Fast 2D Laser-Induced Fluorescence Spectroscopy Mapping of Rare Earth Elements in Rock Samples

 , , ,
, , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Rock Sampling and Preparation
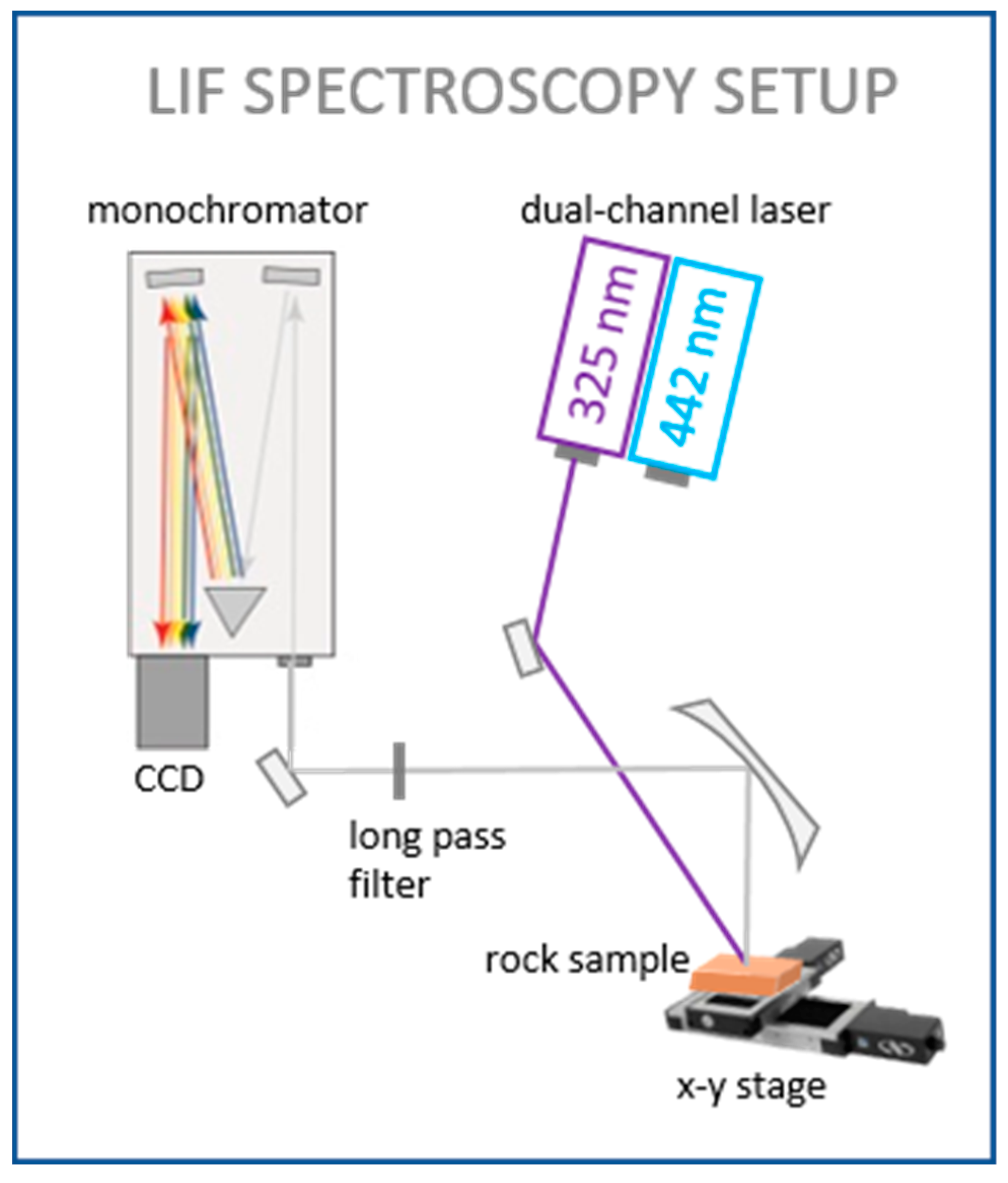
2.2. Laser System and Data Processing
2.3. Further Experimental Methods for Cross-Validation
