Development of Solid-Phase RPA on a Lateral Flow Device for the Detection of Pathogens Related to Sepsis



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Sepsis
1.2. Diagnosis
1.3. Lateral Flow
1.4. RPA Reactions
2. Materials and Methods
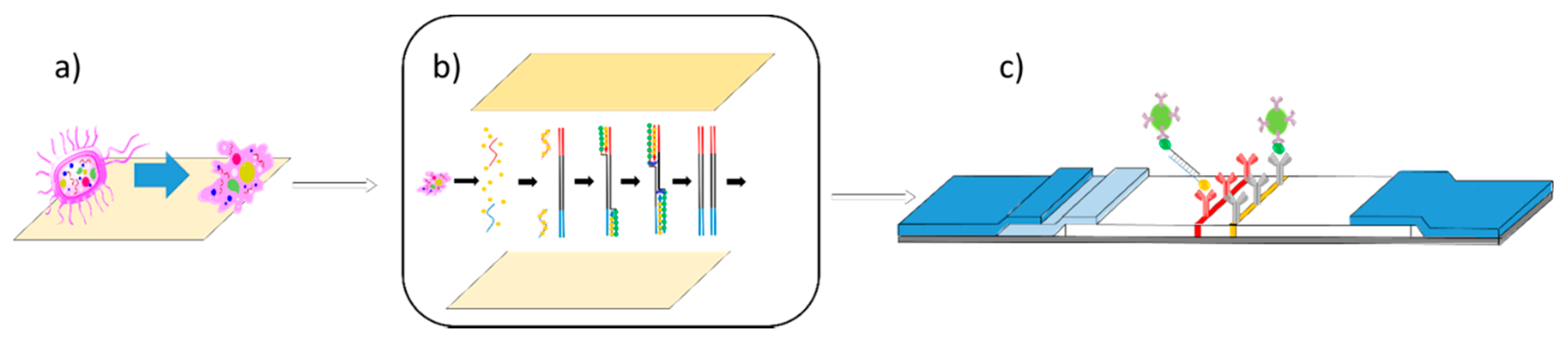
2.1. Fabrication of Lateral Flow
2.2. Development of the Solid Phase RPA Reactions
2.3. Lysis of Pathogens
2.4. Primer Design
2.5. PCR
2.6. RPA
3. Results and Discussion
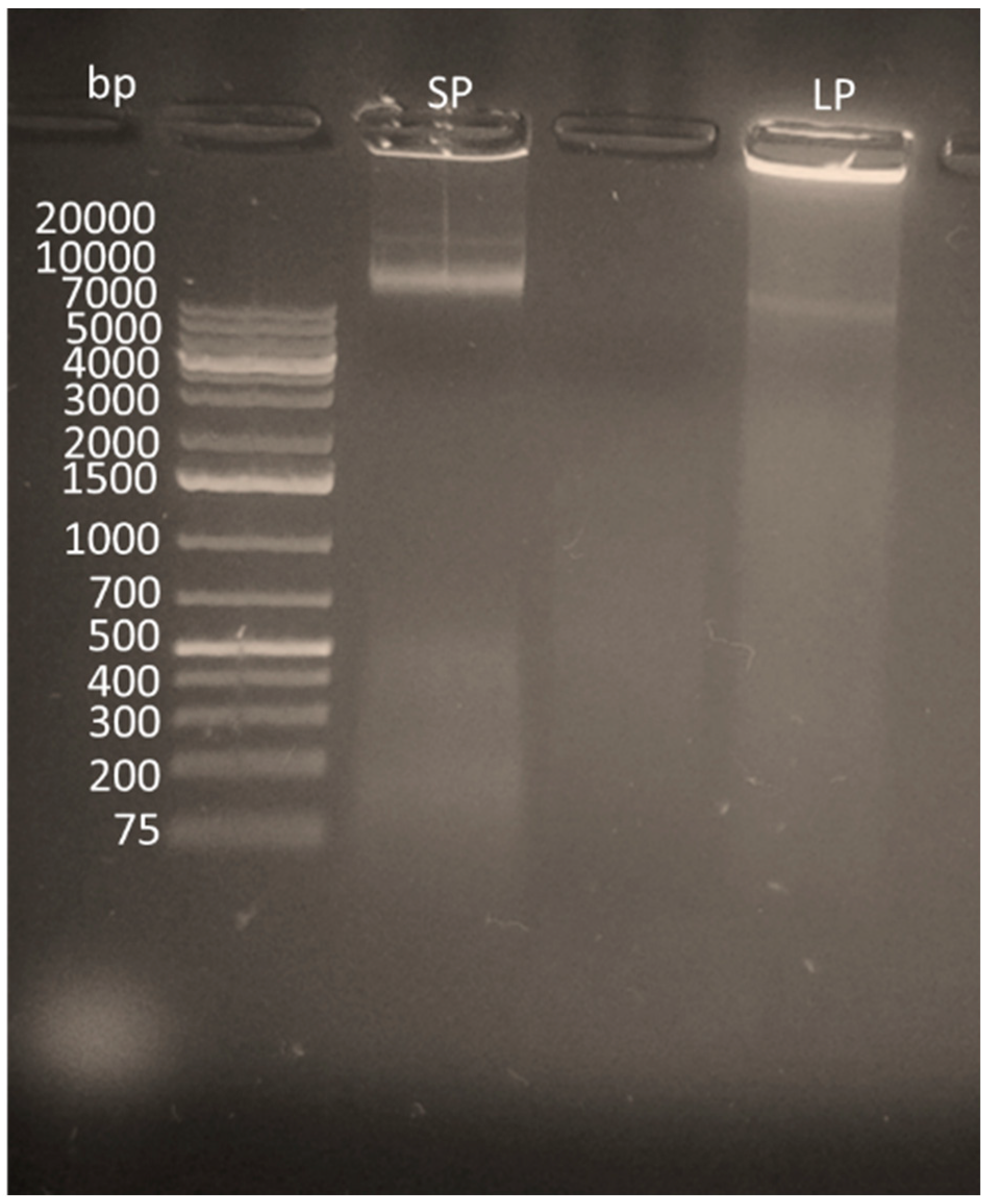
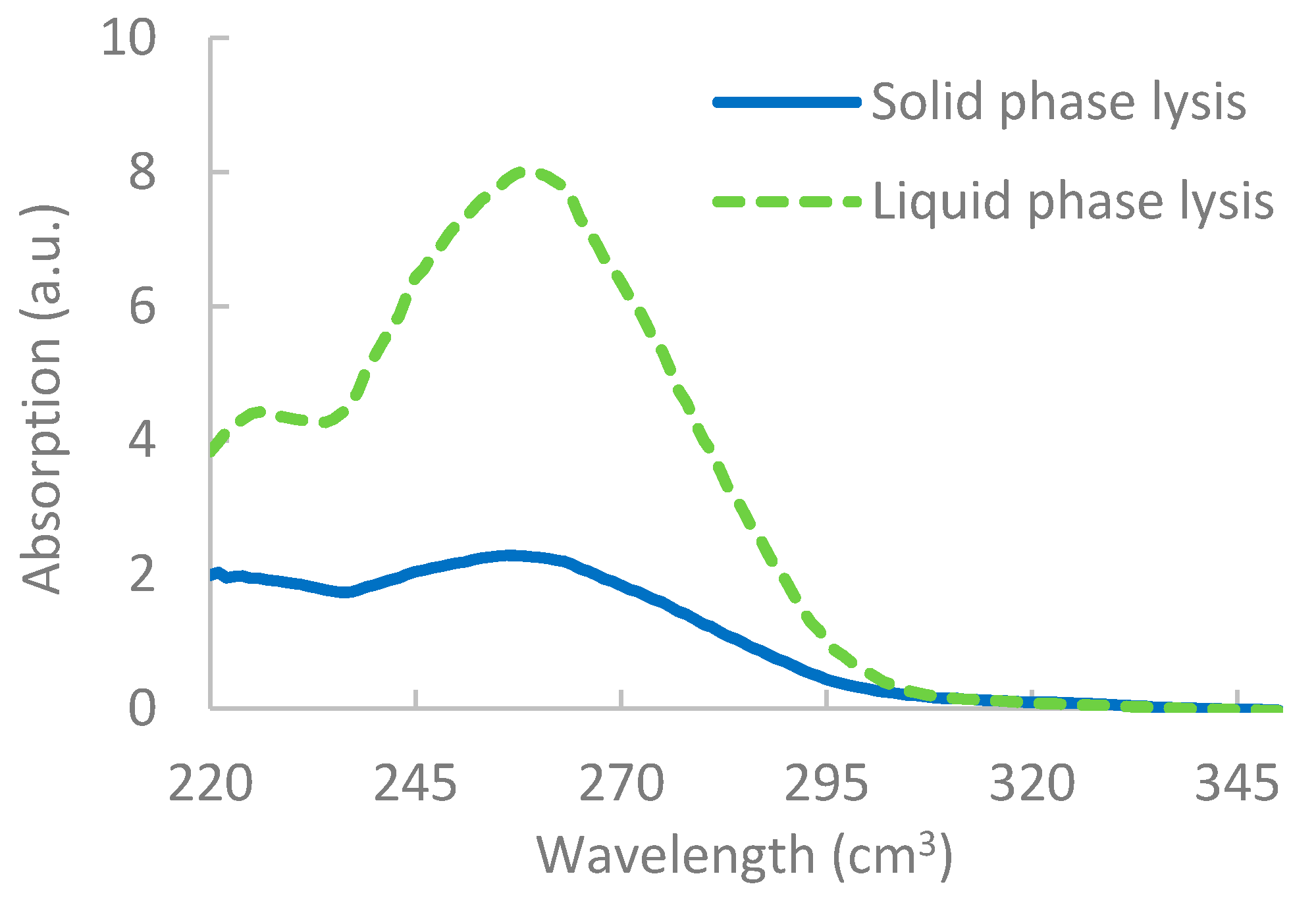
3.1. Lysis Reactions
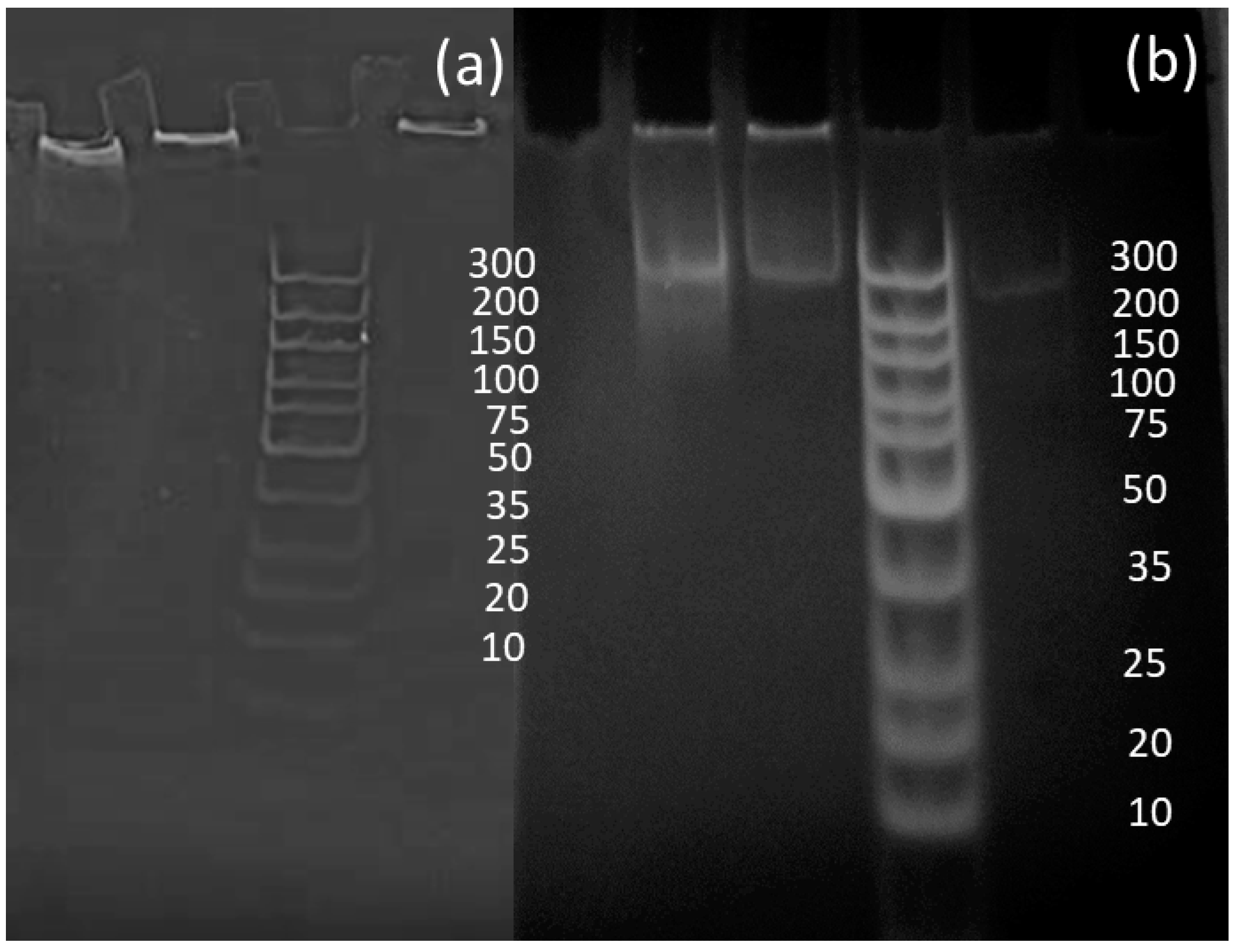
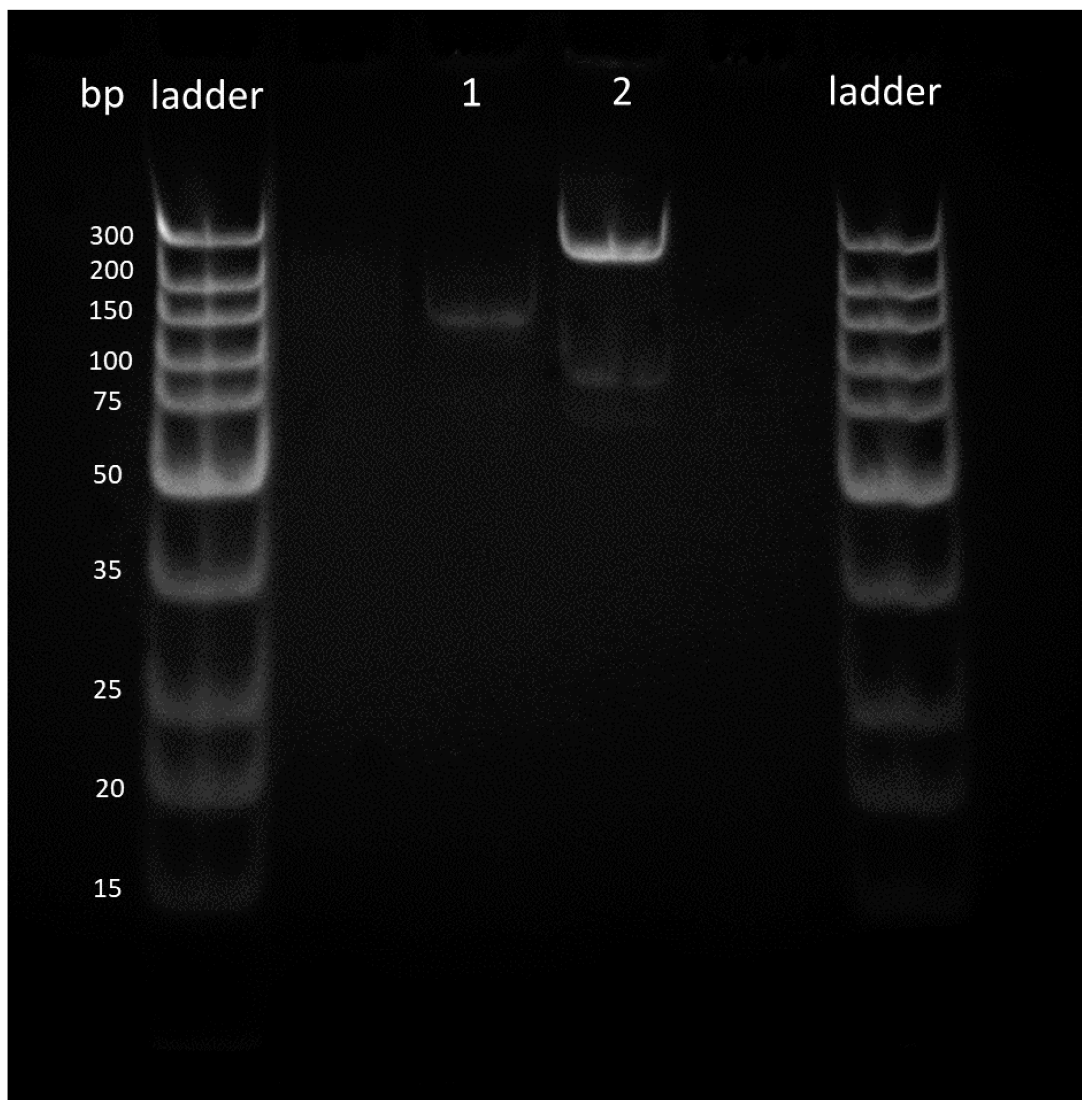
3.2. PCR Reactions
3.3. Liquid RPA Reactions
3.4. RPA Reactions with Contaminates
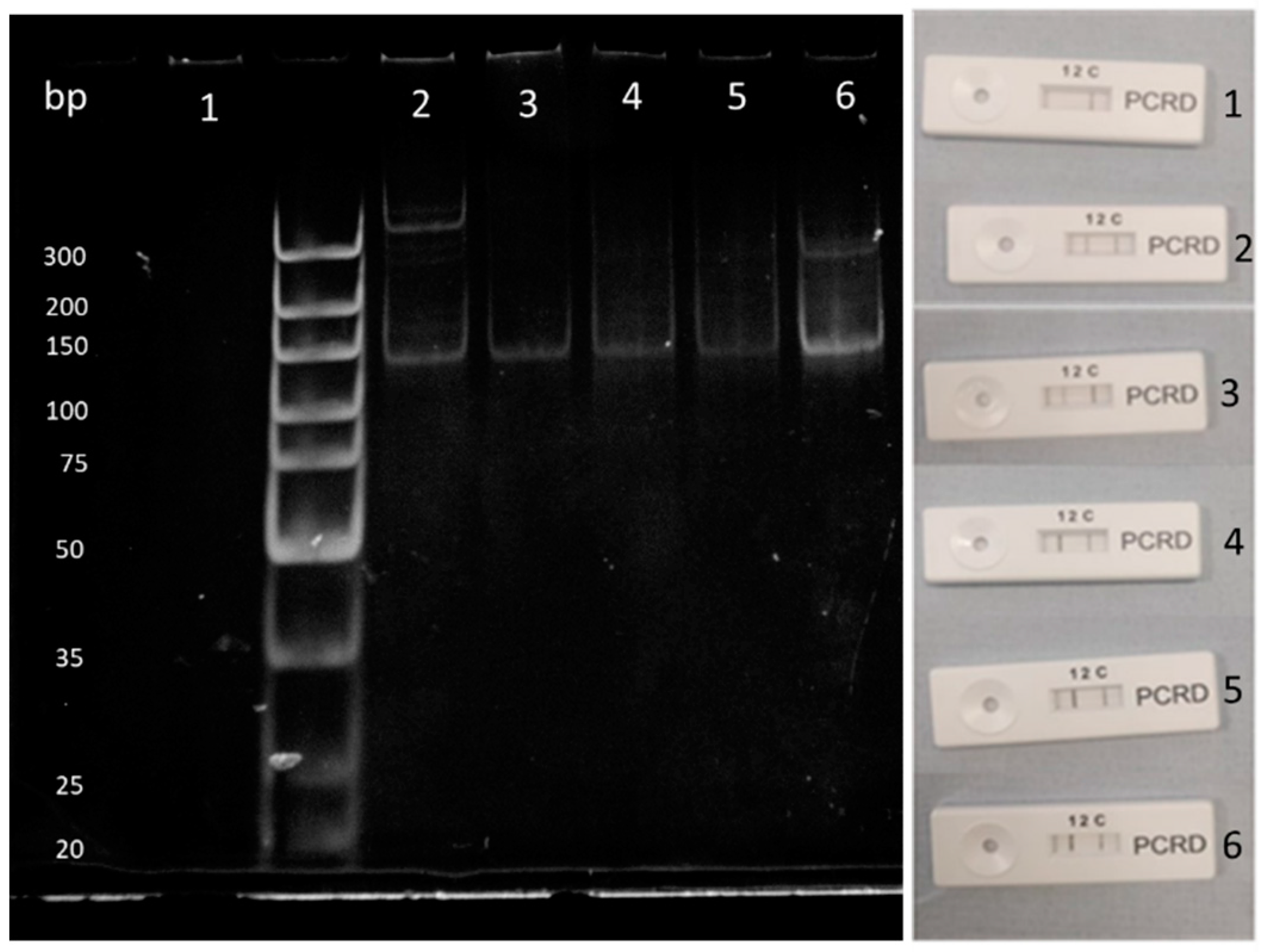
3.5. Solid-Phase RPA Reactions
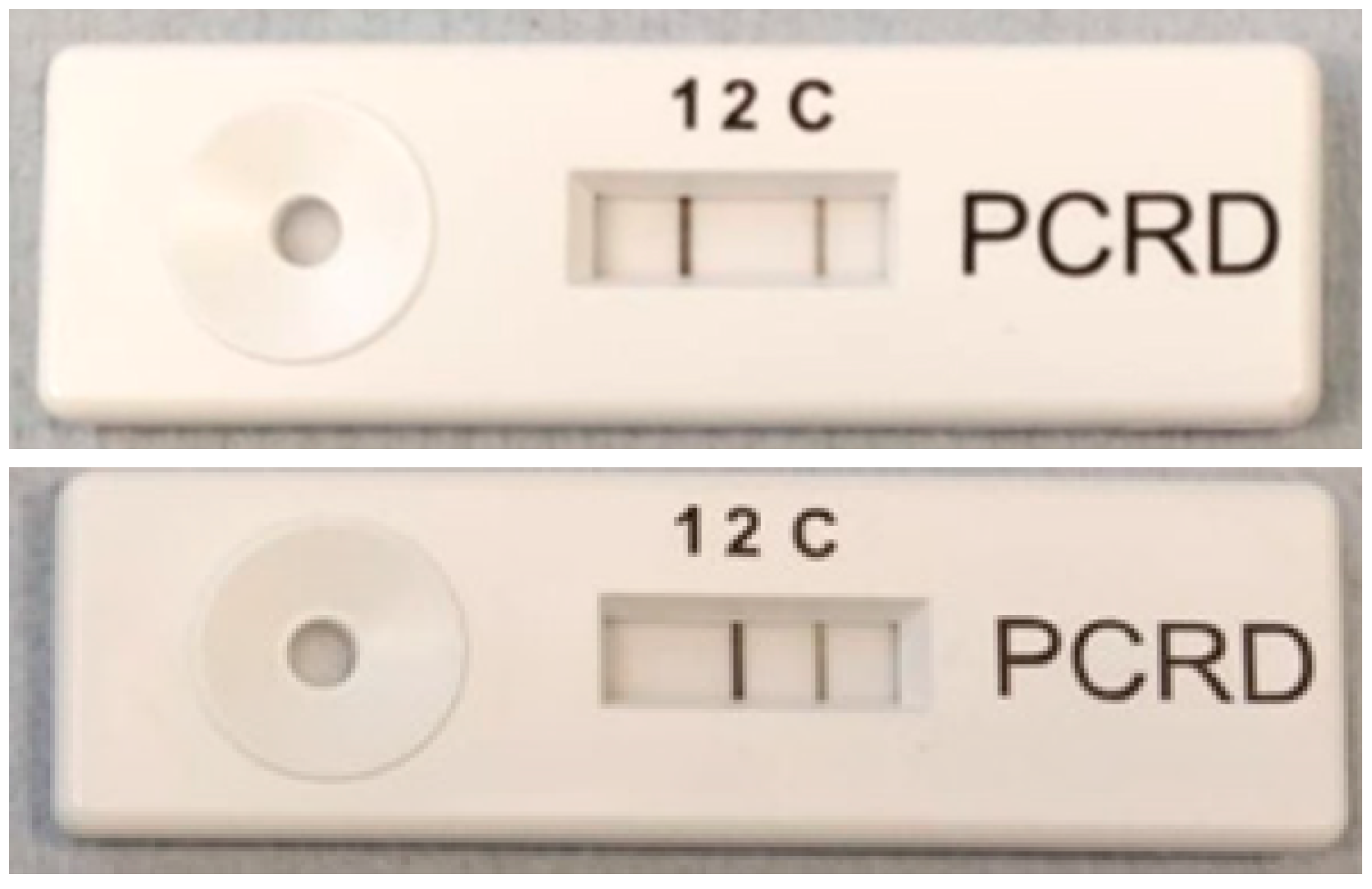
3.6. Integration with Lateral Flow Device
4. Conclusions and Future Work
Author Contributions
Funding
Conflicts of Interest
References
- Gotts, J.E.; Matthay, M.A. Sepsis: Pathophysiology and clinical management. BMJ 2016, 353, 1–20. [Google Scholar] [CrossRef]
- Reinhart, K.; Bauer, M.; Riedemann, N.C.; Hartog, C.S. New approaches to sepsis: Molecular diagnostics and biomarkers. Clin. Microbiol. Rev. 2012, 25, 609–634. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Yu, M.; Chai, Y. Pathological alteration and therapeutic implications of sepsis-induced immune cell apoptosis. Cell Death Dis. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Daniels, R.; Nutbeam, T.; Taylor, G. The sepsis six and the severe sepsis resuscitation bundle: A prospective observational cohort study. Emerg. Med. J. 2017, 28, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Martin-Loeches, I.; Guia, M.C.; Vallecoccia, M.S.; Suarez, D.; Ibarz, M.; Irazabal, M.; Ferrer, R.; Artigas, A. Risk factors for mortality in elderly and very elderly critically ill patients with sepsis: A prospective, observational, multicenter cohort study. Ann. Intensive Care 2019, 9, 26. [Google Scholar] [CrossRef]
