Screen-Printed Soft-Nitrided Carbon Electrodes for Detection of Hydrogen Peroxide


Abstract
1. Introduction
2. Experimental
2.1. Materials
2.2. Preparation of N-Doped Graphite
2.3. Characterization of Carbon Materials
2.4. Preparation of SPCEs and N-SPCEs
2.5. Electrochemical Measurements
2.6. Spectrophotometric Assay for H2O2
3. Results
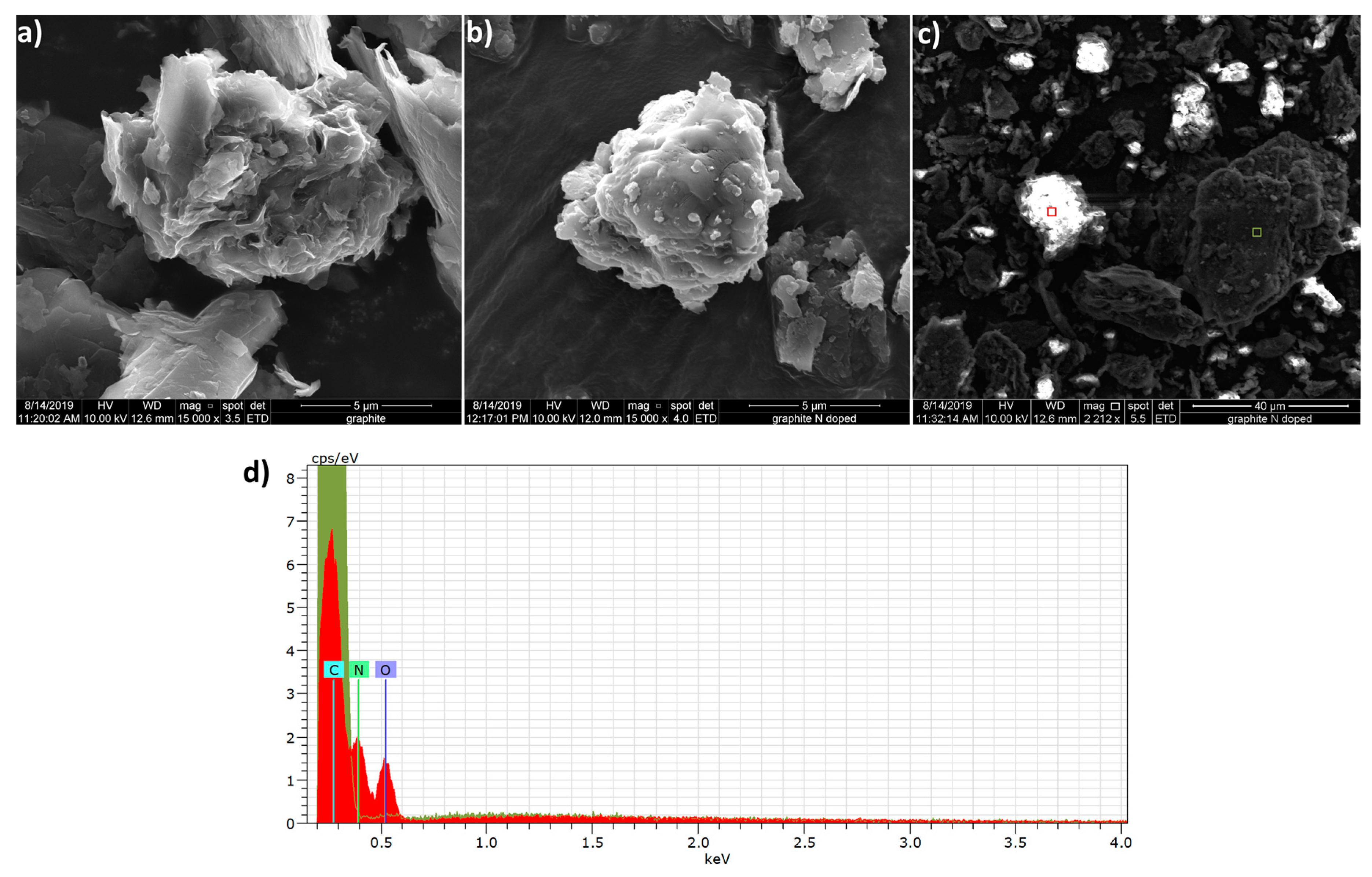
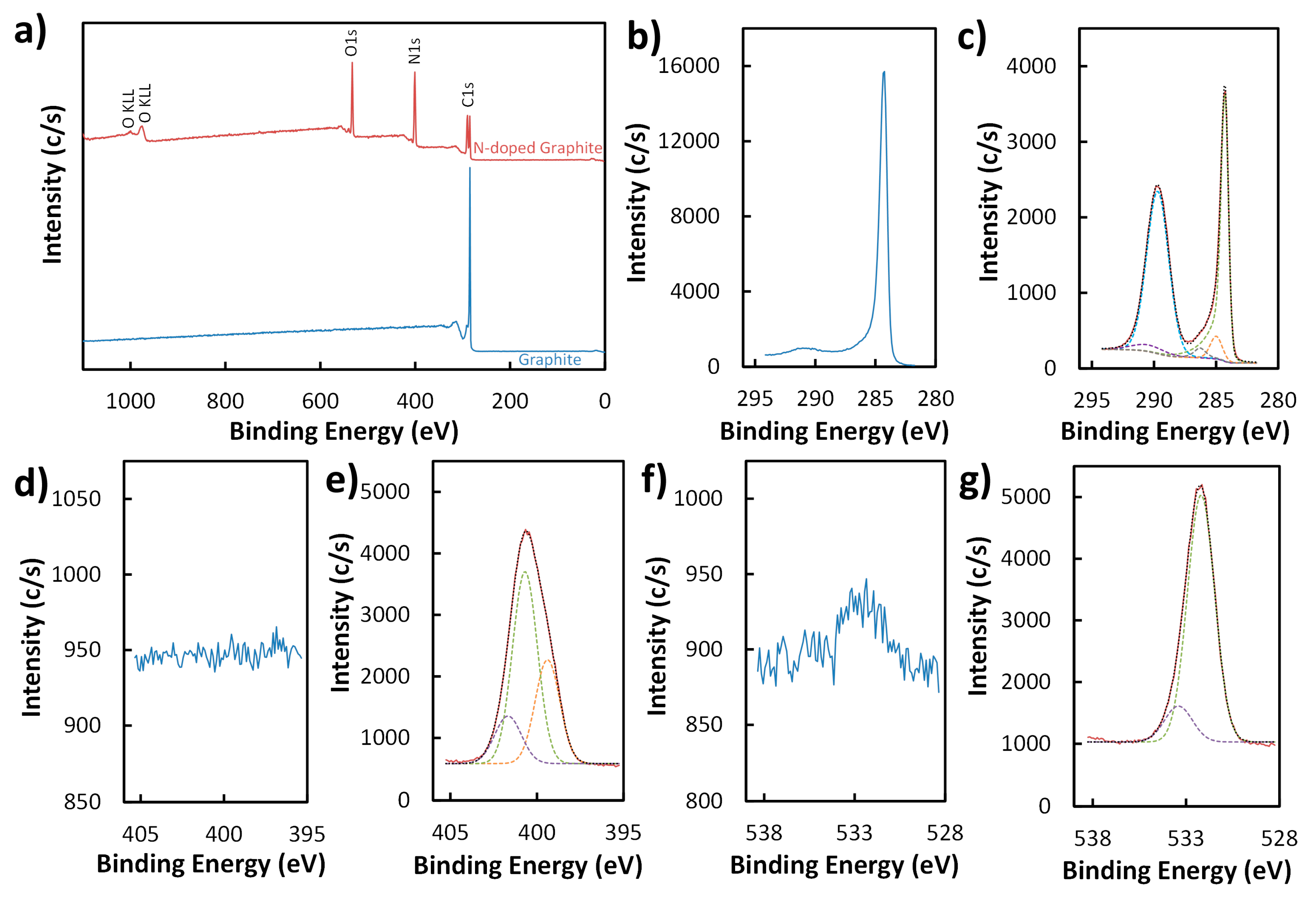
3.1. Characterization of Nitrogen-Doped Graphite
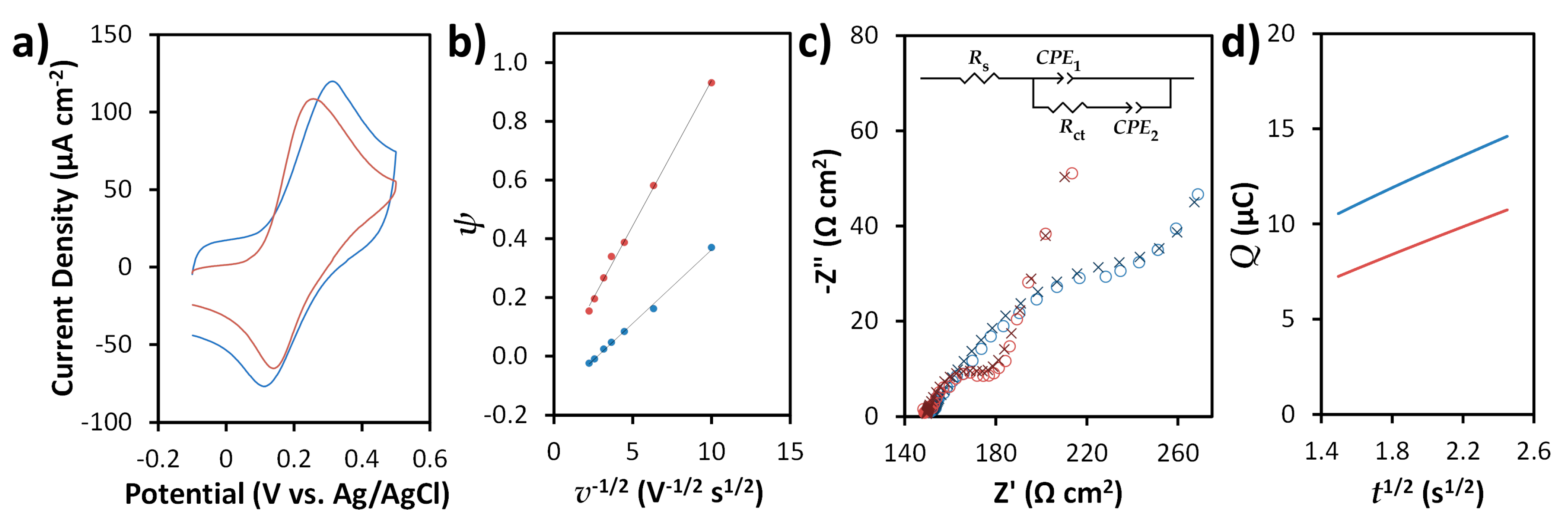
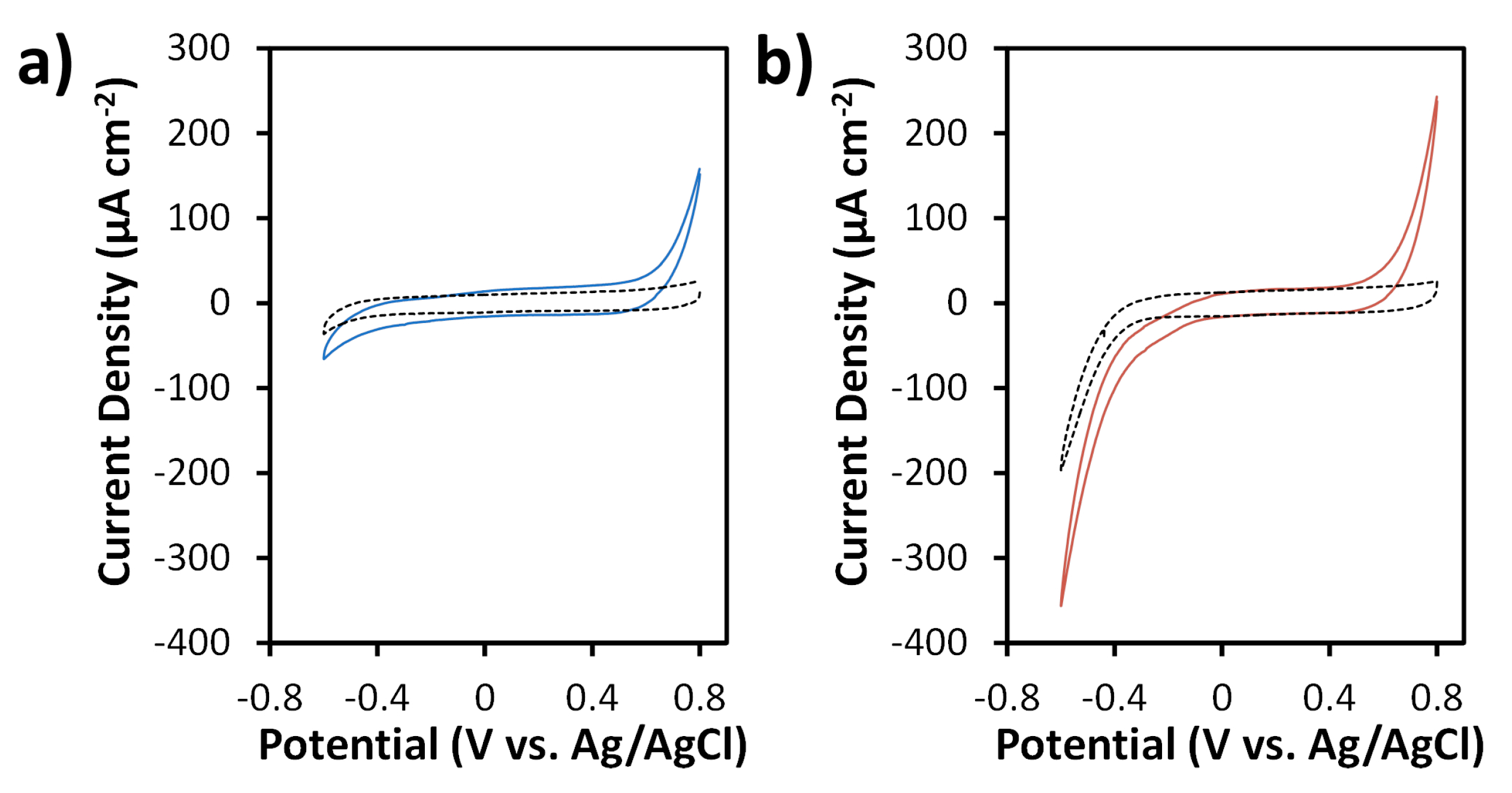
3.2. Electrochemical Characterization and Electrocatalytic Behavior of SPCEs and N-SPCEs
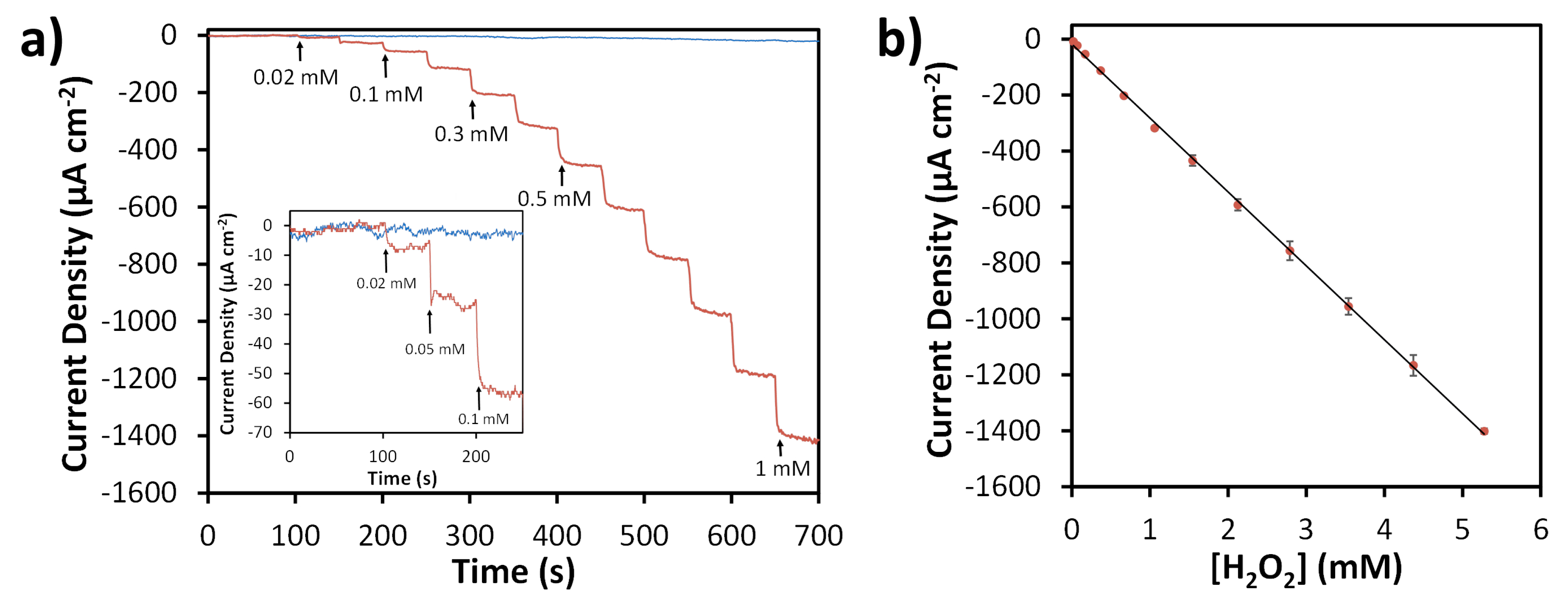
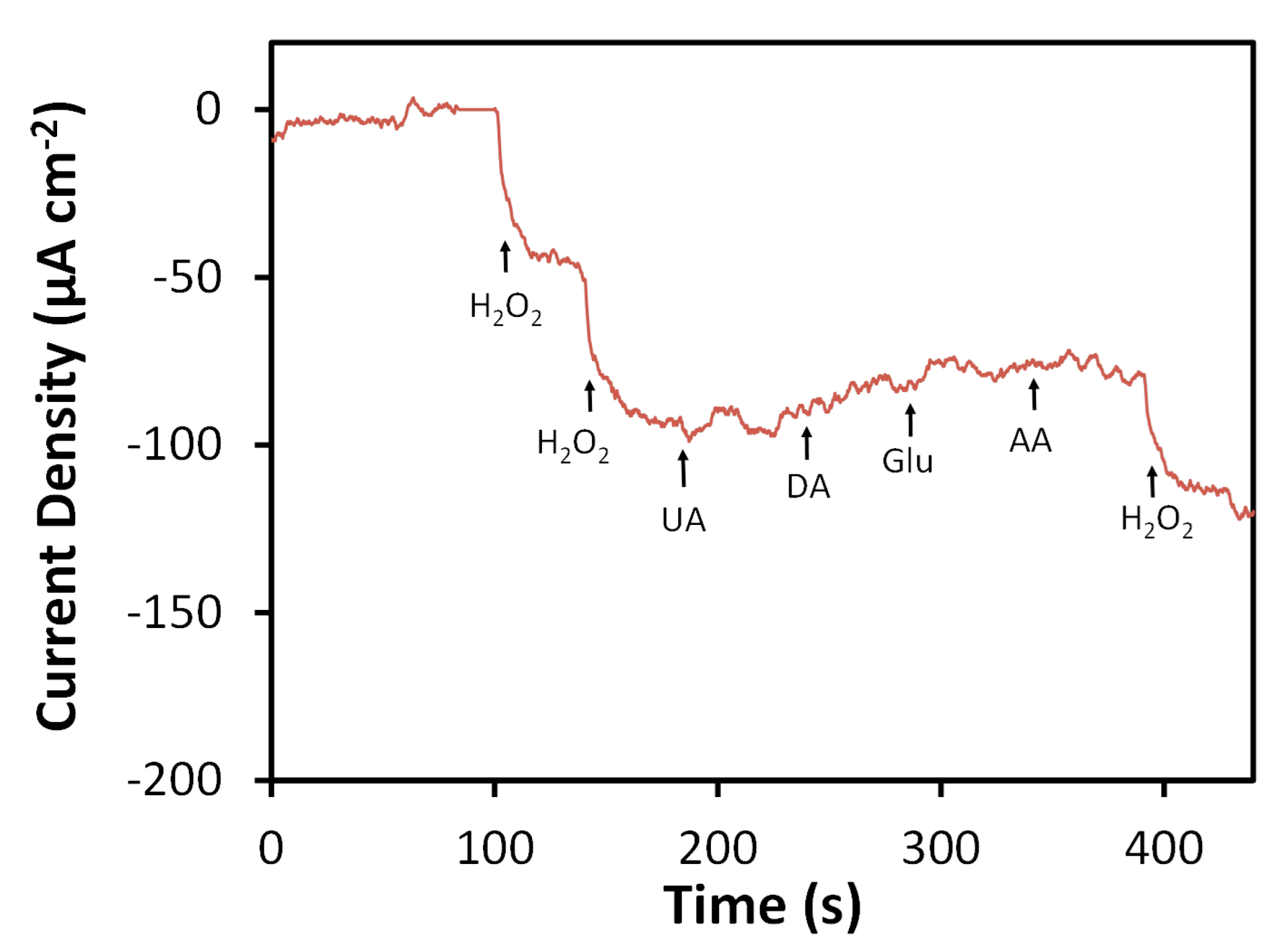
3.3. Amperometric Detection of H2O2 Using N-SPCEs
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tangkuaram, T.; Ponchio, C.; Kangkasomboon, T.; Katikawong, P.; Veerasai, W. Design and Development of a Highly Stable Hydrogen Peroxide Biosensor on Screen Printed Carbon Electrode on Horseradish Peroxidase Bound with Gold Nanoparticles in the Matrix of Chitosan. Biosens. Bioelectron. 2007, 22, 2071–2078. [Google Scholar] [CrossRef] [PubMed]
- Metters, J.P.; Kadara, R.O.; Banks, C.E. New Directions in Screen Printed Electroanalytical Sensors: An Overview of Recent Developments. Analyst 2011, 136, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Cumba, L.R.; Foster, C.W.; Brownson, D.A.C.; Smith, J.P.; Iniesta, J.; Thakur, B.; do Carmo, D.R.; Banks, C.E. Can the Mechanical Activation (Polishing) of Screen-Printed Electrodes Enhance Their Electroanalytical Response? Analyst 2016, 141, 2791–2799. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, Z.; Zhang, M.; Liu, J.; Zou, H.; Wang, L. Improvement of Electrochemical Performance of Screen-Printed Carbon Electrodes by UV/Ozone Modification. Talanta 2019, 192, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.C.; Chang, K.S.; Yuan, C.J. Enhancement of Electrochemical Properties of Screen-Printed Carbon Electrodes by Oxygen Plasma Treatment. Electrochim. Acta 2009, 54, 4937–4943. [Google Scholar] [CrossRef]
- Wang, J.; Pedrero, M.; Sakslund, H.; Hammerich, O.; Pingarron, J. Electrochemical Activation of Screen-Printed Carbon Strips. Analyst 1996, 121, 345–350. [Google Scholar] [CrossRef]
- González-Sánchez, M.I.; Gómez-Monedero, B.; Agrisuelas, J.; Iniesta, J.; Valero, E. Highly Activated Screen-Printed Carbon Electrodes by Electrochemical Treatment with Hydrogen Peroxide. Electrochem. Commun. 2018, 91, 36–40. [Google Scholar] [CrossRef]
- Chikae, M.; Idegami, K.; Kerman, K.; Nagatani, N.; Ishikawa, M.; Takamura, Y.; Tamiya, E. Direct Fabrication of Catalytic Metal Nanoparticles onto the Surface of a Screen-Printed Carbon Electrode. Electrochem. Commun. 2006, 8, 1375–1380. [Google Scholar] [CrossRef]
