Self-Referenced Multifrequency Phase-Resolved Luminescence Spectroscopy

 ,
,  ,
,  and
and
Abstract
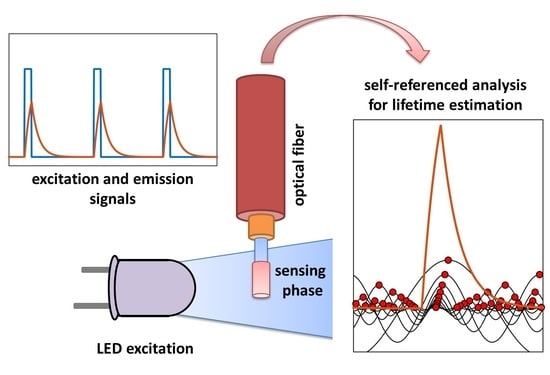
1. Introduction
2. Self-Referenced Estimation of the Luminescence Lifetime
2.1. Response of a Monoexponential Luminescent System
2.2. Conventional Estimation of the Lifetime
2.3. Lifetime Derived from the Amplitudes Referenced to the First Harmonic
2.4. Lifetime Derived from the Delay Referenced to the First Harmonic
2.5. Summary of the Self-Referenced Amplitude- and Delay-Based Lifetime Estimations
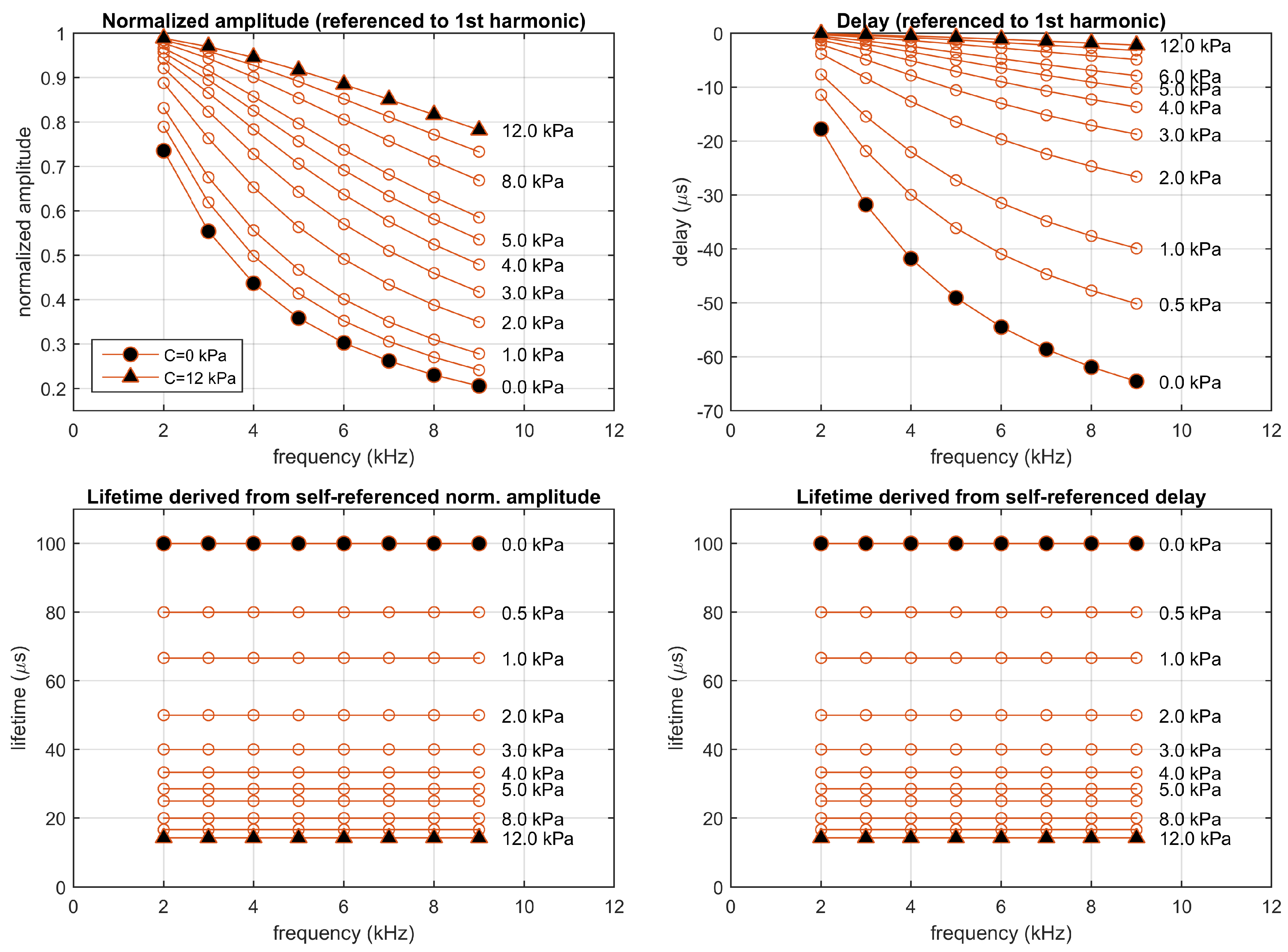
2.6. Self-Referenced Lifetime Determination in a Simulation
2.7. Determination of the Analyte Concentration
3. Experimental Design
4. Results and Discussion
4.1. One-Site and Two-Sites Model Fitting the Experimental Data
4.2. Simulations of the Experiments Using the One-Site Model
4.3. Simulations of the Experiments Using the Two-Sites Model
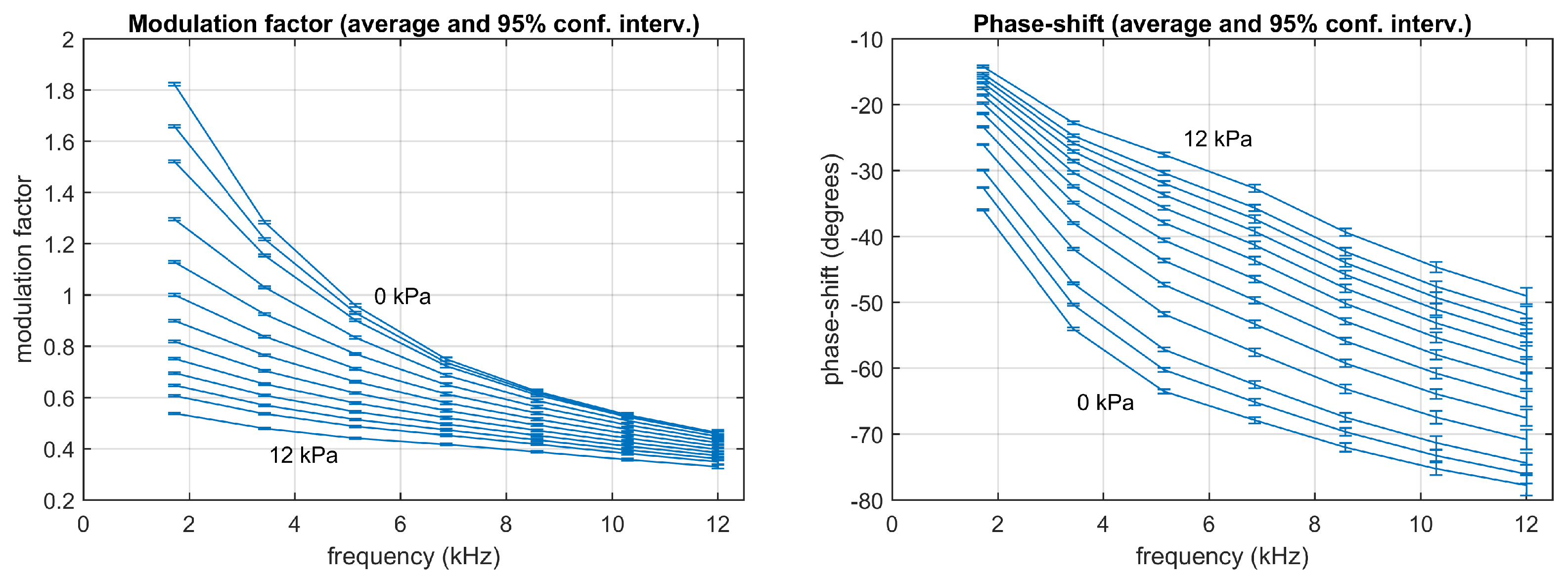
4.4. Experimental Lifetimes Estimated with the Conventional Method
4.5. Experimental Lifetimes Estimated with the Self-Referenced Procedure
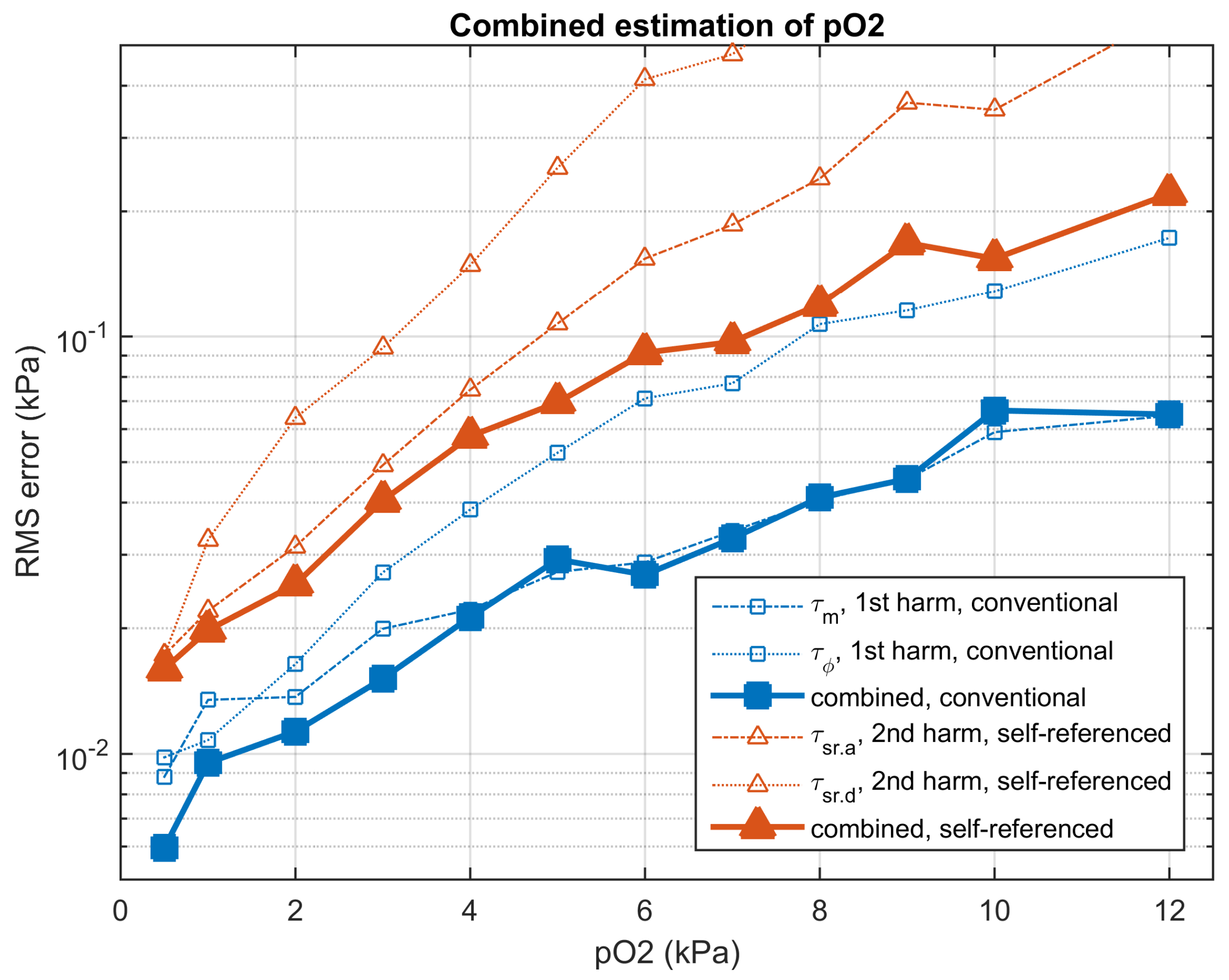
4.6. Accuracy in the Conventional and Self-Referenced Determination
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Trettnak, W.; Gruber, W.; Reininger, F.; Klimant, I. Recent progress in optical oxygen sensor instrumentation. Sens. Actuators B Chem. 1995, 29, 219–225. [Google Scholar] [CrossRef]
- Szmacinski, H.; Lakowicz, J.R. Lifetime-Based Sensing. In Topics in Fluorescence Spectroscopy; Geddes, C.D., Lakowicz, J.R., Eds.; Springer: Berlin/Heidelberg, Germany, 2002; pp. 295–334. [Google Scholar]
- Orellana, G. Luminescent optical sensors. Anal. Bioanal. Chem. 2004, 379, 344–346. [Google Scholar] [CrossRef]
- Wolfbeis, O.S.; Weidgans, B.M.; Baldini, F.; Chester, A.N.; Homola, J.; Martellucci, S. Fiber Optic Chemical Sensors and Biosensors: A View Back Optical Chemical Sensors; Springer: Dordrecht, The Netherlands, 2006; pp. 17–44. [Google Scholar]
