QCL-Based Dual-Comb Spectrometer for Multi-Species Measurements at High Temperatures and High Pressures


Abstract
1. Introduction
2. Materials and Methods
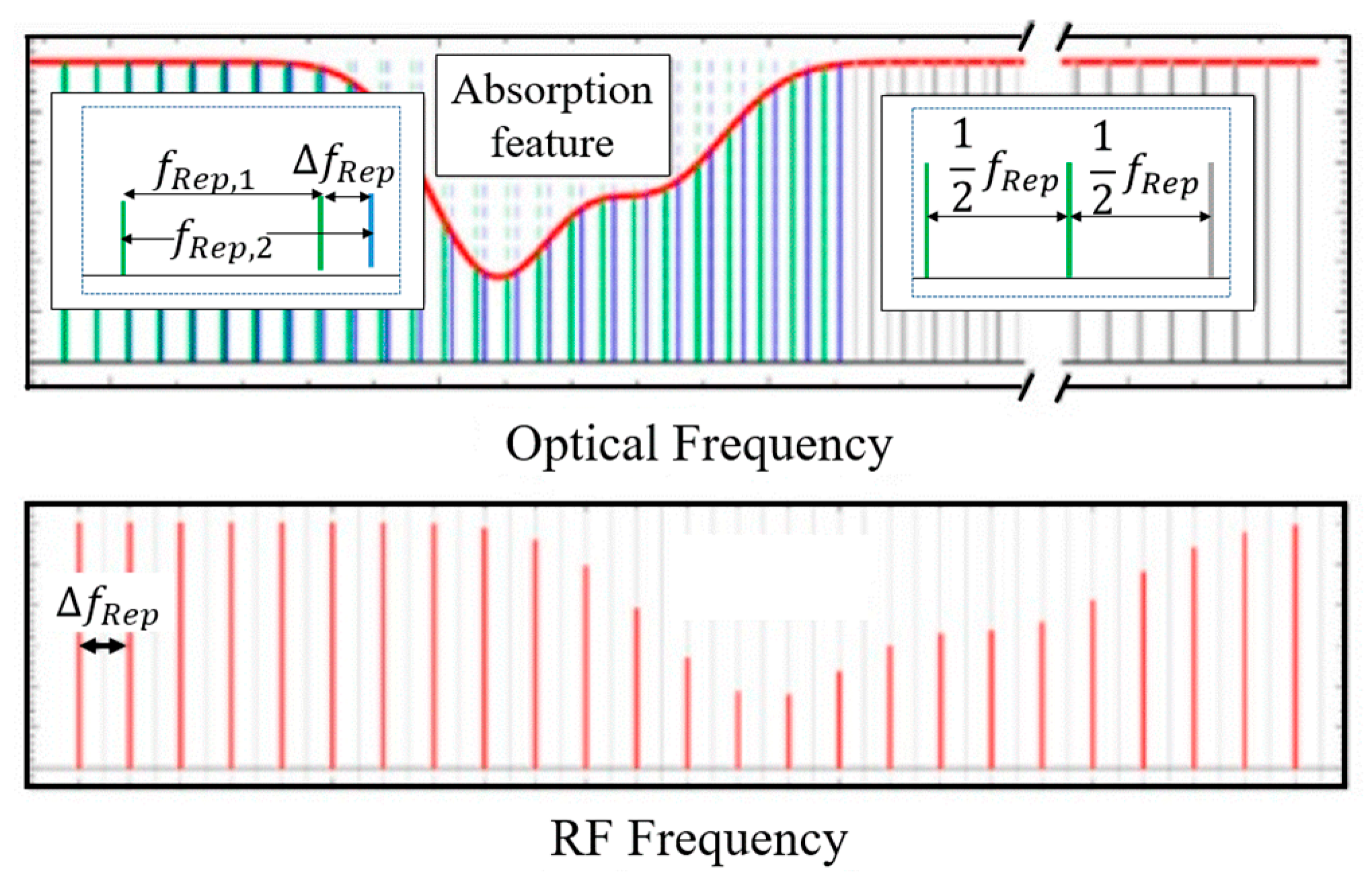
2.1. Dual-Comb Spectroscopy
2.2. Mid-IR Dual-Comb Spectrometer
2.3. Experimental Setup
2.4. Multi-Species Absorption
3. Results and Discussion
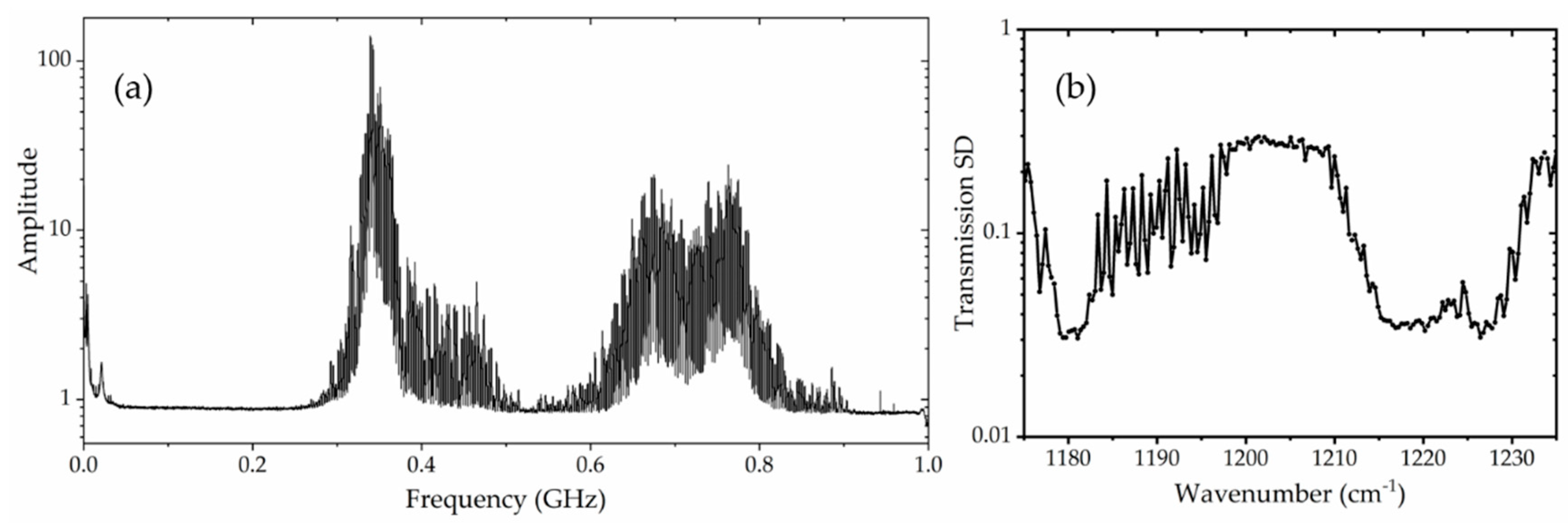
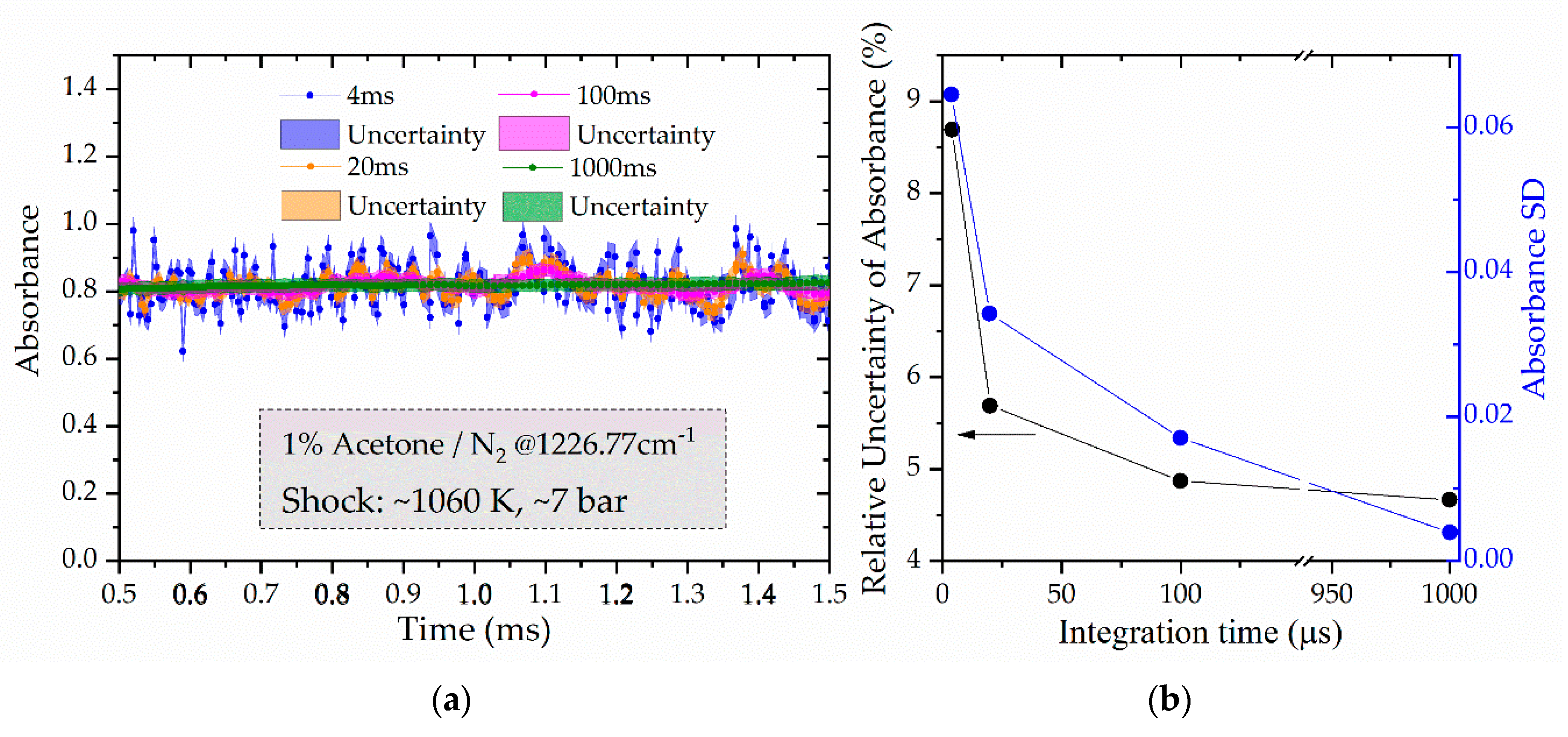
3.1. Transmission Standard Deviation
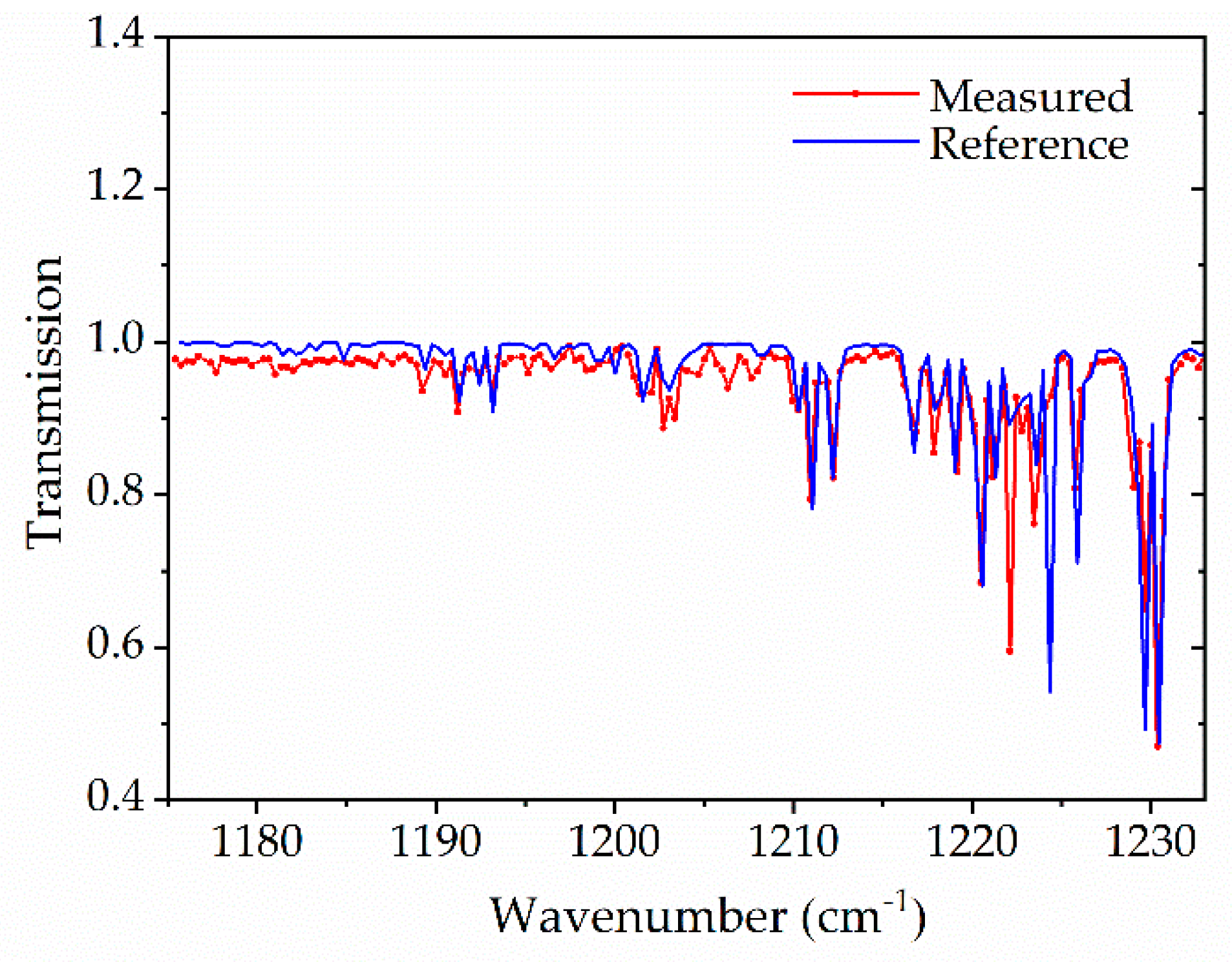
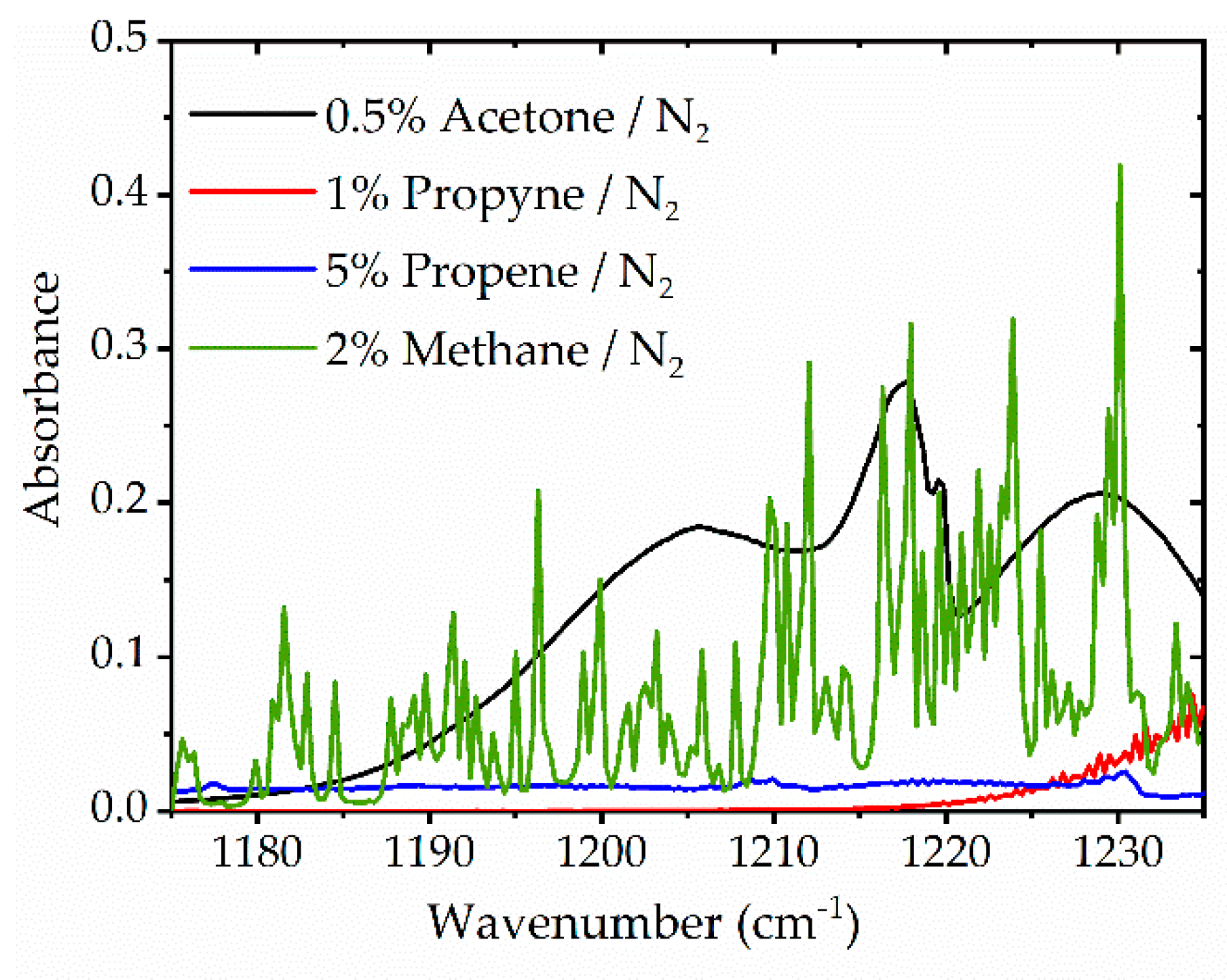
3.2. Multi-Species Detection in Non-Reactive Experiments
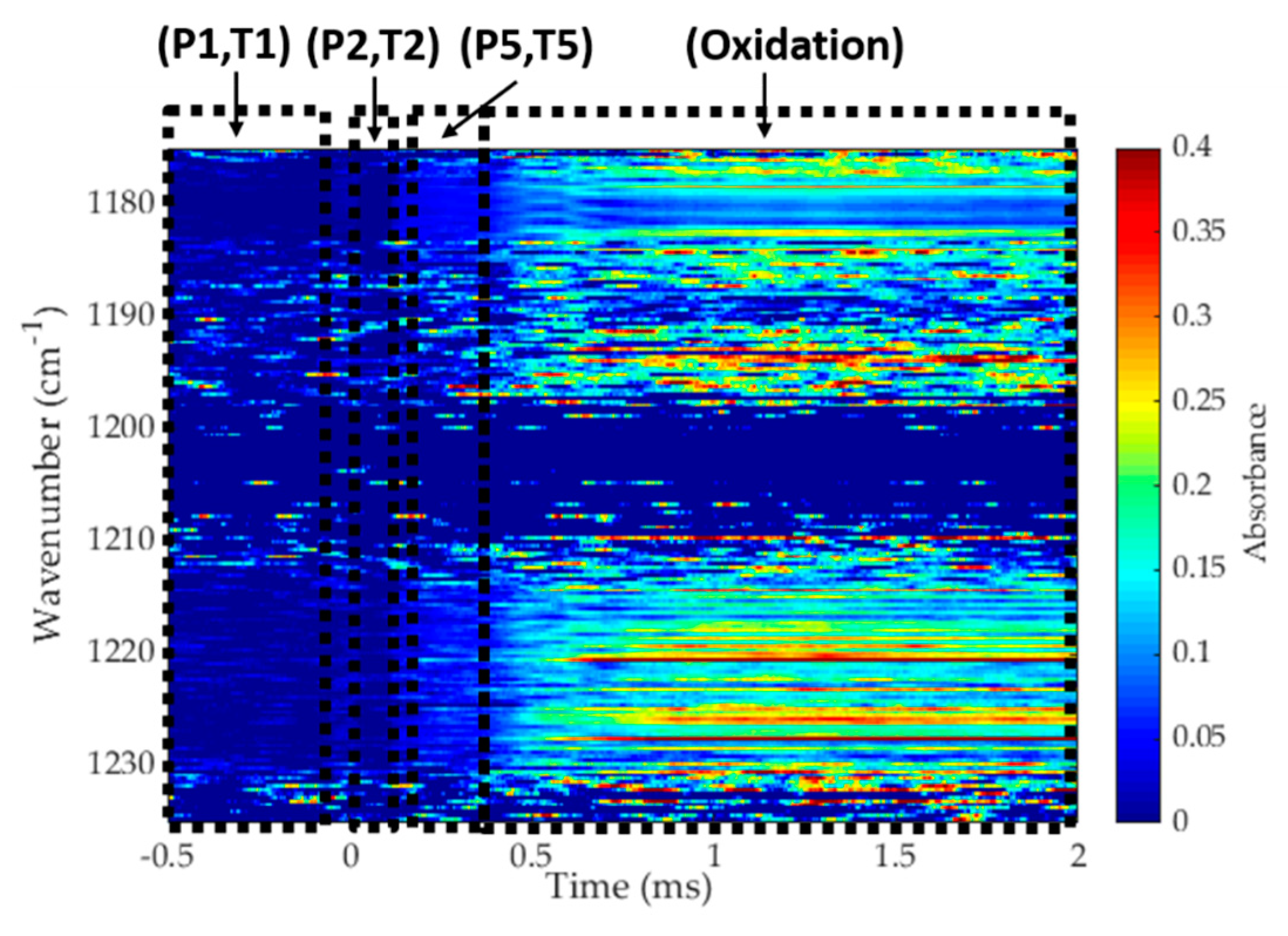
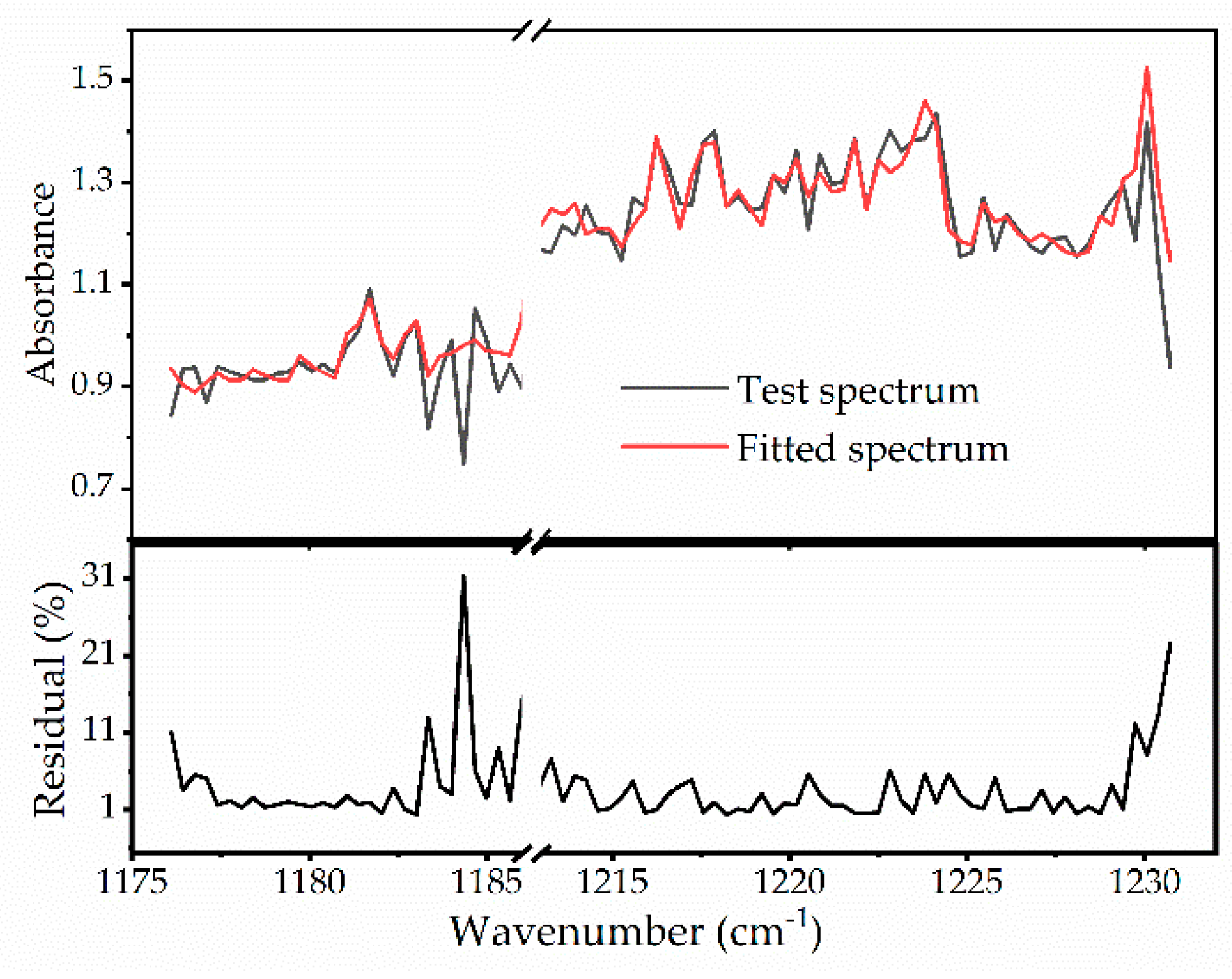
3.3. Application to Reactive Experiments
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A



References
- Griffiths, P.R.; De Haseth, J.A. Fourier Transform Infrared Spectrometry; John Wiley & Sons: Hoboken, NJ, USA, 2007; Volume 171. [Google Scholar]
- Tittel, F.K.; Richter, D.; Fried, A. Mid-infrared laser applications in spectroscopy. In Solid-State Mid-Infrared Laser Sources; Springer: Berlin/Heidelberg, Germany, 2003; pp. 458–529. [Google Scholar]
- Hugi, A.; Maulini, R.; Faist, J. External cavity quantum cascade laser. Semicond. Sci. Technol. 2010, 25, 083001. [Google Scholar] [CrossRef]
- Boyd, R. Nonlinear Optics; Academic: San Diego, CA, USA, 2008; Volume 19922, p. 39. [Google Scholar]
- Manley, J.; Rowe, H. General energy relations in nonlinear reactances. Proc. Inst. Radio Eng. 1959, 47, 2115–2116. [Google Scholar]
- Gmachl, C.; Capasso, F.; Sivco, D.L.; Cho, A.Y. Recent progress in quantum cascade lasers and applications. Rep. Prog. Phys. 2001, 64, 1533. [Google Scholar] [CrossRef]
- Kosterev, A.A.; Tittel, F.K. Chemical sensors based on quantum cascade lasers. IEEE J. Quantum Electron. 2002, 38, 582–591. [Google Scholar] [CrossRef]
- Curl, R.F.; Capasso, F.; Gmachl, C.; Kosterev, A.A.; McManus, B.; Lewicki, R.; Pusharsky, M.; Wysocki, G.; Tittel, F.K. Quantum cascade lasers in chemical physics. Chem. Phys. Lett. 2010, 487, 1–18. [Google Scholar] [CrossRef]