- Zea-Vera, A.; Ochoa, T.J. Challenges in the diagnosis and management of neonatal sepsis. J. Trop. Pediatr. 2015, 61, 1–13. [Google Scholar] [CrossRef]
- Bhatraju, P.K.; Ghassemieh, B.J.; Nichols, M.; Kim, R.; Jerome, K.R.; Nalla, A.K.; Greninger, A.L.; Pipavath, S.; Wurfel, M.M.; Evans, L.; et al. Covid-19 in Critically Ill Patients in the Seattle Region—Case Series. N. Engl. J. Med. 2020, 382, 2012–2022. [Google Scholar] [CrossRef]
- Guo, T.; Fan, Y.; Chen, M.; Chen, M.; Wu, X.; Zhang, L.; He, T.; Wang, H.; Wan, J.; Wang, X.; et al. Cardiovascular Implications of Fatal Outcomes of Patients with Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020, e201017. [Google Scholar] [CrossRef]
- Gingo, M.R.; Morris, A. HIV infection and severe sepsis: A bitter pill to swallow. Crit. Care Med. 2015, 43, 1779–1780. [Google Scholar] [CrossRef]
- Kochanek, M.; Schalk, E.; Bergwelt-Baildon, M.; Beutel, G.; Buchheidt, D.; Hentrich, M.; Henze, L.; Kiehl, M.; Liebregts, T.; Lilienfeld-Toal, M.; et al. Management of sepsis in neutropenic cancer patients: 2018 guidelines from the Infectious Diseases Working Party (AGIHO) and Intensive Care Working Party (iCHOP) of the German Society of Hematology and Medical Oncology (DGHO). Ann. Hematol. 2019, 98, 1051–1069. [Google Scholar] [CrossRef]
- Parker, R.I. Cancer and Sepsis: Adding Insult to Injury-As if Either Alone Were Not Enough? Crit. Care Med. 2019, 47, 1452–1453. [Google Scholar] [CrossRef] [PubMed]