3. Results and Discussion
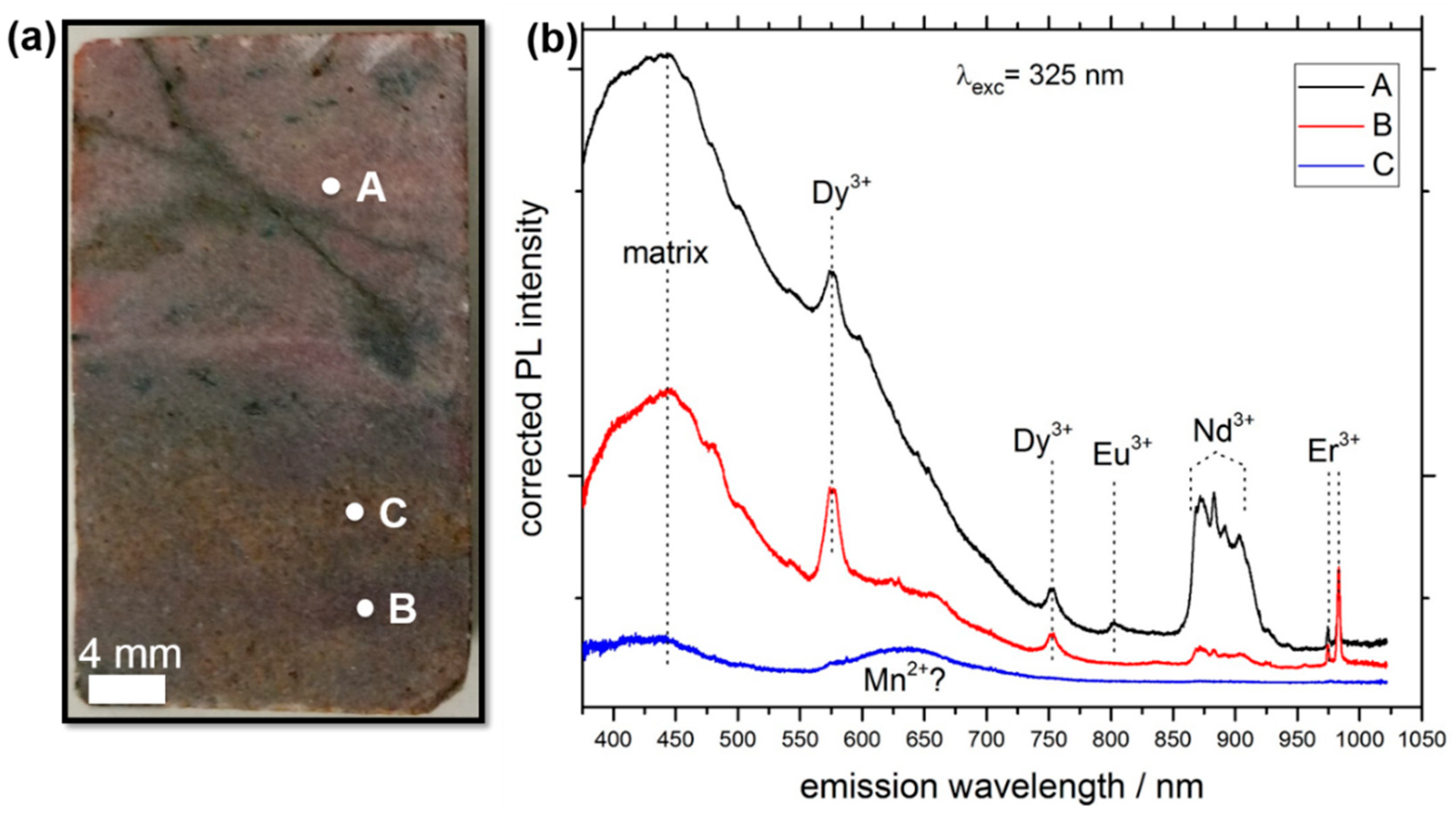
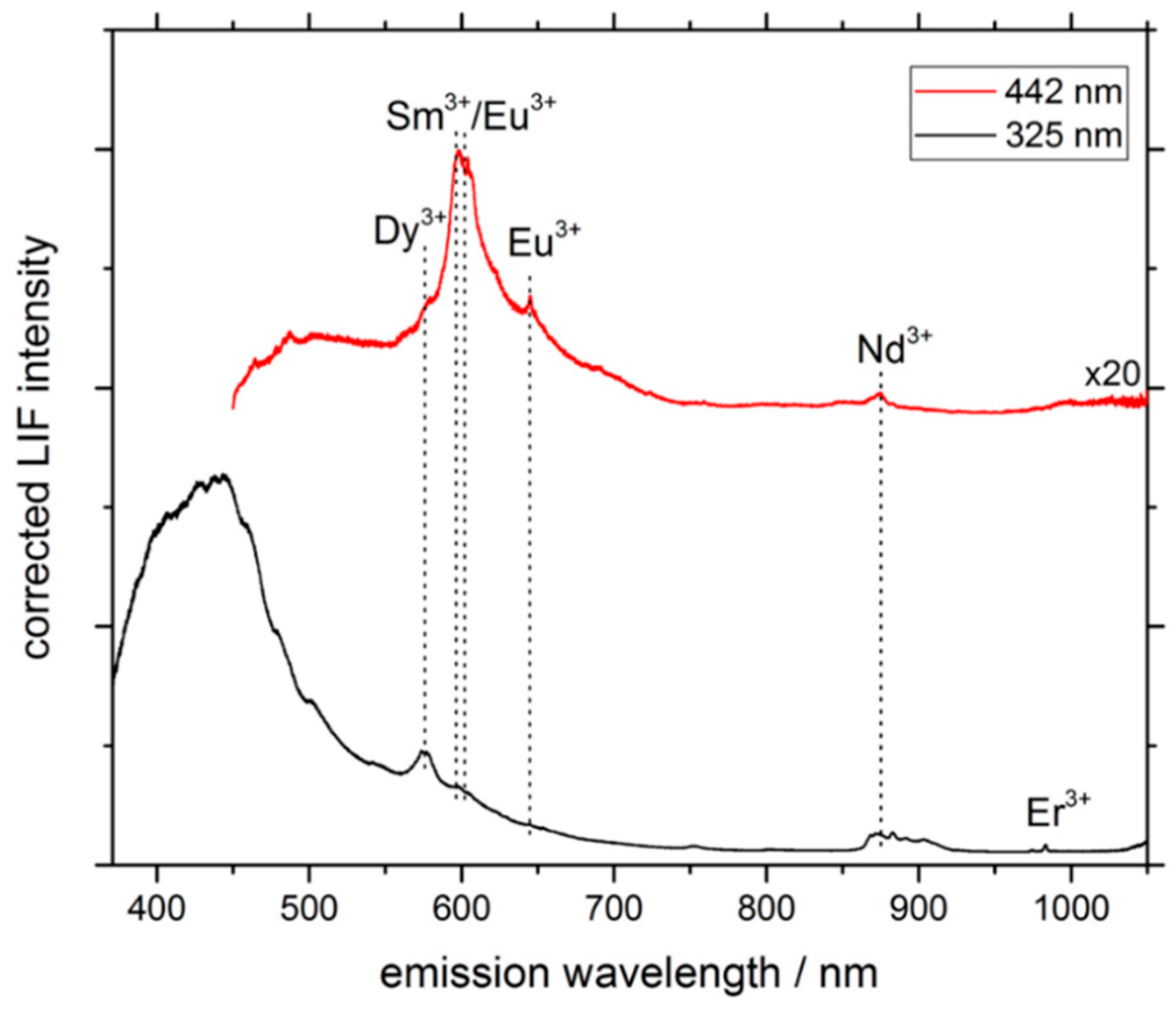
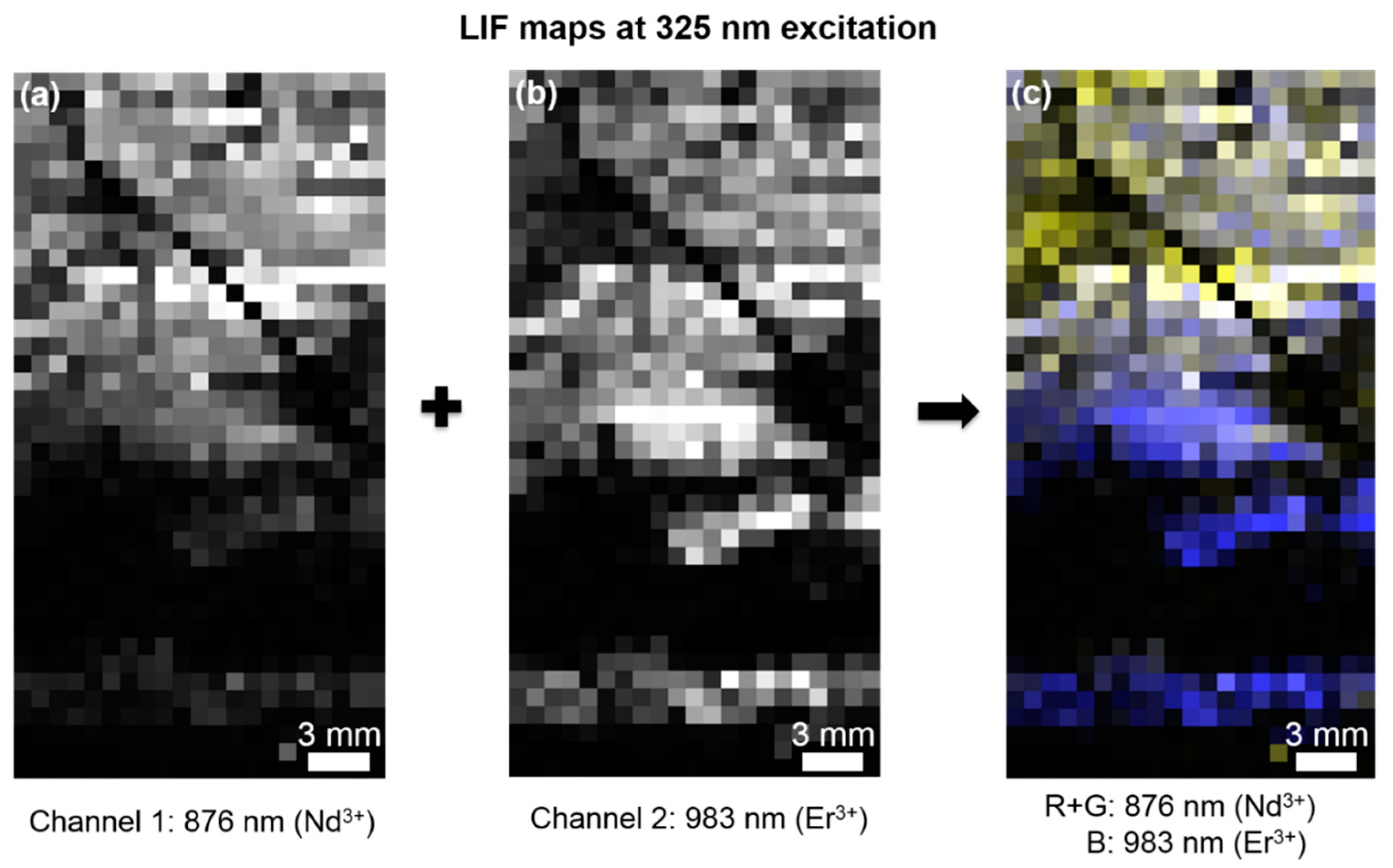
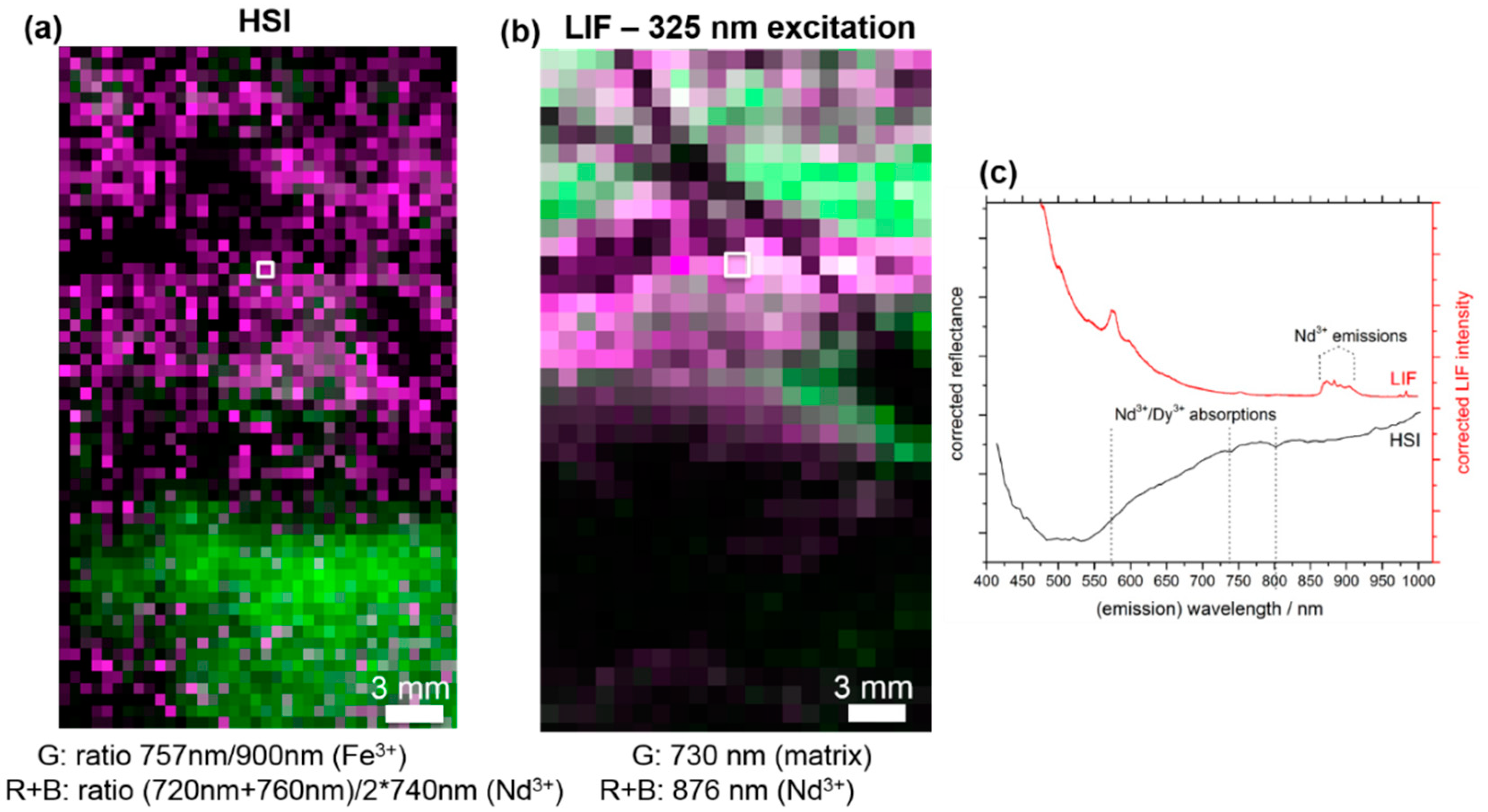