- McManus, J.B.; Zahniser, M.S.; Nelson, D.D.; Shorter, J.H.; Herndon, S.; Wood, E.; Wehr, R. Application of quantum cascade lasers to high-precision atmospheric trace gas measurements. Opt. Eng. 2010, 49, 111124. [Google Scholar] [CrossRef]
- Welzel, S.; Hempel, F.; Hübner, M.; Lang, N.; Davies, P.B.; Röpcke, J. Quantum cascade laser absorption spectroscopy as a plasma diagnostic tool: An overview. Sensors 2010, 10, 6861–6900. [Google Scholar] [CrossRef]
- Patel, I.; Rajamanickam, V.P.; Bertoncini, A.; Pagliari, F.; Tirinato, L.; Laptenok, S.P.; Liberale, C. Quantum Cascade Laser Infrared Spectroscopy of Single Cancer Cells. In Optical Trapping Applications; Optical Society of America: Washington, DC, USA, 2017; p. JTu4A. 21. [Google Scholar]
- Yao, Y.; Hoffman, A.J.; Gmachl, C.F. Mid-infrared quantum cascade lasers. Nat. Photonic 2012, 6, 432. [Google Scholar] [CrossRef]
- Beyer, T.; Braun, M.; Lambrecht, A. Fast gas spectroscopy using pulsed quantum cascade lasers. J. Appl. Phys. 2003, 93, 3158–3160. [Google Scholar] [CrossRef]
- Ren, W.; Farooq, A.; Davidson, D.F.; Hanson, R.K. CO concentration and temperature sensor for combustion gases using quantum-cascade laser absorption near 4.7 μm. Appl. Phys. B 2012, 107, 849–860. [Google Scholar] [CrossRef]
- Sun, K.; Wang, S.; Sur, R.; Chao, X.; Jeffries, J.B.; Hanson, R.K. Time-resolved in situ detection of CO in a shock tube using cavity-enhanced absorption spectroscopy with a quantum-cascade laser near 4.6 µm. Opt. Express 2014, 22, 24559–24565. [Google Scholar] [CrossRef] [PubMed]
- Sajid, M.B.; Es-sebbar, E.; Javed, T.; Fittschen, C.; Farooq, A. Measurement of the rate of hydrogen peroxide thermal decomposition in a shock tube using quantum cascade laser absorption near 7.7 μm. Int. J. Chem. Kinet. 2014, 46, 275–284. [Google Scholar] [CrossRef]
- Sajid, M.; Javed, T.; Farooq, A. High-temperature measurements of methane and acetylene using quantum cascade laser absorption near 8μm. J. Quant. Spectrosc. Radiat. Transf. 2015, 155, 66–74. [Google Scholar] [CrossRef]
- Nasir, E.F.; Farooq, A. Time-resolved temperature measurements in a rapid compression machine using quantum cascade laser absorption in the intrapulse mode. Proc. Combust. Inst. 2017, 36, 4453–4460. [Google Scholar] [CrossRef]