- Gul, F.; Arslantas, M.K.; Cinel, I.; Kumar, A. Changing Definitions of Sepsis. Turkish J. Anesth. Reanim. 2017, 45, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Minasyan, H. Sepsis: Mechanisms of bacterial injury to the patient. Scand. J. Trauma. Resusc. Emerg. Med. 2019, 27, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Natanson, C.; Suflfredini, A.F.; Danner, R.L.; Cunnion, R.E.; Ognibene, F.P. Septic Shock in Humans Advances in the Understanding of Pathogenesis, Cardiovascular. Ann. Intern. Med. 2017, 113, 227–242. [Google Scholar]
- Vincent, J.; Marshall, J.; Anzueto, A.; Martin, C.D.; Gomersall, C. International study of the prevalence and outcomes of infection in intensive care units. JAMA 2009, 302, 2323–2329. [Google Scholar] [CrossRef]
- Zahar, J.-R.; Timsit, J.-F.; Garrouste-Orgeas, M.; Français, A.; Vesim, A.; Descorps-Declere, A.; Dubois, Y.; Souweine, B.; Haouache, H.; Goldgran-Toledano, D.; et al. Outcomes in severe sepsis and patients with septic shock: Pathogen species and infection sites are not associated with mortality. Crit. Care Med. 2011, 39, 1886–1895. [Google Scholar] [CrossRef]
- Lin, G.; McGinley, J.P.; Drysdale, S.B.; Pollard, A.J. Epidemiology and Immune Pathogenesis of Viral Sepsis. Front. Immunol. 2018, 9, 1–21. [Google Scholar] [CrossRef]
- Thompson, K.; Venkatesh, B.; Finfer, S. Sepsis and septic shock: Current approaches to management. Intern. Med. J. 2019, 49, 160–170. [Google Scholar] [CrossRef]
- Marik, P.E.; Taeb, A.M. SIRS, qSOFA and new sepsis definition. J. Thorac. Dis. 2017, 9, 943–945. [Google Scholar] [CrossRef]
- Serafim, R.; Gomes, J.A.; Salluh, J.; Póvoa, P. A Comparison of the Quick-SOFA and Systemic Inflammatory Response Syndrome Criteria for the Diagnosis of Sepsis and Prediction of Mortality. Chest 2018, 153, 646–655. [Google Scholar] [CrossRef]