3.1. LIF Spectroscopy
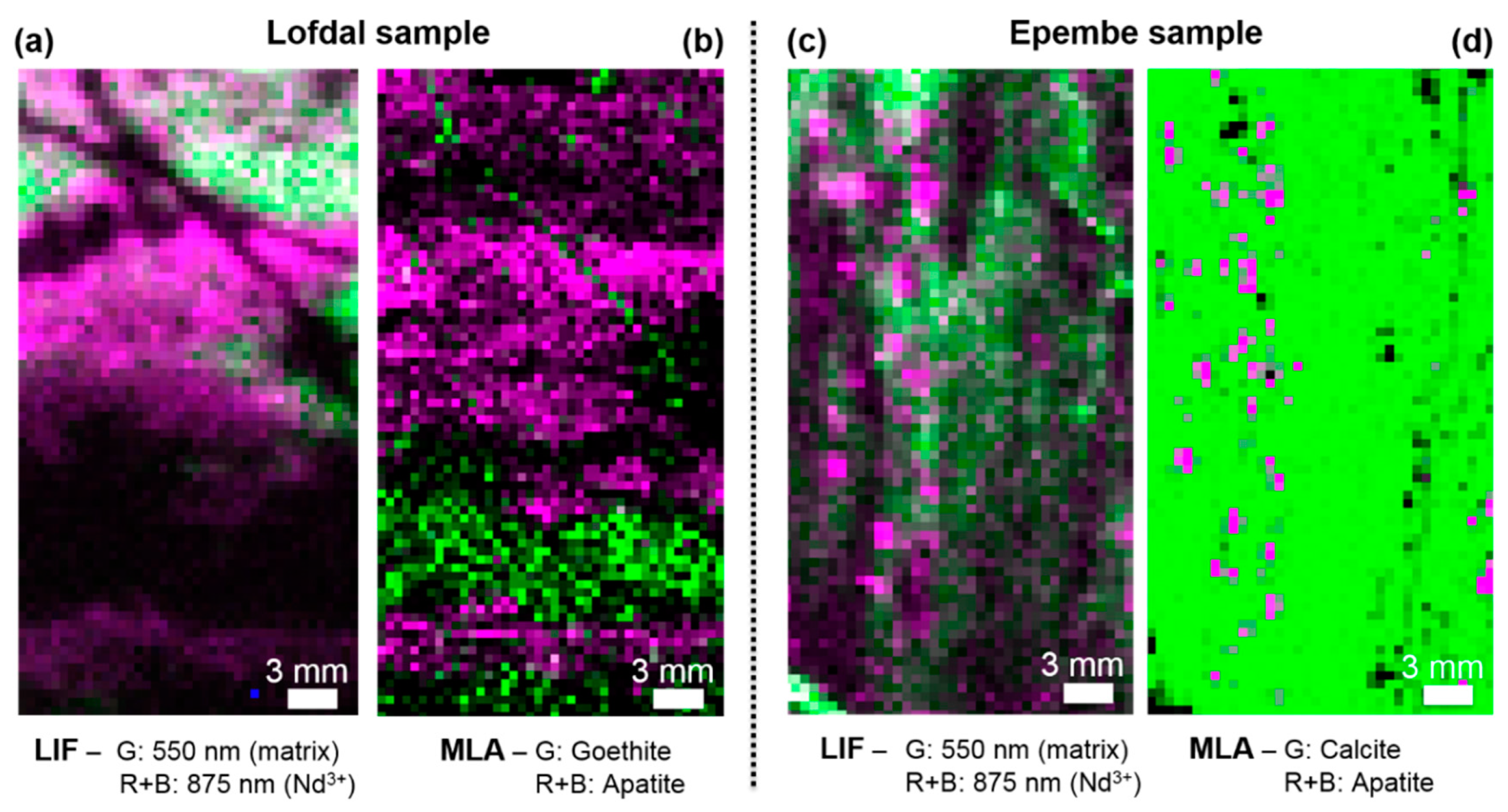
3.2. Comparison of HSI and MLA
3.3. Implications for Mineralogy and Exploration of REEs
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gambogi, J. Rare Earths, Mineral Commodity Summaries 2017; United States Geological Survey: Reston, VG, USA, 2017.