- Fanjul-Bolado, P.; Queipo, P.; Lamas-Ardisana, P.J.; Costa-García, A. Manufacture and Evaluation of Carbon Nanotube Modified Screen-Printed Electrodes as Electrochemical Tools. Talanta 2007, 74, 427–433. [Google Scholar] [CrossRef]
- Cinti, S.; Arduini, F.; Moscone, D.; Palleschi, G.; Killard, A.J. Development of a Hydrogen Peroxide Sensor Based on Screen-Printed Electrodes Modified with Inkjet-Printed Prussian Blue Nanoparticles. Sensors 2014, 14, 14222–14234. [Google Scholar] [CrossRef]
- Shi, L.; Niu, X.; Liu, T.; Zhao, H.; Lan, M. Electrocatalytic Sensing of Hydrogen Peroxide Using a Screen-Printed Carbon Electrode Modified with Nitrogen-Doped Graphene Nanoribbons. Microchim. Acta 2015, 182, 2485–2493. [Google Scholar] [CrossRef]
- Agrisuelas, J.; González-Sánchez, M.I.; Valero, E. Hydrogen Peroxide Sensor Based on In Situ Grown Pt Nanoparticles from Waste Screen-Printed Electrodes. Sens. Actuators B 2017, 249, 499–505. [Google Scholar] [CrossRef]
- Ledru, S.; Ruillé, N.; Boujtita, M. One-Step Screen-Printed Electrode Modified in Its Bulk with HRP Based on Direct Electron Transfer for Hydrogen Peroxide Detection in Flow Injection Mode. Biosens. Bioelectron. 2006, 21, 1591–1598. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.J.; Zuo, S.H.; Lan, M.B. Direct Electron Transfer of Horseradish Peroxidase on Porous Structure of Screen-Printed Electrode. Biosens. Bioelectron. 2009, 24, 1353–1357. [Google Scholar] [CrossRef] [PubMed]
- Wang, J. Electrochemical Glucose Biosensors. Chem. Rev. 2008, 108, 814–825. [Google Scholar] [CrossRef]
- Cinti, S.; Arduini, F.; Vellucci, G.; Cacciotti, I.; Nanni, F.; Moscone, D. Carbon Black Assisted Tailoring of Prussian Blue Nanoparticles to Tune Sensitivity and Detection Limit Towards H2O2 by Using Screen-Printed Electrode. Electrochem. Commun. 2014, 47, 63–66. [Google Scholar] [CrossRef]
- Huang, Y.; Xue, Y.; Zeng, J.; Li, S.; Wang, Z.; Dong, C.; Li, G.; Liang, J.; Zhou, Z. Non-Enzymatic Electrochemical Hydrogen Peroxide Biosensor Based on Reduction Graphene Oxide-Persimmon Tannin-Platinum Nanocomposite. Mater. Sci. Eng. C 2018, 92, 590–598. [Google Scholar] [CrossRef]
- Yuan, C.J.; Wang, Y.C.; Reiko, O. Improving the Detection of Hydrogen Peroxide of Screen-Printed Carbon Paste Electrodes by Modifying with Nonionic Surfactants. Anal. Chim. Acta 2009, 653, 71–76. [Google Scholar] [CrossRef]
- Ricci, F.; Palleschi, G. Sensor and Biosensor Preparation, Optimisation and Applications of Prussian Blue Modified Electrodes. Biosens. Bioelectron. 2005, 21, 389–407. [Google Scholar] [CrossRef]
- Maldonado, S.; Stevenson, K.J. Direct Preparation of Carbon Nanofiber Electrodes via Pyrolysis of Iron (II) Phthalocyanine: Electrocatalytic Aspects for Oxygen Reduction. J. Phys. Chem. B 2004, 108, 11375–11383. [Google Scholar] [CrossRef]