- Weinstein, M.P. Minireview Blood Culture Contamination: Persisting Problems and Partial Progress. J. Clin. Microbiol. 2003, 41, 2275–2278. [Google Scholar] [CrossRef] [PubMed]
- Korber, F.; Zeller, I.; Grunstaudl, M.; Willinger, B.; Apfalter, P.; Hirschl, A.M.; Makristathis, A. SeptiFast versus blood culture in clinical routine—A report on 3 years experience. Wien. Klin. Wochenschr. 2017, 129, 427–434. [Google Scholar] [CrossRef]
- Lambregts, M.M.C.; Bernards, A.T.; Beek, M.T.; Visser, L.G.; Boer, M.G. Time to positivity of blood cultures supports early re-evaluation of empiric broad-spectrum antimicrobial therapy. PLoS ONE 2019, 14, e0208819. [Google Scholar] [CrossRef] [PubMed]
- Nayak, S.; Blumenfeld, N.R.; Laksanasopin, T.; Samuel, K.; Avenue, A.; Program, B.E.; Khru, T. Point-of-Care Diagnostics: Recent Developments in a Connected Age. Anal. Chem. 2018, 89, 102–123. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Xu, T.; Wang, W.; Wen, Y.; Li, K.; Qian, L.; Zhang, X.; Liu, G. Lateral flow biosensors based on the use of micro- and nanomaterials: A review on recent developments. Microchim. Acta 2020, 187, 70. [Google Scholar] [CrossRef]
- Gyawali, B.; Ramakrishna, K.; Dhamoon, A.S. Sepsis: The evolution in definition, pathophysiology, and management. Sage Open Med. 2019, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Shi, L.; Ran, X.; Li, W.; Wang, X.-L.; Wang, F. Development of a Lateral Flow Immunoassay for the Rapid Diagnosis of Invasive Candidiasis. Front. Microbiol. 2016, 7, 1451. [Google Scholar] [CrossRef]
- Shao, X.-Y.; Wang, C.-R.; Xie, C.-M.; Wang, X.-G.; Liang, R.-L.; Xu, W.-W. Rapid and Sensitive Lateral Flow Immunoassay Method for Procalcitonin (PCT) Based on Time-Resolved Immunochromatography. Sensors 2017, 17, 480. [Google Scholar] [CrossRef]
