feig, D.; Kivlighn, S.; Kanellis, J.; Watanabe, S.; Tuttle, K.R.; Rodriguez-Iturbe, B.; Herrera-Acosta, J.; Mazzali, M. Is There a Pathogenetic Role for Uric Acid in Hypertension and Cardiovascular and Renal Disease? Hypertension 2003, 41, 1183–1190. [Google Scholar] [CrossRef]
- Grassi, D.; Ferri, L.; Desideri, G.; Di Giosia, P.; Cheli, P.; Del Pinto, R.; Properzi, G.; Ferri, C. Chronic Hyperuricemia, Uric Acid Deposit and Cardiovascular Risk. Curr. Pharm. Des. 2013, 19, 2432–2438. [Google Scholar] [CrossRef]
- Wortmann, R.L. Gout and hyperuricemia. Curr. Opin. Rheumatol. 2002, 14, 281–286. [Google Scholar] [CrossRef]
- Choi, H.K.; Atkinson, K.; Karlson, E.W.; Willett, W.; Curhan, G. Alcohol intake and risk of incident gout in men: A prospective study. Lancet 2004, 363, 1277–1281. [Google Scholar] [CrossRef]
- Johnson, R.J.; Perez-Pozo, S.E.; Sautin, Y.Y.; Manitius, J.; Sánchez-Lozada, L.G.; Feig, D.I.; Shafiu, M.; Segal, M.; Glassock, R.J.; Shimada, M.; et al. Hypothesis: Could excessive fructose intake and uric acid cause type 2 diabetes? Endocr. Rev. 2009, 30, 96–116. [Google Scholar] [CrossRef]
- Wang, D.D.; Sievenpiper, J.L.; De Souza, R.J.; Chiavaroli, L.; Ha, V.; Cozma, A.I.; Mirrahimi, A.; Yu, M.E.; Carleton, A.J.; Di Buono, M.; et al. The effects of fructose intake on serum uric acid vary among controlled dietary trials. J. Nutr. 2012, 142, 916–923. [Google Scholar] [CrossRef]
- Li, C.; Hsieh, M.-C.; Chang, S.-J. Metabolic syndrome, diabetes, and hyperuricemia. Curr. Opin. Rheumatol. 2013, 25, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Taylor, E.N.; Stampfer, M.J.; Curhan, G.C. Obesity, Weight Gain, and the Risk of Kidney Stones. JAMA J. Am. Med. Assoc. 2005, 293, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.J.; Nakagawa, T.; Sanchez-Lozada, L.-G.; Shafiu, M.; Sundaram, S.; Le, M.; Ishimoto, T.; Sautin, Y.Y.; Lanaspa, M.A. Sugar, Uric Acid, and the Etiology of Diabetes and Obesity. Diabetes 2013, 62, 3307–3315. [Google Scholar] [CrossRef] [PubMed]
- Dawson, J.; Walters, M. Uric acid and xanthine oxidase: Future therapeutic targets in the prevention of cardiovascular disease? Br. J. Clin. Pharmacol. 2006, 62, 633–644. [Google Scholar] [CrossRef]
- Ames, B.N.; Shigenaga, M.K.; Hagen, T.M. Oxidants, antioxidants, and the degenerative diseases of aging. Proc. Natl. Acad. Sci. USA 1993, 90, 7915–7922. [Google Scholar] [CrossRef]
- Cozzi, R.; Ricordy, R.; Aglitti, T.; Gatta, V.; Perticone, P.; De Salvia, R. Ascorbic acid and beta-carotene as modulators of oxidative damage. Carcinogenesis 1997, 18, 223–228. [Google Scholar] [CrossRef][Green Version]
- Wright, A.G.; Fan, J.; Britton, B.; Weissbach, T.; Lee, H.-F.; Kitching, E.A.; Peckham, T.J.; Holdcroft, S. Hexamethyl-p-terphenyl poly(benzimidazolium): A universal hydroxide-conducting polymer for energy conversion devices. Energy Environ. Sci. 2016, 9, 2130–2142. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods, Fundamentals and Applications, 2nd ed.; Wiley: Hoboken, NJ, USA, 2001. [Google Scholar]
- Bertoncello, P.; Ciani, I.; Fei, L.; Unwin, P.R. Measurement of apparent diffusion coefficients within ultrathin nation langmuir-schaefer films: Comparison of a novel scanning electrochemical microscopy approach with cyclic voltammetry. Langmuir 2006, 22, 10380–10388. [Google Scholar] [CrossRef]
- Huang, X.-J.; Im, H.-S.; Yarimaga, O.; Kim, J.-H.; Lee, H.; Kim, H.-S.; Choi, Y.-K. Direct Electrochemistry of Uric Acid at Chemically Assembled Carboxylated Single-Walled Carbon Nanotubes Netlike Electrode. J. Phys. Chem. B 2006, 110, 21850–21856. [Google Scholar] [CrossRef]