- Zhang, G.; Khabibullin, K.; Farooq, A. An IH-QCL based gas sensor for simultaneous detection of methane and acetylene. Proc. Combust. Inst. 2019, 37, 1445–1452. [Google Scholar] [CrossRef]
- Welzel, S.; Gatilova, L.; Röpcke, J.; Rousseau, A. Time-resolved study of a pulsed dc discharge using quantum cascade laser absorption spectroscopy: NO and gas temperature kinetics. Plasma Sources Sci. Technol. 2007, 16, 822. [Google Scholar] [CrossRef]
- Spearrin, R.; Li, S.; Davidson, D.F.; Jeffries, J.; Hanson, R.K. High-temperature iso-butene absorption diagnostic for shock tube kinetics using a pulsed quantum cascade laser near 11.3 μm. Proc. Combust. Inst. 2015, 35, 3645–3651. [Google Scholar] [CrossRef]
- Coddington, I.; Newbury, N.; Swann, W. Dual-comb spectroscopy. Optica 2016, 3, 414. [Google Scholar] [CrossRef]
- Bernhardt, B.; Ozawa, A.; Jacquet, P.; Jacquey, M.; Kobayashi, Y.; Udem, T.; Holzwarth, R.; Guelachvili, G.; Hänsch, T.W.; Picqué, N. Cavity-enhanced dual-comb spectroscopy. Nat. Photonic 2010, 4, 55–57. [Google Scholar] [CrossRef]
- Villares, G.; Hugi, A.; Blaser, S.; Faist, J. Dual-comb spectroscopy based on quantum-cascade-laser frequency combs. Nat. Commun. 2014, 5, 5192. [Google Scholar] [CrossRef]
- Newbury, N.R.; Coddington, I.; Swann, W. Sensitivity of coherent dual-comb spectroscopy. Opt. Express 2010, 18, 7929–7945. [Google Scholar] [CrossRef] [PubMed]
- Rieker, G.B.; Giorgetta, F.R.; Swann, W.C.; Kofler, J.; Zolot, A.M.; Sinclair, L.C.; Baumann, E.; Cromer, C.; Petron, G.; Sweeney, C. Frequency-comb-based remote sensing of greenhouse gases over kilometer air paths. Optica 2014, 1, 290–298. [Google Scholar] [CrossRef]
- Schroeder, P.J.; Wright, R.J.; Coburn, S.; Sodergren, B.; Cossel, K.C.; Droste, S.; Truong, G.W.; Baumann, E.; Giorgetta, F.R.; Coddington, I. Dual frequency comb laser absorption spectroscopy in a 16 MW gas turbine exhaust. Proc. Combust. Inst. 2017, 36, 4565–4573. [Google Scholar] [CrossRef]
- Yan, M.; Luo, P.-L.; Iwakuni, K.; Millot, G.; Hänsch, T.W.; Picqué, N. Mid-infrared dual-comb spectroscopy with electro-optic modulators. Light Sci. Appl. 2017, 6, e17076. [Google Scholar] [CrossRef] [PubMed]