- Medina-Rodríguez, S.; de la Torre-Vega, A.; Fernández-Sánchez, J.F.; Fernández-Gutiérrez, A. An open and low-cost optical-fiber measurement system for the optical detection of oxygen using a multifrequency phase-resolved method. Sens. Actuators B Chem. 2013, 176, 1110–1120. [Google Scholar] [CrossRef]
- Torre-Vega, A.; Medina-Rodríguez, S.; Medina-Rodríguez, C.; Fernández-Sánchez, J.F. CHAPTER 6: Progress in Phosphorescence Lifetime Measurement Instrumentation for Oxygen Sensing. In Quenched-Phosphorescence Detection of Molecular Oxygen: Applications in Life Sciences; Papkovsky, D.B., Dmitriev, R.I., Eds.; RSC Detection Science Series No. 11; The Royal Society of Chemistry: London, UK, 2018; pp. 117–144. [Google Scholar]
- Klimant, I.; Meyer, V.; Kühl, M.M. Fiber-optic oxygen microsensors, a new tool in aquatic biology. Limnol. Oceanogr. 1995, 40, 1159–1165. [Google Scholar] [CrossRef]
- O’Keeffe, G.; MacCraith, B.D.; McEvoy, A.K.; McDonagh, C.M.; McGilp, J.F. Development of a LED-based phase fluorimetric oxygen sensor using evanescent wave excitation of a sol-gel immobilized dye. Sens. Actuators B 1995, 29, 226–230. [Google Scholar] [CrossRef]
- Holst, G.A.; Köster, T.; Voges, E.; Lübbers, D.W. FLOX—An oxygen-flux-measuring system using a phase-modulation method to evaluate the oxygen-dependent fluorescence lifetime. Sens. Actuators B 1995, 29, 231–239. [Google Scholar] [CrossRef]
- Draxler, S.; Lippitsch, M.E.; Klimant, I.; Kraus, H.; Wolfbeis, O.S. Effects of polymer matrixes on the time-resolved luminescence of a ruthenium complex quenched by oxygen. J. Phys. Chem. 1995, 99, 3162–3167. [Google Scholar] [CrossRef]
- Trettnak, W.; Kolle, C.; Reininger, F.; Dolezal, C.; O’Leary, P. Miniaturized luminescence lifetime-based oxygen sensor instrumentation utilizing a phase modulation technique. Sens. Actuators B 1996, 36, 506–512. [Google Scholar] [CrossRef]
- Dakin, J.; Culshaw, B. Optical Fiber Sensors: Applications, Analysis and Future Trends; Artech House: Norwood, MA, USA, 1997. [Google Scholar]