- Fu, S.; Jiang, Y.; Jiang, X.; Zhao, Y.; Chen, S.; Yang, X.; Man, C. Probe-free label system for rapid detection of Cronobacter genus in powdered infant formula. AMB Express 2018, 8, 155. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, L.; Li, J.; Wang, Z.; Jiao, W.; Xiao, J.; Shen, C.; Xu, F.; Qi, H.; Wang, Y.; et al. Label-Free Cross-Priming Amplification Coupled With Endonuclease Restriction and Nanoparticles-Based Biosensor for Simultaneous Detection of Nucleic Acids and Prevention of Carryover Contamination. Front. Chem. 2019, 7, 1–11. [Google Scholar] [CrossRef]
- Zhuang, L.; Ji, Y.; Tian, P.; Wang, K.; Kou, C.; Gu, N.; Zhang, Y. Polymerase chain reaction combined with fluorescent lateral flow immunoassay based on magnetic purification for rapid detection of canine parvovirus 2. BMC Vet. Res. 2019, 15, 30. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Ye, L.; Wang, Z.; Cui, Y.; Wang, J. Utilization of recombinase polymerase amplification combined with a lateral flow strip for detection of Perkinsus beihaiensis in the oyster Crassostrea hongkongensis. Parasit. Vectors 2019, 12, 360. [Google Scholar] [CrossRef]
- Piepenburg, O.; Williams, C.H.; Stemple, D.L.; Armes, N.A. DNA Detection Using Recombination Proteins. PLoS Biol. 2006, 4, 1115–1121. [Google Scholar] [CrossRef] [PubMed]
- Cabada, M.M.; Malaga, J.L.; Castellanos-Gonzalez, A.; Bagwell, K.A.; Naeger, P.A.; Rogers, H.K.; Maharsi, S.; Mbaka, M.; White, A.C. Recombinase Polymerase Amplification Compared to Real-Time Polymerase Chain Reaction Test for the Detection of Fasciola hepatica in Human Stool. Am. J. Trop. Med. Hyg. 2017, 96, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Macdonald, J.; Stetten, F. Review: A comprehensive summary of a decade development of the recombinase polymerase amplification. Analyst 2019, 144, 31–67. [Google Scholar] [CrossRef]