- Gong, K.; Du, F.; Xia, Z.; Durstock, M.; Dai, L. Nitrogen-Doped Carbon Nanotube Arrays with High Electrocatalytic Activity for Oxygen Reduction. Science 2009, 323, 760–764. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, S.; Stevenson, K.J. Influence of Nitrogen Doping on Oxygen Reduction Electrocatalysis at Carbon Nanofiber Electrodes. J. Phys. Chem. B 2005, 109, 4707–4716. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, T.; Hu, G.; Jia, X.; Wågberg, T. Formation of Active Sites for Oxygen Reduction Reactions by Transformation of Nitrogen Functionalities in Nitrogen-Doped Carbon Nanotubes. ACS Nano 2012, 6, 8904–8912. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Zhang, S.; Engelhard, M.H.; Li, G.; Shao, G.; Wang, Y.; Liu, J.; Aksay, I.A.; Lin, Y. Nitrogen-Doped Graphene and Its Electrochemical Applications. J. Mater. Chem. 2010, 20, 7491–7496. [Google Scholar] [CrossRef]
- Wang, Y.; Shao, Y.; Matson, D.W.; Li, J.; Lin, Y. Nitrogen-Doped Graphene and Its Application in Electrochemical Biosensing. ACS Nano 2010, 4, 1790–1798. [Google Scholar] [CrossRef] [PubMed]
- Lyu, Y.P.; Wu, Y.S.; Wang, T.P.; Lee, C.L.; Chung, M.Y.; Lo, C.T. Hydrothermal and Plasma Nitrided Electrospun Carbon Nanofibers for Amperometric Sensing of Hydrogen Peroxide. Microchim. Acta 2018, 185, 371. [Google Scholar] [CrossRef]
- Shui, J.; Wang, M.; Du, F.; Dai, L. N-Doped Carbon Nanomaterials Are Durable Catalysts for Oxygen Reduction Reaction in Acidic Fuel Cells. Sci. Adv. 2015, 1, e1400129. [Google Scholar] [CrossRef]
- Yu, D.; Zhang, Q.; Dai, L. Highly Efficient Metal-Free Growth of Nitrogen-Doped Single-Walled Carbon Nanotubes on Plasma-Etched Substrates for Oxygen Reduction. J. Am. Chem. Soc. 2010, 132, 15127–15129. [Google Scholar] [CrossRef]
- Wiggins-Camacho, J.D.; Stevenson, K.J. Mechanistic Discussion of the Oxygen Reduction Reaction at Nitrogen-Doped Carbon Nanotubes. J. Phys. Chem. C 2011, 115, 20002–20010. [Google Scholar] [CrossRef]
- Dai, L.; Chang, D.W.; Baek, J.B.; Lu, W. Carbon Nanomaterials for Advanced Energy Conversion and Storage. Small 2012, 8, 1130–1166. [Google Scholar] [CrossRef]
- Dai, L.; Xue, Y.; Qu, L.; Choi, H.J.; Baek, J.B. Metal-Free Catalysts for Oxygen Reduction Reaction. Chem. Rev. 2015, 115, 4823–4892. [Google Scholar] [CrossRef] [PubMed]
- Ambrosi, A.; Chua, C.K.; Latiff, N.M.; Loo, A.H.; Wong, C.H.A.; Eng, A.Y.S.; Bonanni, A.; Pumera, M. Graphene and Its Electrochemistry—An Update. Chem. Soc. Rev. 2016, 45, 2458–2493. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Yao, H.; Song, W.; Jin, L.; Mosa, I.M.; Rusling, J.F.; Suib, S.L.; He, J. Ligand-Free Noble Metal Nanocluster Catalysts on Carbon Supports via “Soft” Nitriding. J. Am. Chem. Soc. 2016, 138, 4718–4721. [Google Scholar] [CrossRef] [PubMed]