- Bossi, M.L.; Daraio, M.E.; Aramendía, P.F. Luminescence quenching of Ru(II) complexes in polydimethylsiloxane sensors for oxygen. J. Photochem. Photobiol. A Chem. 1999, 120, 1, 15–21. [Google Scholar] [CrossRef]
- Tsukada, K.; Sakai, S.; Hase, K.; Minamitani, H. Development of catheter-type optical oxygen sensor and applications to bioinstrumentation. Biosens. Bioelectron. 2003, 18, 1439–1445. [Google Scholar] [CrossRef]
- Yeh, T.S.; Chu, C.S.; Lo, Y.L. Highly sensitive optical fiber oxygen sensor using Pt(II) complex embedded in sol-gel matrices. Sens. Actuators B 2006, 119, 701–707. [Google Scholar]
- Ergeneman, O.; Dogangil, G.; Kummer, M.P.; Abbott, J.J.; Nazeeruddin, M.K.; Nelson, B.J. A magnetically controlled wireless optical oxygen sensor for intraocular measurements. IEEE Sens. J. 2008, 8, 29–37. [Google Scholar]
- Ogurtsov, V.I.; Papkovsky, D.B. Application of frequency spectroscopy to fluorescence-based oxygen sensors. Sens. Actuators B 2006, 113, 608–616. [Google Scholar]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed.; Springer: New York, NY, USA, 2006; pp. 278–327. [Google Scholar]
- Medina-Rodríguez, S.; de la Torre-Vega, A.; Sainz-Gonzalo, F.J.; Marín-Suárez, M.; Elosúa, C.; Arregui, F.J.; Matias, I.R.; Fernández-Sánchez, J.F.; Fernández-Gutiérrez, A. Improved Multifrequency Phase-Modulation Method That Uses Rectangular-Wave Signals to Increase Accuracy in Luminescence Spectroscopy. Anal. Chem. 2014, 86, 5245–5256. [Google Scholar]
- Medina-Rodríguez, S.; de la Torre-Vega, A.; Fernández-Sánchez, J.F.; Fernández-Gutiérrez, A. Evaluation of a simple PC-based quadrature detection method at very low SNR for luminescence spectroscopy. Sens. Actuators B Chem. 2014, 192, 334–340. [Google Scholar]
- Medina-Rodríguez, C.; Medina-Rodríguez, S.; de la Torre-Vega, A.; Fernández-Gutiérrez, A.; Fernández-Sánchez, J.F. Direct estimation of the standard error in phase-resolved luminescence measurements: Application to an oxygen measuring system. Sens. Actuators B Chem. 2016, 224, 521–528. [Google Scholar]