- Owens, J. Electrochemical oxidation of uric acid and xanthine An investigation by cyclic voltammetry, double potential step chronoamperometry and thin-layer spectroelectrochemistry. J. Electroanal. Chem. 1978, 91, 231–247. [Google Scholar] [CrossRef]
- Khan, M.I.; Haque, A.-M.J.; Kim, K. Electrochemical determination of uric acid in the presence of ascorbic acid on electrochemically reduced graphene oxide modified electrode. J. Electroanal. Chem. 2013, 700, 54–59. [Google Scholar] [CrossRef]
- Li, Y.; Wang, P.; Li, F.; Huang, X.; Wang, L.; Lin, X. Covalent immobilization of single-walled carbon nanotubes and single-stranded deoxyribonucleic acid nanocomposites on glassy carbon electrode: Preparation, characterization, and applications. Talanta 2008, 77, 833–838. [Google Scholar] [CrossRef]
- Wang, P.; Li, Y.; Huang, X.; Wang, L. Fabrication of layer-by-layer modified multilayer films containing choline and gold nanoparticles and its sensing application for electrochemical determination of dopamine and uric acid. Talanta 2007, 73, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Ngai, K.S.; Tan, W.T.; Zainal, Z.; Zawawi, R.B.M.; Zidan, M. Electrochemical oxidation of ascorbic acid mediated by single-walled carbon nanotube/tungsten oxide nanoparticles modified glassy carbon electrode. Int. J. Electrochem. Sci. 2012, 7, 4210–4222. [Google Scholar]
- Ngai, K.S.; Tan, T.W.; Zainal, Z.; Zawawi, R.M.; Zidan, M. Voltammetry detection of ascorbic acid at glassy carbon electrode modified by single-walled carbon nanotube/zinc oxide. Int. J. Electrochem. Sci. 2013, 8, 10557–10567. [Google Scholar]
- Bilal, S.; Akbar, A.; Shah, A.-U.-H.A. Highly Selective and Reproducible Electrochemical Sensing of Ascorbic Acid Through a Conductive Polymer Coated Electrode. Polymers 2019, 11, 346. [Google Scholar] [CrossRef]
- Olguín, H.J.; Guzmán, D.C.; García, E.H.; Mejía, G.B. The Role of Dopamine and Its Dysfunction as a Consequence of Oxidative Stress. Oxidative Med. Cell. Longev. 2016, 2016, 1–13. [Google Scholar] [CrossRef]
- Da Silva, L.V.; Silva, F.A.S.; Kubota, L.T.; Lopes, C.B.; Lima, P.R.; Costa, E.O.; Júnior, W.P.; Goulart, M.O.F. Amperometric sensor based on carbon nanotubes and electropolymerized vanillic acid for simultaneous determination of ascorbic acid, dopamine, and uric acid. J. Solid State Electrochem. 2016, 20, 2389–2393. [Google Scholar] [CrossRef]
- Manivel, P.; Dhakshnamoorthy, M.; Balamurugan, A.; Ponpandian, N.; Mangalaraj, D.; Viswanathan, C.N.P. Conducting polyaniline-graphene oxide fibrous nanocomposites: Preparation, characterization and simultaneous electrochemical detection of ascorbic acid, dopamine and uric acid. RSC Adv. 2013, 3, 14428–14437. [Google Scholar] [CrossRef]
- Yang, Y.J. One-pot synthesis of reduced graphene oxide/zinc sulfide nanocomposite at room temperature for simultaneous determination of ascorbic acid, dopamine and uric acid. Sens. Actuators B Chem. 2015, 221, 750–759. [Google Scholar] [CrossRef]
- Bao, Y.; Song, J.; Mao, Y.; Han, D.; Yang, F.; Niu, L.; Ivaska, A. Graphene Oxide-Templated Polyaniline Microsheets toward Simultaneous Electrochemical Determination of AA/DA/UA. Electroanalysis 2011, 23, 878–884. [Google Scholar] [CrossRef]
- Zhao, D.; Yu, G.; Tian, K.; Xu, C. A highly sensitive and stable electrochemical sensor for simultaneous detection towards ascorbic acid, dopamine, and uric acid based on the hierarchical nanoporous PtTi alloy. Biosens. Bioelectron. 2016, 82, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S.; Yu, S.H.; Kim, T.H. One-Step Electrochemical Fabrication of Reduced Graphene Oxide/Gold Nanoparticles Nanocomposite-Modified Electrode for Simultaneous Detection of Dopamine, Ascorbic Acid, and Uric Acid. Nanomaterials 2018, 8, 13. [Google Scholar] [CrossRef] [PubMed]