- Chang, Z.; Yang, Y.; He, J.; Rusling, J.F. Gold Nanocatalysts Supported on Carbon for Electrocatalytic Oxidation of Organic Molecules Including Guanines in DNA. Dalton Trans. 2018, 47, 14139–14152. [Google Scholar] [CrossRef] [PubMed]
- Wring, S.A.; Hart, J.P. Chemically Modified, Screen-Printed Carbon Electrodes. Analyst 1992, 117, 1281–1286. [Google Scholar] [CrossRef]
- Miserere, S.; Ledru, S.; Ruillé, N.; Griveau, S.; Boujtita, M.; Bedioui, F. Biocompatible Carbon-Based Screen-Printed Electrodes for the Electrochemical Detection of Nitric Oxide. Electrochem. Commun. 2006, 8, 238–244. [Google Scholar] [CrossRef]
- Bishop, G.W.; Ahiadu, B.K.; Smith, J.L.; Patterson, J.D. Use of Redox Probes for Characterization of Layer-by-Layer Gold Nanoparticle-Modified Screen-Printed Carbon Electrodes. J. Electrochem. Soc. 2017, 164, B23–B28. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, D.; Yu, B.; You, T. A Novel Nonenzymatic Hydrogen Peroxide Sensor Based on Electrospun Nitrogen-Doped Carbon Nanoparticles-Embedded Carbon Nanofibers Film. Sens. Actuators B 2016, 224, 103–109. [Google Scholar] [CrossRef]
- Wu, Y.S.; Liu, Z.T.; Wang, T.P.; Hsu, S.Y.; Lee, C.L. A Comparison of Nitrogen-Doped Sonoelectrochemical and Chemical Graphene Nanosheets as Hydrogen Peroxide Sensors. Ultrason. Sonochem. 2018, 42, 659–664. [Google Scholar] [CrossRef]
- Bondarenko, A.S.; Ragoisha, G.A. Progress in Chemometrics Research; Pomerantsev, A.L., Ed.; Nova Science Publishers: New York, NY, USA, 2005; pp. 89–102. Available online: http://www.abc.chemistry.bsu.by/vi/analyser/ (accessed on 30 July 2019).
- Cheng, X.; Challier, L.; Etcheberry, A.; Noël, V.; Perez, H. The ABTS-HRP System as an Alternative Method to RRDE for the Determination of the Selectivity of the Oxygen Reduction Reaction. Int. J. Electrochem. Sci. 2012, 7, 6247–6262. [Google Scholar]
- León, V.; Quintana, M.; Herrero, M.A.; Fierro, J.L.G.; de la Hoz, A.; Prato, M.; Vázquez, E. Few-Layer Graphenes from Ball-Milling of Graphite with Melamine. Chem. Commun. 2011, 47, 10936–10938. [Google Scholar] [CrossRef] [PubMed]
- Ederer, J.; Janoš, P.; Ecorchard, P.; Tolasz, J.; Štengl, V.; Beneš, H.; Perchacz, M.; Pop-Georgievski, O. Determination of Amino Groups on Functionalized Graphene Oxide for Polyurethane nanomaterials: XPS Quantitation vs. Functional Speciation. RSC Adv. 2017, 7, 12464–12473. [Google Scholar] [CrossRef]
- Kinoshita, H. Synthesis of Melamine from Urea, II. Rev. Phys. Chem. Jpn. 1954, 24, 19–27. [Google Scholar]