- Szmacinski, H.; Lakowicz, J.R. Measurement of the intensity of long-lifetime luminophores in the presence of background signals using phase-modulation fluorometry. Appl. Spectrosc. 1999, 53, 1490–1495. [Google Scholar]
- Andrzejewski, D.; Klimant, I.; Podbielska, H. Method for lifetime-based chemical sensing using the demodulation of the luminescence signal. Sens. Actuators B 2002, 84, 160–166. [Google Scholar]
- Medina-Rodríguez, S.; Marín-Suárez, M.; Fernández-Sánchez, J.F.; de la Torre-Vega, A.; Baranoff, E.; Fernández-Gutiérrez, A. High performance optical sensing nanocomposites for low and ultra-low oxygen concentrations using phase-shift measurements. Analyst 2013, 138, 4607–4617. [Google Scholar]
- Medina-Rodríguez, S.; Orriach-Fernández, F.J.; Poole, C.; Kumar, P.; de la Torre-Vega, A.; Fernández-Sánchez, J.F.; Baranoff, E.; Fernández-Gutiérrez, A. Copper(I) complexes as alternatives to iridium(III) complexes for highly efficient oxygen sensing. Chem. Commun. 2015, 51, 11401–11404. [Google Scholar]
- Medina-Rodríguez, S.; Medina-Rodríguez, C.; de la Torre-Vega, A.; Segura-Luna, J.C.; Mota-Fernández, S.; Fernández-Sánchez, J.F. Real-time optimal combination of multifrequency information in phase-resolved luminescence spectroscopy based on rectangular-wave signals. Sens. Actuators B Chem. 2017, 238, 221–225. [Google Scholar] [CrossRef]
- Martinez-Ferreras, F.; Wolfbeis, O.S.; Gorris, H.H. Dual lifetime referenced fluorometry for the determination of doxorubicin in urine. Anal. Chim. Acta 2012, 729, 62–66. [Google Scholar] [CrossRef]
- Dramićanin, M.D.; Antić, Z.; Ćulubrk, S.; Ahrenkiel, S.P.; Nedeljković, J.M. Self-referenced luminescence thermometry with Sm3+ doped TiO2 nanoparticles. Nanotechnology 2014, 25, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.W.; Lin, J.M.; Mo, Z.W.; He, C.T.; Zhou, H.L.; Zhang, J.P.; Chen, X.M. Mixed-lanthanide porous coordination polymers showing range-tunable ratiometric luminescence for O2 sensing. Inorg. Chem. 2017, 56, 4238–4243. [Google Scholar] [CrossRef]