- Chen, G.; Dong, J.; Yuan, Y.; Li, N.; Huang, X.; Cui, X.; Tang, Z. A general solution for opening double-stranded DNA for isothermal amplification. Sci. Rep. 2016, 6, 34582. [Google Scholar] [CrossRef]
- Zhang, Y.; Tanner, N.A. Isothermal Amplification of Long, Discrete DNA Fragments Facilitated by Single-Stranded Binding Protein. Sci. Rep. 2017, 7, 8497. [Google Scholar] [CrossRef]
- Wang, J.; Liu, L.; Wang, J.; Sun, X.; Yuan, W. Recombinase Polymerase Amplification Assay—A Simple, Fast and Cost-Effective Alternative to Real Time PCR for Specific Detection of Feline Herpesvirus-1. PLoS ONE 2017, 12, e0166903. [Google Scholar] [CrossRef]
- Lobato, I.M.; O’Sullivan, C.K. Recombinase Polymerase Amplification: Basics, applications and recent advances. Trends Anal. Chem. 2018, 98, 19–35. [Google Scholar] [CrossRef]
- Rohrman, B.; Richards-Kortum, R. Inhibition of Recombinase Polymerase Amplification by Background DNA: A Lateral Flow-Based Method for Enriching Target DNA. Anal. Chem. 2015, 87, 1963–1967. [Google Scholar] [CrossRef]
- Adékambi, T.; Drancourt, M.; Raoult, D. The rpoB gene as a tool for clinical microbiologists. Trends Microbiol. 2009, 17, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Ojo, K.K.; Tung, D.; Luis, H.; Bernardo, M.; Leitao, J.; Roberts, M.C. Gram-positive merA gene in gram-negative oral and urine bacteria. FEMS Microbiol. Lett. 2004, 238, 411–416. [Google Scholar] [PubMed]
- Li, Q.Q.; Skinner, J.; Bennett, J.E. Evaluation of reference genes for real-time quantitative PCR studies in Candida glabrata following azole treatment. BMC Mol. Biol. 2012, 13, 1–13. [Google Scholar] [CrossRef]