- Schaber, P.M.; Colson, J.; Higgins, S.; Thielen, D.; Anspach, B.; Brauer, J. Thermal Decomposition (Pyrolysis) of Urea in an Open Reaction Vessel. Thermochim. Acta 2004, 424, 131–142. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, T.; Wang, Z.; Dawson, G.; Chen, W. Simple Pyrolysis of Urea into Graphitic Carbon Nitride with Recyclable Adsorption and Photocatalytic Activity. J. Mater. Chem. 2011, 21, 14398–14401. [Google Scholar] [CrossRef]
- Shi, L.; Liang, L.; Wang, F.; Liu, M.; Chen, K.; Sun, K.; Zhang, N.; Sun, J. Higher Yield Urea-Derived Polymeric Graphitic Carbon Nitride with Mesoporous Structure and Superior Visible-Light-Responsive Activity. ACS Sustain. Chem. Eng. 2015, 3, 3412–3419. [Google Scholar] [CrossRef]
- Mahalingam, P.; Loganathan, A.; Selvaraju, M.; Sivakumar, P. Simultaneous Exfoliation and Reduction of Urea Intercalated Graphite Oxide Using Microwave Radiation. Graphene 2015, 3, 40–43. [Google Scholar] [CrossRef]
- León, V.; Rodriguez, A.M.; Prieto, P.; Prato, M.; Vázquez, E. Exfoliation of Graphite with Triazine Derivatives under Ball-Milling Conditions: Preparation of Few-Layer Graphene via Selective Noncovalent Interactions. ACS Nano 2014, 8, 563–571. [Google Scholar] [CrossRef]
- Wang, Y.; Wei, B.; Wang, Q. Crystal Structure of Melamine Cyanuric Acid Complex (1:1) Trihydrochloride, MCA·3HCl. J. Crystallogr. Spectrosc. Res. 1990, 20, 79–84. [Google Scholar] [CrossRef]
- Seto, C.T.; Whitesides, G.M. Self-Assembly Based on the Cyanuric Acid–Melamine Lattice. J. Am. Chem. Soc. 1990, 112, 6409–6411. [Google Scholar] [CrossRef]
- Jun, Y.S.; Lee, E.Z.; Wang, X.; Hong, W.H.; Stucky, G.D.; Thomas, A. From Melamine-Cyanuric Acid Supramolecular Aggregates to Carbon Nitride Hollow Spheres. Adv. Funct. Mater. 2013, 23, 3661–3667. [Google Scholar] [CrossRef]
- Jun, Y.S.; Park, J.; Lee, S.U.; Thomas, A.; Hong, W.H.; Stucky, G.D. Three-Dimensional Macroscopic Assemblies of Low-Dimensional Carbon Nitrides for Enhanced Hydrogen Evolution. Angew. Chem. Int. Ed. 2013, 52, 11083–11087. [Google Scholar] [CrossRef] [PubMed]
- Roy, B.; Bairi, P.; Nandi, A.K. Supramolecular Assembly of Melamine and Its Derivatives: Nanostructures to Functional Materials. RSC Adv. 2014, 4, 1708–1734. [Google Scholar] [CrossRef]
- Guo, H.; Ren, Y.; Chen, Q.; Wang, D.; Liu, Y. Gold Nanoparticles on Cyanuric Acid-Based Support: A Highly Active Catalyst for the Reduction of 4-Nitrophenol in Water. Catal. Commun. 2017, 102, 136–140. [Google Scholar] [CrossRef]
- Liang, X.; Pu, X.; Zhou, H.; Wong, N.B.; Tian, A. Keto-Enol Tautomerization of Cyanuric Acid in the Gas Phase and in Water and Methanol. J. Mol. Struct.: THEOCHEM 2007, 816, 125–136. [Google Scholar] [CrossRef]
- Pérez-Manríquez, L.; Cabrera, A.; Sansores, L.E.; Salcedo, R. Aromaticity in Cyanuric Acid. J. Mol. Model. 2011, 17, 1311–1315. [Google Scholar] [CrossRef] [PubMed]