- Sekulić, M.; Dordević, V.; Ristić, Z.; Midić, M.; Dramićanin, M.D. Highly sensitive dual self-referencing temperature readout from the Mn4+/Ho3+ binary luminescence thermometry probe. Adv. Opt. Mater. 2018, 6, 1–7. [Google Scholar]
- Potter, M.; Lodge, T.A.; Morten, K.J. CHAPTER 8: Monitoring of Extracellular and Intracellular O2 on a Time-resolved Fluorescence Plate Reader. In Quenched-Phosphorescence Detection of Molecular Oxygen: Applications in Life Sciences; Papkovsky, D.B., Dmitriev, R.I., Eds.; RSC Detection Science Series No. 11; The Royal Society of Chemistry: London, UK, 2018; pp. 175–192. [Google Scholar]
- Tang, J.; Huang, Y.; Sun, M.; Gou, J.; Zhang, Y.; Li, G.; Kang, Y.; Yang, J.; Xiao, Z. Spectroscopic characterization and temperature-dependent upconversion behavior of Er3+ and Yb3+ co-doped zinc phosphate glass. J. Lumin. 2018, 197, 153–158. [Google Scholar] [CrossRef]
- Borisov, S.M.; Pommer, R.; Svec, J.; Peters, S.; Novakova, V.; Klimant, I. New red-emitting Schiff base chelates: Promising dyes for sensing and imaging of temperature and oxygen via phosphorescence decay time. J. Mater. Chem. C 2018, 6, 8999–9009. [Google Scholar] [CrossRef] [PubMed]
- Radunz, S.; Andresen, E.; Würth, C.; Koerdt, A.; Tschiche, H.R.; Resch-Genger, U. Simple self-referenced luminescent pH sensors based on upconversion nanocrystals and pH-sensitive fluorescent BODIPY dyes. Anal. Chem. 2019, 91, 7756–7764. [Google Scholar] [CrossRef]
- Brites, C.D.S.; Martinez, E.D.; Urbano, R.R.; Rettori, C.; Carlos, L.D. Self-calibrated double luminescent thermometers through upconverting nanoparticles. Front. Chem. 2019, 7, 267. [Google Scholar] [CrossRef]
- Wolfbeis, O.S. Fiber-Optic Chemical Sensors and Biosensors; CRC Press: Boca Raton, MA, USA, 1991. [Google Scholar]
- Schäferling, M. The Art of Fluorescence Imaging with Chemical Sensors. Angew. Chem. Int. Ed. Engl. 2012, 51, 3532–3554. [Google Scholar] [CrossRef]
- Proakis, J.G.; Manolakis, D.G. Digital Signal Processing-Principles, Algorithms and Applications, 4th ed.; Pearson Prentice Hall: Upper Saddle River, NJ, USA, 2007. [Google Scholar]