- Link, S.M.; Maas, D.J.; Waldburger, D.; Keller, U. Dual-comb spectroscopy of water vapor with a free-running semiconductor disk laser. Science 2017, 356, 1164–1168. [Google Scholar] [CrossRef]
- Cossel, K.C.; Waxman, E.M.; Giorgetta, F.R.; Cermak, M.; Coddington, I.R.; Hesselius, D.; Ruben, S.; Swann, W.C.; Truong, G.-W.; Rieker, G.B.; et al. Open-path dual-comb spectroscopy to an airborne retroreflector. Optica 2017, 4, 724–728. [Google Scholar] [CrossRef]
- Ycas, G.; Giorgetta, F.; Baumann, E.; Coddington, I.; Herman, D.; Diddams, S.; Newbury, N. Mid-Infrared Dual Comb Spectroscopy of Propane. In Optics and Photonics for Energy and the Environment; Optical Society of America: Washington, DC, USA, 2017; p. ETu1B. 3. [Google Scholar]
- Mandon, J.; Cristescu, S.M.; Harren, F.J. Mid-infrared dual-comb spectroscopy for real-time gas analysis with an optical parametric oscillator. In Proceedings of the 2017 Conference on Lasers and Electro-Optics Europe & European Quantum Electronics Conference (CLEO/Europe-EQEC), Munich, Germany, 25–29 June 2017; p. 1. [Google Scholar]
- Schroeder, P.J. Dual Comb Spectroscopy of High Temperature Environments. Ph.D. Thesis, University of Colorado, Boulder, CO, USA, 2017. [Google Scholar]
- Yu, M.; Okawachi, Y.; Griffith, A.G.; Picqué, N.; Lipson, M.; Gaeta, A.L. Silicon-chip-based mid-infrared dual-comb spectroscopy. Nat. Commun. 2018, 9, 1–6. [Google Scholar] [CrossRef]
- Coburn, S.; Alden, C.B.; Wright, R.; Cossel, K.; Baumann, E.; Truong, G.-W.; Giorgetta, F.; Sweeney, C.; Newbury, N.R.; Prasad, K.; et al. Regional trace-gas source attribution using a field-deployed dual frequency comb spectrometer. Optica 2018, 5, 320–327. [Google Scholar] [CrossRef]
- Klocke, J.L.; Mangold, M.; Allmendinger, P.; Hugi, A.; Geiser, M.; Jouy, P.; Faist, J.; Kottke, T. Single-shot sub-microsecond mid-infrared spectroscopy on protein reactions with quantum cascade laser frequency combs. Anal. Chem. 2018, 90, 10494–10500. [Google Scholar] [CrossRef]