- Malitesta, C.; Losito, I.; Sabbatini, L.; Zambonin, P.G. New Findings on Polypyrrole Chemical Structure by XPS Coupled to Chemical Derivatization Labelling. J. Electron Spectrosc. Relat. Phenom. 1995, 75, 629–634. [Google Scholar] [CrossRef]
- Mao, J.; Peng, T.; Zhang, X.; Li, K.; Ye, L.; Zan, L. Effect of Graphitic Carbon Nitride Microstructures on the Activity and Selectivity of Photocatalytic CO2 Reduction under Visible Light. Catal. Sci. Technol. 2013, 3, 1253–1260. [Google Scholar] [CrossRef]
- Newman, R.; Badger, R.M. Infrared Spectra of Cyanuric Acid and Deutero Cyanuric Acid. J. Am. Chem. Soc. 1952, 74, 3545–3548. [Google Scholar] [CrossRef]
- Zhou, J.; Yang, Y.; Zhang, C. A Low-Temperature Solid-Phase Method to Synthesize Highly Fluorescent Carbon Nitride Dots with Tunable Emission. Chem. Commun. 2013, 49, 8605–8607. [Google Scholar] [CrossRef]
- Wang, J.; Tian, B.; Nascimento, V.B.; Angnes, L. Performance of Screen-Printed Carbon Electrodes Fabricated from Different Carbon Inks. Electrochim. Acta 1998, 43, 3459–3465. [Google Scholar] [CrossRef]
- Kadara, R.O.; Jenkinson, N.; Banks, C.E. Characterisation of Commercially Available Electrochemical Sensing Platforms. Sens. Actuators B 2009, 138, 556–562. [Google Scholar] [CrossRef]
- Choudry, N.A.; Kampouris, D.K.; Kadara, R.O.; Banks, C.E. Disposable Highly Ordered Pyrolytic Graphite-Like Electrodes: Tailoring the Electrochemical Reactivity of Screen Printed Electrodes. Electrochem. Commun. 2010, 12, 6–9. [Google Scholar] [CrossRef]
- Washe, A.P.; Lozano-Sánchez, P.; Bejarano-Nosas, D.; Katakis, I. Facile and Versatile Approaches to Enhancing Electrochemical Performance of Screen Printed Electrodes. Electrochim. Acta 2013, 91, 166–172. [Google Scholar] [CrossRef]
- Randviir, E.P. A Cross Examination of Electron Transfer Rate Constants for Carbon Screen-Printed Electrodes using Electrochemical Impedance Spectroscopy and Cyclic Voltammetry. Electrochim. Acta 2018, 286, 179–186. [Google Scholar] [CrossRef]
- Nicholson, R.S.; Shain, I. Theory of Stationary Electrode Polarography. Single Scan and Cyclic Methods Applied to Reversible, Irreversible, and Kinetic Systems. Anal. Chem. 1964, 36, 706–723. [Google Scholar] [CrossRef]
- Nicholson, R.S. Theory and Application of Cyclic Voltammetry for Measurement of Electrode Reaction Kinetics. Anal. Chem. 1965, 37, 1351–1355. [Google Scholar] [CrossRef]
- Lavagnini, I.; Antiochia, R.; Magno, F. An Extended Method for Practical Evaluation of the Standard Rate Constant from Cyclic Voltammetric Data. Electroanalysis 2004, 16, 505–506. [Google Scholar] [CrossRef]
- Chen, P.; McCreery, R.L. Control of Electron Transfer Kinetics at Glassy Carbon Electrodes by Specific Surface Modification. Anal. Chem. 1996, 68, 3958–3965. [Google Scholar] [CrossRef]