- Qing, S.; XinLei, Z.; Yan, Z.; Wenbao, J.; Yongsheng, L.; DaQian, H.; ShengNan, C. Development of an Online X-Ray Fluorescence Analysis System for Heavy Metals Measurement in Cement Raw Meal. Spectrosc. Lett. 2015, 49, 188–193. [Google Scholar] [CrossRef]
- Khajehzadeh, N.; Kauppinen, T.K. Fast mineral identification using elemental LIBS technique. IFAC-PapersOnLine 2015, 48, 119–124. [Google Scholar] [CrossRef]
- Kruse, F.; Weatherbee, O.; Peppin, W.; Bedell, R.; Calvin, W.; Taranik, J.V. HSI Mineral Mapping from Airborne, Outcrop, and Drill-Core Perspectives. Proc. SPIE 2010, 7687. [Google Scholar] [CrossRef]
- Zimmermann, R.; Brandmeier, M.; Andreani, L.; Mhopjeni, K.; Gloaguen, R. Remote Sensing Exploration of Nb-Ta-LREE-Enriched Carbonatite (Epembe/Namibia). Remote Sens. 2016, 8, 620. [Google Scholar] [CrossRef]
- Turner, D.; Rivard, B.; Groat, L.A. Visible to shortwave infrared reflectance spectroscopy of rare earth element minerals. Br. Columbia Geol. Surv. Pap. 2015, 3, 219–229. [Google Scholar]
- Neave, D.A.; Black, M.; Riley, T.R.; Gibson, S.A.; Ferrier, G.; Wall, F.; Broom-Fendley, S. On the feasibility of imaging carbonatite-hosted rare earth element deposits using remote sensing. Econ. Geol. 2016, 111, 641–665. [Google Scholar] [CrossRef]
- Lenz, C.; Nasdala, L.; Talla, D.; Hauzenberger, C.; Seitz, R.; Kolitsch, U. Laser-induced REE3+ photoluminescence of selected accessory minerals—An “advantageous artefact” in Raman spectroscopy. Chem. Geol. 2015, 415, 1–16. [Google Scholar] [CrossRef]
- Gaft, M.; Reisfeld, R.; Panczer, G. Modern Luminescence Spectroscopy of Minerals and Materials, 2nd ed.; Springer: Cham, Switzerland, 2015. [Google Scholar]
- Fuchs, M.C.; Gloaguen, R.; Beyer, J.; Jakob, S.; Heitmann, J. Emission spectroscopy for the identification of rare earth elements using laser-induced photoluminescence. In Proceedings of the 2016 8th Workshop on Hyperspectral Image and Signal Processing: Evolution in Remote Sensing (WHISPERS), Los Angeles, CA, USA, 21–24 August 2016; pp. 1–5. [Google Scholar]
- Lorenz, S.; Beyer, J.; Fuchs, M.; Seidel, P.; Turner, D.; Heitmann, J.; Gloaguen, R. The Potential of Reflectance and Laser Induced Luminescence Spectroscopy for Near-Field Rare Earth Element Detection in Mineral Exploration. Remote Sens. 2019, 11, 21. [Google Scholar] [CrossRef]
- Wall, F.; Niku-Paavola, V.N.; Storey, C.; Müller, A.; Jeffries, T. Xenotime-(Y) from carbonatite dykes at Lofdal, Namibia: Unusually low LREE:HREE ratio in carbonatite, and the first dating of xenotime overgrowths on zircon. Can. Mineral. 2008, 46, 861–877. [Google Scholar] [CrossRef]
- Williams-Jones, A.E.; Wollenberg, R.; Bodeving, S. Hydrothermal Fractionation of the Rare Earth Elements and the Genesis of the Lofdal REE Deposit. In Proceedings of the Symposium on Strategic and Critical Materials 2015, Victoria, BC, Canada, 13–14 November 2015; pp. 125–130. [Google Scholar]
- Bodeving, S.; Williams-Jones, A.E.; Swinden, S. Carbonate–silicate melt immiscibility, REE mineralising fluids, and the evolution of the Lofdal Intrusive Suite, Namibia. Lithos 2017, 268, 383–398. [Google Scholar] [CrossRef]
- Broom-Fendley, S.; Styles, M.T.; Appleton, J.D.; Gunn, G.; Wall, F. Evidence for dissolution-reprecipitation of apatite and preferential LREE mobility in carbonatite-derived late-stage hydrothermal processes. Am. Mineral. 2016, 101, 596–611. [Google Scholar] [CrossRef]
- Unger, G.; Zimmermann, R.; Gloaguen, R. 3D Modeling of the Epembe (Namibia) Nb-Ta-P-(LREE) Carbonatite Deposit: New Insights into Geometry Related to Rare Metal Enrichment. Minerals 2018, 8, 600. [Google Scholar] [CrossRef]