- Ycas, G.; Giorgetta, F.R.; Baumann, E.; Coddington, I.; Herman, D.; Diddams, S.A.; Newbury, N.R. High-coherence mid-infrared dual-comb spectroscopy spanning 2.6 to 5.2 μm. Nat. Photonics 2018, 12, 202–208. [Google Scholar] [CrossRef]
- Hoghooghi, N.; Wright, R.J.; Swann, W.C.; Coddington, I.; Newbury, N.R.; Rieker, G.B. Broadband Cavity-Enhanced Dual-Comb Spectroscopy of Multiple Trace Gas Species. In Proceedings of the 2018 Conference on Lasers and Electro-Optics (CLEO), San Jose, CA, USA, 13–18 May 2018; pp. 1–2. [Google Scholar]
- Bergevin, J.; Wu, T.-H.; Yeak, J.; Brumfield, B.E.; Harilal, S.S.; Phillips, M.C.; Jones, R.J. Dual-comb spectroscopy of laser-induced plasmas. Nat. Commun. 2018, 9, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Sterczewski, L.A.; Westberg, J.; Bagheri, M.; Frez, C.; Vurgaftman, I.; Canedy, C.L.; Bewley, W.W.; Merritt, C.D.; Kim, C.S.; Kim, M. Mid-infrared dual-comb spectroscopy with interband cascade lasers. Opt. Lett. 2019, 44, 2113–2116. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Hänsch, T.W.; Picqué, N. Mid-infrared feed-forward dual-comb spectroscopy. Proc. Natl. Acad. Sci. USA 2019, 116, 3454–3459. [Google Scholar] [CrossRef]
- Draper, A.D.; Cole, R.K.; Makowiecki, A.S.; Mohr, J.; Zdanowicz, A.; Marchese, A.; Hoghooghi, N.; Rieker, G.B. Broadband dual-frequency comb spectroscopy in a rapid compression machine. Opt. Express 2019, 27, 10814–10825. [Google Scholar] [CrossRef]
- Ycas, G.; Giorgetta, F.R.; Cossel, K.C.; Waxman, E.M.; Baumann, E.; Newbury, N.R.; Coddington, I. Mid-infrared dual-comb spectroscopy of volatile organic compounds across long open-air paths. Optica 2019, 6, 165–168. [Google Scholar] [CrossRef]
- Scalari, G.; Faist, J.; Picqué, N. On-Chip Mid-Infrared and THz Frequency Combs for Spectroscopy; AIP Publishing LLC: Melville, NY, USA, 2019. [Google Scholar]
- Picqué, N.; Hänsch, T.W. Frequency comb spectroscopy. Nat. Photonics 2019, 13, 146–157. [Google Scholar] [CrossRef]
- Ycas, G.; Giorgetta, F.R.; Friedlein, J.T.; Herman, D.; Cossel, K.C.; Baumann, E.; Newbury, N.R.; Coddington, I. Compact mid-infrared dual-comb spectrometer for outdoor spectroscopy. Opt. Express 2020, 28, 14740–14752. [Google Scholar] [CrossRef]