- Bernalte, E.; Marín-Sánchez, C.; Pinilla-Gil, E.; Brett, C.M.A. Characterisation of Screen-Printed Gold and Gold Nanoparticle-Modified Carbon Sensors by Electrochemical Impedance Spectroscopy. J. Electroanal. Chem. 2013, 709, 70–76. [Google Scholar] [CrossRef]
- Morrin, A.; Killard, A.J.; Smyth, M.R. Electrochemical Characterization of Commercial and Home-Made Screen-Printed Carbon Electrodes. Anal. Lett. 2003, 36, 2021–2039. [Google Scholar] [CrossRef]
- Fanjul-Bolado, P.; Hernández-Santos, D.; Lamas-Ardisana, P.J.; Martín-Pernía, A.; Costa-García, A. Electrochemical Characterization of Screen-Printed and Conventional Carbon Paste Electrodes. Electrochim. Acta 2008, 53, 3635–3642. [Google Scholar] [CrossRef]
- Fragkou, V.; Ge, Y.; Steiner, G.; Freeman, D.; Bartetzko, N.; Turner, A.P.F. Determination of the Real Surface Area of a Screen-Printed Electrode by Chronocoulometry. Int. J. Electrochem. Sci. 2012, 7, 6214–6220. [Google Scholar]
- Ferrari, A.G.M.; Foster, C.W.; Kelly, P.J.; Brownson, D.A.C.; Banks, C.E. Determination of the Electrochemical Area of Screen-Printed Electrochemical Sensing Platforms. Biosensors 2018, 8, 53. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Bo, X.; Nsabimana, A.; Luhana, C.; Wang, G.; Wang, H.; Li, M.; Guo, L. Fabrication of 2D Ordered Mesoporous Carbon Nitride and Its Use as Electrochemical Sensing Platform for H2O2, Nitrobenzene, and NADH Detection. Biosens. Bioelectron. 2014, 53, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Yu, B.; Fei, T.; Zhang, T. Low Temperature Thermal Treatment of Hexamethylenetetramine to Synthesize Nitrogen-Doped Carbon for Non-Enzymatic H2O2 Sensing. Sens. Actuators B 2014, 201, 240–245. [Google Scholar] [CrossRef]
- Pollack, B.; Holmberg, S.; George, D.; Tran, I.; Madou, M.; Ghazinejad, M. Nitrogen-Rich Polyacrylonitrile-Based Graphitic Carbons for Hydrogen Peroxide Sensing. Sensors 2017, 17, 2407. [Google Scholar] [CrossRef] [PubMed]
- Otieno, B.A.; Krause, C.E.; Latus, A.; Chikkaveeraiah, B.V.; Faria, R.C.; Rusling, J.F. On-line Protein Capture on Magnetic Beads for Ultrasensitive Microfluidic Immunoassays of Cancer Biomarkers. Biosens. Bioelectron. 2014, 53, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Hayes, W.I.; Lubarsky, G.; Li, M.; Papakonstantinou, P. Mechanical Exfoliation of Graphite in 1-Butyl-Methylimidazolium Hexafluorophosphate (BMIM-PF6) Providing Graphene Nanoplatelets that Exhibit Enhanced Electrocatalysis. J. Power Sources 2014, 271, 312–325. [Google Scholar] [CrossRef]
- Yang, X.; Haubold, L.; DeVivo, G.; Swain, G.M. Electroanalytical Performance of Nitrogen-Containing Tetrahedral Amorphous Carbon Thin-Film Electrodes. Anal. Chem. 2012, 84, 6240–6248. [Google Scholar] [CrossRef]
- Kamata, T.; Kato, D.; Hirono, S.; Niwa, O. Structure and Electrochemical Performance of Nitrogen-Doped Carbon Film Formed by Electron Cyclotron Resonance Sputtering. Anal. Chem. 2013, 85, 9845–9851. [Google Scholar] [CrossRef] [PubMed]