- Rahfeld, A.; Wiehl, N.; Dressler, S.; Möckel, R.; Gutzmer, J. Major and trace element geochemistry of the European Kupferschiefer—An evaluation of analytical techniques. Geochem. Explor. Env. A 2018, 18, 132–141. [Google Scholar] [CrossRef]
- Gu, Y. Automated scanning electron microscope based mineral liberation analysis an introduction to JKMRC/FEI mineral liberation analyser. J. Miner. Mater. Char. Eng. 2003, 2, 33–41. [Google Scholar] [CrossRef]
- Fandrich, R.; Gu, Y.; Burrows, D.; Moeller, K. Modem SEM-based mineral liberation analysis. Int. J. Miner. Process. 2007, 84, 310–320. [Google Scholar] [CrossRef]
- Bachmann, K.; Frenzel, M.; Krause, J.; Gutzmer, J. Advanced Identification and Quantification of In-Bearing Minerals by Scanning Electron Microscope-Based Image Analysis. Microsc. Microanal. 2017, 23, 527–537. [Google Scholar] [CrossRef]
- Reisfeld, R. Radiative and non-radiative transitions of rare-earth ions in glasses. In Rare Earths. Structure and Bonding; Springer: Berlin/Heidelberg, Germany, 1975; Volume 22, ISBN 978-038-707-268-3. [Google Scholar]
- Hull, R.; Parisi, J.; OsgoodJr, R.M.; Warlimont, H.; Liu, G.; Jacquier, B. Spectroscopic Properties of Rare Earths in Optical Materials; Springer: Berlin/Heidelberg, Germany, 2005; ISBN 978-3-540-28209-9. [Google Scholar]
- Lüthi, B. Crystal field effects in rare earth systems. J. Magn. Magn. Mater. 1980, 15, 1–8. [Google Scholar] [CrossRef]
- Elliott, R.J. Crystal Field Theory in the Rare Earths. Rev. Mod. Phys. 1953, 25, 167. [Google Scholar] [CrossRef]
- Marfunin, S.A. Spectroscopy, Luminescence and Radiation Centers in Minerals; Springer: Berlin/Heidelberg, Germany, 1979; ISBN 978-3-642-67112-8. [Google Scholar]
- Habermann, D.; Niklas, J.R.; Meijer, J.; Stephan, A.; Götte, T. Structural point defects in “Iceland spar” calcite. Nucl. Instrum. Methods Phys. Res. B 2001, 181, 563–569. [Google Scholar] [CrossRef]
- Cazenave, S.; Chapoulie, R.; Villeneuve, G. Cathodoluminescence of synthetic and natural calcite: The effects of manganese and iron on orange emission. Miner. Petrol. 2003, 78, 243–253. [Google Scholar] [CrossRef]
- Czaja, M.; Bodył-Gajowska, S.; Lisiecki, R.; Meijerink, A.; Mazurak, Z. The luminescence properties of rare-earth ions in natural fluorite. Phys. Chem. Minerals 2012, 39, 639–648. [Google Scholar] [CrossRef]
- Seferis, I.E.; Zeler, J.; Michail, C.; David, S.; Valais, I.; Fountos, G.; Kalyvas, N.; Bakas, A.; Kandarakis, I.; Zych, E.; et al. Grains size and shape dependence of luminescence efficiency of Lu2O3:Eu thin screens. Results Phys. 2017, 7, 980–981. [Google Scholar] [CrossRef]
- Burruss, R.C.; Ging, T.G.; Eppinger, R.G.; Samson, M. Laser-excited fluorescence of rare earth elements in fluorite: Initial observations with a laser Raman microprobe. Geochim. Cosmochim. Acta 1992, 56, 2713–2723. [Google Scholar] [CrossRef]
- Gaft, M.; Reisfeld, R.; Panczer, G.; Boulon, G.; Shoval, S.; Champagnon, B. Accommodation of rare-earths and manganese by apatite. Opt. Mat. 1997, 8, 149–156. [Google Scholar] [CrossRef]
- Lu, Z.; Chen, L.; Tang, Y.; Li, Y. Preparation and luminescence properties of Eu3+-doped MSnO3 (M = Ca, Sr and Ba) perovskite materials. J. Alloys Compd. 2005, 387, L1–L4. [Google Scholar] [CrossRef]
- Reisfeld, R.; Gaft, M.; Boulon, G.; Panczer, C.; Jørgensen, C.K. Laser-induced luminescence of rare-earth elements in natural fluor-apatites. J. Lumin. 1996, 69, 343–353. [Google Scholar] [CrossRef]
- Foley, N.; De Vivo, B.; Salminen, R. Rare Earth Elements: The role of geology, exploration, and analytical geochemistry in ensuring diverse sources of supply and a globally sustainable resource. J. Geochem. Explor. 2013, 133, 1–5. [Google Scholar] [CrossRef]