- Pinkowski, N.H.; Ding, Y.; Strand, C.L.; Hanson, R.K.; Horvath, R.; Geiser, M. Dual-comb spectroscopy for high-temperature reaction kinetics. Meas. Sci. Technol. 2020, 31, 055501. [Google Scholar] [CrossRef]
- Hugi, A.; Villares, G.; Blaser, S.; Liu, H.; Faist, J. Mid-infrared frequency comb based on a quantum cascade laser. Nature 2012, 492, 229. [Google Scholar] [CrossRef]
- Bidaux, Y.; Sergachev, I.; Wuester, W.; Maulini, R.; Gresch, T.; Bismuto, A.; Blaser, S.; Muller, A.; Faist, J. Plasmon-enhanced waveguide for dispersion compensation in mid-infrared quantum cascade laser frequency combs. Opt. Lett. 2017, 42, 1604–1607. [Google Scholar] [CrossRef]
- Khurgin, J.; Dikmelik, Y.; Hugi, A.; Faist, J. Coherent frequency combs produced by self frequency modulation in quantum cascade lasers. Appl. Phys. Lett. 2014, 104, 081118. [Google Scholar] [CrossRef]
- Burghoff, D.; Yang, Y.; Hayton, D.J.; Gao, J.-R.; Reno, J.L.; Hu, Q. Evaluating the coherence and time-domain profile of quantum cascade laser frequency combs. Opt. Express 2015, 23, 1190–1202. [Google Scholar] [CrossRef]
- Cappelli, F.; Villares, G.; Riedi, S.; Faist, J. Intrinsic linewidth of quantum cascade laser frequency combs. Optica 2015, 2, 836–840. [Google Scholar] [CrossRef]
- Villares, G.; Riedi, S.; Wolf, J.; Kazakov, D.; Süess, M.J.; Jouy, P.; Beck, M.; Faist, J. Dispersion engineering of quantum cascade laser frequency combs. Optica 2016, 3, 252–258. [Google Scholar] [CrossRef]
- Faist, J.; Villares, G.; Scalari, G.; Rösch, M.; Bonzon, C.; Hugi, A.; Beck, M. Quantum cascade laser frequency combs. Nanophotonics 2016, 5, 272–291. [Google Scholar] [CrossRef]
- Hillbrand, J.; Andrews, A.M.; Detz, H.; Strasser, G.; Schwarz, B. Coherent injection locking of quantum cascade laser frequency combs. Nat. Photonics 2019, 13, 101–104. [Google Scholar] [CrossRef]
- Cundiff, S.T.; Ye, J. Colloquium: Femtosecond optical frequency combs. Rev. Mod. Phys. 2003, 75, 325. [Google Scholar] [CrossRef]
- Newbury, N.R.; Swann, W.C. Low-noise fiber-laser frequency combs. JOSA B 2007, 24, 1756–1770. [Google Scholar] [CrossRef]