- Behan, J.A.; Stamatin, S.N.; Hoque, M.K.; Ciapetti, G.; Zen, F.; Esteban-Tejeda, L.; Colavita, P.E. Combined Optoelectronic and Electrochemical Study of Nitrogenated Carbon Electrodes. J. Phys. Chem. C 2017, 121, 6596–6604. [Google Scholar] [CrossRef]
- Tian, J.; Liu, Q.; Ge, C.; Xing, Z.; Asiri, A.M.; Al-Youbi, A.O.; Sun, X. Ultrathin Graphitic Carbon Nitride Nanosheets: A Low-Cost, Green, and Highly Efficient Electrocatalyst Toward the Reduction of Hydrogen Peroxide and Its Glucose Biosensing Application. Nanoscale 2013, 5, 8921–8924. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Spiked (mM) | Measured (mM) | % Recovery | ABTS-HRP Assay (mM) |
|---|---|---|---|
| 0.015 | 0.0137 (±0.00019) | 91 (±1.3) | 0.016 (±0.0011) |
| 0.025 | 0.024 (±0.0010) | 97 (±4.0) | 0.024 (±0.0012) |
| 0.080 | 0.074 (±0.0021) | 93 (±2.7) | 0.080 (±0.0012) |
| 0.40 | 0.39 (±0.026) | 98 (±6.5) | 0.39 (±0.012) |
| 2.50 | 2.49 (±0.062) | 100 (±2.5) | 2.41 (±0.040) |
| Electrode | E (V) 1 | Linear Range (mM) | Sensitivity 2 (µA mM−1 cm−2) | LOD (µM) | Ref. |
|---|---|---|---|---|---|
| N-Graphene/GCE | −0.2 | 10−5–2.8 | NR 3 | NR 3 | [24] |
| N-GrNR 4/SPCE | −0.4 | 0.005–0.085 0.135–1.385 | 2180 640 | 1.72 | [11] |
| g-C3N4 NS 5/GCE | −0.3 | 0.1–90 | NR 3 | 2.0 | [84] |
| N-Carbon/GCE | −0.3 | 0.1–40 | NR 3 | 90 | [77] |
| N-CNFht 6/GCE | −0.4 | 0.01–0.71 0.71–2.91 | 357 203 | 0.62 | [26] |
| N-CNFp 7/GCE | −0.4 | 0.01–0.21 0.21–2.21 | 257 180 | 1.84 | [26] |
| OMCN 8/GCE | −0.19 | 0.004–0.04 0.04–12.4 | 642 9 288 9 | 1.52 | [76] |
| N-SEGN 10/GCE | −0.4 | 0.01–2.225 | 231.3 | 0.88 | [39] |
| N-rGO 11/GCE | −0.4 | 0.01–4.625 | 57.3 | 0.94 | [39] |
| N-CNF mat 12 | −0.5 | 0.5–2.5 | 28.7 9 | 0.609 | [78] |
| N-CNPF 13/GCE | −0.4 | 0.005–27 | 383.9 | 1.5 | [38] |
| N-SPCE | −0.4 | 0.02–5.3 | 264 | 2.5 | This work |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ogbu, C.I.; Feng, X.; Dada, S.N.; Bishop, G.W. Screen-Printed Soft-Nitrided Carbon Electrodes for Detection of Hydrogen Peroxide. Sensors 2019, 19, 3741. https://doi.org/10.3390/s19173741
Ogbu CI, Feng X, Dada SN, Bishop GW. Screen-Printed Soft-Nitrided Carbon Electrodes for Detection of Hydrogen Peroxide. Sensors. 2019; 19(17):3741. https://doi.org/10.3390/s19173741
Chicago/Turabian StyleOgbu, Chidiebere I., Xu Feng, Samson N. Dada, and Gregory W. Bishop. 2019. "Screen-Printed Soft-Nitrided Carbon Electrodes for Detection of Hydrogen Peroxide" Sensors 19, no. 17: 3741. https://doi.org/10.3390/s19173741
APA StyleOgbu, C. I., Feng, X., Dada, S. N., & Bishop, G. W. (2019). Screen-Printed Soft-Nitrided Carbon Electrodes for Detection of Hydrogen Peroxide. Sensors, 19(17), 3741. https://doi.org/10.3390/s19173741