- Adams, J.W. The visible region absorption spectra of rare earth minerals. Am. Mineral. 1965, 50, 356–366. [Google Scholar]
- Rowan, L.C.; Kingston, M.J.; Crowley, J.K. Spectral reflectance of carbonatites and related alkalic igneous rocks; selected samples from four North American localities. Econ. Geol. 1986, 81, 857–871. [Google Scholar] [CrossRef]
- Turner, D.J.; Rivard, B.; Groat, L.A. Visible and short-wave infrared reflectance spectroscopy of REE fluorocarbonates. Am. Mineral. 2014, 99, 1335–1346. [Google Scholar] [CrossRef]
- Turner, D.; Rivard, B.; Groat, L. Rare earth element ore grade estimation of mineralized drill core from hyperspectral imaging spectroscopy. In Proceedings of the 2014 IEEE Geoscience and Remote Sensing Symposium, Quebec City, QC, Canada, 13–18 July 2014. [Google Scholar]
- Nagasawa, H. Rare earth concentrations in zircons and apatites and their host dacites and granites. Earth Planet. Sci. Lett. 1970, 9, 359–364. [Google Scholar] [CrossRef]
- Frietsch, R.; Perdahl, J.-A. Rare earth elements in apatite and magnetite in Kiruna-type iron ores and some other iron ore types. Ore Geol. Rev. 1995, 9, 489–510. [Google Scholar] [CrossRef]
- Filippelli, G.M.; Delaney, M.L. The effects of Manganese(II) and Iron(II) on the cathodoluminescence signal in synthetic apatite. J. Sedimen. Res. 1993, 63, 167–173. [Google Scholar] [CrossRef]
- Pierson, B.J. The control of cathodoluminescence in dolomite by iron and manganese. Sedimentology 1981, 28, 601–610. [Google Scholar] [CrossRef]
- Boesche, N.; Rogass, C.; Lubitz, C.; Brell, M.; Herrmann, S.; Mielke, C.; Tonn, S.; Appelt, O.; Altenberger, U.; Kaufmann, H. Hyperspectral REE (Rare Earth Element) Mapping of Outcrops—Applications for Neodymium Detection. Remote Sens. 2015, 7, 5160–5186. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Excitation Wavelength | Condition | Spot Diameter/nm | Power Density/(W/cm2) |
|---|---|---|---|
| 325 nm–UV | focused | 0.16 | 24.9 |
| unfocused | 0.71 | 1.3 | |
| 442 nm–blue | focused | 0.18 | 78.6 |
| unfocused | 0.79 | 4.1 |
| Parameter | HSI | MLA | LIF |
|---|---|---|---|
| Acquisition time | ~1 s | ~10,000 s | ~1500 s |
| Spatial resolution | >300 µm | >2 µm | >50 µm |
| REE detection Limit | >0.03 wt% | >0.01 wt% | >0.01 wt% |
| Sample preparation | flat surface preferred | flat surface + C-coating + transfer to high vacuum | flat surface |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seidel, P.; Lorenz, S.; Heinig, T.; Zimmermann, R.; Booysen, R.; Beyer, J.; Heitmann, J.; Gloaguen, R. Fast 2D Laser-Induced Fluorescence Spectroscopy Mapping of Rare Earth Elements in Rock Samples. Sensors 2019, 19, 2219. https://doi.org/10.3390/s19102219
Seidel P, Lorenz S, Heinig T, Zimmermann R, Booysen R, Beyer J, Heitmann J, Gloaguen R. Fast 2D Laser-Induced Fluorescence Spectroscopy Mapping of Rare Earth Elements in Rock Samples. Sensors. 2019; 19(10):2219. https://doi.org/10.3390/s19102219
Chicago/Turabian StyleSeidel, Peter, Sandra Lorenz, Thomas Heinig, Robert Zimmermann, René Booysen, Jan Beyer, Johannes Heitmann, and Richard Gloaguen. 2019. "Fast 2D Laser-Induced Fluorescence Spectroscopy Mapping of Rare Earth Elements in Rock Samples" Sensors 19, no. 10: 2219. https://doi.org/10.3390/s19102219
APA StyleSeidel, P., Lorenz, S., Heinig, T., Zimmermann, R., Booysen, R., Beyer, J., Heitmann, J., & Gloaguen, R. (2019). Fast 2D Laser-Induced Fluorescence Spectroscopy Mapping of Rare Earth Elements in Rock Samples. Sensors, 19(10), 2219. https://doi.org/10.3390/s19102219