- Rohrman, B.A.; Richards-Kortum, R.R. A paper and plastic device for performing recombinase polymerase amplification of HIV DNA. Lab. Chip. 2012, 12, 3082–3088. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.; Tiwari, S.; Gomes, A.V.; Biology, M. Protein purification and analysis: The next generation Western Blotting techniques. Expert Rev. Proteom. 2019, 14, 1037–1053. [Google Scholar] [CrossRef] [PubMed]
- Hauk, A. Precipitation of nucleic acids. Biol. Unserer Zeit. 2016, 46, 344. [Google Scholar] [CrossRef]
- Chomczynski, P.; Sacchi, N. The single-step method of RNA isolation by acid guanidinium thiocyanate–phenol–chloroform extraction: Twenty-something years on. Nat. Protoc. 2006, 1, 581–585. [Google Scholar] [CrossRef]
- Kaufman, D.; Fairchild, K.D. Clinical Microbiology of Bacterial and Fungal Sepsis in Very-Low-Birth-Weight Infants. Clin. Microbiol. Rev. 2004, 17, 638–680. [Google Scholar] [CrossRef]
- Westh, H.; Lisby, G.; Breysse, F.; Böddinghaus, B.; Chomarat, M.; Gant, V.; Goglio, A.; Raglio, A.; Schuster, H.; Stuber, F.; et al. Multiplex real-time PCR and blood culture for identification of bloodstream pathogens in patients with suspected sepsis. Clin. Microbiol. Infect. 2009, 15, 544–551. [Google Scholar] [CrossRef]
- Marko, M.A.; Chipperfield, R.; Birnboim, H.C. A procedure for the large-scale isolation of highly purified plasmid DNA using alkaline extraction and binding to glass powder. Anal. Biochem. 1982, 121, 382–387. [Google Scholar] [CrossRef]
- Song, C.S.; Nam, J.H.; Kim, C.-J.; Ro, S.T. Temperature distribution in a vial during freeze-drying of skim milk. J. Food Eng. 2005, 67, 467–475. [Google Scholar] [CrossRef]
- Dobbs, L.J.; Madigan, M.N.; Carter, A.B.; Earls, L. Use of FTA Gene Guard filter paper for the storage and transportation of tumor cells for molecular testing. Arch. Pathol. Lab. Med. 2002, 126, 56–63. [Google Scholar] [PubMed]