- Zou, C.; Zhong, J.; Li, S.; Wang, H.; Wang, J.; Yan, B.; Du, Y. Fabrication of reduced graphene oxide-bimetallic PdAu nanocomposites for the electrochemical determination of ascorbic acid, dopamine, uric acid and rutin. J. Electroanal. Chem. 2017, 805, 110–119. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| [HMT-PMBI] % | Φ/(10−4) cm | Γ/(10−9) mol cm−2 | Cp/(10−2) mol dm−3 | m/(10−10) moles | Dapp/RS(10−10) cm2s−1 | S/(10−4) Cs−1/2 | Dapp/Anson(10−10) Cm2s−1 |
|---|---|---|---|---|---|---|---|
| K4Fe(CN)6 | |||||||
| 0.5 | 0.49 ± 0.03 | 1.95 ± 0.40 | 3.92 ± 0.79 | 1.39 ± 0.28 | 2.51 ± 0.74 | 1.25 ± 0.08 | 2.61 ± 0.42 |
| 0.75 | 0.69 ± 0.21 | 8.50 ± 0.72 | 10.38 ± 1.04 | 6.03 ± 0.51 | 4.72 ± 0.91 | 1.11 ± 0.07 | 1.12 ± 0.13 |
| 1 | 1.09 ± 0.13 | 10.15 ± 0.80 | 9.35 ± 0.74 | 7.21 ± 0.57 | 6.44 ± 1.06 | 1.21 ± 0.07 | 2.42 ± 0.61 |
| K2IrCl6 | Dapp/RS(10−9) cm2s−1 | Dapp/Anson(10−9) Cm2s−1 | |||||
| 0.5 | 0.49 ± 0.03 | 3.08 ± 0.48 | 6.19 ± 0.96 | 2.18 ± 0.34 | 1.53 ± 0.53 | 2.19 ± 0.14 | 1.11 ± 0.38 |
| 0.75 | 0.69 ± 0.21 | 3.04 ± 0.27 | 4.40 ± 0.39 | 2.16 ± 0.19 | 3.71 ± 0.72 | 1.74 ± 0.02 | 1.28 ± 0.26 |
| 1 | 1.09 ± 0.13 | 6.23 ± 1.14 | 5.74 ± 1.05 | 4.43 ± 0.81 | 2.72 ± 0.85 | 1.65 ± 0.08 | 3.11 ± 1.24 |
| Electrode | Linear Range UA (µM) | LoD UA (µM) | Linear Range AA (µM) | LoD AA (µM) | Technique | Reference |
|---|---|---|---|---|---|---|
| MWCNT/poly VA GCE | 5–120 | 1.5 | 5–120 | 3.5 | CV | [69] |
| PANi/rGO GCE | 2–18 | 0.2 | 25–200 | 20 | DPV | [70] |
| CTAB/rGO/ZnS/GCE | 1–500 | 0.4 | 1–500 | 30 | DPV | [71] |
| GO-PAN GCE | 3–26 | 1 | 150–1050 | 50 | DPV | [72] |
| HNP-PtTi | 100–1000 | 5.3 | 200–1000 | 24.2 | DPV | [73] |
| rGO/AuNPs GCE | 0.1–100 | 2.2 | 0.01–1 | 5.7 | DPV | [74] |
| PdAu@RGO/GCE | 2.5–66.25 | 2.5 | 12.5–700 | 12.5 | DPV | [75] |
| HMT-PMBI GCE in NaCl | 5–200 | 25 | 20–750 | 33 | DPV | This work |
| HMT-PMBI GCE in Surine® | 5–200 | 7.7 | - | - | DPV | This work |
| HMT-PMBI GCE in Vit. C tablet | - | - | 25–450 | 41 | DPV | This work |
| Sample | Added (µM) | Found (µM) | Recovery % |
|---|---|---|---|
| Vitamin C | - | 50 ± 4.39 | - |
| 100 | 147.93 ± 6.41 | 98.6% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rees, M.; Wright, A.G.; Holdcroft, S.; Bertoncello, P. Voltammetry at Hexamethyl-P-Terphenyl Poly(Benzimidazolium) (HMT-PMBI)-Coated Glassy Carbon Electrodes: Charge Transport Properties and Detection of Uric and Ascorbic Acid. Sensors 2020, 20, 443. https://doi.org/10.3390/s20020443
Rees M, Wright AG, Holdcroft S, Bertoncello P. Voltammetry at Hexamethyl-P-Terphenyl Poly(Benzimidazolium) (HMT-PMBI)-Coated Glassy Carbon Electrodes: Charge Transport Properties and Detection of Uric and Ascorbic Acid. Sensors. 2020; 20(2):443. https://doi.org/10.3390/s20020443
Chicago/Turabian StyleRees, Matthew, Andrew G. Wright, Steven Holdcroft, and Paolo Bertoncello. 2020. "Voltammetry at Hexamethyl-P-Terphenyl Poly(Benzimidazolium) (HMT-PMBI)-Coated Glassy Carbon Electrodes: Charge Transport Properties and Detection of Uric and Ascorbic Acid" Sensors 20, no. 2: 443. https://doi.org/10.3390/s20020443
APA StyleRees, M., Wright, A. G., Holdcroft, S., & Bertoncello, P. (2020). Voltammetry at Hexamethyl-P-Terphenyl Poly(Benzimidazolium) (HMT-PMBI)-Coated Glassy Carbon Electrodes: Charge Transport Properties and Detection of Uric and Ascorbic Acid. Sensors, 20(2), 443. https://doi.org/10.3390/s20020443