- Sundararajan, D. The Discrete Fourier Transform: Theory, Algorithms and Applications; World Scientific Publishing Co. Pte. Ltd.: Singapore, 2001. [Google Scholar]
- Cajlakovic, M.; Bizzarri, A.; Konrad, C.; Voraberger, H. Optochemical Sensors Based on Luminescence. In Encyclopedia of Sensors; Grimes, C.A., Dickey, E.C., Pishko, M.V., Eds.; American Scientific Publishers: Valencia, CA, USA, 2006; Volume 7, pp. 291–313. [Google Scholar]
- de la Torre, A.; Medina-Rodriguez, S.; Segura, J.C.; Fernandez-Sanchez, J.F. A polynomial-exponent model for calibrating the frequency response of photoluminescence-based sensors. Sensors 2020, 20, 4635. [Google Scholar] [CrossRef] [PubMed]
- Papkovsky, D.B. Oxygen Sensing. In Methods in Enzymology; Sen, C.K., Semenza, G.L., Eds.; Academic Press: Cambridge, MA, USA, 2004; Volume 381, pp. 1–824. [Google Scholar]
- Demas, J.N.; DeGraff, B.A.; Xu, W. Modeling of Luminescence Quenching-Based Sensors: Comparison of Multisite and Nonlinear Gas Solubility Models. Anal. Chem. 1995, 67, 1377–1380. [Google Scholar]
- Ogurtsov, V.I.; Papkovsky, D.B.; Papkovskaia, N.Y. Approximation of calibration of phase-fluorimetric oxygen sensors on the basis of physical models. Sens. Actuators B Chem. 2001, 81, 17–24. [Google Scholar]
- Ogurtsov, V.I.; Papkovsky, D.B. Modeling of luminescence-based oxygen sensors with non-uniform distribution of excitation and quenching characteristics inside active medium. Sens. Actuators B Chem. 2003, 88, 89–100. [Google Scholar] [CrossRef]
- Ogurtsov, V.I.; Papkovsky, D.B. Modelling of phase-fluorometric oxygen sensors: Consideration of temperature effects and operational requirements. Sens. Actuators B Chem. 2006, 113, 917–929. [Google Scholar] [CrossRef]
- Demas, J.N.; DeGraff, B.A. Luminescence-based sensors: Microheterogeneous and temperature effects. Sens. Actuators B Chem. 1993, 11, 35–41. [Google Scholar]