- Desjardins, P.; Conklin, D. NanoDrop Microvolume Quantitation of Nucleic Acids. J. Vis. Exp. 2010, e2565. [Google Scholar] [CrossRef] [PubMed]
- García, M.E.; Blanco, J.L.; Caballero, J.; Gargallo-Viola, D. Rapid detection of Plasmodium falciparum with isothermal recombinase polymerase amplification and lateral flow analysis. J. Clin. Microbiol. 2002, 40, 1567–1568. [Google Scholar] [PubMed]
- Low, S.C.; Shaimi, R.; Thandaithabany, Y.; Lim, J.K.; Ahmad, A.L.; Ismail, A. Electrophoretic interactions between nitrocellulose membranes and proteins: Biointerface analysis and protein adhesion properties. Colloids Surf. B Biointerfaces 2013, 110, 248–253. [Google Scholar] [CrossRef]
- Farid, M.; Butcher, S. A Generalized Correlation for Heat and Mass Transfer in Freezing, Drying, Frying, and Freeze Drying. Dry. Technol. 2003, 21, 231–247. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heeroma, A.J.; Gwenin, C. Development of Solid-Phase RPA on a Lateral Flow Device for the Detection of Pathogens Related to Sepsis. Sensors 2020, 20, 4182. https://doi.org/10.3390/s20154182
Heeroma AJ, Gwenin C. Development of Solid-Phase RPA on a Lateral Flow Device for the Detection of Pathogens Related to Sepsis. Sensors. 2020; 20(15):4182. https://doi.org/10.3390/s20154182
Chicago/Turabian StyleHeeroma, Alice Jane, and Christopher Gwenin. 2020. "Development of Solid-Phase RPA on a Lateral Flow Device for the Detection of Pathogens Related to Sepsis" Sensors 20, no. 15: 4182. https://doi.org/10.3390/s20154182
APA StyleHeeroma, A. J., & Gwenin, C. (2020). Development of Solid-Phase RPA on a Lateral Flow Device for the Detection of Pathogens Related to Sepsis. Sensors, 20(15), 4182. https://doi.org/10.3390/s20154182