- Coddington, I.; Swann, W.C.; Newbury, N.R. Coherent multiheterodyne spectroscopy using stabilized optical frequency combs. Phys. Rev. Lett. 2008, 100, 013902. [Google Scholar] [CrossRef]
- Grudinin, I.S.; Yu, N.; Maleki, L. Generation of optical frequency combs with a CaF 2 resonator. Opt. Lett. 2009, 34, 878–880. [Google Scholar] [CrossRef]
- Kippenberg, T.J.; Holzwarth, R.; Diddams, S.A. Microresonator-based optical frequency combs. Science 2011, 332, 555–559. [Google Scholar] [CrossRef]
- Matsko, A.; Savchenkov, A.; Liang, W.; Ilchenko, V.; Seidel, D.; Maleki, L. Mode-locked Kerr frequency combs. Opt. Lett. 2011, 36, 2845–2847. [Google Scholar] [CrossRef] [PubMed]
- Schliesser, A.; Picqué, N.; Hänsch, T.W. Mid-infrared frequency combs. Nat. Photonics 2012, 6, 440. [Google Scholar] [CrossRef]
- Burghoff, D.; Kao, T.-Y.; Han, N.; Chan, C.W.I.; Cai, X.; Yang, Y.; Hayton, D.J.; Gao, J.-R.; Reno, J.L.; Hu, Q. Terahertz laser frequency combs. Nat. Photonics 2014, 8, 462–467. [Google Scholar] [CrossRef]
- QCL Frequency Combs. Available online: http://www.alpeslasers.ch/?a=28,126,191 (accessed on 10 April 2020).
- Jouy, P.; Wolf, J.M.; Bidaux, Y.; Allmendinger, P.; Mangold, M.; Beck, M.; Faist, J. Dual comb operation of λ∼ 8.2 μ m quantum cascade laser frequency comb with 1 W optical power. Appl. Phys. Lett. 2017, 111, 141102. [Google Scholar] [CrossRef]
- Strand, C.; Ding, Y.; Johnson, S.; Hanson, R. Measurement of the mid-infrared absorption spectra of ethylene (C2H4) and other molecules at high temperatures and pressures. J. Quant. Spectrosc. Radiat. Transf. 2019, 222, 122–129. [Google Scholar] [CrossRef]
- Szczepaniak, U.; Schneider, S.H.; Horvath, R.; Kozuch, J.; Geiser, M. Vibrational Stark Spectroscopy of Fluorobenzene Using Quantum Cascade Laser Dual Frequency Combs. Appl. Spectrosc. 2020, 74, 347–356. [Google Scholar] [CrossRef]
- Gordon, I.E.; Rothman, L.S.; Hill, C.; Kochanov, R.V.; Tan, Y.; Bernath, P.F.; Birk, M.; Boudon, V.; Campargue, A.; Chance, K. The HITRAN2016 molecular spectroscopic database. J. Quant. Spectrosc. Radiat. Transf. 2017, 203, 3–69. [Google Scholar] [CrossRef]