- Medina-Rodríguez, S.; de la Torre-Vega, A.; Medina-Rodríguez, C.; Fernández-Sánchez, J.F.; Fernández-Gutiérrez, A. On the calibration of chemical sensors based on photoluminescence: Selecting the appropriate optimization criterion. Sens. Actuators B Chem. 2015, 212, 278–286. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RMS Error in Determination (kPa); in Parenthesis, Relative RMS Error (%) | |||
|---|---|---|---|
| Conventional | Self-Referenced | Self-ref./conv. | |
| (kPa) | Harmonics 1–7 | Harmonics 2–7 | Ratio |
| 0.50 | 0.0060 kPa (1.20%) | 0.0160 kPa (3.20%) | 2.68 |
| 1.00 | 0.0095 kPa (0.95%) | 0.0198 kPa (1.98%) | 2.08 |
| 2.00 | 0.0113 kPa (0.56%) | 0.0255 kPa (1.27%) | 2.25 |
| 3.00 | 0.0152 kPa (0.51%) | 0.0405 kPa (1.35%) | 2.67 |
| 4.00 | 0.0213 kPa (0.53%) | 0.0577 kPa (1.44%) | 2.71 |
| 5.00 | 0.0292 kPa (0.58%) | 0.0695 kPa (1.39%) | 2.38 |
| 6.00 | 0.0269 kPa (0.45%) | 0.0915 kPa (1.52%) | 3.41 |
| 7.00 | 0.0328 kPa (0.47%) | 0.0971 kPa (1.39%) | 2.96 |
| 8.00 | 0.0412 kPa (0.51%) | 0.1194 kPa (1.49%) | 2.90 |
| 9.00 | 0.0456 kPa (0.51%) | 0.1680 kPa (1.87%) | 3.69 |
| 10.00 | 0.0665 kPa (0.66%) | 0.1541 kPa (1.54%) | 2.32 |
| 12.00 | 0.0651 kPa (0.54%) | 0.2202 kPa (1.83%) | 3.38 |
| average | 0.0309 kPa (0.62%) | 0.0899 kPa (1.69%) | 2.79 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de la Torre, A.; Medina-Rodríguez, S.; Segura, J.C.; Fernández-Sánchez, J.F. Self-Referenced Multifrequency Phase-Resolved Luminescence Spectroscopy. Sensors 2020, 20, 5482. https://doi.org/10.3390/s20195482
de la Torre A, Medina-Rodríguez S, Segura JC, Fernández-Sánchez JF. Self-Referenced Multifrequency Phase-Resolved Luminescence Spectroscopy. Sensors. 2020; 20(19):5482. https://doi.org/10.3390/s20195482
Chicago/Turabian Stylede la Torre, Angel, Santiago Medina-Rodríguez, Jose C. Segura, and Jorge F. Fernández-Sánchez. 2020. "Self-Referenced Multifrequency Phase-Resolved Luminescence Spectroscopy" Sensors 20, no. 19: 5482. https://doi.org/10.3390/s20195482
APA Stylede la Torre, A., Medina-Rodríguez, S., Segura, J. C., & Fernández-Sánchez, J. F. (2020). Self-Referenced Multifrequency Phase-Resolved Luminescence Spectroscopy. Sensors, 20(19), 5482. https://doi.org/10.3390/s20195482