- Badra, J.; Elwardany, A.E.; Khaled, F.; Vasu, S.S.; Farooq, A. A shock tube and laser absorption study of ignition delay times and OH reaction rates of ketones: 2-Butanone and 3-buten-2-one. Combust. Flame 2014, 161, 725–734. [Google Scholar] [CrossRef]
- Hanson, R.K.; Spearrin, R.M.; Goldenstein, C.S. Spectroscopy and Optical Diagnostics for Gases; Springer: Berlin/Heidelberg, Germany, 2016; Volume 1. [Google Scholar]
- Kosterev, A.A.; Curl, R.F.; Tittel, F.K.; Gmachl, C.; Capasso, F.; Sivco, D.L.; Baillargeon, J.N.; Hutchinson, A.L.; Cho, A.Y. Effective utilization of quantum-cascade distributed-feedback lasers in absorption spectroscopy. Appl. Opt. 2000, 39, 4425–4430. [Google Scholar] [CrossRef]
- Sharpe, S.W.; Johnson, T.J.; Sams, R.L.; Chu, P.M.; Rhoderick, G.C.; Johnson, P.A. Gas-phase databases for quantitative infrared spectroscopy. Appl. Spectrosc. 2004, 58, 1452–1461. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Mole Fraction (Measured) | Mole Fraction (Expected) | Relative Error (%) |
|---|---|---|---|
| Acetone | 0.55% | 0.5% | 9.1% |
| Propyne | 0.93% | 1% | 7.0% |
| Propene | 4.70% | 5% | 6.1% |
| Methane | 2.19% | 2% | 9.5% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, G.; Horvath, R.; Liu, D.; Geiser, M.; Farooq, A. QCL-Based Dual-Comb Spectrometer for Multi-Species Measurements at High Temperatures and High Pressures. Sensors 2020, 20, 3602. https://doi.org/10.3390/s20123602
Zhang G, Horvath R, Liu D, Geiser M, Farooq A. QCL-Based Dual-Comb Spectrometer for Multi-Species Measurements at High Temperatures and High Pressures. Sensors. 2020; 20(12):3602. https://doi.org/10.3390/s20123602
Chicago/Turabian StyleZhang, Guangle, Raphael Horvath, Dapeng Liu, Markus Geiser, and Aamir Farooq. 2020. "QCL-Based Dual-Comb Spectrometer for Multi-Species Measurements at High Temperatures and High Pressures" Sensors 20, no. 12: 3602. https://doi.org/10.3390/s20123602
APA StyleZhang, G., Horvath, R., Liu, D., Geiser, M., & Farooq, A. (2020). QCL-Based Dual-Comb Spectrometer for Multi-Species Measurements at High Temperatures and High Pressures. Sensors, 20(12), 3602. https://doi.org/10.3390/s20123602